 René Gaupmann1*
René Gaupmann1* Saskia Gruber1
Saskia Gruber1 Klara Schmidthaler1
Klara Schmidthaler1 Bianca Pauger1Brigitte Mersi1
Bianca Pauger1Brigitte Mersi1 Alexandra Graf2
Alexandra Graf2 Zsolt Szépfalusi1Sabine Renner1Eleonora Dehlink1
Zsolt Szépfalusi1Sabine Renner1Eleonora Dehlink1
- 1Department of Pediatrics and Adolescent Medicine, Division of Pediatric Pulmonology, Allergology and Endocrinology, Medical University of Vienna, Vienna, Austria
- 2Institute for Medical Statistics, Centre for Medical Data Science, Medical University of Vienna, Vienna, Austria
Introduction: Maintaining good lung function is a primary goal in managing Cystic Fibrosis (CF). As spirometry lacks sensitivity for detecting mild lung disease, early progression often remains unrecognized. To overcome this limitation, more sensitive monitoring tools are needed. We evaluated arterialized oxygen tension (pO2) as an easily accessible, and widely applicable surveillance method.
Methods: In this retrospective observational single-center cohort study, arterialized gas exchange was assessed in 103 young people with CF (47 females, 56 males, aged 5–18 years). Trends from baseline (age 5 years) to early adulthood and performance relative to annual best pulmonary function (FEV1) and lung clearance index were examined, along with baseline oxygen tension's predictive value on future FEV1 decline and the occurrence of CF-related complications.
Results: pO2 correlated significantly with FEV1 (p < 0.001) and inversely with lung clearance index (p < 0.001). Higher pO₂ was associated with pancreatic sufficiency (p = 0.069) and dual CFTR modulator use (p < 0.05), with no differences by sex or chronic Pseudomonas aeruginosa infection. By age 5, 19.8% of young individuals with CF had pO₂ below 80 mmHg (5th percentile), of whom 73.7% had normal FEV1. A linear mixed model showed a steeper FEV1 decline in those with abnormal pO2 at baseline [estimate = 0.06 (Z-score*year−1), p < 0.001]. Early low pO2 was significantly associated with a higher probability of allergic bronchopulmonary aspergillosis (HR = 7.69, p = 0.016) and a trend towards early CF-related diabetes (HR = 2.78, p = 0.06) and early chronic Pseudomonas aeruginosa infection (HR = 2.38, p = 0.09).
Conclusions: Early abnormal pO2 at age 5 significantly correlated with accelerated FEV1 decline and a greater probability for CF-related complications. Implementing arterialized oxygen tension may offer valuable insights beyond spirometry alone in identifying high-risk patients.
1 Introduction
Cystic Fibrosis (CF) is an autosomal recessive disorder affecting approximately 1 in 3,500 Caucasian newborns (1). It results from a dysfunction in the CF transmembrane conductance regulator (CFTR) protein, causing thickened mucus and impaired airway clearance (2). Structural lung changes begin early, even in infancy, well before clinical symptoms appear or spirometry can detect them (3, 4). Early diagnosis and intervention are crucial to interrupt the cycle of inflammation, infection, and lung damage, thereby mitigating disease progression (3–5). Early identification of high-risk individuals allows early intervention to optimize long-term outcomes (5).
Spirometry is the preferred method for monitoring CF-related lung disease progression (6, 7). However, spirometry has certain limitations. Firstly, it can only be performed from a certain age, typically school age and older, as it relies on adequate cooperation (8). Secondly, spirometry lacks sensitivity in detecting very early or mild lung disease (3). Alternative techniques, such as computed tomography or magnetic resonance imaging of the chest or multiple breath washout, may identify structural or functional lung changes earlier in the disease course than spirometry. However, these alternatives have associated concerns, including radiation exposure, the need for anesthesia to prevent motion artifacts, and limitations in availability and costs (9). Thus, the optimal methods for detecting early lung parenchymal and functional changes remain unclear (3).
As the lungs' primary role is oxygen uptake, measuring arterial oxygen tension appears to be a viable option for further exploration (10). While pulse oximetry is non-invasive, its correlation with arterial oxygen tension is poor (11). In pediatric settings, arterial puncture is impractical, but arterialized capillary blood sampling is a reliable alternative. Arterialized capillary oxygen tension (pO2) offers a highly accurate estimation, as a suitable substitute for true arterial oxygen levels (11–13). Kraemer et al. investigated longitudinal pO2 progression in young people with CF (PwCF) and found that pO2 trends paralleled those of FEV1, with both showing a decline until early adulthood (14, 15). Since low FEV1 at a young age is associated with a more severe clinical course in PwCF, we hypothesized that pO2 might follow a similar pattern (5).
This retrospective observational study evaluated the role of pO2 as an additional marker for pediatric CF lung disease and its potential for risk stratification, focusing on whether low pO2 at a young age is associated with a worse clinical course.
2 Methods
2.1 Study design and population
This retrospective observational single-center cohort study included individuals newly diagnosed with CF after the nationwide newborn screening program's implementation in October 1997, until the analysis in December 2020. Eligible PwCF were under the care of the pediatric CF center at the Medical University of Vienna.
PwCF had to be over 5 years of age at enrolment, with their annual best pulmonary function tests (best FEV1), concurrently sampled arterialized capillary blood gases and multiple breath washout maneuvers (MBW) reviewed up to the age of 18. PwCF under 5 years of age who might be unable to perform spirometry accurately, and those after solid organ transplantation were excluded from analysis. Associations were analyzed for pO2, FEV1, and LCI2.5 when available (introduced in 2015). To evaluate the potential for risk stratification and prediction of future clinical outcomes, we analyzed the longitudinal progression of FEV1 and the time to onset of CF-related complications—previously shown to be associated with an unfavorable disease course—stratified by dichotomized pO₂ at age 5 (normal vs. abnormal). All necessary data were extracted from medical records. An explanatory flowchart is shown in Supplementary Figure 1 in the online supplement.
The study protocol was approved by the local ethics committee, adhering to the Declaration of Helsinki and Good Clinical Practice. Due to the retrospective study design, no informed consent was required before the medical records were reviewed.
2.2 Pulmonary function tests
Spirometry followed the European Respiratory Society guidelines using the MasterScreen Body (Vyaire Medical, Mettawa, Illinois, USA, Software: Sentry Suite) (16). Forced expiratory volume in the first second (FEV1) was reported in Z-scores using the Global Lung Initiative normative dataset (17).
The lung clearance index (LCI2.5) was derived from nitrogen multiple breath washout using the ExhalyzerD system (EcoMedics, Duernten, Switzerland, Software: Spiroware Version 3.2.2), adhering to the standardized protocol outlined in the consensus statement (18). LCI2.5 has been routinely obtained at our CF center since 2015. As introduced by Anagnostopoulou et al., 7.91 was considered the upper limit of normal (19, 20).
2.3 Capillary blood sampling and blood gas analysis
Blood gas sampling from the earlobe preceded pulmonary function tests at every outpatient visit starting from the third spirometry, typically around the age of 5 years. Local capillary blood circulation was increased using topical nonyl vanillamide and nicotinic acid-β-butoxyethyl ester containing ointment (Finalgon®, Sanofi, Paris, France). After 15 min incubation time, capillary blood was collected in heparinized plastic capillaries and immediately analyzed for oxygen tension (pO2) in mmHg at room temperature using an ABL800 FLEX blood gas analyzer (Radiometer Medical ApS, Copenhagen, Denmark). The analyzer was located in the same room, ensuring a short transfer time. pO2 values were converted into Z-scores based on pediatric normative data from Gaultier et al. (13). The lower limit of normal (LLN) for pO2 was defined as the 5th percentile of a healthy pediatric population, corresponding to a Z-score of −1.64 (5–8 years: 80 mmHg; ≥8 years: 84.3 mmHg).
The sampling technique was highly accepted, with a 100% acceptance rate among all 5-year-olds included in the study.
2.4 Definition of CF-related complications
The modified Leeds criteria were applied to define chronic Pseudomonas aeruginosa (PsA) airway infection using surveillance microbiology airway samples (21). CF-related diabetes (CFRD) was defined by a pathological oral glucose tolerance test (OGTT), routinely performed annually from the age of 10 during routine follow-ups when patients were in a stable health condition (6). Allergic bronchopulmonary Aspergillosis (ABPA) episodes were diagnosed based on the Cystic Fibrosis Foundation Consensus, as described elsewhere (22).
2.5 Statistics
Data analysis was conducted using R software, version 4.3.2 (The R Foundation for Statistical Computing, Vienna, Austria). Descriptive statistics included absolute numbers and percentages for categorical data, while quantitative parameters (ages 5–18) were presented as mean ± SD. Linear mixed models with a random patient effect were used to assess differences and the progression of serial measured variables. To evaluate CFTR modulator effects, a binary factor was included to compare measurements before vs. after modulator initiation.
To assess the impact of pO2 at age 5 on the future clinical course, the dataset was split into two groups: pO2 Z-score ≤1.64 vs. pO2 Z-score ≥−1.64. Baseline data were compared using t-tests for continuous variables and Fisher's exact tests for categorical data. The future FEV1 time course was analyzed using a two-step approach: (1) simple linear mixed-effect models with random patient effects for our baseline grouping variable and potential confounders [age, year of birth, sex, pancreatic insufficiency, genotype, baseline FEV1, body mass index (BMI) at age 5, first and chronic PsA infection before age 5, chronic Staphylococcus aureus infection before age 5 and CFTR modulator therapy], followed by (2) a multivariable linear mixed-effect model, including all significant factors from the simple models (more detailed in the online supplements' statistics section) (5). To investigate the impact of early hypoxemic states on the probability for CF-related diseases (ABPA, CFRD, chronic PsA infection), Kaplan–Meier and univariable Cox regression models were employed. The level of significance was set to <0.05.
3 Results
3.1 Study population
Among 150 eligible young PwCF, 47 were excluded. Of the remaining 103 (47 females and 56 males) aged 5–18, 923 arterialized blood gases were reviewed. Patient numbers and examinations by age group are detailed in Supplementary Table 1 in the online supplement. The mean observational period was 8.1 ± 4.0 years. Of the 103 patients, 87 (84.5%) were pancreatic-insufficient, 29 (28.1%) received CFTR modulator therapy at some point during the observational period; none received modulators before the age of 6 years. 10/103 (9.7%) patients were considered chronically infected with PsA and one patient was diagnosed with ABPA as early as age 5. A detailed overview of patients' characteristics at baseline is provided in Table 1 (23).
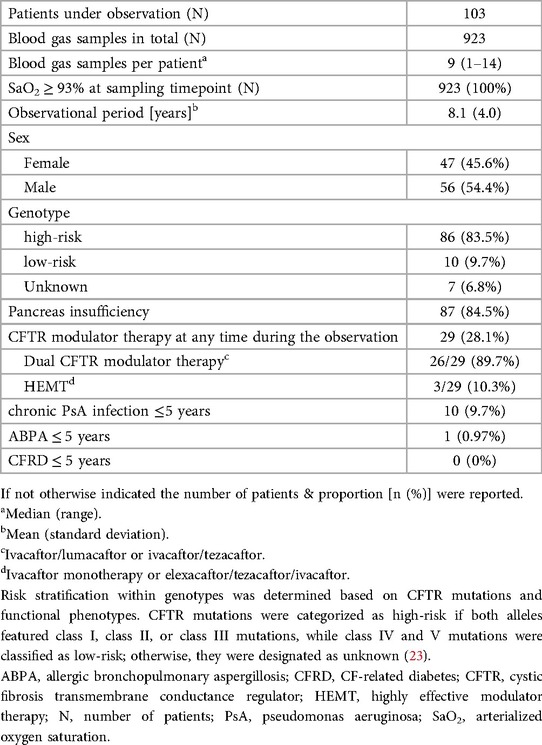
Table 1. Patients’ characteristics.
3.2 Pulmonary function and gas exchange during childhood and adolescence
Within the study cohort, pulmonary function (FEV1) showed a significant decline from early childhood to adulthood (p < 0.001, Figure 1A). While the mean arterialized pO2 remained stable above the lower limit of normal (Z-score of −1.64) across all age groups, lower pO2 values were observed in PwCF with lower FEV1 values (p < 0.001, Figure 1B). For further analysis, PwCF with pO2 below a Z-score of −1.64 were classified as “hypoxemic”, while those with values equal to or above −1.64 were classified as “normoxemic”.
![Two line graphs (A and B) depict FEV1 and pO₂ Z-scores against age for patients. Graph A shows FEV1 Z-score trends, while Graph B illustrates pO₂ Z-scores. Each graph contains three lines representing patient groups: pO₂ [Z-score] ≥ -1.64 (light gray), all patients (medium gray), and pO₂ [Z-score] < -1.64 (black). Error bars indicate variability.](https://www.frontiersin.org/files/Articles/1653323/fped-13-1653323-HTML/image_m/fped-13-1653323-g001.jpg)
Figure 1. Progression of arterialized oxygen tension (pO2) and FEV1 during childhood and adolescence. Mean values (points) and standard deviations (vertical lines) for FEV1 (A), and pO2 (B) are shown across age groups for all patients (grey) and stratified by whether the corresponding pO2 (A) or FEV1 (B) falls above (light grey) or below (black) −1.64 Z-scores. Mean values were calculated based on the actual number of patients in each age group. The number of patients is detailed in Supplementary Table 1 in the online supplement.
Pancreatic sufficient PwCF tended to have higher average pO₂ values across all age groups (p = 0.069), while levels did not differ by sex or chronic PsA airway infection (defined for each age group in Supplementary Table 1, online supplement), as illustrated in Supplementary Figure 2 (online supplement).
3.3 pO2 in relation to LCI2.5 and FEV1
A total of 188 nitrogen multiple breath washout maneuvers from 85 young PwCF, spanning ages 6–18 years and obtained concurrently with blood gas sampling and spirometry, were analyzed (more detailed in Supplementary Table 1 in the online supplement). Lower pO2 values were associated with higher LCI2.5 across all ages (Figure 2, linear mixed model: estimate −1.09, 95%−CI: −1.38 to −0.81, p < 0.001). LCI2.5 was pathologically high in 133 out of 188 measurements, with 45 (33.8%) showing hypoxemia in arterialized blood gases, while FEV1 Z-score being below −1.64 in only 21 (15.8%) cases (Figure 2). Forty-four out of 45 PwCF (98%) with hypoxemia had an LCI2.5 exceeding 7.91, while only one individual hat a normal LCI2.5 (2%).
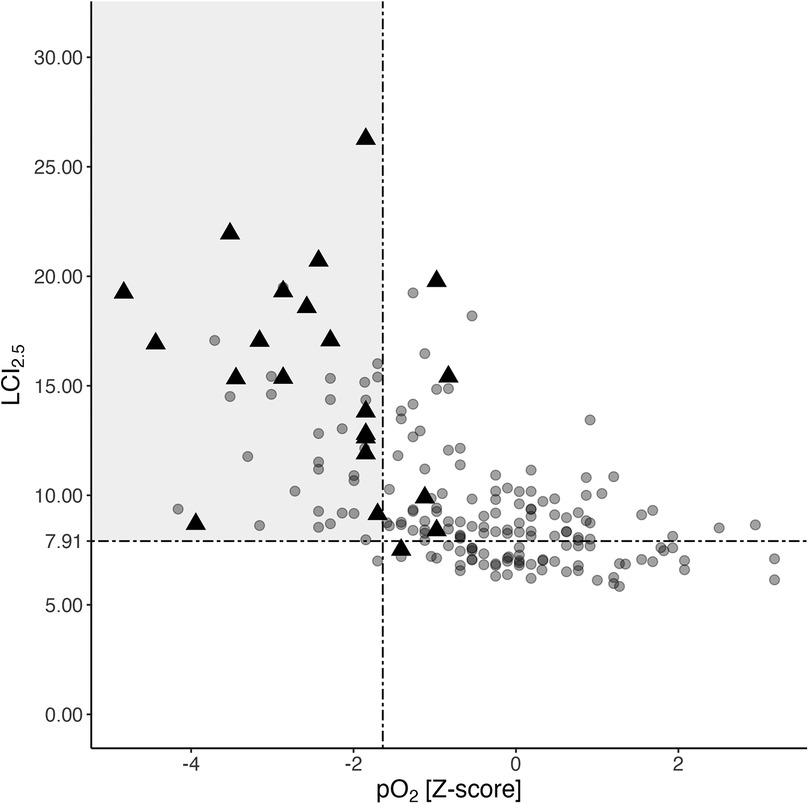
Figure 2. Association of corresponding LCI2.5, pO2, and FEV1 values. Distribution of LCI2.5 and pO2 values from a total of 188 N2BMW maneuvers and corresponding blood gas analyses from 85 PwCF aged 6 to 18 years (see Supplementary Table 1 in the online supplement for age distribution). Black triangles represent individuals with poor pulmonary function (FEV1 Z-scores <−1.64), whereas the grey circles indicate individuals with normal FEV1 at the time of assessment. The grey rectangle indicates a pathologically high LCI2.5 (>7.91) combined with a concurrently low pO2 Z-score (<−1.64). FEV1, forced expiratory volume in the first second; LCI2.5, lung clearance index; N2MBW, nitrogen multiple breath washout; pO2, arterialized oxygen tension; PwCF, people with CF.
3.4 pO2 at the age of 5 years
Seven out of the total cohort of 103 PwCF lacked sufficient data before the age of 5 years, either due to late diagnosis or because they transferred to our center at later ages. Among the remaining 96 patients at 5 years of age, the mean FEV1 Z-score was −0.09 ± 1.056 (SD), corresponding to 98.8 ± 13.5% predicted. Supplementary Table 2 in the online data supplement provides a more detailed overview of patients' characteristics at the age of 5 years.
Five of 96 (5.2%) had FEV1 values below the lower limit of normal, while 19 of 96 (19.8%) were already hypoxemic at this early age (as depicted in Figure 3). Compared to normoxemic PwCF, the hypoxemic group tended to be female, with 12 out of 19 (63.2%, p = 0.091), and had significantly lower FEV1 values (p = 0.021, see Supplementary Table 2 in the online data supplement).
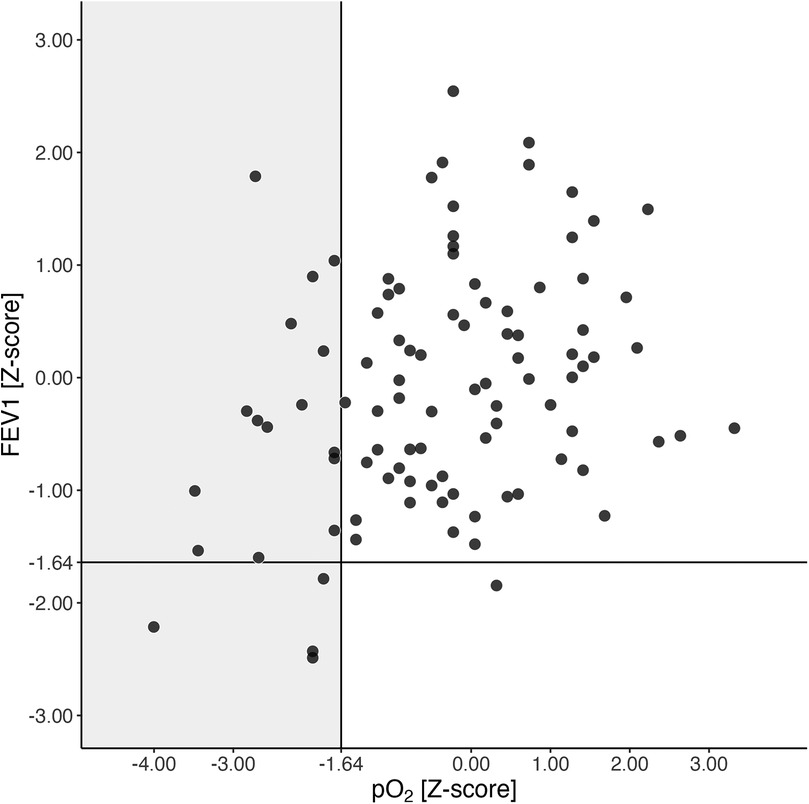
Figure 3. FEV1 and pO2 at the age of 5 years. pO2 and FEV1 Z-scores at the age of 5 years are shown. The black lines represent the lower limits of normal (LLN) for both pO2 and FEV1 Z-scores. The grey rectangle indicates hypoxemic 5-year-olds (pO2 Z-score <−1.64, equivalent to 80 mmHg). Abbreviations used: FEV1 (forced expiratory volume in the first second), N (number of patients), pO2 (arterialized oxygen tension), PwCF (people with CF).
3.5 Impact of CFTR modulator therapy on FEV1 and pO2 trend
The proportion of PwCF receiving CFTR modulator therapy increased with age, ranging from 7% to 25% among patients aged 10–18 years (Supplementary Table 1, online supplement). Figure 4 shows FEV1 and pO₂ trends before and after CFTR modulator initiation. Among individuals on dual CFTR modulators (ivacaftor/lumacaftor or ivacaftor/tezacaftor), no significant change in FEV1 was observed (estimate: −0.56; 95% CI: −4.23 to 3.11; n.s.), whereas pO₂ showed a slight improvement (estimate: 0.46; 95% CI: 0.01–0.91; p = 0.044). For the three PwCF on Highly Effective Modulator Therapy (HEMT), statistical analysis was not performed due to the small sample size; these patients were followed only until modulator initiation in the prediction model.
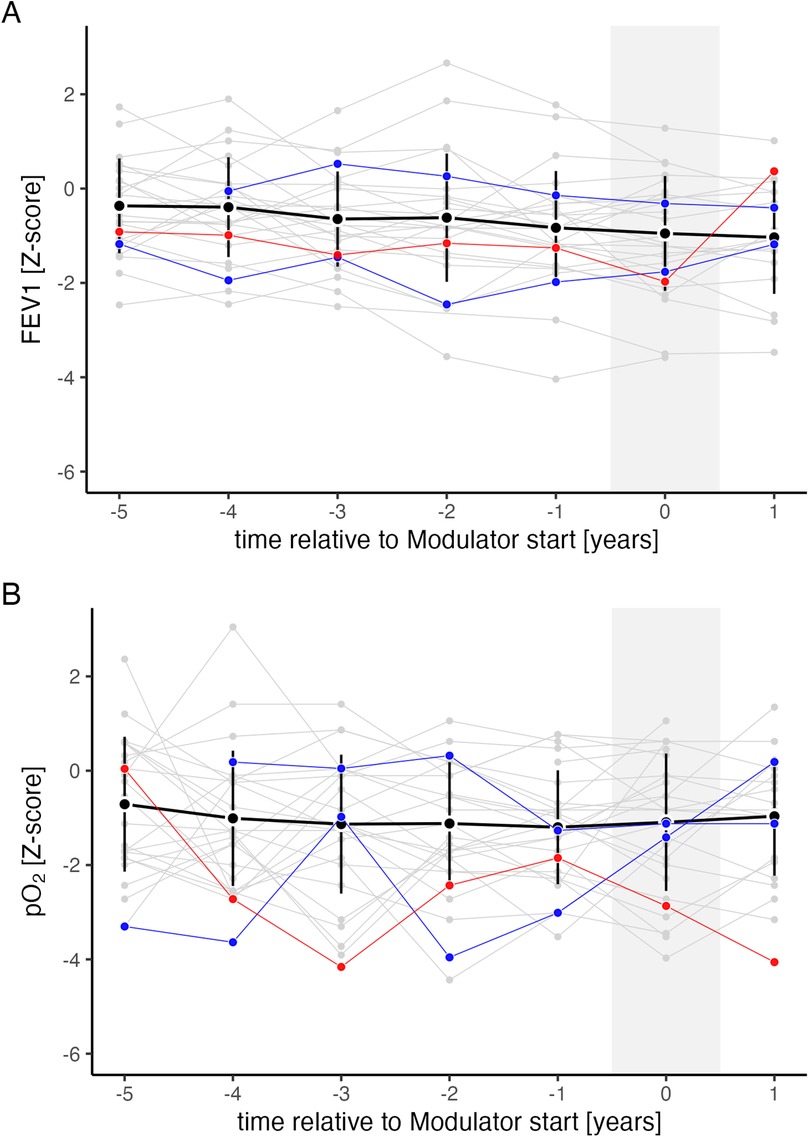
Figure 4. FEV1 and pO2 trend relative to CFTR modulator introduction. Progression of FEV1 (A) and pO₂ (B) relative to CFTR modulator introduction, expressed in years before (−) and after () initiation. Shown are mean values (black points) and standard deviations (black vertical lines) for all individuals on ivacaftor/lumacaftor or ivacaftor/tezacaftor. Individual trajectories are shown separately for each person with CF (PwCF) on ivacaftor/lumacaftor or ivacaftor/tezacaftor (grey, n = 26), ivacaftor monotherapy (blue, n = 2), and the triple combination of elexacaftor/tezacaftor/ivacaftor (red, n = 1).
3.6 Association between early low pO2 and clinical course
Being hypoxemic at the age of 5 years was significantly associated with future FEV1 loss within our study. Compared to individuals with normal pO2, Z-scores below −1.64 (equivalent to 80 mmHg) at this early stage were associated with a subsequent steeper decline in FEV1 (p < 0.001, estimate = −0.06, 95%−CI: −0.09 to −0.02, Figure 5). This association remained significant even in the multivariable model, adjusted for other significant influencing factors on the FEV1 course from the simple models, such as genotype, pancreatic insufficiency, chronic PsA infection before age 5, FEV1 at baseline, year of birth, and CFTR modulator use (calculations and estimates are provided in the statistics part of the online supplement and Supplementary Table 4).
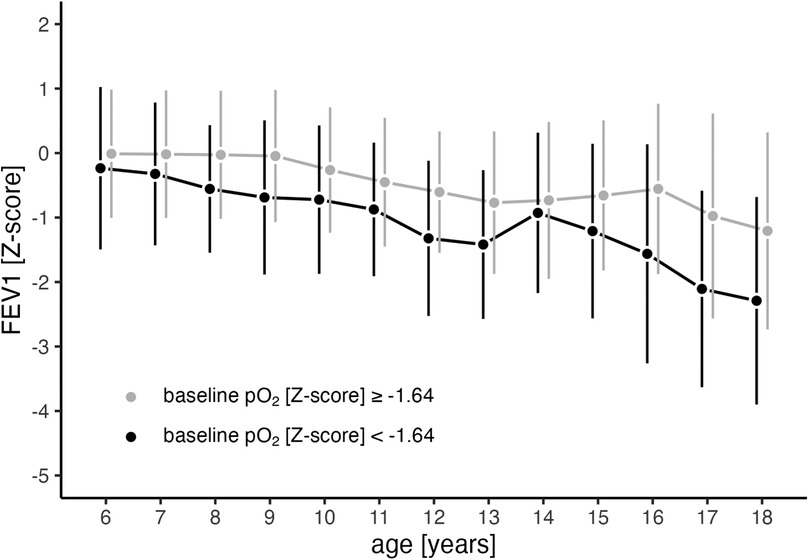
Figure 5. Trajectory of FEV1 in relation to pO2 at 5 years (baseline). Mean values (points) and standard deviations (vertical lines) for FEV1 progression from ages 6 to 18 years are shown separately for individuals with abnormal pO2 Z-score (<−1.64, black line) and those with normal pO2 Z-score (≥−1.64, grey line) at baseline (age 5). At age 5, a pO2 Z-score of −1.64 corresponds to 80 mmHg. FEV1 (forced expiratory volume in the first second), pO2 (arterialized oxygen tension).
The number of observed CF-related complications during the observational period is provided in Supplementary Table 3 in the online supplement. Five-year-olds with hypoxemia in arterialized blood gases were significantly more likely to experience an ABPA episode compared to those with normoxemia [HR: 7.69, 95% CI: 2.22–25, p = 0.001; see Supplementary Figure 3A in the online supplement]. A similar trend was observed for the onset of CFRD and the acquisition of chronic PsA infections in hypoxemic states at age 5 [CFRD: HR: 2.78, 95%−CI: 0.96–7.69, p = 0.06; PsA: HR: 2.38, 95%−CI: 0.87–6.67, p = 0.09; see Supplementary Figures 3B, C in the online supplement].
4 Discussion
In this study, we assessed pO2 in arterialized blood as an easily accessible, and widely applicable method for monitoring lung disease and identifying patients at risk of a more severe disease course. In the analyzed CF cohort, pO2 remained stable from 5 to 18 years, which contrasts with the longitudinal decline in FEV1 observed in young PwCF. Dual CFTR modulators positively influenced the pO₂ trajectory, whereas its levels were independent of pancreatic function, sex, and chronic Pseudomonas aeruginosa infection. While most children (94.8%) had normal lung function at age 5, 19.8% already showed abnormally low pO2. These children experienced a greater decline in FEV1 during childhood and adolescence and faced a higher risk of CF-related complications such as ABPA, CFRD, and chronic PsA infections.
The longitudinal pO2 progression in our study diverges from previously published data on pO2 (14, 15). In the study of pediatric PwCF by Kraemer and colleagues, pO2 values were notably lower and exhibited a linear decline during childhood, starting at 80.7 ± 1.9 mmHg at 5 years of age and decreasing to a mean of 69.9 ± 1.6 mmHg at age 18 years (14). This discrepancy may be attributed mainly to the inclusion of pre-symptomatic children through newborn screening in the present study, the generally improved overall health of individuals with CF in recent years, and the use of CFTR modulators. The latter may also partially explain the slightly increasing pO₂ trend observed in our older adolescents. To our knowledge, this is the first study to evaluate the impact of any generation of CFTR modulator therapy on pO₂.
In pediatrics, hypoxemia is rarely defined by arterial or arterialized pO2; instead, it is more commonly characterized as transcutaneous oxygen saturation falling below 90% or 93%, depending on the child's age (24). Notably, within our cohort, even PwCF having hypoxemic pO2 values exhibited arterialized oxygen saturation above 93%. We did not measure transcutaneously, but there is a strong correlation between arterial oxygen saturation and transcutaneous oxygen saturation (25). Abnormal pO₂ despite normal oxygen saturation aligns with the physiology of the oxygen-hemoglobin dissociation curve, which flattens at higher pO₂ levels. Even when pO₂ falls below the lower limit of normal (5th percentile), oxygen saturation may remain within the normal range (26, 27). We therefore propose arterialized pO₂ as a more accurate surrogate marker of oxygenation than oxygen saturation.
Predicting disease progression is crucial for identifying PwCF who may benefit from more aggressive surveillance strategies or therapeutic interventions (5, 28). FEV1 remains the most significant predictor of mortality and early CF lung disease progression (5). However, especially in the era of highly potent CFTR modulators, the proportion of patients with pulmonary function within the normal range is increasing (29). FEV1 alone may not provide a comprehensive assessment of lung damage, particularly in cases of milder CF lung disease or during the subclinical period from birth to school age, when disease progression can occur without noticeable symptoms and spirometry is unreliable (3, 4, 30, 31). While we cannot draw conclusions for the subclinical period of infancy as children under 5 years were excluded from this study, pO₂ may offer significant advantages as an adjunct to traditional spirometry, particularly in mild CF lung disease.
Blood oxygen tension primarily depends on oxygen uptake and the lung's ability to transfer oxygen into the bloodstream independent of factors such as sex, age, anthropometrics and ethnicity (10, 32). Ventilatory inhomogeneity and the simultaneous increase in dead space ventilation may play a significant role in gas exchange abnormalities among pediatric PwCF (14, 31). Over the last decade, multiple breath washout, serving as a marker for ventilatory inhomogeneity, has emerged as a promising method for monitoring early or mild CF lung disease (33). Not only does it serve as a more sensitive marker for early structural lung damage compared to FEV1, but abnormal LCI2.5 in early childhood have also been demonstrated to predict poor pulmonary function during school years (3, 34, 35). In our study, low pO2 was associated with high LCI2.5, suggesting a potential indication of poor oxygenation due to ventilatory inhomogeneity. Moreover, low pO2 showed greater sensitivity in detecting LCI2.5 values above 7.9 compared to low FEV1, with sensitivity increasing from 0.18 for FEV1 to 0.45 for pO2. This suggests that blood gas analysis could be a more sensitive method for detecting mild CF lung disease than spirometry.
Limitations of our study are the limited generalizability of our results due to the retrospective design of the study. Additionally, outcomes may have been influenced by the evolving landscape of new treatment approaches, particularly the widespread availability of Highly Effective Modulator Therapy over the last years, especially among young children. Our findings must be confirmed in prospective studies involving PwCF on triple CFTR modulator therapy. This is particularly relevant as most PwCF on triple therapy have normal FEV1 (29).
While we do not advocate the use of pO2 as a direct substitute for LCI2.5 or spirometry, it is worth noting that multiple breath washouts can be laborious and resource-intensive, particularly in very young children (35). Our data suggest that pO2 is a more sensitive marker than FEV1 for mild CF lung disease and, hence, could serve as an easily accessible, and widely applicable addition to established monitoring tools to identify young PwCF at increased risk of a more severe disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Medical University of Vienna. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants' legal guardians/next of kin, as the retrospective study design did not require informed consent prior to the review of medical records.
Author contributions
RG: Methodology, Software, Conceptualization, Data curation, Investigation, Writing – original draft, Visualization. SG: Supervision, Writing – review & editing. KS: Writing – review & editing, Validation. BP: Writing – review & editing. BM: Writing – review & editing. AG: Software, Formal analysis, Writing – review & editing. ZS: Conceptualization, Writing – review & editing, Methodology. SR: Conceptualization, Methodology, Writing – review & editing. ED: Writing – review & editing, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We extend our gratitude to the entire nursing staff at the pediatric CF clinic at the Medical University of Vienna for their invaluable support. Special thanks go to the children and adolescents with CF, whose enthusiastic participation in the demanding CF surveillance program has significantly contributed to the progress of our research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. During the preparation of this work the author(s) used ChatGPT and Grammarly in order to check grammar, spelling, and rephrase sections. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1653323/full#supplementary-material
References
1. Frischer T, Eber E, Ellemunter H, Zacharasiewicz A, Kaluza I, Riedler J, et al. Cystic fibrosis in Austria. Wien Klin Wochenschr. (2017) 129(15–16):527–32. doi: 10.1007/s00508-017-1179-x
2. Brown SD, White R, Tobin P. Keep them breathing: cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA. (2017) 30(5):23–7. doi: 10.1097/01.JAA.0000515540.36581.92
3. Grasemann H, Ratjen F. Early lung disease in cystic fibrosis. Lancet Respir Med. (2013) 1(2):148–57. doi: 10.1016/S2213-2600(13)70026-2
4. Ramsey KA, Ranganathan S, Park J, Skoric B, Adams AM, Simpson SJ, et al. Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am J Respir Crit Care Med. (2014) 190(10):1111–6. doi: 10.1164/rccm.201407-1277OC
5. Breuer O, Caudri D, Stick S, Turkovic L. Predicting disease progression in cystic fibrosis. Expert Rev Respir Med. (2018) 12(11):905–17. doi: 10.1080/17476348.2018.1519400
6. Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. (2018) 17(2):153–78. doi: 10.1016/j.jcf.2018.02.006
7. Burgel PR, Southern KW, Addy C, Battezzati A, Berry C, Bouchara JP, et al. Standards for the care of people with cystic fibrosis (CF); recognising and addressing CF health issues. J Cyst Fibros. (2024) 23(2):187–202. doi: 10.1016/j.jcf.2024.01.005
8. Ranganathan S, Linnane B, Nolan G, Gangell C, Hall G. Early detection of lung disease in children with cystic fibrosis using lung function. Paediatr Respir Rev. (2008) 9(3):160–7. doi: 10.1016/j.prrv.2008.05.002
9. Lahiri T, Hempstead SE, Brady C, Cannon CL, Clark K, Condren ME, et al. Clinical practice guidelines from the cystic fibrosis foundation for preschoolers with cystic fibrosis. Pediatrics. (2016) 137(4):e20151784. doi: 10.1542/peds.2015-1784
10. Soni R, Dobbin CJ, Milross MA, Young IH, Bye PPT. Gas exchange in stable patients with moderate-to-severe lung disease from cystic fibrosis. J Cyst Fibros. (2008) 7(4):285–91. doi: 10.1016/j.jcf.2007.11.003
11. Zavorsky GS, Cao J, Mayo NE, Gabbay R, Murias JM. Arterial versus capillary blood gases: a meta-analysis. Respir Physiol Neurobiol. (2007) 155(3):268–79. doi: 10.1016/j.resp.2006.07.002
12. Pitkin AD, Roberts CM, Wedzicha JA. Arterialised earlobe blood gas analysis: an underused technique. Thorax. (1994) 49(4):364–6. doi: 10.1136/thx.49.4.364
13. Gaultier C, Boule M, Allaire Y, Clement A, Buvry A, Girard F. Determination of capillary oxygen tension in infants and children: assessment of methodology and normal values during growth. Bull Eur Physiopathol Respir. (1979) 14(3):287–97.476326
14. Kraemer R, Latzin P, Pramana I, Ballinari P, Gallati S, Frey U. Long-term gas exchange characteristics as markers of deterioration in patients with cystic fibrosis. Respir Res. (2009) 10(1):106. doi: 10.1186/1465-9921-10-106
15. Fuger M, Aupiais C, Thouvenin G, Taytard J, Tamalet A, Escudier E, et al. Gas exchanges in children with cystic fibrosis or primary ciliary dyskinesia: a retrospective study. Respir Physiol Neurobiol. (2018) 251:1–7. doi: 10.1016/j.resp.2018.01.010
16. Graham BL, Steenbruggen I, Miller MR, Barjaktarevic IZ, Cooper BG, Hall GL, et al. Standardization of spirometry 2019 update. An official American thoracic society and European respiratory society technical statement. Am J Respir Crit Care Med. (2019) 200(8):e70–88. doi: 10.1164/rccm.201908-1590ST
17. Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. (2012) 40(6):1324–43. doi: 10.1183/09031936.00080312
18. Robinson PD, Latzin P, Verbanck S, Hall GL, Horsley A, Gappa M, et al. Consensus statement for inert gas washout measurement using multiple- and single- breath tests. Eur Respir J. (2013) 41(3):507–22. doi: 10.1183/09031936.00069712
19. Anagnostopoulou P, Latzin P, Jensen R, Stahl M, Harper A, Yammine S, et al. Normative data for multiple breath washout outcomes in school-aged Caucasian children. Eur Respir J. (2020) 55(4):1901302. doi: 10.1183/13993003.01302-2019
20. Kentgens AC, Latzin P, Anagnostopoulou P, Jensen R, Stahl M, Harper A, et al. Normative multiple-breath washout data in school-aged children corrected for sensor error. Eur Respir J. (2022) 60(2):2102398. doi: 10.1183/13993003.02398-2021
21. Proesmans M, Balinska-Miskiewicz W, Dupont L, Bossuyt X, Verhaegen J, Hoiby N, et al. Evaluating the “Leeds criteria” for pseudomonas aeruginosa infection in a cystic fibrosis centre. Eur Respir J. (2006) 27(5):937–43. doi: 10.1183/09031936.06.00100805
22. Stevens DA, Moss RB, Kurup VP, Knutsen AP, Greenberger P, Judson MA, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis–state of the art. Cystic fibrosis foundation consensus conference. Clin Infect Dis. (2003) 37(Suppl 3):S225–64. doi: 10.1086/376525
23. McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest. (2006) 130(5):1441–7. doi: 10.1378/chest.130.5.1441
24. Krivchenia K, Hawkins SM, Iyer NP, Hayes D Jr, Deterding RR, Ruminjo J, et al. 2019 clinical practice guideline summary for clinicians: home oxygen therapy for children. Ann Am Thorac Soc. (2019) 16(7):781–5. doi: 10.1513/AnnalsATS.201902-136CME
25. Tittle M, Flynn MB. Correlation of pulse oximetry and co-oximetry. Dimens Crit Care Nurs. (1997) 16(2):88–95. doi: 10.1097/00003465-199703000-00004
26. Collins JA, Rudenski A, Gibson J, Howard L, O’Driscoll R. Relating oxygen partial pressure, saturation and content: the haemoglobin-oxygen dissociation curve. Breathe (Sheff). (2015) 11(3):194–201. doi: 10.1183/20734735.001415
27. Nitzan M, Romem A, Koppel R. Pulse oximetry: fundamentals and technology update. Med Devices (Auckl). (2014) 7:231–9.25031547
28. VanDevanter DR, Wagener JS, Pasta DJ, Elkin E, Jacobs JR, Morgan WJ, et al. Pulmonary outcome prediction (POP) tools for cystic fibrosis patients. Pediatr Pulmonol. (2010) 45(12):1156–66. doi: 10.1002/ppul.21311
29. Orenti A, Zolin A, Jung A, van Rens J, Prasad V, Fox A, et al. ECFSPR annual report 2021. (2023). Available online at: https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf (Accessed August 26, 2023).
30. Vilozni D, Bentur L, Efrati O, Minuskin T, Barak A, Szeinberg A, et al. Spirometry in early childhood in cystic fibrosis patients. Chest. (2007) 131(2):356–61. doi: 10.1378/chest.06-1351
31. Kraemer R, Blum A, Schibler A, Ammann RA, Gallati S. Ventilation inhomogeneities in relation to standard lung function in patients with cystic fibrosis. Am J Respir Crit Care Med. (2005) 171(4):371–8. doi: 10.1164/rccm.200407-948OC
32. Urquhart DS, Montgomery H, Jaffe A. Assessment of hypoxia in children with cystic fibrosis. Arch Dis Child. (2005) 90(11):1138–43. doi: 10.1136/adc.2005.071795
33. Aurora P, Bush A, Gustafsson P, Oliver C, Wallis C, Price J, et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am J Respir Crit Care Med. (2005) 171(3):249–56. doi: 10.1164/rccm.200407-895OC
34. Aurora P, Stanojevic S, Wade A, Oliver C, Kozlowska W, Lum S, et al. Lung clearance index at 4 years predicts subsequent lung function in children with cystic fibrosis. Am J Respir Crit Care Med. (2011) 183(6):752–8. doi: 10.1164/rccm.200911-1646OC
Keywords: blood gas monitoring, respiratory function test, disease progression, secondary prevention, risk stratification
Citation: Gaupmann R, Gruber S, Schmidthaler K, Pauger B, Mersi B, Graf A, Szépfalusi Z, Renner S and Dehlink E (2025) Arterialized oxygen tension and unfavorable clinical outcomes in pediatric cystic fibrosis. Front. Pediatr. 13:1653323. doi: 10.3389/fped.2025.1653323
Received: 24 June 2025; Accepted: 19 September 2025;
Published: 8 October 2025.
Edited by:
Burkhard Tümmler, Hannover Medical School, GermanyReviewed by:
Anna Dittrich, Hannover Medical School, GermanyOlaf Eickmeier, University Hospital Frankfurt, Germany
Copyright: © 2025 Gaupmann, Gruber, Schmidthaler, Pauger, Mersi, Graf, Szépfalusi, Renner and Dehlink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: René Gaupmann, cmVuZS5nYXVwbWFubkBtZWR1bml3aWVuLmFjLmF0