 Ning Li
Ning Li Chen Chen
Chen Chen- 1Pediatric Department, Liuyang People Hospital, Changsha, China
- 2Children’s Medical Center, Xiangya Hospital, Central South University, Changsha, China
Background: Congenital disorders of glycosylation (CDG) are a group of multi-systemic genetic disorders. Over 100 monogenic human diseases were known related with defects in glycosylation process. Defects of SSR4 gene lead to a rare X linked pattern of CDG which has been rarely reported.
Method: We reported a Chinese boy with developmental delay, microcephaly, and epileptic seizures. Whole exome sequencing and Sanger sequencing were performed in the family.
Result: A novel maternal splice variant c.351+1del in SSR4 gene was identified by trio-exome sequencing, and confirmed by Sanger sequencing. The functional effect of the variant was further investigated by minigene. The minigene results showed three abnormal splice forms: (1) 1 bp deletion in 3′ end of exon 4; (2) 42 bp deletion in 3′ end of exon 4; (3) skipping of exon 4. All three forms resulted in truncated proteins. c.351+1del in SSR4 gene causes congenital disorder of glycosylation, type Iy, consisted with the proband's phenotype. Up to date, all of the pathogenic SSR4 gene variants were null variants. The most variants were reported in exon 4. Patients (within or between families) carrying the same variants exhibited phenotypic heterogeneity.
Conclusion: The current study expanded the pathogenic variant spectrum of SSR4 gene and revealed the impact of c.351+1del on SSR4 splicing. Standardizing the transcript and naming conventions of variants were crucial for the study of SSR4 genotypes and phenotypes.
1 Introduction
N-linked glycosylation is a post-translational modification essential for the folding, stability, and other cellular functions of membrane proteins (1, 2). In recent years, literatures had reported alterations in the N-glycans of membrane proteins are associated with various pathological conditions (3, 4). Signal sequence receptor protein 4 (SSR4, OMIM 300090) on chromosome Xq28, encodes a subunit of a transmembrane protein complex involved in the transport of proteins across the endoplasmic reticulum membrane, enhancing the efficiency of N-linked glycosylation (5). Hemizygous variants in the SSR4 gene cause congenital disorder of glycosylation, type Iy (CDG1Y, OMIM 300934), also called SSR4-CDG. SSR4-CDG is characterized by developmental delay, speech delay, impaired intellectual development, muscular hypotonia, microcephaly, seizures and distinctive facial features (5). Extended phenotype has cardiomyopathy (6) and connective tissue disorder (redundant skin, joint laxity, blue sclerae, and vascular tortuosity) (7). Up to date, more than 20 patients with loss-of-function variants were reported with CDG1Y, including total gene deletions (6 patients), nonsense (5 patients), frameshift (7 patients) and splice variants (5 patients) (5–13). However, the relationship between SSR4 genotype and phenotype was not yet clear.
We reported a Chinese boy with a maternal splice variant of SSR4 gene and verified the impact on mRNA coding. To conduct accurate analysis of genotypes, we summarized the characteristics of the SSR4 genotypes and provided theoretical foundation for the study of the genotype and phenotype of the SSR4.
2 Materials and methods
2.1 Clinical course
The proband, a 4-year-old boy (G1P1), was delivered naturally with a birth weight of 2.7 kg. He underwent developmental delay, microcephaly, and epileptic seizures twice before. He was admitted to the Pediatric of the People's Hospital of Liuyang due to epileptic seizure accompanied by high fever one day ago. He had a high fever with unknown reason (up to 39℃) before seizure. The seizure type was tonic-clonic seizure, characterized by unresponsiveness, eyes rolling up, dark complexion, trembling limbs, and lasting for about 2 min. Further examination after admission revealed swollen tonsils (Ⅰ°) and coarse breath sounds in both lungs. Routine electrolyte test results suggest hypokalemia (potassium, 3.14 mmol/L). No obvious brain structural abnormalities were found on cranial MRI. Urine organic acid test results were normal. Blood amino acid and acylcarnitine profile analysis showed a slight decrease in glutamine (Gln) level (0.89, normal range from 1.00 to 55.00). Facial features included deep set eyes, large ears and mouth. The child's general developmental level was equivalent to 30-month-old according to the Gesell Developmental Diagnostic Scale, with a Developmental Quotient (DQ) of 54, indicating moderate developmental delay. His parents were healthy, non-consanguineous, and had no family history of genetic diseases. After adequate genetic counseling, we performed trios whole exome sequencing (WES) on the proband and his parents.
2.2 Whole exome sequencing
Genomic DNA were extracted from peripheral blood samples using Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH, Hilden, Germany). 50 ng genomic DNA was interrupted to approximately 250 bp around by fragmentation enzymes. The DNA fragments were then end repaired, and the 3′end was added one A base. The DNA fragments were ligated with barcoded sequencing adaptors and collected by XP beads. The DNA fragments were hybridized and captured by probe named Nano WES V2 (Berry Genomics, Beijing, China). The products were eluted and collected, and subjected to PCR amplification and the purification. The libraries were quantified by qPCR and sequenced by Novaseq6000 platform (Illumina, San Diego, USA) with 150 bp pair-end sequencing mode.
The raw sequencing reads were aligned to the human reference genome (GRCh38) using Burrows Wheeler Aligner (14) tool and PCR duplicates were removed by using Picard v1.57 (http://picard.sourceforge.net/). Verita Trekker® Variants Detection System by Berry Genomics and GATK (https://software.broadinstitute.org/gatk/) were employed for variant calling. Variant annotation and interpretation were conducted by ANNOVAR (15) and the Enliven® Variants Annotation Interpretation System authorized by Berry Genomics. Annotation databases mainly included gnomAD (http://gnomad.broadinstitute.org/), EXAC database (https://gnomad.broadinstitute.org/), the 1,000 Genome Project (http://browser.1000genomes.org), Berry inhouse population database, dbSNP (http://www.ncbi.nlm.nih.gov/snp), SIFT (http://sift.jcvi.org), FATHMM (http://fathmm.biocompute.org.uk), MutationAssessor (http://mutationassessor.org), CADD (http://cadd.gs.washington.edu), SPIDEX (16), spliceAI (17), FF (https://www.fruitfly.org/seq_tools/splice.html), OMIM (http://www.omim.org), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), HGMD (http://www.hgmd.org), HPO (https://hpo.jax.org/app/) etc. The variants were classified to five categories (pathogenic, likely pathogenic, uncertain significance, likely benign and benign) according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of genetic variants (18).
2.3 Sanger sequencing
The candidate variant was verified by Sanger sequencing. The primers (Forward: CAGAAGGTGACCCTGCCTTT; Reverse: AAACAGAGGCGGGATGATGG) were designed by Primer 5 software. DNA fragment containing the candidate variant was amplified by polymerase chain reaction: initial denaturation at 95°C for 5 min, followed by 34 cycles at 95°C for 30 s, 58°C for 30 s and 72°C for 20 s and hold at 72°C for 10 min. PCR product was purified and sequenced using an ABI 3730XL DNA Analyzer with the BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems, Foster, CA, USA). The results of Sanger sequencing were analyzed by Chromas software according to NM_006280.
2.4 In vitro minigene assays
We conducted in vitro minigene assays to verify the impact of candidate variant. To enhance the reliability of the experimental results, we transfected two types of recombinant plasmids into two types of tool cells respectively.
Using normal human gDNA as a template, nested PCRs were carried out by Nested primer 1 and 2. Then using nested PCR products as template to generate the wide and mutant SSR4 fragments of pcDNA3.1 and pcMINI-C. Pair of pcDNA3.1-SSR4-KpnI-F and pcDNA3.1-SSR4-EcoRI-R were used to generate fragment of pcDNA3.1-SSR4-wt. Using the mixture of PCR reaction products (pcDNA3.1-SSR4-KpnI-F and SSR4-mut-R, SSR4-mut-F and pcDNA3.1-SSR4-EcoRI-R) as template to generate fragment of pcDNA3.1-SSR4-mut by pair of pcDNA3.1-SSR4-KpnI-F and pcDNA3.1-SSR4-EcoRI-R. Pair of pcMINI-C-SSR4-KpnI-F and pcMINI-C-SSR4-EcoRI-R were used to generate fragment of pcMINI-C-SSR4-wt. Using the mixture of PCR reaction products (pcMINI-C-SSR4-KpnI-F and SSR4-mut-R, SSR4-mut-F and pcMINI-C-SSR4-EcoRI-R) as template to generate fragment of pcMINI-C-SSR4-mut by pair of pcMINI-C-SSR4-KpnI-F and pcMINI-C-SSR4-EcoRI-R. All of primers (Table 1) were designed by Primer5 software.
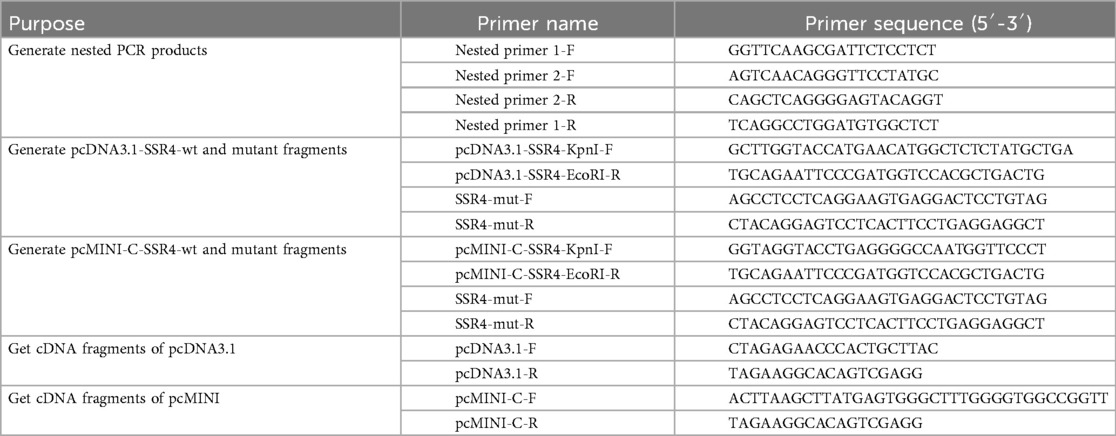
Table 1. Sequences and purposes of the primers used in minigene assay.
The wild and mutant fragments were constructed in pcDNA3.1 and pcMINI-C plasmids (Figures 2A,B) and confirmed by Sanger sequencing. The vector constructions were supplied in the Supplementary Figure. The primers sequences used in vector constructions were show in Table 1. The recombinant plasmids were transfected into HEK293T and HeLa cells separately using Hieff Trans™ Liposomal Transfection Reagent (YEASEN, Shanghai, China). Total RNA was extracted from cells cultured for 48 h with Trizol Reagent (TaKaRa, Kusatsu, Japan) and reverse-transcribed with the Superscript III reverse transcriptase (HifairTM 1st Strand cDNA Synthesis SuperMix for qPCR) (YEASEN, Shanghai, China). PCRs were performed by primers (pcDNA3.1-F and pcDNA3.1-R; pcMINI-C-F and pcMINI-C-R) in the target region of cDNA. The PCR products were analyzed on 1% agarose gel electrophoresis and using an ABI 3730XL DNA Analyzer with the BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems, Foster, CA, USA). Compare the sequencing results of wild and mutant-type in the cells and analysis the splice form of the mutant type.
2.5 The three-dimensional (3D) structure
The three-dimensional (3D) structure of SSR4 was analyzed by PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC), using AF-P51571-F1-model_v4 as the template.
2.6 Analysis of the SSR4 genotype
Using “SSR4 gene”, “SSR4-CDG” and “congenital disorder of glycosylation, type Iy” as the key words, conduct a search in the PUBMED database. Using “SSR4” as the key word to search the transcripts (Homo sapiens) in NCBI database. According to Human Genome Variation Society (HGVS) nomenclature rules (https://hgvs-nomenclature.org/), we checked and corrected the variants reported. We drew the gene structure of SSR4 based on the transcript structure provided by ProteinPaint (https://proteinpaint.stjude.org/) and performed localization and analysis of the SSR4 SNVs and indels.
3 Results
3.1 Results of WES
A novel hemizygous splice variant NM_006280: c.351+1del in SSR4 gene (OMIM 300090) was identified in the proband by WES. Both of WES and sanger sequencing confirmed it inherited from the mother (Figure 1). c.351+1del was in the intron 4 of SSR4 gene. It was a splice donor variant, predicted to undergo nonsense-mediated mRNA decay (NMD)(PVS1) (19). Multiple software programs predict the variant impact the splicing of SSR4 (spliceAI: donor loss 1.00, donor gain 0.98; FF: donor loss 0.99). It wasn't reported in HGMD (https://www.hgmd.cf.ac.uk/ac/index.php) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases previously. The frequency of the variant was not found in gnomAD, EXAC, 1000G and Berry inhouse database (PM2_Supporting). c.351+1del was classified as “likely pathogenic” (PVS1+PM2_Supporting) according to ACMG guidelines. SSR4 gene was related with Congenital disorder of glycosylation, type Iy (CDG1Y, OMIM 300934). Developmental delay, microcephaly, and epileptic seizures were consisted with the clinical phenotype of CDG1Y. The normal mother carried the heterozygote variant c.351+1del, while the proband carried the hemizygous variant. The co-segregation pattern of the family was consistent with X-linked recessive inheritance.
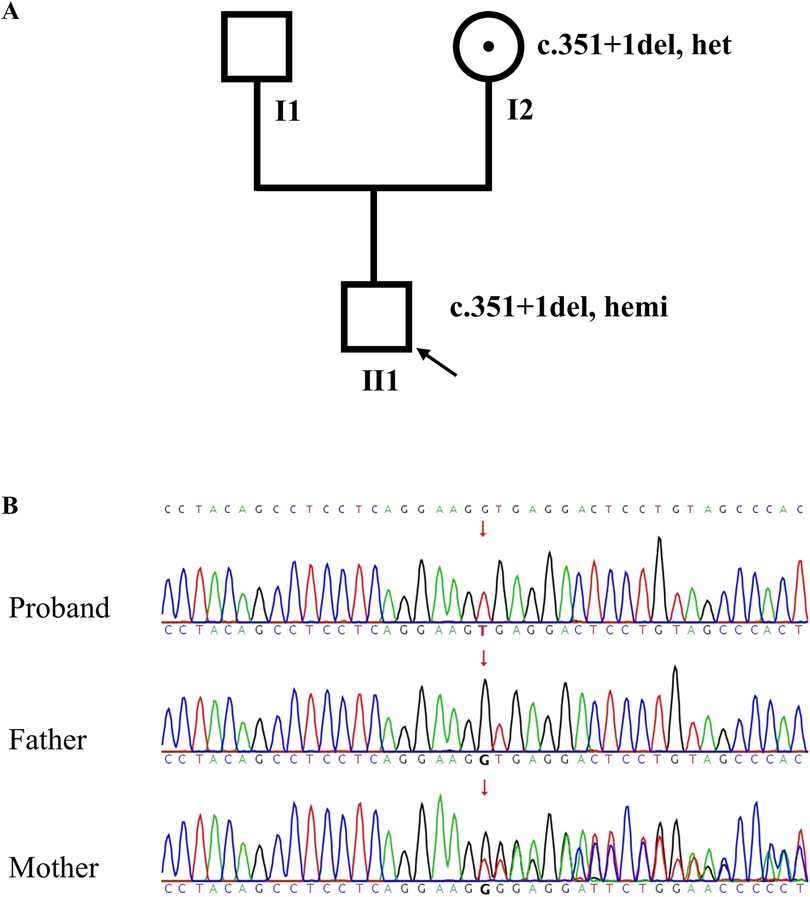
Figure 1. Pedigree and SSR4 gene sequencing results. (A) The pedigree of the family; (B) Sanger sequence chromatogram of SSR4 gene. The results showed that c.351+1del in SSR4 was hemizygous in the proband (Ⅱ1) and heterozygote in the mother (Ⅰ2).
3.2 Results of minigene splicing assay (SSR4 gene c.351+1del)
The functional effect of the variant was investigated by in vitro minigene assay. The minigene results of two systems were consisten. There were three abnormal splice forms: (1) 1 bp deletion in 3′ end of exon 4 (c.351del, p.Ala118Leufs*42); (2) 42 bp deletion in 3′ end of exon 5 (c.310_351del, p.Val104_Lys117del); (3) skipping of exon 4 (c.262_351del, p.Val88_Lys117del) (Figures 2, 3). All three forms resulted in the production of truncated proteins.
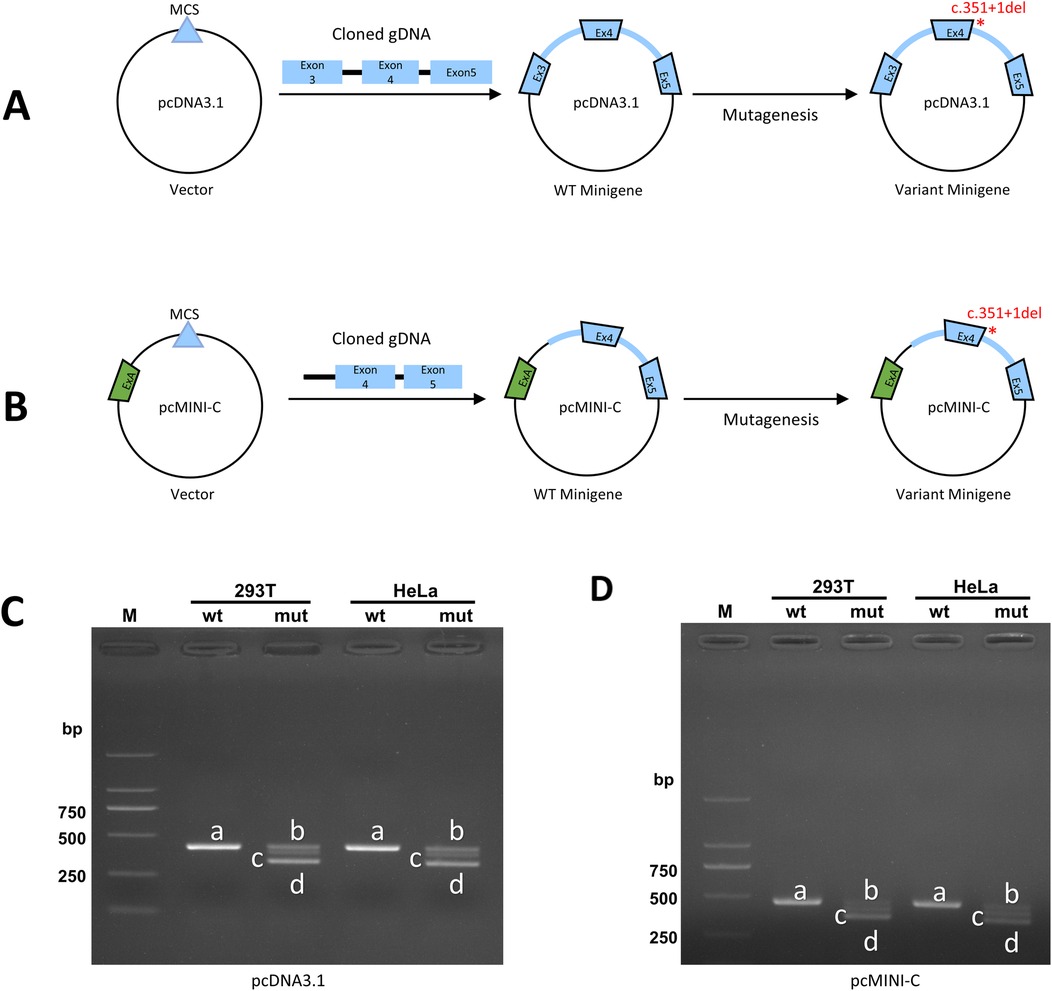
Figure 2. The in vitro minigene assay of pcDNA3.1 and pcMINI-C. (A) Strategies for plasmid pcDNA3.1 construction; (B) Strategies for plasmid pcMINI-C construction; (C,D) The Gel electrophoresis of RT-PCR products displayed a single band (a) from the wild type (wt) and three different bands (b–d) in the mutant type (mut).
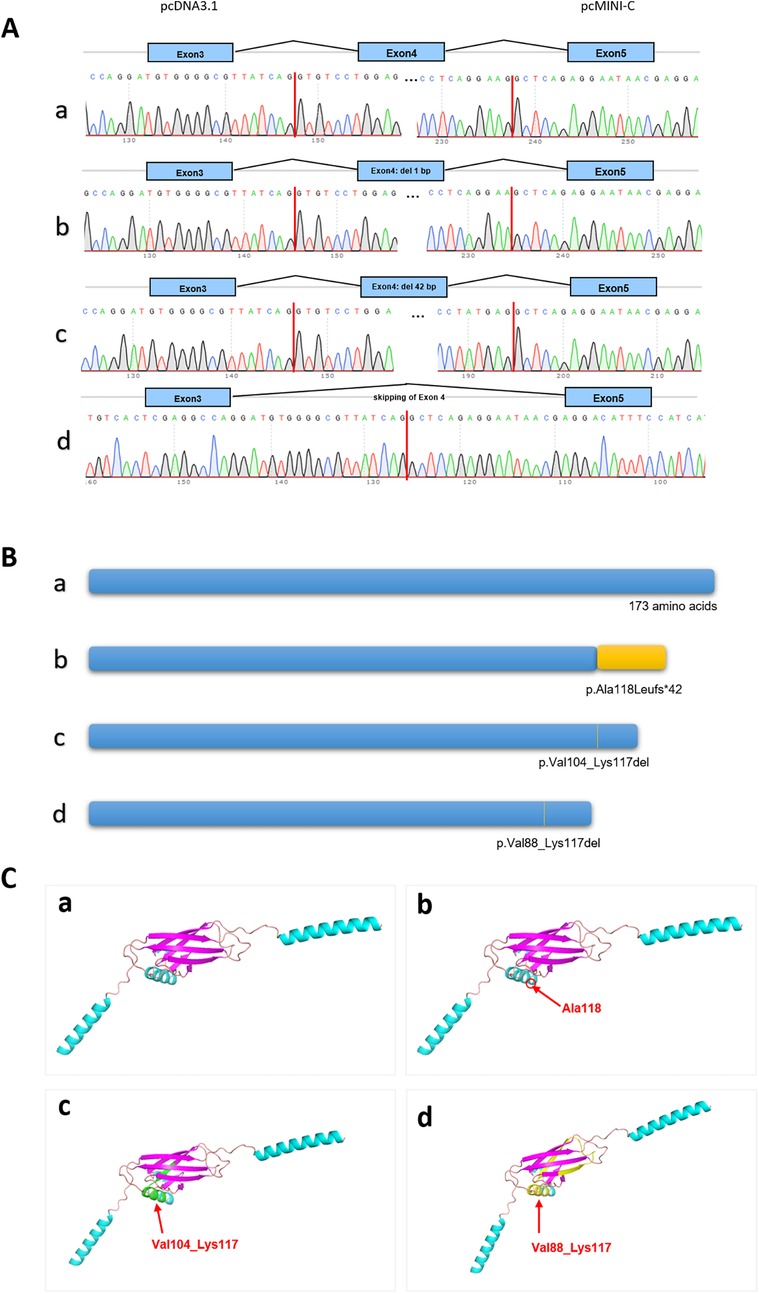
Figure 3. The results of minigene assay of SSR4 gene c.351+1del. (A) The cDNA Sanger sequencing of RT-PCR products in pcDNA3.1 was consistent in pcMINI-C. Band a led to a shorter transcript with 1 bp deletion of exon 4; Band b led to a shorter transcript with 42 bp deletion of exon 4; Band c led to a shorter transcript with skipping of exon 4; (B) Schematic diagram of three types of protein length. (C) The positions of three different variants on the three-dimensional protein structure diagram.
3.3 Genotype of SSR4
There were three transcripts of SSR4 gene in the NCBI database: NM_006280 (MANE select), NM_001204526 and NM_001204527 (Figure 4). According to the MANE transcript and the HGVS Nomenclature, we standardized the variants of 16 previously reported SSR4-CDG patients and patient in this work (see Supplementary Table). The distribution of variants on the SSR4 gene (MANE transcript) was summarized in Figure 4. All of the variants distributed in the coding exons and canonical splice sites of NM_006280. The most cases and variants were reported in exon 4 (3 variants in 4 patients), including the recurrent variant NM_006280: c.269G>A (p.Trp90Ter) reported in 2 unrelated patients [NM_006280: c.269G>A (p.Trp90Ter) equivalent to NM_001204527:c.302G>A (p.Trp101Ter)]. While no variant reported in exon 1 of NM_001204526 or NM_001204527.
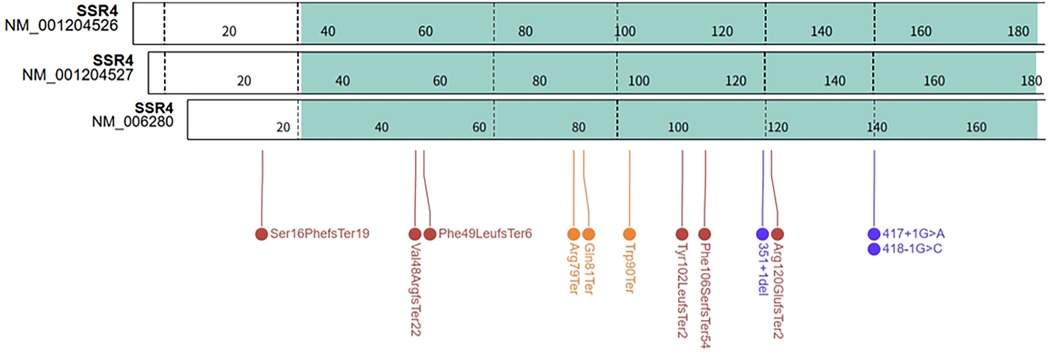
Figure 4. SSR4 gene transcripts and distribution of reported variants in SSR4 gene.
3.4 Follow-up
After six months of language rehabilitation training at our hospital, the child underwent the Gesell Developmental Schedules assessment at the age of four years and ten months. The results indicated that his general developmental level was equivalent to 35-month-old, with a DQ of 56, falling within the range of mild developmental delay. The child later received acupuncture and massage rehabilitation treatment at another hospital. At the age of five years and three months, he was assessed again using the Gesell Developmental Schedules, which showed his general developmental level to be equivalent to 39-month-old, with a DQ of 57, still indicating mild developmental delay.
4 Discussion and conclusions
Glycosylation can regulate enzyme activity or its interaction with other proteins to play roles in various cellular processes, including cell recognition, cell signaling, resistance to proteases, adhesion, migration, and host defense mechanisms (20). An important process required for N-glycosylation is the co-translational translocation of proteins from the cytoplasm to the endoplasmic reticulum, which is facilitated by the translocon-associated protein (TRAP) complex. The transmembrane subunit SSR4 is a member of the signal sequence receptor (SSR) family in the structure of the translocon complex (5, 21). When SSR4 is defective, the addition of glycan precursors to proteins in the endoplasmic reticulum is impaired, potentially leading to disordered N-linked glycosylation of many proteins and the complete loss of glycans (7, 9, 22).
SSR4-CDG is a multisystem disorder characterized by neurological phenotypes (9, 23). Unlike most autosomal recessive CDG disorders, SSR4 is located on the X chromosome and the pathogenic variants maybe de novo or maternal inheritance (5). In our work, the mother was a heterozygote of SSR4 gene c.351+1del without typical clinical symptoms. Carbohydrate-deficient transferrin (CDT) is a reliable and cost-effective screening method for identifying CDG cases (24). However, it cannot pinpoint specific genetic defects. Previously reported cases of SSR4-CDG had mostly shown Type I transferrin (Tf) patterns (5, 9, 10). Due to the lack of specific clinical features associated with CDG-related disorders, the initial presentation is often global developmental delay (GDD). Most CDG cases were identified through genetic testing and subsequently confirmed CDT. As seen in our work, patients often refuse CDT testing after receiving the positive genetic results. This highlights the importance of considering SSR4-CDG as a potential diagnosis in multisystem disorders with predominant neurological manifestations and conducting genetic testing.
Regarding the clinical phenotype of SSR4-CDG patients, three previous publications have organized and presented cohort samples (5, 9, 10). Our study referenced previously reported cases and standardized the nomenclature of SNVs and indels. After standardizing the variants, we found that two variants were recurrent in two unrelated families: c.269G>A, (p.Trp90Ter) in exon 4 and c.417+1G>A in intron 5. The study by Johnsen C et al (5) showed that P14 and P22 were found to carry c.269G>A, p.(Trp90Ter), exhibiting the same core phenotypes (developmental delay, intellectual disability, muscular hypotonia and abnormal facial features), different the other phenotypes (feeding difficulties, connective tissue, gastrointestinal Symptoms, skeletal abnormalities, behavioral issues). The study by Wang et al (10) showed that c.269G>A leaded to mRNA expression of the SSR4 gene approaching zero. The study by Johnsen C et al (5) that P6 and P19 carry c.417+1G>A, presenting with a consistent core phenotype but differing in other phenotypic features. c.417+1G>A were predicted might result in the skipping of exon 5. Similar to many other types of CDG, clinical phenotyping revealed heterogeneity among different individuals (25).
Our study conducted minigene to clarify the impact of splice variants on mRNA encoding. The shortest transcript products, as shown in Figures 2C,D, result from the skipping of exon 4, leading to the deletion of amino acids within the reading frame. In the agarose gel electrophoresis results (Figures 2C,D), this product was found to have the highest expression level. All indications suggest that the integrity of the protein encoded by the SSR4 gene is crucial for its function. Current, the structure of the protein encoded by SSR4 and the function of the region encoded by exon 4 are not well understood and require further investigation. No variations have been reported in the last exon, and its pathogenicity requires further observation.
In conclusion, the current study expanded the pathogenic variant spectrum of SSR4 gene and revealed the impact of c.351+1del on SSR4 splicing. Standardizing the transcript and naming conventions of variants were crucial for the correct study of SSR4 genotypes and phenotypes.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of LiuYang People's Hospital [2023(006)]. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
NL: Funding acquisition, Writing – original draft. CC: Investigation, Methodology, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Health Research Project of Hunan Provincial Health Commission (grant number: D202306016324).
Acknowledgments
I am very grateful for the patient and parents' support and cooperation with the genetic testing and the publication of the article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1651524/full#supplementary-material
References
1. Huang Z, Lai PF, Cocker ATH, Haslam SM, Dell A, Brady HJM, et al. Roles of N-linked glycosylation and glycan-binding proteins in placentation: trophoblast infiltration, immunomodulation, angiogenesis, and pathophysiology. Biochem Soc Trans. (2023) 51(2):639–53. doi: 10.1042/BST20221406
2. Esmail S, Manolson MF. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. Eur J Cell Biol. (2021) 100(7–8):151186. doi: 10.1016/j.ejcb.2021.151186
3. Conroy LR, Hawkinson TR, Young LEA, Gentry MS, Sun RC. Emerging roles of N-linked glycosylation in brain physiology and disorders. Trends Endocrinol Metab. (2021) 32(12):980–93. doi: 10.1016/j.tem.2021.09.006
4. Cawley NX, Lyons AT, Abebe D, Luke R, Yerger J, Telese R, et al. Complex N-linked glycosylation: a potential modifier of Niemann-Pick disease, type C1 pathology. Int J Mol Sci. (2022) 23(9):5082. doi: 10.3390/ijms23095082
5. Johnsen C, Tabatadze N, Radenkovic S, Botzo G, Kuschel B, Melikishvili G, et al. SSR4-CDG, an ultra-rare X-linked congenital disorder of glycosylation affecting the TRAP complex: review of 22 affected individuals including the first adult patient. Mol Genet Metab. (2024) 142(3):108477. doi: 10.1016/j.ymgme.2024.108477
6. Zemet R, Hope KD, Edmondson AC, Shah R, Patino M, Yesso AM, et al. Cardiomyopathy, an uncommon phenotype of congenital disorders of glycosylation: recommendations for baseline screening and follow-up evaluation. Mol Genet Metab. (2024) 142(4):108513. doi: 10.1016/j.ymgme.2024.108513
7. Castiglioni C, Feillet F, Barnerias C, Wiedemann A, Muchart J, Cortes F, et al. Expanding the phenotype of X-linked SSR4-CDG: connective tissue implications. Hum Mutat. (2021) 42(2):142–9. doi: 10.1002/humu.24151
8. Wu R, Tang W, Qiu K, Li X, He Z. [Analysis of SSR4 gene variant in a child with congenital glycosylation type 1y in conjunct with congenital dysplasia of external auditory canal]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2022) 39(7):727–30. Chinese. doi: 10.3760/cma.j.cn511374-20210205-0011
9. Ng BG, Raymond K, Kircher M, Buckingham KJ, Wood T, Shendure J, et al. Expanding the molecular and clinical phenotype of SSR4-CDG. Hum Mutat. (2015) 36(11):1048–51. doi: 10.1002/humu.22856
10. Wang J, Gou X, Wang X, Zhang J, Zhao N, Wang X. Case report: the novel hemizygous mutation in the SSR4 gene caused congenital disorder of glycosylation type Iy: a case study and literature review. Front Genet. (2022) 13:955732. doi: 10.3389/fgene.2022.955732
11. Sun W, Jin X, Zhu X. A novel SSR4 variant associated with congenital disorder of glycosylation: a case report and related analysis. Front Genet. (2024) 15:1402883. doi: 10.3389/fgene.2024.1402883
12. Weng L, Deng Q, Chen X, Wang K, Shao J. A case of congenital disorder of glycosylation due to SSR4 gene deletion. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2023) 40(3):364–7. doi: 10.3760/cma.j.cn511374-20210918-00762
13. Losfeld ME, Ng BG, Kircher M, Buckingham KJ, Turner EH, Eroshkin A, et al. A new congenital disorder of glycosylation caused by a mutation in SSR4, the signal sequence receptor 4 protein of the TRAP complex. Hum Mol Genet. (2014) 23(6):1602–5. doi: 10.1093/hmg/ddt550
14. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
15. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38(16):e164. doi: 10.1093/nar/gkq603
16. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. (2015) 347(6218):1254806. doi: 10.1126/science.1254806
17. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176(3):535–48.e24. doi: 10.1016/j.cell.2018.12.015
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
19. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. (2018) 39(11):1517–24. doi: 10.1002/humu.23626
20. He M, Zhou X, Wang X. Glycosylation: mechanisms, biological functions and clinical implications. Signal Transduct Target Ther. (2024) 9(1):194. doi: 10.1038/s41392-024-01886-1
21. Huang Y, Xu X, Arvan P, Liu M. Deficient endoplasmic reticulum translocon-associated protein complex limits the biosynthesis of proinsulin and insulin. FASEB J. (2021) 35(5):e21515. doi: 10.1096/fj.202002774R
22. Ng BG, Lourenco CM, Losfeld ME, Buckingham KJ, Kircher M, Nickerson DA, et al. Mutations in the translocon-associated protein complex subunit SSR3 cause a novel congenital disorder of glycosylation. J Inherit Metab Dis. (2019) 42(5):993–7. doi: 10.1002/jimd.12091
23. Francisco R, Brasil S, Poejo J, Jaeken J, Pascoal C, Videira PA, et al. Congenital disorders of glycosylation (CDG): state of the art in 2022. Orphanet J Rare Dis. (2023) 18(1):329. doi: 10.1186/s13023-023-02879-z
24. Sierra T, Crevillen AG, Escarpa A. Electrochemical sensor for the assessment of carbohydrate deficient transferrin: application to diagnosis of congenital disorders of glycosilation. Biosens Bioelectron. (2021) 179:113098. doi: 10.1016/j.bios.2021.113098
Keywords: SSR4, congenital disorder of glycosylation, whole exome sequencing, minigene, transcript
Citation: Li N and Chen C (2025) Novel SSR4 gene splice variant leads to congenital disorder of glycosylation, type Iy. Front. Pediatr. 13:1651524. doi: 10.3389/fped.2025.1651524
Received: 21 June 2025; Accepted: 6 October 2025;
Published: 24 October 2025.
Edited by:
Anjana Munshi, Central University of Punjab, IndiaReviewed by:
Zheng Jin Tu, Cleveland Clinic, United StatesWenhao Ouyang, Sun Yat-sen University, China
Roberta Salinas, Universidad Autónoma del Estado de Morelos, Mexico
Copyright: © 2025 Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Chen, YWxsZXlDQ0Bjc3UuZWR1LmNu