 Bowen Zhao
Bowen Zhao Fengjuan Ding
Fengjuan Ding Fei Hou
Fei Hou Hua Jin
Hua Jin- Department of Prenatal Diagnosis, Jinan Maternity and Child Care Hospital Affiliated Shandong First Medical University (Jinan Maternity and Child Care Hospital), Jinan, Shandong, China
Objective: Bainbridge-Ropers syndrome (BRS) is a neurodevelopmental disorder predominantly caused by pathogenic variants in the ASXL3 gene, which have been conventionally considered to occur de novo. This study aimed to investigate the potential role of parental mosaicism in BRS inheritance and its clinical implications for genetic counseling.
Methods: Trio-based whole-exome sequencing (WES) was performed on the proband and both parents to identify candidate variants, which were subsequently validated by Sanger sequencing. ASXL3-targeted ultra-deep sequencing of paternal semen DNA was then carried out to detect low-level mosaicism. Prenatal diagnosis via amniocentesis was used to evaluate transmission of the familial variant.
Results: We definitively diagnosed this family by WES and found the lowest level of paternal mosaicism reported to date, with a peripheral blood variant allele frequency (VAF) of 8.17% and a semen VAF of 15.03%. Prenatal diagnosis at 18 weeks of gestation confirmed that the variant was not detected in this pregnancy.
Conclusion: This study establishes parental chimerism as an important genetic mechanism for ASXL3-associated disorders and emphasizes the need for ultrasensitive testing in genetic counseling. The findings redefine genetic risk stratification for BRS and provide a basis for accurate family planning based on high-depth sequencing.
1 Introduction
Bainbridge-Ropers syndrome (BRPS)[OMIM #615,485], a rare autosomal dominant neurodevelopmental disorder first identified in 2013 (1), has become a focus of growing research interest.BRPS is caused by heterozygous loss-of-function mutations in ASXL3 (18q12.1).BRPS is characterized by multisystemic involvement, including (1) neurodevelopmental manifestations (global developmental delay, moderate to severe intellectual disability, autistic traits, language impairment/absent speech, etc.); (2) neuromuscular/nutritional features (hypotonia, feeding difficulties with failure to thrive, etc.); and (3) dysmorphic features (frontal bossing, arched eyebrows, hypertelorism, down slanting palpebral fissures) (2, 3). These characteristic phenotypes facilitate preliminary clinical identification. While the exact prevalence of BRPS is unknown, the Deciphering Developmental Disorders study (n = 9,625 ID trios) identified de novo ASXL3 variants in 50 probands (1:193), placing ASXL3 among the top 10 most frequently mutated genes in neurodevelopmental disorders (4).
Current literature on this disorder primarily consists of case reports, with most cases caused by de novo mutations. Mosaicism in ASXL3 has emerged as a significant phenomenon in BRPS research, profoundly influencing disease pathogenesis, progression, and inheritance patterns (5). This study presents a detailed characterization of paternally inherited ASXL3 mosaicism at low variant allele fractions (VAFs) in phenotypically normal fathers leading to BRPS offspring. These findings have critical implications for accurate genetic counseling, informed reproductive decision-making, and prenatal diagnostic strategies.
2 Materials and methods
2.1 Patients and clinical information
The proband was an 8-year-old girl (II1) who presented with a global developmental delay characterized by profound speech and motor impairments, accompanied by distinctive craniofacial features (including frontal bossing, everted lower lip, and dental crowding), feeding difficulties, generalized hypotonia, and autism spectrum behaviors (notably poor eye contact and stereotypic hand movements) (Figure 1). She was born at term by vaginal delivery complicated by birth asphyxia (Apgar scores unavailable) and required 10 days of respiratory support in the neonatal intensive care unit. During infancy, she showed failure to thrive, necessitating nutritional supplementation, and her gross-motor milestones were substantially delayed, with head control achieved at 12 months, independent sitting at 18 months, and ambulation at 36 months. At age 3 years, a brain MRI revealed delayed white matter myelination and mild ventriculomegaly. At the current evaluation (8 years), she remained nonverbal, with persistent global delay (height 120 cm, 25th percentile; weight 17.5 kg, <5th percentile). The parents denied consanguinity and reported no significant family history of inherited disorders. The father (I2), a mosaic carrier of the pathogenic variant, underwent comprehensive clinical assessment. No neurodevelopmental abnormalities or facial dysmorphism were observed. The proband's 33-year-old mother(I1), who previously underwent pregnancy termination after detection of an ASXL3 heterozygous variant by external amniocentesis two years ago (II2), is now at 18 weeks’ gestation(II3). She seeks genetic counseling and prenatal diagnostic evaluation to ensure a healthy offspring. First-trimester nuchal translucency ultrasound screening demonstrated normal findings (1.1 mm, within the 50th percentile for gestational age), with no detectable structural anomalies on subsequent detailed fetal anatomical survey.

Figure 1. (A) Pedigree of the family with genotype results for ASXL3 (c.1534_1535del, p.Leu512Alafs*4) variant. (I-1) Mother of the proband; (I-2) Father with ASXL3 (c.1534_1535del, p.Leu512Alafs*4) variant in his sperm; (II-1) Proband; II-2 Pregnancy termination performed due to an ASXL3 (c.1534_1535del, p.Leu512Alafs*4) variant;(II- 3) fetus. (B) Clinical characteristics of the proband:global developmental delay,frontal bossing, everted lower lip, dental crowding, hypotonia, feeding difficulties, and autism spectrum disorder.
2.2 DNA extraction
Following signing the informed consent, peripheral blood samples (2 ml each) were collected from the proband and biological parents. For the pregnant mother, amniocentesis was performed under ultrasound guidance at 18 weeks of gestation, with 30 ml of amniotic fluid obtained. Genomic DNA was extracted from peripheral blood and amniotic fluid using the MagPure Blood DNA TL Kit (Magen Biotech, China) following the manufacturer's protocol.
2.3 Whole-exome sequencing (WES) and pathogenicity analysis
Library preparation and hybridization-based capture were performed, followed by high-throughput sequencing on the NovaSeq 6000 platform (Illumina, San Diego, CA), achieving an average sequencing depth of 200×. Variant annotation and filtering were conducted using public genomic databases, including OMIM (Online Mendelian Inheritance in Man, https://omim.org); GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/); GHR (Genetics Home Reference, https://ghr.nlm.nih.gov); ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); HGMD (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/); PubMed (https://pubmed.ncbi.nlm.nih.gov). The pathogenicity of identified variants was classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (6).
2.4 Sanger sequencing
Sanger sequencing verified that the candidate variant was verified for the proband, the amniotic fluid of the second fetus, and the parents. Primer 3.0 designed the primers with a forward primer sequence -GTGAAGCTCACTACTGGACCAA- and a reverse primer sequence -TGGGCTCTCAGAAGAAAAGGAC-. The PCR products were sequenced on the ABI 3730XL DNA analyzer (Applied Biosystems, Foster City, CA, USA).
2.5 Target capture-based deep sequencing
PCR amplification of the father's candidate gene was subsequently performed, and the primers were designed by Primer 3.0 with forward primer sequence -AGGATATCTTGATCCCTGAAGA- and reverse primer sequence-AGTCACAGACTTCTAACTGATCGA-. Subsequently, sequencing was performed on the AmCareSeq-2000 sequencer (AmCare Genomics Lab, Guangzhou, China) with a read length of PEx150. Bioinformatics analysis and annotation were the same as for clinical exome sequencing. The depth of coverage was around 10,000 × per coding base. Variants at each position were reported as percentage values to quantify the change in each base, where the sensitivity of detection of the mutational load of coding region variants was more significant than 0.5%. The sequencing data were visualized using the Integrative Genomics Viewer (IGV) version 2.8.13.
3 Results
3.1 Clinical genetic findings
Through integrated phenotype-genotype analysis of clinical exome sequencing data, we identified a heterozygous pathogenic variant in ASXL3 (c.1534_1535del, p.Leu512Alafs*4) in the proband. Sanger sequencing confirmed this variant in the proband but not in parental samples or amniotic fluid (Figure 2). Given the limited sensitivity of Sanger sequencing for low-level mosaicism, we performed targeted ultra-deep sequencing (10,000× coverage), which detected paternal gonosomal mosaicism at 8.17% VAF in peripheral blood, 15.03% VAF in paternal sperm (Table 1).
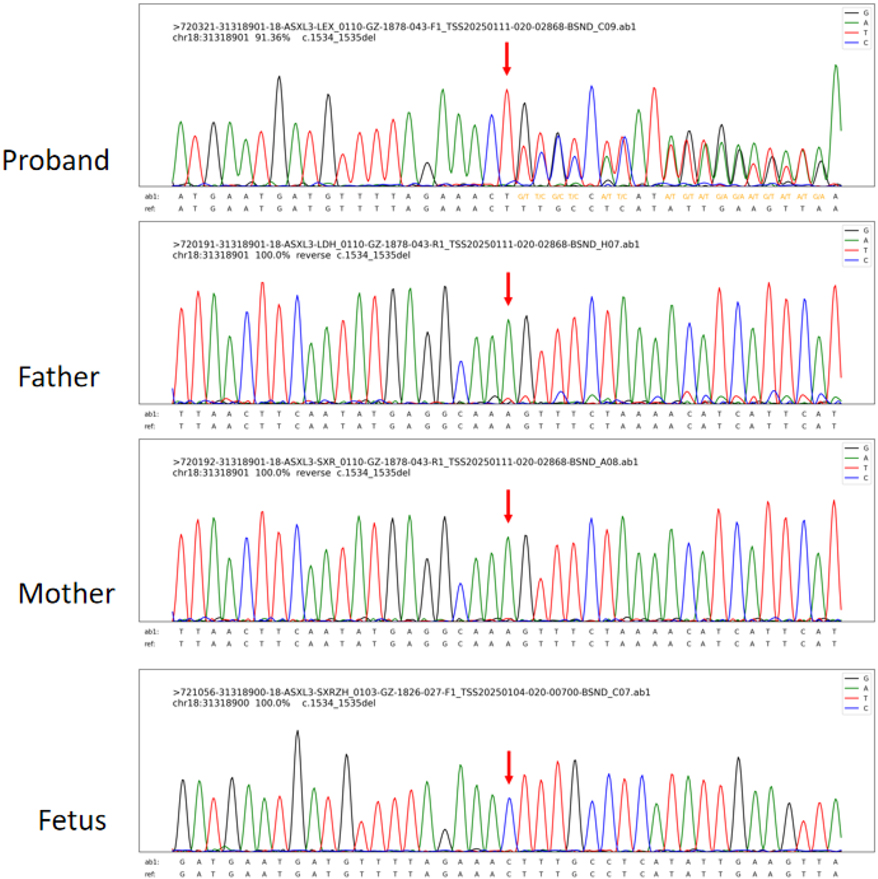
Figure 2. Sanger sequencing results of the proband, the fetus, and their parents [the proband was detected with hemizygous ASXL3 c.1534_1535del (p.Leu512Alafs*4) variants, while by this method, no variants were detected in the fetus and their parents].
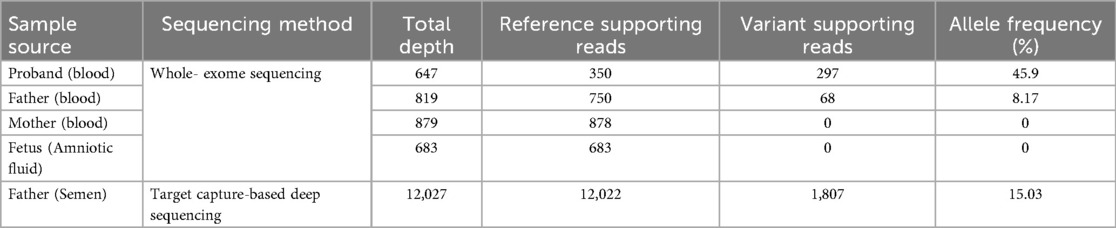
Table 1. Variant allele frequencies and read counts for parents, Fetus, and proband.
3.2 Candidate variant pathogenicity ratings
The c.1534_1535del (p.Leu512Alafs*4) variant in ASXL3 was classified as likely pathogenic based on ACMG/AMP guidelines with the following evidence: (1) PM2-P: the total population variant database frequency of this variant in the database is 0; (2) PVS1: This frameshift variant is predicted to introduce a premature termination codon, potentially triggering nonsense-mediated mRNA decay as it occurs within the terminal exon of all biologically relevant transcripts, thereby meeting PVS1 criteria; (3) PS2-M: confirmed de novo occurrence in published cases with matching phenotypes including intellectual disability, language delay, and hypotonia (7) and (4) PS2: paternal mosaicism confirmed by both Sanger sequencing and ultra-deep sequencing (8.17% VAF in blood;15.03% VAF in sperm). This variant meets the criteria for likely pathogenic classification through its combination of population frequency data, predicted molecular consequences, established disease association, and inheritance pattern.
4 Discussion
ASXL3 gene, the largest member of the ASXL gene family (12 exons encoding 2,248 amino acids), orchestrates critical neurodevelopmental processes through its conserved domains (ASXN, ASXH, ASXM1, ASXM2, PHD) (8). Studies demonstrate that ASXL3 governs the fate specification of the hindbrain, neural crest, and primary neurons during early neurodevelopment. Loss of ASXL3 function results in aberrant neural plate patterning, phenocopying the neural and craniofacial defects observed in BRS patients (9). Current evidence establishes that ASXL3 regulates the level of histone H2A lysine 119 monoubiquitination (H2AK119Ub1) through the Polycomb repressive deubiquitination (PR-DUB) complex, directly participating in the transcriptional regulation of brain development genes (such as HOX gene clusters) and neurodevelopmental pathways. Loss of ASXL3 function leads to abnormal chromatin states and dysregulation of key gene expression, ultimately causing structural and functional brain defects, which provides a molecular mechanism for neurodevelopmental disorders such as BRS (10).
The previously reported ASXL3 gene variants were predominantly de novo mutations, with only speculative mentions of mosaicism. This study presents the first comprehensive analysis of ASXL3 mosaicism (Table 2). Koboldt et al. (11) first proposed germline mosaicism as a potential mechanism for familial BRPS through their report of affected sisters sharing an identical de novo nonsense variant in ASXL3 (p.Tyr392*). Schirwani (12) and colleagues subsequently identified five affected individuals across three families, with two families exhibiting germline mosaicism patterns. Their cohort included an 11-year-old female presenting with hypotonia, feeding difficulties, intellectual disability, global developmental delay, and behavioral abnormalities. WES demonstrated 30%–35% VAF in matched blood and saliva samples. In their 2021 follow-up study, Schirwani's team characterized three inheritance patterns: the p.(Gln1512*) in P2 and the p.(Gln931fs) in P10 were inherited from the affected parents, and the p.(Leu1481fs) in P22 was inherited from the apparently asymptomatic but highly educated mother. No chimerism was detected in the blood samples tested. However, chimerism in other tissues could not be excluded (7). Additionally, a recent study reported a novel case involving two non-twin siblings carrying a likely pathogenic variant. This variant was inherited from an unaffected parent who exhibited mosaicism, which was detected in approximately 30% of the analyzed cells in peripheral blood DNA samples (5). Our study reports the lowest-level paternal mosaicism documented to date in BRPS (8.17% VAF in peripheral blood; 15.03% VAF in sperm). Prenatal diagnosis via amniocentesis at 18 gestational weeks yielded negative results.

Table 2. Summary of clinical cases of presumed ASXL3 gene mosaicism.
Although sperm mosaicism has minimal impact on sperm function, the offspring of affected individuals can inherit germline mutations, which frequently result in severe genetic disorders. Sperm mosaicism represents a major source of de novo mutations (DNMs) (13). The distribution of mosaic mutations is largely determined by the developmental timing of the mutational event: (1) Early mutations (pre-gastrulation, 2–4 cell stage): These mutations may propagate into multiple tissues, including germ cells, leading to gonadal-somatic mosaicism. (2) Mid-stage mutations (post-gastrulation, pre-organogenesis): Such mutations are typically restricted to tissues derived from a specific germ layer. (3) Late-stage mutations (post-organogenesis): These usually result in single-organ-specific mosaicism (or chimerism) (14–16). Sperm mosaicism can be divided into three types: Type I arises during sperm meiosis and is nonage dependent; Type II arises in spermatogonia and increases as men age;and Type III arises during paternal embryogenesis, spreads throughout the body, and contributes stably to sperm throughout life. Where Types I and II confer little risk of recurrence, Type III may confer identifiable risk to future offspring.These mutations are likely to be the single largest contributor to human genetic diversity (17). The underlying reason for the predominance of paternal mutation is attributable to the fact that oocytes undergo prolonged arrest following meiosis, while spermatogonial stem cells undergo continuous mitosis throughout their lifespan. This results in a significantly higher accumulation of replication errors and DNA damage in spermatogonial stem cells during division compared to oocytes. During the process of embryonic development, cells that carry different mutations exhibit varying capabilities with respect to survival and proliferation. Cell competition plays a pivotal role in embryonic development, influencing the distribution and proportion of mutated cells across different tissues. This, in turn, affects embryonic development and the onset and progression of diseases. The process of cell competition is generally accomplished via several different pathways, including apoptosis, proliferation regulation, mitochondrial function, and the dynamics of stem cells and progenitor cells (18).
Mosaicism complicates the inheritance pattern of genetic disorders in genetic counseling, posing challenges for accurate recurrence risk assessment. Conventional sequencing approaches often fail to detect mosaicvariants, necessitating the integration of high-depth sequencing and genetic pattern analysis for precise risk evaluation. The identification of mosaicism has broadened the genetic spectrum of BRPS, demonstrating that overlooking the possibility of gonadal mosaicism may lead to underestimation of disease recurrence risk in familial cases. The presence of mosaicism underscores the need for heightened clinical vigilance, particularly in multiplex families, and the implementation of more comprehensive and precise testing strategies to accurately characterize genetic variants.
In genetic counseling practice, potential mosaicism must be thoroughly considered. Parents should undergo detailed genetic testing and evaluation to ensure accurate risk stratification and reproductive guidance, enabling informed decision-making. While this family pursued naturally conceived pregnancy with prenatal diagnosis, preimplantation genetic testing (PGT-M) could have enabled earlier selection of unaffected embryos (19). Future studies should further elucidate the mechanisms underlying mosaicism and its phenotypic modulation, providing a foundation for personalized medicine.
In conclusion, this study significantly advances our understanding of ASXL3-related disorders by systematically characterizing paternal mosaicism in BRS, reporting the lowest-level germline mosaicism to date (8.17% VAF in peripheral blood; 15.03% VAF in sperm). This work redefines the genetic architecture of BRS, emphasizing that comprehensive parental testing and advanced sequencing technologies are indispensable for precise genetic counseling and family planning.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by The Ethics Committee of Jinan Maternal and Child Health Hospital (Approval No. KYR-25-014). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
BZ: Methodology, Data curation, Writing – original draft. FD: Methodology, Writing – original draft, Data curation. FH: Supervision, Writing – review & editing, Data curation, Methodology. HJ: Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Jinan Municipal Health Commission Science and Technology Development Plan Project (No.202328017).
Acknowledgments
We thank all the volunteers, medical staff who participated in this study, and colleagues who provided technical support and advice. The ASXL3 gene analysis was conducted at the Prenatal Diagnosis Center (Jinan Maternal and Child Health Hospital, Jinan, Shandong Province) and Amcare Genomics Laboratory (Guangzhou, Guangdong Province).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issue please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, et al. de novo truncating mutations in asxl3 are associated with a novel clinical phenotype with similarities to bohring-opitz syndrome. Genome Med. (2013) 5(2):11. doi: 10.1186/gm415
2. Woods E, Holmes N, Albaba S, Evans IR, Balasubramanian M. Asxl3-related disorder: molecular phenotyping and comprehensive review providing insights into disease mechanism. Clin Genet. (2024) 105(5):470–87. doi: 10.1111/cge.14506
3. Balasubramanian M, Schirwani S. ASXL3-Related disorder. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2020).
4. Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the ddd study: a scalable analysis of genome-wide research data. Lancet. (2015) 385(9975):1305–14. doi: 10.1016/S0140-6736(14)61705-0
5. Trujillano L, Valenzuela I, Costa-Roger M, Cusco I, Fernandez-Alvarez P, Cueto-Gonzalez A, et al. Comprehensive clinical and genetic characterization of a Spanish cohort of 22 patients with bainbridge-ropers syndrome. Clin Genet. (2025) 107(6):646–62. doi: 10.1111/cge.14701
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
7. Schirwani S, Albaba S, Carere DA, Guillen SM, Milan ZF, Si Y, et al. Expanding the phenotype of ASXL3-related syndrome: a comprehensive description of 45 unpublished individuals with inherited and de novo pathogenic variants in asxl3. Am J Med Genet A. (2021) 185(11):3446–58. doi: 10.1002/ajmg.a.62465
8. Katoh M, Katoh M. Identification and characterization of ASXL3 gene in silico. Int J Oncol. (2004) 24(6):1617–22.15138607
9. Lichtig H, Artamonov A, Polevoy H, Reid CD, Bielas SL, Frank D. Modeling bainbridge-ropers syndrome in xenopus laevis embryos. Front Physiol. (2020) 11:75. doi: 10.3389/fphys.2020.00075
10. Srivastava A, Ritesh KC, Tsan YC, Liao R, Su F, Cao X, et al. de novo dominant asxl3 mutations alter h2a deubiquitination and transcription in bainbridge-ropers syndrome. Hum Mol Genet. (2016) 25(3):597–608. doi: 10.1093/hmg/ddv499
11. Koboldt DC, Mihalic Mosher T, Kelly BJ, Sites E, Bartholomew D, Hickey SE, et al. A de novo nonsense mutation in ASXL3 shared by siblings with Bainbridge-ropers syndrome. Cold Spring Harb Mol Case Stud. (2018) 4(3):a002410. doi: 10.1101/mcs.a002410
12. Schirwani S, Hauser N, Platt A, Punj S, Prescott K, Canham N, et al. Mosaicism in asxl3-related syndrome: description of five patients from three families. Eur J Med Genet. (2020) 63(6):103925. doi: 10.1016/j.ejmg.2020.103925
13. Jonsson H, Sulem P, Arnadottir GA, Palsson G, Eggertsson HP, Kristmundsdottir S, et al. Multiple transmissions of de novo mutations in families. Nat Genet. (2018) 50(12):1674–80. doi: 10.1038/s41588-018-0259-9
14. Aluri J, Cooper MA. Genetic mosaicism as a cause of inborn errors of immunity. J Clin Immunol. (2021) 41(4):718–28. doi: 10.1007/s10875-021-01037-z
15. Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. (2015) 31(7):382–92. doi: 10.1016/j.tig.2015.03.013
16. Martinez-Glez V, Tenorio J, Nevado J, Gordo G, Rodriguez-Laguna L, Feito M, et al. A six-attribute classification of genetic mosaicism. Genet Med. (2020) 22(11):1743–57. doi: 10.1038/s41436-020-0877-3
17. Breuss MW, Yang X, Gleeson JG. Sperm mosaicism: implications for genomic diversity and disease. Trends Genet. (2021) 37(10):890–902. doi: 10.1016/j.tig.2021.05.007
18. Waldvogel SM, Posey JE, Goodell MA. Human embryonic genetic mosaicism and its effects on development and disease. Nat Rev Genet. (2024) 25(10):698–714. doi: 10.1038/s41576-024-00715-z
Keywords: bainbridge-ropers syndrome1, ASXL3 gene, whole-exome sequencing, paternal mosaicism, prenatal diagnosis
Citation: Zhao B, Ding F, Hou F and Jin H (2025) Paternal mosaicism in ASXL3-related bainbridge-ropers syndrome: implications for genetic counseling and prenatal diagnosis. Front. Pediatr. 13:1655021. doi: 10.3389/fped.2025.1655021
Received: 7 July 2025; Accepted: 25 August 2025;
Published: 4 September 2025.
Edited by:
Tudor Constantin Badea, Transilvania University of Brașov, RomaniaReviewed by:
Adela Chirita-Emandi, Victor Babes University of Medicine and Pharmacy, RomaniaJiexue Pan, Fudan University, China
Copyright: © 2025 Zhao, Ding, Hou and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Jin, cHJlbmF0YWwxMjNAMTYzLmNvbQ==
†These authors have contributed equally to this work