 Qi Yang1,2,3,†
Qi Yang1,2,3,† Qiang Zhang1,2,3,†Sheng Yi1,2,3Gaojie Huang4Xunzhao Zhou1,2,3Shang Yi1,2,3
Qiang Zhang1,2,3,†Sheng Yi1,2,3Gaojie Huang4Xunzhao Zhou1,2,3Shang Yi1,2,3 Jingsi Luo1,2,3,5*
Jingsi Luo1,2,3,5*
- 1Guangxi Key Laboratory of Birth Defects Research and Prevention, Guangxi Key Laboratory of Reproductive Health and Birth Defects Prevention, Maternal and Child Health Hospital, Nanning, China
- 2Department of Genetic and Metabolic Central Laboratory, Maternal and Child Health Hospital, Nanning, China
- 3Guangxi Clinical Research Center for Birth Defects, Maternal and Child Health Hospital, Nanning, China
- 4Neonatal Surgery, Maternal and Child Health Hospital, Nanning, China
- 5Guangxi Clinical Research Center for Pediatric Diseases, Maternal and Child Health Hospital, Nanning, China
Intellectual developmental disorder type 61 (MRD61) is an extremely rare autosomal dominant disorder caused by variants in the MED13 gene. This gene encodes a subunit of the mediator complex, which is also known as TRAP, SMCC, DRIP or ARC. This complex functions as a transcriptional coactivator and is essential for the expression of almost all genes. To date, only 26 cases of MRD61 have been reported worldwide. The main symptoms are intellectual disability (ID) of varying degrees, developmental delay (DD), hypotonia during infancy, facial dysmorphism, language impairment, restricted growth, skeletal and limb abnormalities, and behavioral abnormalities. In this study, we recruited a Chinese family in which two individuals were diagnosed with MRD61. Whole-exome sequencing of the proband revealed a novel heterozygous frameshift variant in the MED13 gene (NM_005121.3): c.5641delinsTC (p.R1882Sfs*9). This variant was inherited from the affected mother and was subsequently confirmed in both the proband and her other family members using Sanger sequencing. The dysmorphology profile of both patients described here is similar to that associated with MRD61. Compared with previously reported cases of MRD61, the proband presented with congenital megacolon, a previously unreported complication. Additionally, skeletal and limb deformities, eye or vision abnormalities, behavioral issues and brain abnormalities were absent in our patients. This is the first report of MED13-associated intellectual developmental disorder-61 in China. This molecular diagnosis expands the known genetic spectrum of MRD61. Furthermore, the specific manifestations observed in these patients with this condition provide valuable additional clinical insight into the syndrome.
Introduction
Intellectual developmental disorder type 61 (MRD61, MIM 618009) is a very rare autosomal dominant disorder characterized by global developmental delay apparent in infancy, mild to moderate intellectual disability, facial dysmorphism, language impairment, growth restriction, and behavioral abnormalities, including autism spectrum disorder (ASD) and attention-deficit hyperactivity disorder (ADHD) (1). The disease is caused by heterozygous mutations in the MED13 gene (NM_005121.3; OMIM 603808), which is located on chromosome 17q23.2. This 30-exon gene encodes a subunit of the mediator complex, which is also referred to as TRAP, SMCC, DRIP, or ARC (2). The Mediator complex, a large multiprotein complex universally expressed in eukaryotes, plays a crucial role in transcription regulation by stabilizing the preinitiation complex and communicating transcription-activating signals to RNA Polymerase II, thus promoting the transcription of all protein-coding and most nonprotein-coding genes (3–5). A key component of this complex is the four-subunit kinase module, composed of MED13, MED12, and cyclin-dependent kinase 8 (CDK8), along with their respective paralogues MED13l, MED12l, and CDK19, which function together with Cyclin C (3, 4). This kinase module, also known as the “Module”, is involved in both transcriptional activation and repression. It has been reported to prevent the association of the Mediator with the RNA Polymerase II preinitiation complex, thereby inhibiting transcription initiation and/or re-initiation (6). Additionally, the Module may activate transcription (7). MED13, in particular, facilitates the reversible physical interaction between the Module and the Mediator in both humans and yeast (4, 8, 9). Given its critical role in transcription, MED13 is a compelling candidate gene for neurodevelopmental disabilities, which are often associated with variants affecting the Module genes and are broadly termed “transcriptomopathies” (10, 11).
To date, only 26 patients carrying MED13 variants have been documented in the literature (1, 12–20). The mechanisms by which distinct MED13 mutations produce divergent clinical phenotypes remain unknown. Additional case reports documenting MED13 mutations and their phenotypic manifestations will clarify both the disease pathology and its genotype-phenotype correlations. In this study, we identified a novel frameshift mutation in the MED13 gene in a Chinese family with MRD61. The patient inherited this variant from her affected mother. The clinical phenotypes observed in both the patient and her mother were thoroughly characterized. This report expands the spectrum of MED13 gene variants and provides additional molecular and clinical information, which helps to further enhance the understanding of MRD61 syndrome.
Materials and methods
Patients
A Chinese family with aganglionic megacolon and developmental delay was referred to the Paediatric Surgery Department of Guangxi Maternal and Child Health Hospital for genetic evaluation (Figure 1A). The Institutional Review Board and Ethics Committee of Guangxi Maternal and Child Health Hospital approved the study protocol, which adhered to the principles of the Declaration of Helsinki. The parents of the affected individuals provided written informed consent for publishing clinical data and photographs.
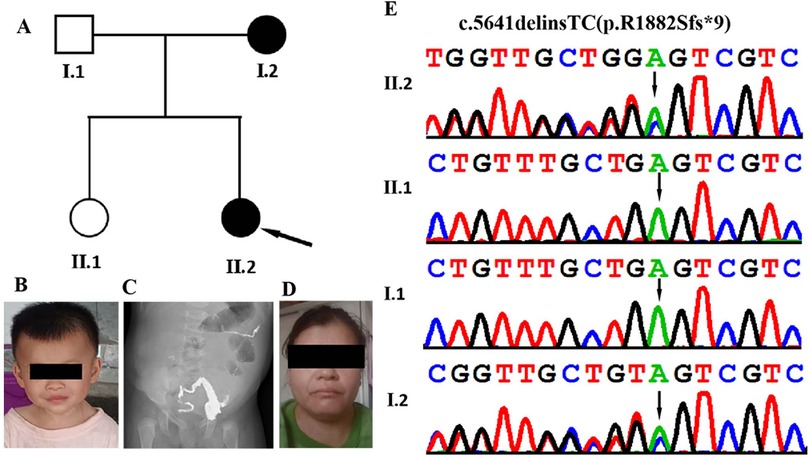
Figure 1. Clinical and genetic features. (A) Family pedigree showing that both the proband and her mother and affected. (B) Facial appearance of the proband (II-2) at the age of 2 years and 3 months, showing hypertelorism, everted lower eyelids, depressed nasal bridge, broad nasal tipand, and broad philtrum. (C) Abdominal x-ray acquired at 2 months and 23 days in the proband (II-2) show congenital megacolon. (D) Facial appearance of the proband's motherexhibiting epicanthal folds, a long philtrum, and a wide mouth. (E) DNA sequence chromatograms from Sanger sequencing of MED13, showing showing a heterozygous frameshift variant MED13 (NM_005121.3): c.5641delinsTC (p.R1882Sfs*9) in the proband. Sanger sequencing further revealed that his affected mother was heterozygous for the same variant and that his father and older sister were normal.
Whole exome sequencing and sanger sequencing
Peripheral blood lymphocytes (2 mL) were collected from the patient and his family members. Genomic DNA was subsequently extracted from these samples with a Lab Aid DNA kit (Zeshan Biotechnology Co., Ltd., Xiamen, China) according to the manufacturer's instructions. Whole-exome sequencing (WES) was achieved using the Agilent SureSelect V5 enrichment capture kit (Agilent Technologies, Santa Clara, CA, USA) and analyzed using an Illumina HiSeq 2000 with 100 bp paired-end sequencing (Illumina, San Diego, California, USA). The sequencing reads were aligned to the human reference genome (hg19/GRCh37) using the Genome Analysis Toolkit (GATK, version 3.4; Broad Institute, Cambridge, MA, USA). The TGex software (http://tgex.genecards.org/) was used for variant calling and annotation. Variants with a minor allele frequency (MAF) of ≤0.001 in public databases (the 1000 Genomes Project, the Exome Sequencing Project and the Exome Aggregation Consortium) were prioritized. In silico tools (REVEL, PolyPhen2, SIFT, LRT, Mutation Assessor, CADD, and MutationTaster) were used to predict the functional impact of candidate variants. The pathogenicity of these variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines and the ClinGen Sequence Variant Interpretation (SVI) Working Group recommendations (21).
Result
The proband was a Chinese girl who was the second child of non-consanguineous parents (Figures 1A,B). The patient was born vaginally at 39 weeks gestation following an uncomplicated spontaneous labor. Birth measurements included a weight of 3.3 kg (75th percentile), length of 50 cm (80th percentile), and head circumference of 34 cm (50th percentile). APGAR scores were 9 at 1 min, and 10 at both 5 and 10 min. At two months of age, the infant was admitted to a local hospital with recurrent fever and diarrhea. After showing no improvement following one week of initial treatment, the patient was transferred to our hospital for further evaluation and management. She was subsequently diagnosed with congenital megacolon and enteritis (Figure 1C). She underwent radical surgery for the megacolon and reversal of the intestinal ostomy. Subsequently, she was admitted to hospital several times due to intestinal obstruction and constipation. During her first year, she exhibited feeding difficulties, failure to thrive and hypotonia. Her developmental milestones were also slightly delayed. She achieved head control at 5 months, started crawling at 9 months, and began walking with support at 17 months. Her language development has been delayed; she did not speak her first word until she was around 20 months old, and she is currently unable to speak in complete sentences. At 10 months of age, the Gesell Developmental Diagnostic Scale measured her Developmental Quotient (DQ; scores <70 indicate delay), yielding the following domain scores: gross motor (70), fine motor (68), adaptive (65), language (48), and personal-social (67). Her most recent examination, at the age of 2 years and 3 months, revealed postnatal growth retardation, short stature (height: 80 cm, <−2 SD) and dysmorphic facial features, which included a hypertelorism, everted lower eyelids, depressed nasal bridge, broad nasal tip, and broad philtrum (Figure 1B).
The patient's family history reveals that her mother experienced delayed motor and language development during childhood. Specifically, she began walking at 17 months and speaking simple words at 20 months. Currently, at the age of 36, the patient's mother exhibits short stature, with a height of 154 cm (<−2.5 SD). Additionally, she has a borderline intellectual disability (IQ: 78–83), learning difficulties and presents with mild facial dysmorphism, including epicanthal folds, a long philtrum, and a wide mouth (Figure 1D).
To identify the genetic basis of the disease, we conducted whole-exome sequencing on the proband. The sequencing yielded 6.9 Gb of data, covering 98.8% of the target region with 98.5% of targets exceeding 20X read depth. We detected 29,046 single nucleotide variants (SNVs) and insertion/deletion (indel) variants in coding regions and splice sites (within 10 bp of exon boundaries). After excluding synonymous variants and those with a minor allele frequency (MAF) >1% in public databases (1000 Genomes Project, gnomAD, ESP, ExAC) and our internal datasets, 610 variants remained. TGex analysis (LifeMap Sciences, USA) prioritized 9 candidate variants across 7 genes (MED13, POGZ, SETBP1, SCN9A, SCO1, APTX, TIE1) associated with phenotypes including congenital megacolon, intellectual disability, motor delay, and developmental delays. Heterozygous variants in SCO1 and APTX were excluded as their associated disorders follow autosomal recessive inheritance. Variants in POGZ, SETBP1, SCN9A, and TIE1 were paternally inherited from the unaffected father. The analysis revealed a heterozygous frameshift variant in MED13 exon 25 (c.5641delinsTC, p.R1882Sfs*9) in the proband, which Sanger sequencing confirmed as maternally inherited (Figure 1E). This variant is a novel variant that has not been found in major public databases, including the Exome Sequencing Project, gnomAD database, and 1000 Genomes Project, as well as disease-specific databases such as ClinVar and the Human Gene Mutation Database. This variant (c.5641delinsTC; p.R1882Sfs*9) was located in the exon 25 of the MED13 gene. Its functional impact likely resembles other reported MED13 loss-of-function variants, such as the known c.4198C>T(p.Arg400*) mutation. This alteration leads to complete protein loss accompanied by markedly reduced mRNA levels through nonsense-mediated decay. According to the ACMG/AMP standards and guidelines, the variant is classified as ‘likely pathogenic’ based on the PVS1 and PM2 supporting criteria (Table 1). XHMM software and depth-of-coverage analysis detected no pathogenic or likely pathogenic copy number variants in the patient's WES data.

Table 1. Predicted pathogenicity of novel MED13 variant.
Discussion
Intellectual developmental disorder type 61 (MRD61) is a very rare autosomal dominant disorder (1). The MED13 variant was initially detected through whole exome sequencing (WES) in a large group of patients with autism, for whom a detailed clinical description was unavailable (12, 13). Subsequently, Snijders Blok et al. provided a detailed description of the clinical phenotype and molecular genetic features of 13 patients with MED13-related disease (1). To date, only 26 individuals with MRD61 have been reported (Table 2). The clinical manifestations of these patients include facial dysmorphism, global developmental delay in infancy, varying degrees of intellectual disability, language developmental delay, growth restriction, short stature, skeletal abnormalities, and behavioral abnormalities, including autism spectrum disorder and attention-deficit/hyperactivity disorder (1, 12–20). In this study, we conducted WES and detected a heterozygous frameshift variant in the MED13 gene in a Chinese patient diagnosed with MRD61.
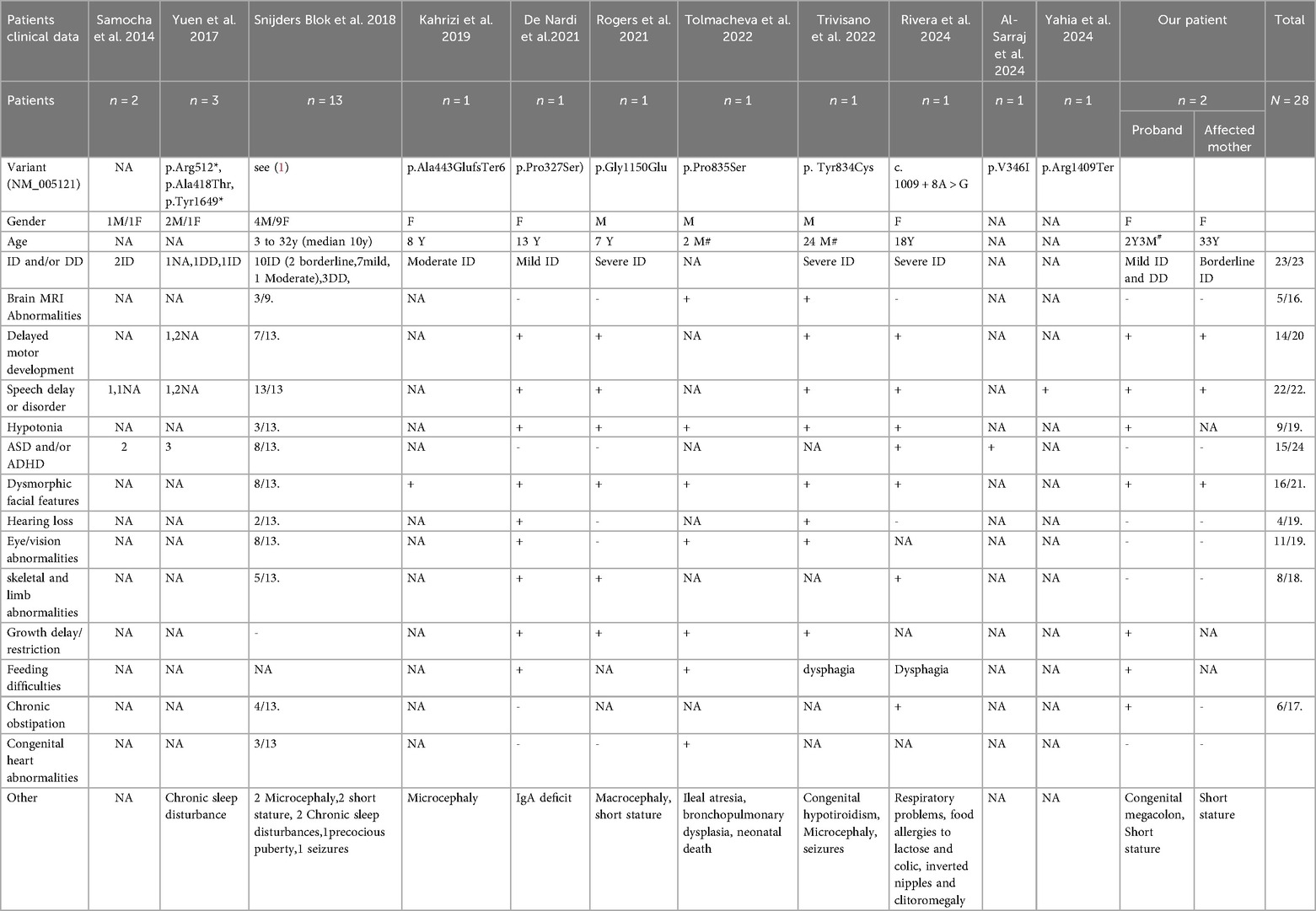
Table 2. Clinical characteristic of patients with MED13 novel variants.
To explore the genetic and clinical characteristics of MRD61, we conducted a comprehensive review of literature on the clinical manifestations and genetic characteristics of patients with MED13 variants (Table 2). Statistical analysis revealed that, to date, only 28 affected individuals from 26 unrelated families (including our patients) have been reported to carry MED13 gene variants worldwide (1, 12–20). These patients exhibit highly heterogeneous clinical manifestations across multiple domains. However, certain common features (>50% of cases) can still be identified. All patients (23/23) have intellectual disability or and developmental delay. Notably, the severity of these conditions is relatively mild, with only five patients showing moderate to severe intellectual impairment. Our patients also present with mild intellectual disability and motor developmental delay. In the language domain, all patients (22/22) exhibit language developmental delay, with about half of them having significant language disorders. This finding indicates that language disorders are a typical feature of MRD61. Most patients (14/20) experience motor delay or impairment. Although some patients do not learn to walk until after 4.5 years of age, all patients eventually acquire the ability to walk. Hypotonia in infancy is also relatively common (9/19). Almost all patients (16/21) show facial dysmorphic features, but there is a lack of consistency—some patients only present with wide-set eyes, narrow eyelids, and a wide mouth, while others exhibit more pronounced facial deformities. In our patients, the proband shows a marked dysmorphic face, while the affected mother has a milder presentation. Fifteen of the 24 patients reported behavioral problems, including autistic behavior and attention deficit hyperactivity disorder (ADHD). Notably, two patients in this study had no behavioral problems. Skeletal and limb abnormalities were observed in eight patients (8/18), including high-arched feet, spinal deformities (scoliosis/kyphosis), hip dysplasia/joint hypermobility, and toe deformities. Another point of concern is eye/vision abnormalities; more than half of the patients have such impairments, and early corrective treatment can improve the prognosis of children. In addition, these patients present a wide variety of symptoms. Among these patients, five exhibited growth restriction, which may be partly attributed to feeding difficulties and gastrointestinal issues. All five of these patients experienced feeding difficulties, with one patient diagnosed with ileal atresia and the proband in this study presenting with a previously unreported case of congenital megacolon. Some patients had brain abnormalities (5/16), chronic constipation (6/17 cases), deafness (4/19 cases), and congenital heart abnormalities (4/18 cases). Other dysmorphic features included IgA deficiency, seizures, congenital hypothyroidism, microcephaly, macrocephaly, short stature, chronic sleep disorders, ileal atresia, bronchopulmonary dysplasia, respiratory problems, lactose intolerance/food allergies, colic, and congenital megacolon. In contrast, the patients in this group did not present with these diverse dysmorphic features, including skeletal and limb deformities, eye/vision abnormalities, behavioral problems, brain abnormalities, and other rare dysmorphic features.
To date, a total of 25 MED13 variants (including ours) associated with MRD61 have been reported (1, 12–20). These variants include 14 missense and in-frame variants, as well as 12 truncating variants (e.g., nonsense, frameshift, and splicing variants that result in protein frameshifts) (Table 2). The type of genetic variant is often linked to the severity of the associated clinical phenotype. According to the literature, missense variants in the MED13 homolog gene MED13l have been reported to correlate with more severe phenotypes, including a higher incidence of seizures, MRI abnormalities, autism spectrum features, and cardiac abnormalities (22). Consistent with the literature on MED13l, we observed that severe intellectual disability was only present in two MRD61 patients with missense mutations and one MRD61 patient with a splicing site mutation (potentially in-frame). However, this observation has not reached statistical significance. Furthermore, the presence of other clinical features, such as delayed language development, skeletal and limb abnormalities, seizures, congenital heart defects, and brain abnormalities, does not appear to be dependent on the type of mutation. Additionally, patients carrying the same genetic mutation may also exhibit a variety of phenotypes. For example, two unrelated patients carrying the same mutation p.Pro327Ser show significant phenotypic differences. Although both patients have intellectual disability, delayed language and motor development, deafness, skeletal and limb abnormalities, and hypotonia, the patient described by De Nardi et al. has prominent facial dysmorphism similar to Kabuki syndrome and more extensive skeletal and limb abnormalities, while the patient reported by Snijders Blok et al. does not have significant facial or skeletal and limb abnormalities but presents with additional cardiac phenotypes, manifested as aortic root and pulmonary artery dilation (1, 16). Moreover, within the same family, the clinical manifestations of different patients are not the same, and it seems that the condition is more severe in the next generation. In the cases reported by Snijders Blok et al. and in this study, the affected mothers have normal or borderline intelligence, with mild facial dysmorphism and skeletal and limb abnormalities; whereas the affected children exhibit mild intellectual disability, feeding difficulties, megacolon, more extensive skeletal and limb abnormalities, and more pronounced facial dysmorphism (1). Therefore, patients with heterozygous mutations in the MED13 gene exhibit highly diverse clinical manifestations, and no clear correlation between genotype and phenotype has been established so far. The exact mechanisms by which these mutations lead to MRD61 remain unclear. According to Snijders Blok et al., the disease may be caused by multiple mechanisms (1). On the one hand, loss-of-function mutations may lead to haploinsufficiency of the MED13 gene. On the other hand, missense mutations in MED13 may disrupt or introduce new phosphorylation sites (e.g., casein kinase 1 phosphorylation sites), thereby affecting the protein-protein interactions of the forkhead-associated domain involved in various important cellular functions. Further case reports and functional studies are crucial for our understanding of the disease and its potential molecular mechanisms.
In summary, we identified a novel frameshift mutation in the MED13 gene in a Chinese family with MRD61. This is the first report of a Chinese family with the MED13 variant. The proband in this family exhibited a clinical phenotype similar to MRD61, including mild intellectual disability, developmental delay, delayed language development, growth restriction, short stature, facial dysmorphism, feeding difficulties, hypotonia during infancy, chronic constipation, and congenital megacolon. The affected mother only presented with borderline intellectual disability, mild language impairment, mild facial dysmorphism, and short stature. The phenotypic diversity caused by MED13 variants suggests that in-depth investigation of the specific functions of these variants will help deepen the understanding of the disease and its pathogenesis. Moreover, these data further expand the mutational and phenotypic spectrum of MRD61.
Data availability statement
All data that support the findings of the current study are available from the corresponding author upon reasonable request.
Ethics statement
The studies involving humans were approved by Institutional Review Board and Ethics Committee of Guangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
QY: Resources, Conceptualization, Project administration, Writing – review & editing, Investigation, Validation, Funding acquisition, Methodology, Writing – original draft, Formal analysis, Data curation, Supervision, Software, Visualization. QZ: Conceptualization, Investigation, Formal analysis, Methodology, Writing – review & editing. SheY: Investigation, Software, Writing – review & editing, Methodology, Validation. GH: Supervision, Software, Methodology, Writing – review & editing, Data curation, Investigation. XZ: Software, Writing – review & editing, Data curation, Methodology, Conceptualization. ShaY: Methodology, Data curation, Software, Writing – review & editing, Conceptualization. JL: Methodology, Writing – original draft, Investigation, Resources, Data curation, Funding acquisition, Conceptualization, Supervision, Formal analysis, Software, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Guangxi Key Laboratory of reproductive health and birth defect prevention (21-220-22), Guangxi Key Laboratory of Birth Defects and Stem Cell Biobank (ZTJ2020002), the Health Department of Guangxi Province (Grant No. Z-A20220256 and Z20190311), and the Guangxi Clinical Research Center for Pediatric Diseases (Guike AD22035121).
Acknowledgments
We are grateful to the all the patients and their families participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Blok LS, Hiatt SM, Bowling KM, Prokop JW, Engel KL, Cochran JN, et al. de novo mutations in MED13, a component of the mediator complex, are associated with a novel neurodevelopmental disorder. Hum Genet. (2018) 137(5):375–88. doi: 10.1007/s00439-018-1887-y
2. Sato S, Tomomori-Sato C, Parmely TJ, Florens L, Zybailov B, Swanson SK, et al. A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol Cell. (2004) 14(5):685–91. doi: 10.1016/j.molcel.2004.05.006
3. Calpena E, Hervieu A, Kaserer T, Swagemakers SMA, Goos JAC, Popoola O, et al. de novo missense substitutions in the gene encoding CDK8, a regulator of the mediator complex, cause a syndromic developmental disorder. Am J Hum Genet. (2019) 104(4):709–20. doi: 10.1016/j.ajhg.2019.02.006
4. Harper TM, Taatjes DJ. The complex structure and function of mediator. J Biol Chem. (2018) 293(36):13778–85. doi: 10.1074/jbc.R117.794438
5. Soutourina J. Transcription regulation by the mediator complex. Nat Rev Mol Cell Biol. (2018) 19(4):262–74. doi: 10.1038/nrm.2017.115
6. Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls mediator coactivator function. Genes Dev. (2009) 23(4):439–51. doi: 10.1101/gad.1767009
7. Conaway RC, Conaway JW. Function and regulation of the mediator complex. Curr Opin Genet Dev. (2011) 21(2):225–30. doi: 10.1016/j.gde.2011.01.013
8. Fant CB, Taatjes DJ. Regulatory functions of the mediator kinases CDK8 and CDK19. Transcription. (2019) 10(2):76–90. doi: 10.1080/21541264.2018.1556915
9. Tsai KL, Sato S, Tomomori-Sato C, Conaway RC, Conaway JW, Asturias FJ. A conserved mediator-CDK8 kinase module association regulates mediator-RNA polymerase II interaction. Nat Struct Mol Biol. (2013) 20(5):611–9. doi: 10.1038/nsmb.2549
10. Caro-Llopis A, Rosello M, Orellana C, Oltra S, Monfort S, Mayo S, et al. de novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy? Pediatr Res. (2016) 80(6):809–15. doi: 10.1038/pr.2016.162
11. Poot M. Mutations in mediator complex genes CDK8, MED12, MED13, and MEDL13 mediate overlapping developmental syndromes. Mol Syndromol. (2020) 10(5):239–42. doi: 10.1159/000502346
12. Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. (2017) 20(4):602–11. doi: 10.1038/nn.4524
13. Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. (2014) 46(9):944–50. doi: 10.1038/ng.3050
14. Trivisano M, De Dominicis A, Micalizzi A, Ferretti A, Dentici ML, Terracciano A, et al. MED13 Mutation: a novel cause of developmental and epileptic encephalopathy with infantile spasms. Seizure. (2022) 101:211–7. doi: 10.1016/j.seizure.2022.09.002
15. Rogers AP, Friend K, Rawlings L, Barnett CP. A de novo missense variant in MED13 in a patient with global developmental delay, marked facial dysmorphism, macroglossia, short stature, and macrocephaly. Am J Med Genet A. (2021) 185(8):2586–92. doi: 10.1002/ajmg.a.62238
16. De Nardi L, Faletra F, D'Adamo AP, Bianco AMR, Athanasakis E, Bruno I, et al. Could the MED13 mutations manifest as a kabuki-like syndrome? Am J Med Genet A. (2021) 185(2):584–90. doi: 10.1002/ajmg.a.61994
17. Tolmacheva E, Bolshakova AS, Shubina J, Rogacheva MS, Ekimov AN, Podurovskaya JL, et al. Expanding phenotype of MED13-associated syndrome presenting novel de novo missense variant in a patient with multiple congenital anomalies. BMC Med Genomics. (2024) 17(1):130. doi: 10.1186/s12920-024-01857-z
18. Rivera MD, Aponte SN, Rivera F, Arciniegas NJ, Carlo S. MED13 gene mutation related to autism Spectrum disorder: a case report. Cureus. (2024) 16(5):e59904. doi: 10.7759/cureus.59904
19. Al-Sarraj Y, Taha RZ, Al-Dous E, Ahram D, Abbasi S, Abuazab E, et al. The genetic landscape of autism spectrum disorder in the middle eastern population. Front Genet. (2024) 15:1363849. doi: 10.3389/fgene.2024.1363849
20. Yahia A, Li D, Lejerkrans S, Rajagopalan S, Kalnak N, Tammimies K. Whole exome sequencing and polygenic assessment of a Swedish cohort with severe developmental language disorder. Hum Genet. (2024) 143(2):169–83. doi: 10.1007/s00439-023-02636-z
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
Keywords: intellectual developmental disorder type 61, MED13 gene, novel variant, whole-exome sequencing, unreported complication
Citation: Yang Q, Zhang Q, Yi S, Huang G, Zhou X, Yi S and Luo J (2025) A novel frameshift variant in the MED13 gene causing intellectual developmental disorder-61 in a Chinese family. Front. Pediatr. 13:1699544. doi: 10.3389/fped.2025.1699544
Received: 5 September 2025; Accepted: 6 October 2025;
Published: 21 October 2025.
Edited by:
Giulia Pascolini, Institute of Immaculate Dermatology (IRCCS), ItalyReviewed by:
Simon Carlo, Ponce Health Sciences University, Puerto RicoGongao Wu, The Third Affiliated Hospital of Zhengzhou University, China
Copyright: © 2025 Yang, Zhang, Yi, Huang, Zhou, Yi and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingsi Luo, eWFuZ3Fpc2tsbWcxMjZAMTI2LmNvbQ==
†These authors have contributed equally to this work