 Cuicui Jiang1,†
Cuicui Jiang1,† Ying Zhou
Ying Zhou- 1Obstetrics Department, Quzhou Maternal and Child Health Care Hospital, Quzhou, Zhejiang, China
- 2Laboratory of Prenatal Diagnosis Center, Quzhou Maternal and Child Health Care Hospital, Quzhou, Zhejiang, China
Background: The heterozygous STAG1 gene (OMIM*604358) variants are associated with autosomal dominant intellectual developmental disorder 47, known as mental retardation autosomal dominant 47 (MRD47, OMIM#617635). Although more than 10 STAG1 variants have been reported, functional studies in vitro have not been performed. Our functional studies of a novel frameshift STAG1 variant in a Chinese boy have provided preliminary evidence confirming that the underlying pathogenic mechanism of MRD47 may be associated with STAG1 haploinsufficiency.
Methods: Trio-based whole-exome sequencing (trio-WES) was performed on genomic DNA (gDNA) of peripheral blood samples from the boy and his parents. Mutant STAG1 expression vectors pcDNA3.1(+)-FLAG-STAG1-mut and control pcDNA3.1(+)-FLAG-STAG1-WT mammalian expression vectors were constructed. Both vectors were transformed into HEK293T cells. The assays of relative STAG1 gene mRNA expression and STAG1 protein expression were adopted.
Results: Trio-WES identified a novel heterozygous frameshift STAG1 gene variant (NM_005862.3) c.500dup (p.Gly168TrpfsTer13). Our in vitro functional findings revealed that this variant resulted in a dramatic reduction in the formation of STAG1 protein due to the decay of mutant STAG1 mRNA. The underlying pathogenic mechanism of MRD47 may be related to STAG1 haploinsufficiency.
Conclusion: MRD47 exhibits non-specific characteristics and diverse clinical phenotypes. Our functional studies have provided preliminary evidence confirming the haploinsufficiency of the STAG1 gene as the underlying pathogenic mechanism of MRD47. This study also expanded the mutational spectrum of the STAG1 gene and the clinical spectrum of MRD47.
Introduction
Autosomal dominant intellectual developmental disorder 47, alias mental retardation autosomal dominant 47 (MRD47, OMIM #617635) is caused by heterozygous variants in the STAG1 gene (OMIM*604358) on chromosome 3q22.3. The heterozygous intragenic deletions within the STAG1 gene, de novo heterozygous missense and frameshift STAG1 gene variants in 17 unrelated patients with similar phenotypes (intellectual disability/developmental delay, growth retardation, feeding difficulties, facial dysmorphism, epilepsy, autistic features) were first reported by Lehalle et al. who considered the STAG1 gene could be a novel gene responsible for non-specific syndromic intellectual disability (1). For now, more than twenty cases with STAG1 variations have been described in the literature, but functional studies of these variants in vitro have not been performed. These authors postulated that the neurodevelopmental phenotypes were caused by loss-of-function (LoF) effects of STAG1 variants. Herein, we first reported a Chinese patient with a novel heterozygous frameshift STAG1 variant, and performed a serial of functional studies on this variant. This study preliminarily suggested the pathogenic mechanism of MRD47 may be associated with STAG1 haploinsufficiency.
Materials and methods
Clinical features
The patient was a 3-year-old boy who first presented at the age of 2 years for evaluation of developmental delay. There was no family history of congenital anomalies or intellectual disability/neurodevelopmental disorders (NDD). He was born at 38 weeks’ gestation with normal birth weight and length following an uncomplicated pregnancy. His developmental milestones were delayed; he raised his head at 5 months, sat independently at 10 months, stood up at 18 months and walked at 19 months. The boy was followed up until the age of 3 years and 10 months. At the last follow-up, he still could not say any words. His height was 92 cm (<10th centile) and weight was 12 kg (<10th centile). His occipitofrontal circumference was within the normal range. The electrocardiogram and cerebral magnetic resonance imaging were normal. His vision and hearing evaluations were both normal. Facial dysmorphisms, limb anomalies, autistic features and other behavioral anomalies have not been observed.
Whole-exome sequencing (WES)
Trio-based whole-exome sequencing (trio-WES) was performed on genomic DNA (gDNA) of peripheral blood samples from this boy and his patients using the Blood Genome Column Medium Extraction Kit (Kangweishiji, China) according to the manufacturer's instructions. The extracted DNA samples were subjected to quality control using a Qubit 2.0 fluorimeter and electrophoresis with a 0.8% agarose gel for further protocols. The xGen™ Exome Research Panel v2 (designed by Integrated DNA Technologies) was used for WES. Quality control (QC) of the DNA library was performed using an Agilent 2100 Bioanalyzer System (Agilent, USA). DNA nanoball (DNB) preps of clinical samples were sequenced on ultra high throughput DNBSEQ-T7 platform (MGI, Shenzhen, China) with paired-end 150 nt strategy following manufacturer's protocol.
Bioinformatic analysis
Sequencing data was analyzed according to our in-house (Chigene Translational Medicine Research Center) procedures. Raw data were processed quickly for adapter removal and low-quality read filtering, and then data quantity and data quality was statistics. The trimmed reads were then mapped to the University of California at Santa Cruz (UCSC) GRCh37/hg19 reference genome using Burrows-Wheeler Aligner (BWA) software (version 0.7.17). The Genome Analysis ToolKit (GATK) software (version 4.2.1.0) was used for single nucleotide polymorphisms/variants (SNPs/SNVs) and short insertions/deletions (indels) (<50 bp) calling. Samtools (version 1.22.1) and Picard software (version 2.22.1) packages were used to generate clean binary alignment map (BAM) data by removing duplicate data. Variants were annotated for analysis using the single nucleotide polymorphism database (dbSNP, version 5.3), gnomAD exomes database (version 4.1.0) and Chigene in-house minor allele frequency (MAF) database. Tools of pathogenicity prediction like REVELAL and AlphaMissense were used for predicting possible impact of variants. As a prioritized pathogenicity annotation to the American College of Medical Genetics and Genomics (ACMG) guidelines, Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD) and ClinVar databases were used as conferences of pathogenicity of every variant.
Variants classification
As per the guidelines of ACMG for interpreting sequence variants, variants were classified. Classification considered the position of the variant in the human genome, MAF, the pathogenicity prediction of variants, disease mechanism, clinical phenotypes, literature evidence, evolutionary conservation.
Variants verification
gDNA samples was used for the verification of this variant. All reactions were carried out in a total volume of 20 μl using the Taq DNA polymerase. After confirmation of the size of the amplicons, the PCR products were purified by standard protocol and Sanger sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (ABI 3730 Applied Biosystem, USA) for the verification of candidate variants. The STAG1 gene primers used for PCR amplification were as follows: forward 5′-TTTATCCAGTGTTCAGGA-3′, reverse 5′-AGGGTACTTGTATGCCTAA-3′. The identification of variants was performed using Chromas software (version2.2.6, Technelysium, Australia).
Construction of the STAG1 variant in vitro and transfection
Mutant STAG1 gene pcDNA3.1(+)-FLAG-STAG1-mut and control pcDNA3.1(+)-FLAG-STAG1-WT mammalian expression vectors were constructed and purchased from Wuhan Biorun Biosciences Co., Ltd. Human embryonic kidney (HEK)-293T cells were transfected with expression vectors in two tubes (one for the mutant expression vectors and one for the wild-type expression vectors). The constructed expression vectors were verified using Sanger sequencing. Then, Six hours after the transfection, we gently removed the lipofectamine-containing medium, replaced the medium with Dulbecco's modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS), and incubated the slide at 37 ℃ in a 5% CO2 incubator until day 2. On day 3, the samples were collected and used in a reverse transcription-polymerase chain reaction (RT-PCR) to quantify the STAG1 mRNA expression and a Western blot (WB) assay to quantify the STAG1 protein expression. Each cell line underwent three independent replicates of the RT-PCR assay. The RT-PCR and Western blotting experiments were conducted in accordance with the manufacturer's instructions.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
After transfecting HEK293T cells with wild-type (STAG1-WT) and mutant (STAG1-mut) expression vectors for 24 h, we centrifuged the transfected cells to collect them, and then extract the total RNA from the cells using the Trizol method. Total RNA concentration and quality were checked using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Complementary DNA (cDNA) was synthesized using NovoScript® Plus All-in-one 1st Strand cDNA Synthesis SuperMix (Novoprotein, China) following the manufacturer's instructions. The cDNA concentration was about 1,200 ng/μl. The total reaction system was 20 µl, and the total RNA template was 500 ng. Quantitative PCR was performed using the Maxima SYBR Green/ROX qPCR Master Mix (2×) (Thermo Fisher Scientific, USA). Relative STAG1 mRNA expression levels were determined using 2−ΔΔCt method, and the GAPDH gene was used as the reference gene. The qRT-PCR reaction was performed in triplicate, and mean values and standard deviations were calculated from the obtained data. The primers used for PCR amplification of STAG1 gene were as follows: forward 5′-AGAATTTGATGAGGACAGTGGTGA-3′, reverse 5′-TCAGGACTCCAATAAATTCACAAAA-3′.
Western blot (WB)
After transfecting HEK293T cells with wild-type (STAG1-WT) and mutant (STAG1-mut) expression vectors for 24 h, western blot was performed using standard protocol. Equal amounts of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% non-fat milk in a tris buffered saline with tween-20 (TBST) solution for 2 h at room temperature. Anti-Flag (at a dilution of 1:1,000) and anti-glyceraldehyde phosphate dehydrogenase (anti-GAPDH) dilutions (at a dilution of 1:1,000) were added respectively and incubated at 4 ℃ overnight. Horseradish peroxidase (HRP)-labeled secondary antibodies were added and incubated at room temperature for 1 h. GAPDH served as the loading control and internal reference protein. The loading amount of WB protein was 10 μg/well. Blot bands were quantified by densitometry using ImageJ software (ImageJ2). Protein levels were normalized to GAPDH. The relative STAG1 protein expression level is quantified by the fold change (FC) in the ratio of the grayscale between the STAG1 protein band and the internal reference protein band.
Statistical analysis
All data statistical comparisons between two groups (STAG1-WT and STAG1-mut) were evaluated by Student's t-test using Prism software (GraphPad Prism 10.5.0). P < 0.05 was considered statistically significant.
Literature review
We searched the PubMed database using “STAG1 gene” as keywords. The search time was from the establishment of the databases to February 31, 2025. We reviewed STAG1-related cases with availability of clinical data including relevant genetic testing results, prenatal/postnatal manifestations and pregnancy outcomes.
Results
Genetic analysis and confirmation
Trio-WES identified a novel heterozygous frameshift variant in the STAG1 gene (NM_005862.3) c.500dup (p.Gly168TrpfsTer13). This variant was further verified by Sanger sequencing, confirming that it was a de novo variant (Figure 1). The allele frequency of this heterozygous variant has not been registered in population databases (1,000 Genomes Project, gnomAD, and dbSNP) or reported in disease databases (ClinVar, HGMD, OMIM) (PM2_Supporting). This patient was the only de novo occurrence of this variant (PS2_Supporting). The probability of being LoF intolerant (pLI) value of STAG1 gene in the gnomAD v4.1.0 was 1.0, which indicated that STAG1 gene was extremely intolerant to loss-of-function variants (pLI > 0.9). The frameshift STAG1 gene variant was a presumed LoF variant, LoF was a putative mechanism of STAG1-related disease (PVS1). As per the interpretation guidelines by ACMG (2), this novel variant was classified as “likely pathogenic”. We have submitted this variant to a public database [Leiden Open Variation Database (LOVD)], and the accession number can be found at the following URL: https://databases.lovd.nl/shared/individuals/00464708.
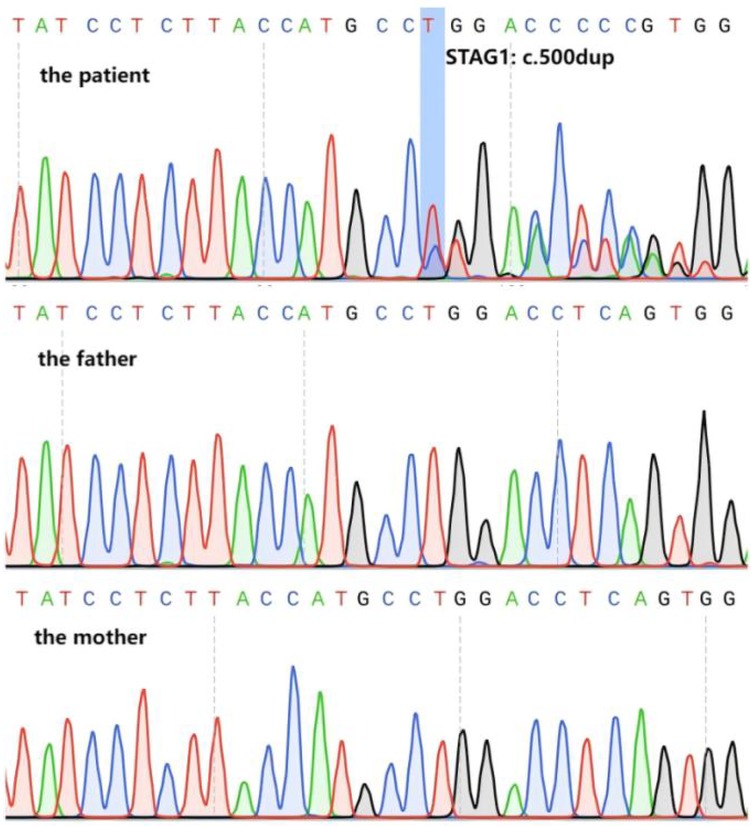
Figure 1. Variant verification of Sanger sequencing. Sanger sequencing confirmed that the patient carried the heterozygous frameshift variant in STAG1 gene (NM_005862.3) c.500dup (marked by the blue box), the parents were normal.
Functional analysis of the frameshift variant
To evaluate the effect of the frameshift STAG1 variant, our functional studies revealed that this STAG1 variant, c.500dup, markedly decreased mRNA expression of STAG1 gene (Figure 2), leading to a lack of normal STAG1 protein (Figures 3, 4), compared to the STAG1-WT cell lines.

Figure 2. The relative mRNA expression of the STAG1 gene. The results of RT-PCR analysis showed that the mRNA expression of mutant STAG1 cell lines was significantly reduced compared to the STAG1-WT cell lines. Each cell line underwent three independent replicates of the RT-PCR assay, The difference between STAG1-WT and STAG1-mut was evaluated by Student's t-test. ***P < 0.001.
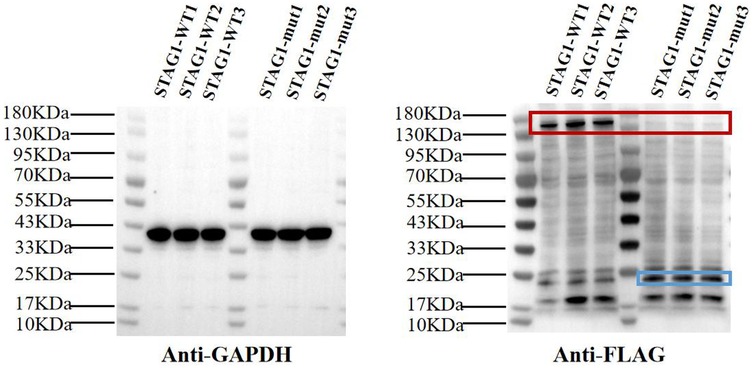
Figure 3. Western blot analysis of STAG1 protein. The results of WB analysis showed that mutant STAG1 cell lines could not produce the normal STAG1 protein compared to the STAG1-WT cell lines (marked by the red box). The molecular mass of normal STAG1 (NM_005862.3) was about 144 kDa. The theoretical molecular mass of the truncated STAG1 protein was approximately 22.9 kDa (marked by the blue box). Each cell line underwent three independent replicates of WB analysis.
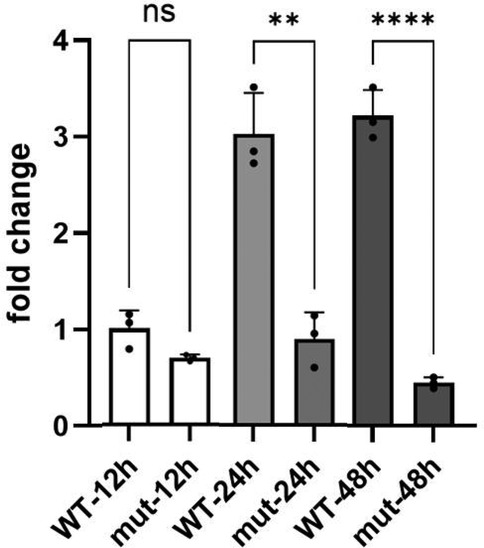
Figure 4. The relative STAG1 protein expression. The relative STAG1 protein expression was quantified by the fold change (FC). WB analysis indicated that the relative expression level of STAG1 protein in the STAG1-mut group was significantly lower than that in the STAG1-WT group. From 12 to 24 h, FC showed a slow upward trend (increasing from 0.69 to 0.90), while from 24 to 48 h, it exhibited a more significant downward trend (decreasing from 0.90 to 0.45). Each cell line underwent three independent replicates of WB analysis. The difference between STAG1-WT and STAG1-mut was evaluated by Student's t-test. **P < 0.01, ***P < 0.001, ns: not significant.
Discussion
The STAG1 gene (OMIM*604358), known as “stromal antigen 1 (SA1)”, is located on chromosome 3q22.3. The transcript of STAG1 (NM_005862.3) has 34 exons with the transcript length of 6,062 base pairs. Cohesin subunit SA-1 (STAG1 protein) also named as SCC3 homolog 1, is a 1258 amino acid protein (UniProt database accession Q8WVM7), which belongs to the SCC3 family (3). STAG1 is a subunit component of the cohesin complex (a ring-shaped structure is composed of four subunits: SMC1A, SMC3, RAD21/REC8, STAG1/2/3) required for sister chromatids cohesion along the length of a chromosome from DNA replication through prophase and prometaphase (4). At anaphase, the cohesin complex is cleaved and dissociates from chromatin, allowing sister chromatids to segregate (5). A spectrum of human developmental syndromes (such as Cornelia de Lange syndrome, Roberts-SC phocomelia syndrome, Warsaw breakage syndrome, chronic atrial and intestinal dysrhythmia syndrome, CHOPS syndrome, and alpha-thalassemia/impaired intellectual development syndrome), collectively termed “cohesinopathies”, refers to disorders resulting from variants in genes encoding the cohesin complex and its regulators (such as NIPBL, ESCO2, ANKRD11, HDAC8, DDX11, SGO1, AFF4, ATRX and others) (6). STAG1-related MRD47 (OMIM#617635) can also be classified as one of the cohesinopathies.
The STAG1-related cohesinopathy has been reported in seven studies, encompassing a total of 28 patients aged 0 to 29 years. We retrospectively reviewed a total of 18 cases with heterozygous missense/frameshift/nonsense STAG1 variants (SNVs/indels) (1, 7–12) (Table 1). All these STAG1 SNVs/indels were de novo. Among them, 2 were nonsense variants, 4 were frameshift variants, and 12 were missense variants. An additional 10 patients with copy-number deletions or intragenic deletions affecting the STAG1 gene [regarded as “structural variations (SVs)”] have been reported, three of whom lacked detailed clinical documentation (1, 7). The most prevalent shared characteristics observed in these 25 patients included developmental delay/intellectual disability (DD/ID) (100%, 25/25), speech delay (100%, 25/25), and dysmorphic facial features (92%, 23/25). The earliest reported age of first spoken words among patients in the literature was 18 months, while the latest was 15 years. In our case, the patient remained non-verbal at 3 years of age, and the onset of language function recovery could not be determined. Other common clinical phenotypes of MRD47 included autism spectrum disorder (32%, 8/25) and epilepsy (32%, 8/25). During the evaluation period, this patient did not exhibit any signs of autistic behavior or seizure. Other uncommon clinical phenotypes of MRD47 included congenital heart disease (2/25), hearing loss (3/25), visual impairment (2/25), scoliosis (2/25), cryptorchidism (4/25), fifth finger clinodactyly (2/15), and syndactyly (2/15). None of these phenotypic features were observed in the present case. It has been reported that the STAG1-related clinical manifestations overlapped with the phenotype of Cornelia de Lange syndrome (12). The study by Lehalle et al. suggested that there was no notable disparity in clinical manifestations caused by STAG1 deletions or STAG1 variants apart from microcephaly (4/7 cases with SVs of STAG1 had microcephaly, whereas 10 cases with STAG1 SNVs/indels didn't have microcephaly) (1). Although the STAG1-related clinical manifestations showed the low specificity and clinical diversity, various degrees of DD/ID have been found in all STAG1-related cases. At the last clinical evaluation, our case didn't show facial dysmorphisms, epilepsy, autism, limb anomalies or behavioral anomalies. Among these cases with reported prenatal examination records (a total of 16 cases), we found the pregnancies of 14/16 cases (including our case) with STAG1 SNVs/indels were uneventful, other two cases presented with abnormal genitalia and intrauterine growth retardation (IUGR). Therefore, it is warranted to further deliberate on the necessity of prenatal diagnosis for fetuses in low-risk pregnancies. Strikingly, the pregnancies of 66.7% cases (4/6) with SVs of STAG1 were abnormal (such as IUGR, increased nuchal translucency, hydramnios, heart defects) (1). It can be inferred that the majority of MRD47 fetuses could not be diagnosed prenatally and are only identified postnatally. Consequently, it is worth further deliberation whether prenatal whole-exome sequencing is warranted in low-risk pregnancies.

Table 1. Summary of the clinical phenotypes of children with previously reported STAG1 gene variants.
Based on these cases with frameshift/nonsense STAG1 variants and copy-number deletions/intragenic deletions of STAG1, Lehalle et al. (1), Di Muro et al. (8) and Bregvadze et al. (11) postulated that a pathogenic mechanism known as haploinsufficiency (loss of one of two functional alleles) was caused by STAG1 LoF variants, which may result in partial or complete knockdown of protein activity or product. Our functional studies in vitro of this variant have been performed, RT-PCR showed that the mRNA expression of mutant STAG1 cell lines was extremely lower than wild-type mRNA (Figure 2). Theoretically, the heterozygous frameshift variant c.500dup produced a premature termination codon (PTC), leading to a truncated protein consisting of 180 amino acids.
The result of WB analysis showed mutant STAG1 cell lines could not produce the normal STAG1 protein compared to the STAG1-WT cell lines (Figure 3). Based on the these results, it could be inferred that the heterozygous frameshift variant c.500dup resulted in a partial absence of the biologically relevant transcript of the STAG1 gene (NM_005862.3) attributed to an mRNA quality-control mechanism of nonsense-mediated mRNA decay (NMD). NMD surveys newly synthesized mRNAs and degrades those that harbor a PTC (13), leading to complete absence of the normal STAG1 protein (Figure 4 showed that from 12 to 24 h, the level of the mutant protein progressively declined due to mutant mRNA decay, whereas the wild-type protein remained stable due to mutant mRNA decay). Thereby, this variant c.500dup prevented the production of normal STAG1 protein, which caused the STAG1-related cohesinopathy.
In conclusion, there was no significant difference in the clinical manifestations observed between patients with STAG1 deletions and those with STAG1 SNVs/indels, except for microcephaly and abnormal pregnancies. The STAG1-related cohesinopathy exhibited nonspecific characteristics and diverse presentations, but various degrees of ID/DD have been observed in all cases. Our functional studies have provided preliminary evidence confirming the haploinsufficiency of the STAG1 gene as the underlying pathogenic mechanism of the STAG1-related cohesinopathy.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethical Committee of Quzhou Maternal And Child Health Care Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the participants' legal guardians/next of kin for the publication of this case report.
Author contributions
CJ: Supervision, Writing – original draft, Formal analysis, Investigation. KW: Writing – original draft, Writing – review & editing, Methodology, Supervision, Investigation, Visualization. YZ: Writing – review & editing, Methodology.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are grateful to the patient and their families for participation in this study, as well as for the help of all the physicians in the course of the medical treatment. We wish to thank the staff of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with sequencing data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lehalle D, Mosca-Boidron AL, Begtrup A, Boute-Benejean O, Charles P, Cho MT, et al. STAG1 Mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J Med Genet. (2017) 54(7):479–88. doi: 10.1136/jmedgenet-2016-104468
2. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
3. Carramolino L, Lee BC, Zaballos A, Peled A, Barthelemy I, Shav-Tal Y, et al. SA-1, a nuclear protein encoded by one member of a novel gene family: molecular cloning and detection in hemopoietic organs [published correction appears in Gene 1998 Jan 12;206(2):283]. Gene. (1997) 195(2):151–9. doi: 10.1016/s0378-1119(97)00121-2
4. Piché J, Van Vliet PP, Pucéat M, Andelfinger G. The expanding phenotypes of cohesinopathies: one ring to rule them all!. Cell Cycle. (2019) 18(21):2828–48. doi: 10.1080/15384101.2019.1658476
5. Sumara I, Vorlaufer E, Gieffers C, Peters BH, Peters JM. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol. (2000) 151(4):749–62. doi: 10.1083/jcb.151.4.749
6. Banerji R, Skibbens RV, Iovine MK. How many roads lead to cohesinopathies? Dev Dyn. (2017) 246(11):881–8. doi: 10.1002/dvdy.24510
7. Yuan B, Neira J, Pehlivan D, Santiago-Sim T, Song X, Rosenfeld J, et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet Med. (2019) 21(3):663–75. doi: 10.1038/s41436-018-0085-6
8. Di Muro E, Palumbo P, Benvenuto M, Accadia M, Di Giacomo MC, Manieri S, et al. Novel STAG1 frameshift mutation in a patient affected by a syndromic form of neurodevelopmental disorder. Genes (Basel). (2021) 12(8):1116. doi: 10.3390/genes12081116
9. Funato M, Uehara T, Okada Y, Kaneko H, Kosaki K. Cohesinopathy presenting with microtia, facial palsy, and hearing loss caused by STAG1 pathogenic variant. Congenital Anom. (2022) 62(2):82–3. doi: 10.1111/cga.12454
10. Cipriano L, Russo R, Andolfo I, Manno M, Piscopo R, Iolascon A, et al. A novel de novo STAG1 variant in monozygotic twins with neurodevelopmental disorder: new insights in clinical heterogeneity. Genes (Basel). (2024) 15(9):1184. doi: 10.3390/genes15091184
11. Bregvadze K, Sukhiashvili A, Lartsuliani M, Melikidze E, Tkemaladze T. A novel STAG1 variant associated with congenital clubfoot and microphthalmia: a case report. SAGE Open Med Case Rep. (2024) 12:2050313X241277123. doi: 10.1177/2050313X241277123
12. Seymour H, Feben C, Nevondwe P, Kerr R, Spencer C, Mudau M, et al. Mutation profiling in South African patients with Cornelia de Lange syndrome phenotype. Mol Genet Genomic Med. (2024) 12(1):e2342. doi: 10.1002/mgg3.2342
Keywords: intellectual disability, STAG1 gene, haploinsufficiency, frameshift, functional studies
Citation: Jiang C, Wu K and Zhou Y (2025) A novel frameshift STAG1 variant exhibiting haploinsufficiency due to the nonsense-mediated mRNA decay: a case report and literature review. Front. Pediatr. 13:1648430. doi: 10.3389/fped.2025.1648430
Received: 19 June 2025; Accepted: 10 October 2025;
Published: 24 October 2025.
Edited by:
Giulia Pascolini, Institute of Immaculate Dermatology (IRCCS), ItalyReviewed by:
Giuseppina Covello, University of Padua, ItalyXilong Du, Beijing Chigene Translational Medical Research Center Co., Ltd., China
Copyright: © 2025 Jiang, Wu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhou, MTM5MDU3MDgyMDVAMTYzLmNvbQ==
†These authors have contributed equally to this work