 Martina Filipič1,†
Martina Filipič1,† Špela Stangler Herodež2,†
Špela Stangler Herodež2,† Mirjam Močnik1*
Mirjam Močnik1* Sonja Golob Jančič1
Sonja Golob Jančič1 Nataša Marčun Varda1
Nataša Marčun Varda1 Danijela Krgović2
Danijela Krgović2
- 1Department of Pediatrics, University Medical Centre Maribor, Maribor, Slovenia
- 2Laboratory of Medical Genetics, University Clinical Centre Maribor, Maribor, Slovenia
Introduction: Genetic causes of chronic kidney disease present a diverse group. Some of them are associated with extrarenal malformations, especially ear anomalies. Genetic diagnosis is essential to confirm the diagnosis, search for additional potential manifestations, and predict the prognosis.
Case presentation: We report a case of a 10-year-old girl with elevated creatinine in whom the presence of auricular appendices led to clinical suspicion of a genetic cause of chronic kidney disease. After investigation, Townes-Brocks syndrome was confirmed with the absence of anorectal and limb abnormalities. Therefore, it can be less apparent and needs to be suspected of.
Conclusion: Rare causes of renal diseases can be suspected already after examination. Malformations of the ears are particularly associated with chronic kidney disease, which should be screened for in such cases. Genetic confirmation allows for the establishment of diagnosis and comprehensive management.
Summary: Townes-Brocks syndrome highlights the importance of chronic kidney disease suspicion in a child when malformations of the ears are present in a child.
Introduction
Genetic diseases and syndromes often affect the kidneys and lead to chronic kidney disease (CKD), which is present in approximately 10% of the world's adult population and, according to recent data, in 1% of the child population (1, 2). The general opinion that genetic and environmental factors are important in the development of CKD has been reinforced in recent years by the rapid progress in understanding the genetic basis of kidney function and disease (1). Today, more than 600 genes are known whose mutations are involved in the development of monogenic kidney disease (3). In the pediatric population, excluding diabetic nephropathy and hypertension, these genes are responsible for 20%–50% of CKD cases (1, 4). In the European study of congenital anomalies of the kidneys and urinary tract (CAKUT), in cases with extrarenal manifestations, the latter were part of known syndromes or chromosomal abnormalities in more than half of the cases (5).
Abnormalities affecting both the kidneys and the ears are among the most frequently observed associations and were first recognized around 80 years ago. The association is not clear in all cases, but mostly it includes mutations in transcription factors, growth factors, and their receptors affecting the differentiation of both kidneys and ears (6).
Individuals with ear malformations have a higher incidence of clinically significant structural renal anomalies compared to the general population (6). Kidney disorders linked to ear abnormalities encompass a broad spectrum, including glomerulopathies, CAKUT, ciliopathies, and tubulopathies (7). Therefore, whenever ear abnormalities are present, kidney disease should be searched for.
Case description
A 10-year-old girl came to our department for an additional diagnostic workup of suspected CKD. Before that, she was found to have an elevated creatinine level (88 μmol/L) a few days after an episode of gastroenteritis. However, with additional medical documentation review, we found that her creatinine levels were elevated at a similar level (85 μmol/L) already two years before that episode with no apparent cause. CKD was suspected, and additional diagnostics was performed.
Her family members were unaffected concerning kidney disease. She did have problems with a cough that might be of gastroesophageal origin. Also, she had a history of allergies (cat hair, pollen, grass) and lactose intolerance. In her physical examination, we noticed that she wore glasses; otherwise, no significant findings were seen at the general examination, such as anorectal, limb, or ear anomalies. Her physical growth was unaffected; at 10 years of age, she was 146 centimeters high reaching 90th percentile and weighed 37 kilograms reaching 75th percentile, both percentiles evaluated according to CDC growth charts.
In the laboratory work-up, we confirmed elevated serum creatinine level (94 μmol/L) with elevated cystatin C (1.45 mg/L) and normal urea and electrolytes. In the urine, there was no significant daily proteinuria (less than 0.17 g/day), no hematuria, only mild microalbuminuria (urinary albumin/creatinine was 4.3 g/mol), and a slightly increased amount of low-molecular-weight proteins in the morning urine sample (urinary ɑ-1-microglobulin/creatinine was 2.24 g/mol). Ultrasonographically, both kidneys were hypoplastic with preserved corticomedullary differentiation and without other morphological pathology such as obstructive uropathy or vesicoureterorenal reflux. Her right kidney was measured at 6.8 centimeters and left at 7.2 centimeters, both under 1st percentile for her age.
Glomerular filtration rate was assessed using iohexol, which showed glomerular filtration of 61 ml/min/1.73 m2, confirming stage 2 CKD. In accordance with her stage, other complications, such as anemia, high blood pressure, proteinuria, and poor nutrition, were searched for and excluded at the time. Further follow-up is needed to monitor her CKD progress.
Meanwhile, we also performed a more thorough medical history, where we found out that the girl's periauricular appendages were removed at the age of 5, and hypoplastic ears (photo documentation from the mother before the operation presented in Figure 1) were operated on. Her audiometry showed mild bilateral sensorineural hearing loss with hearing threshold 20–30 dB. According to her presentation we suspected a genetic origin of her CKD with extra-renal manifestations in the branchio-oto-renal (BOR) spectrum. Her blood sample was sent for genetic testing using targeted panel sequencing (next-generation sequencing (NGS) of genes EYA1, SIX1, SIX5) coupled with multiplex ligation dependent probe amplification (MLPA) using the SALSA MLPA Probemix P153 EYA1 (https://www.mrcholland.com), which yielded no genetic mutations.
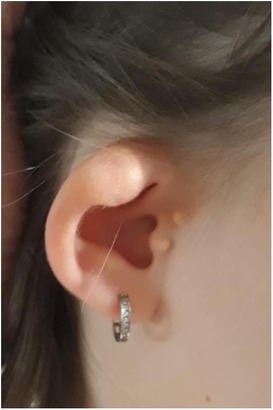
Figure 1. Preoperative view of the patient's hypoplastic ear with appendages.
Additionally, whole exome sequencing (WES) of the patient and parents was performed, which showed the presence of a de novo pathogenic heterozygous insertion NM_002968.3:c.1148_1149insTTTA in the SALL1 gene that caused the insertion of four nucleotides, resulting in a frameshift change from 383. amino acid. This variant has not been described previously and is considered pathogenic (class 5) according to American College of Medical Genetics and Genomics (ACMG) guidelines (8).
Therefore, we conclude that in our patient, a novel mutation in the SALL1 gene is a cause of Townes-Brocks syndrome, diagnosed in our patient.
Discussion
More than 20 disorders encompass both kidney and ear abnormalities (7). Commonly, the ears are affected mainly by sensorineural hearing loss, as in the most commonly associated Alport syndrome; however, in some cases, external ear malformations are present, such as in branchio-oto-renal (BOR) syndrome, CHARGE syndrome, and Townes-Brocks syndrome (7). These are usually recognized soon as several other malformations are present as well. The least obvious is BOR syndrome, usually characterized by branchial fistulas, hearing impairment, renal malformations, and auricular anomalies (9). It has a high penetrance with variable expressivity (9); therefore, it was also the first differential diagnostic option in our case.
CHARGE and Townes-Brocks syndrome were less likely due to the absence of other manifestations. CHARGE syndrome is a genetic condition characterized by a specific and recognizable pattern of features: coloboma, heart defects, choanal atresia, retardation of growth or development, genitourinary malformations, and ear anomalies. Consequently, it was less likely in our case; however, the phenotypic spectrum associated with variants in the chromodomain helicase DNA-binding protein 7 (CHD7) as the major cause of CHARGE syndrome has been broadened. Several predicted pathogenic CHD7 variants have been identified in individuals with isolated features of CHARGE syndrome (10); therefore, this was also possible in our case. Townes-Brocks syndrome is characterized by the triad of anorectal malformations, dysplastic ears, with or without hearing impairment, and hand or thumb anomalies (11), which seemed less likely, since in our case there was no anorectal or hand and thumb abnormalities.
After exclusion of BOR syndrome, we therefore proceeded with WES testing of patient and parents, which revealed a de novo pathogenic mutation in SALL1 gene, usually consistent with Townes-Brocks syndrome, however with absence of cardinal features, such as anorectal and limb malformations. Mutations in SALL1 gene with atypical presentation were first described 25 years ago (12), followed by other similar, but rare, reports (13). Both reports present similar clinical manifestations as in our case. The entity was also named Townes-Brocks like syndrome in older publications or Townes-Brocks syndrome variant or SALL1 variant in recent publications and includes hypoplastic kidneys, kidney function impairment, gastroesophageal reflux, ear abnormalities, mild developmental delay, hearing loss and vision impairment, with the absence of rectal and limb abnormalities, as usually typical for Townes-Brocks syndrome. Our patient showed almost all manifestations of Townes-Brocks syndrome or SALL1 variant; even her need for glasses, and history of persistent cough that was explained with gastroesophageal reflux, as part of her genetic disease, leading to limiting further diagnostic work-up and burden in persistent cough evaluation. Further studies reveal that in individuals undergoing broad-based genetic testing with a kidney gene panel, variants in SALL1 are rare, yielding a prevalence of 1:1,592 among patients tested for monogenic kidney disease. Usually, these patients (in 91%) progressed to CKD with decreased glomerular filtration rate and included diverse kidney features (agenesis/hypoplasia, focal segmental glomerulosclerosis, kidney cysts). Hearing loss/ear anomalies or anorectal malformations were present in about a third (not all having both), and only 18% had hand/thumb abnormalities. Only 14% of patients had the traditional “triad” called Townes-Brocks syndrome (11). As the variants are rare, this data is based on a relatively small set of patients. Nevertheless, they are consistent with findings in our patient.
On the contrary, auricular anomalies and appendages are more common, with an estimated incidence of external ear malformations in 1 per 6,000 to 6,830 newborns, associated with syndromes involving other organ systems in 30% (14). Most ear anomalies are therefore acquired and originate from exposures in utero (infections, medications, malnutrition, Rh incompatibility, hypoxia, bleeding, disturbances of metabolism) (14). Therefore, in the absence of cardinal features of a syndrome, the clinical suspicion for associated kidney disease is low.
Our case emphasizes the need for increased clinical suspicion of apparently isolated anomalies to think of other clinical manifestations, especially in external ear anomalies and kidney disease. Early diagnosis is important for comprehensive evaluation of potentially affected organ systems, genetic testing of family members and genetic counseling, early renal management, which can be crucial in slowing of CKD progression (15). Concerning CKD, ongoing monitoring and renal protective interventions are needed.
Otherwise, management of Townes-Brocks syndrome depends on clinical presentations and is usually based on multidisciplinary approach with involvement of several specialists, such as pediatric nephrologist for management of CKD, an ear-nose-throat (ENT) specialist for hearing aids and cosmetic surgical procedures, and orthopedic or general surgeons when limb or anorectal abnormalities are present. According to other manifestations, other specialists might be included as well, such as ophthalmologist for any eye-related manifestations, or neurologist when developmental delay is present etc.
Conclusion
We confirmed Townes-Brocks syndrome in a 10-years-old girl. The suspicion of the genetic cause of the disease was established already with bilateral kidney hypoplasia; however, the history of auricular hypoplasia and appendages directed us to a specific genetic origin. We encourage kidney disease suspicion whenever ear anomalies are present despite absence of other possible malformations.
Patient perspective
Children with apparently isolated ear anomalies should be screened for kidney disease as this is a possible association. Identification of a specific gene mutation allows comprehensive management of a patient with a reduction of unnecessary diagnostic procedures. Timely diagnosis of kidney disease allows early management of CKD in children, possibly prolonging their normal glomerular filtration rate and postponing the development of complications.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MF: Data curation, Writing – review & editing, Supervision. ŠS: Data curation, Investigation, Supervision, Formal analysis, Writing – review & editing. MM: Project administration, Conceptualization, Writing – original draft. SGJ: Investigation, Writing – review & editing, Data curation. NMV: Data curation, Supervision, Writing – review & editing. DK: Data curation, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Köttgen A, Cornec-Le Gall E, Halbritter J, Kiryluk K, Mallett AJ, Parekh RS, et al. Genetics in chronic kidney disease: conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2022) 101:1126–41. doi: 10.1016/j.kint.2022.03.019
2. Harambat J, Madden I, Hogan J. Épidémiologie de la maladie rénale chronique chez l’enfant. Néphrol Thér. (2021) 17:476–84. doi: 10.1016/j.nephro.2021.06.001
3. Rasouly HM, Groopman EE, Heyman-Kantor R, Fasel DA, Mitrotti A, Westland R, et al. The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med. (2019) 170:11–21. doi: 10.7326/M18-1241
4. Cunha MFMD, Sevignani G, Pavanelli GM, Carvalho MD, Barreto FC. Rare inherited kidney diseases: an evolving field in nephrology. Braz J Nephrol. (2020) 42:219–30. doi: 10.1590/2175-8239-jbn-2018-0217
5. Pal A, Reidy KJ. Genetic syndromes affecting kidney development. Results Probl Cell Differ. (2017) 60:257–79. doi: 10.1007/978-3-319-51436-9_10
6. Izzedine H, Tankere F, Launay-Vacher V, Deray G. Ear and kidney syndromes: molecular versus clinical approach. Kidney Int. (2004) 65:369–85. doi: 10.1111/j.1523-1755.2004.00390.x
7. Phelan PJ, Rheault MN. Hearing loss and renal syndromes. Pediatr Nephrol. (2018) 33:1671–83. doi: 10.1007/s00467-017-3835-9
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Wen YY, Sun Y, Kong WJ. Emphasizing the application of genetic diagnosis in branchio-oto-renal syndrome. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. (2018) 32:1226–31.30282165
10. Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. (2016) 170:344–54. doi: 10.1002/ajmg.a.37435
11. Stein Q, Vostrizansky A, Magay Y, Jandeska S, Westemeyer M, Hendricks E, et al. Townes-brocks syndrome revealed by kidney gene panel testing. Kidney Int Rep. (2024) 9:1810–6. doi: 10.1016/j.ekir.2024.03.030
12. Engels S. A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet. (2000) 37:458–60. doi: 10.1136/jmg.37.6.458
13. Albrecht B, Liebers M, Kohlhase J. Atypical phenotype and intrafamilial variability associated with a novel SALL1 mutation. Am J Med Genet A. (2004) 125A:102–4. doi: 10.1002/ajmg.a.20484
14. Acosta-Rodríguez A, Reza-López SA, Aguilar-Torres CR, Hinojos-Gallardo LC, Chávez-Corral DV. A systematic review of congenital external ear anomalies and their associated factors. Front Pediatr. (2025) 13:1520200. doi: 10.3389/fped.2025.1520200
Keywords: chronic kidney disease, child, genetics, rare variants, Townes-Brocks syndrome
Citation: Filipič M, Stangler Herodež Š, Močnik M, Golob Jančič S, Marčun Varda N and Krgović D (2025) Case Report: Wide spectrum of SALL1 variants—a rare cause of pediatric chronic kidney disease. Front. Pediatr. 13:1649707. doi: 10.3389/fped.2025.1649707
Received: 18 June 2025; Accepted: 18 August 2025;
Published: 8 September 2025.
Edited by:
Jakub Zieg, University Hospital in Motol, CzechiaReviewed by:
Zhilang Lin, Sun Yat-sen University, ChinaMaria Malarska, Polish Mother’s Memorial Hospital Research Institute, Poland
Copyright: © 2025 Filipič, Stangler Herodež, Močnik, Golob Jančič, Marčun Varda and Krgović. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Močnik, bWlyamFtbW9jbmlrOTFAZ21haWwuY29t
†These authors have contributed equally to this work