
- Department of Pharmacology, Physiology and Neuroscience, School of Medicine, University of South Carolina, Columbia, SC, USA
G protein-coupled inward rectifier K+ (GIRK) channels represent novel targets for the development of new therapeutic agents. GIRK channels are activated by a large number of G protein-coupled receptors (GPCRs) and regulate the electrical activity of neurons, cardiac myocytes, and β-pancreatic cells. Abnormalities in GIRK channel function have been implicated in the patho-physiology of neuropathic pain, drug addiction, cardiac arrhythmias, and other disorders. However, the pharmacology of these channels remains largely unexplored. In this paper we describe the development of a screening assay for identifying new modulators of neuronal and cardiac GIRK channels. Pituitary (AtT20) and cardiac (HL-1) cell lines expressing GIRK channels were cultured in 96-well plates, loaded with oxonol membrane potential-sensitive dyes and measured using a fluorescent imaging plate reader. Activation of the endogenous GPCRs in the cells caused a rapid, time-dependent decrease in the fluorescent signal; indicative of K+ efflux through the GIRK channels (GPCR stimulation versus control, Z′-factor = 0.5–0.7). As expected this signal was inhibited by addition of Ba2+ and the GIRK channel toxin tertiapin-Q. To test the utility of the assay for screening GIRK channel blockers, cells were incubated for 5 min with a compound library of Na+ and K+ channel modulators. Ion transporter inhibitors such as 5-(N,N-hexamethylene)-amiloride and SCH-28080 were identified as blockers of the GIRK channel at sub-micromolar concentrations. Thus, the screening assay will be useful for expanding the limited pharmacology of the GIRK channel and in developing new agents for the treatment of GIRK channelopathies.
Introduction
G protein-coupled inward rectifier K+ (GIRK) channels function as cellular mediators of a wide range of hormones and neurotransmitters and are expressed in the brain, heart, skeletal muscle, and endocrine tissue (Hibino et al., 2010; Lusscher and Slesinger, 2010). GIRK channels are members of the super-family of proteins known as inward rectifier K+ (Kir) channels (Hibino et al., 2010; Lusscher and Slesinger, 2010) that function to stabilize the cell resting membrane potential near the K+ equilibrium potential (EK). Four GIRK channel subunits are expressed in mammals: GIRK1 (Kir3.1), GIRK2 (Kir3.2), GIRK3 (Kir3.3), and GIRK4 (Kir3.4; Hibino et al., 2010; Lusscher and Slesinger, 2010). The GIRK1, GIRK2, and GIRK3 subunits are expressed in a number of regions of the brain including the hippocampus, cerebellum, substantia nigra, locus coeruleus, and ventral tegmental area (Lusscher and Slesinger, 2010). In addition, GIRK1 and GIRK2 subunits are expressed in the superficial layers of the spinal dorsal horn and are enriched in the postsynaptic membranes of substantia gelatinosa neurons (Lusscher and Slesinger, 2010). In neuronal tissue GIRK subunits combine to form both heterotetrameric (e.g., GIRK1/GIRK2, GIRK1/GIRK3, etc.) and homotetrameric (GIRK2/GIRK2) channels. In the atrial myocardium and sinus and atrioventricular nodes of the heart, GIRK channels are composed of heteromers of GIRK1 and GIRK4 subunits (Hibino et al., 2010).
G protein-coupled inward rectifier K+ channels become activated following the binding of ligands to their cognate G protein-coupled receptors (GPCRs). This binding causes the dissociation of the βγ subunits of pertussis toxin-sensitive G proteins which subsequently bind to and activate the GIRK channel. Once opened the GIRK channel allows the movement of K+ out of the cell causing the resting membrane potential to become more negative. As a consequence, GIRK channel activation in neurons decreases spontaneous action potential formation and inhibits the release of excitatory neurotransmitters. In the heart, activation of the GIRK channel inhibits pacemaker activity thereby slowing the heart rate.
G protein-coupled inward rectifier K+ channels represent novel targets for the development of new therapeutic agents for the treatment neuropathic pain, drug addiction, cardiac arrhythmias, and other disorders. However, the pharmacology of these channels remains largely unexplored. Although a number of drugs including anti-arrhythmic agents, antipsychotic drugs, and antidepressants block the GIRK channel (Kobayashi and Ikeda, 2006), this inhibition is non-selective and occurs at relatively high drug concentrations. In this paper we describe the development of a screening assay for identifying new modulators of neuronal and cardiac GIRK channels. Pituitary (AtT20) and cardiac (HL-1) cell lines expressing GIRK channels were analyzed using patch clamp recording procedures and fluorescent imaging plate reader measurements. Application of the GPCR ligands somatostatin (AtT20 cells) and carbachol (HL-1 cells) activated whole-cell GIRK currents that were blocked by the Kir channel selective toxin tertiapin-Q. In cells loaded with the membrane potential-sensitive fluorescent dye HLB 021-152, GPCR stimulation caused a rapid, time-dependent decrease in the fluorescent signal. This GPCR-induced decrease in fluorescence was block by BaCl2 and tertiapin-Q. A preliminary screen of the GIRK channel fluorescent assay identified several ion transporter inhibitors as blockers of the channel. Thus, the GIRK channel screening assay will be useful for expanding the limited pharmacology of the channel and for developing new agents for the treatment of GIRK channelopathies.
Materials and Methods
Cell Culture and Plating
The immortalized cardiac HL-1 cell line was generously supplied by Dr. William Claycomb (LSU Medical Center, New Orleans, LA, USA). HL-1 cells were maintained in Claycomb/DMEM media supplemented with 10% fetal bovine serum, penicillin/streptomycin, and L-glutamine (Sigma-Aldrich). The AtT20 pituitary cell line was obtained from Dr. Ken Mackie (Indiana University) and grown in DMEM media with 10% horse serum. Cells were platted on gelatin- (HL-1 cells) or poly L-lysine- (AtT20 cells) coated glass coverslips (5,000 cells per coverslip; patch clamp recording) and in black 96-well plates (Corning; 30,000 cells per well; plate reader measurements). Cells were stored in an incubator at 37°C (5% O2/95% CO2) and used on days 2–4 after plating.
Patch Clamp Recording of GIRK Currents
The patch clamp method (Hamill et al., 1981) was used to record the whole-cell, GIRK currents using L/M EPC-7 (Adams and List Associates) and Axopatch 200 (Axon Instruments) amplifiers. Our procedures for measuring K+ channels have been described (Walsh and Zhang, 2008; Walsh, 2010). Pipettes were made from borosilicate glass capillaries (World Precision Instruments) and had resistances of 1–2 Mohm when filled with internal solution. All experiments were conducted on isolated, non-coupled cells at room temperature (22–24°C). For the measurement of GIRK currents, cells were placed in a normal buffer solution consisting of; 132 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM dextrose, 5 mM HEPES, pH 7.4 (with NaOH; 280 mOsm). The internal solution consisted of; 50 mM KCl, 60 mM K+–glutamate, 2 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, 3 mM ATP, 0.1 mM GTP, 10 mM HEPES, pH 7.3 (with KOH; 280 mOsm). Upon establishment of the whole-cell configuration, the cell was perfused with a high K+ buffer solution containing; 107 mM NaCl, 30 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM dextrose, 5 mM HEPES, pH 7.4. Following the measurement of the cell background current, GIRK channels were activated by the addition of the GPCR ligands carbachol (10 μM; HL-1 cells) or somatostatin (200 nM; AtT20 cells) using a rapid perfusion system. In each case the GIRK current was defined as the BaCl2-sensitive current (see Figure 1).
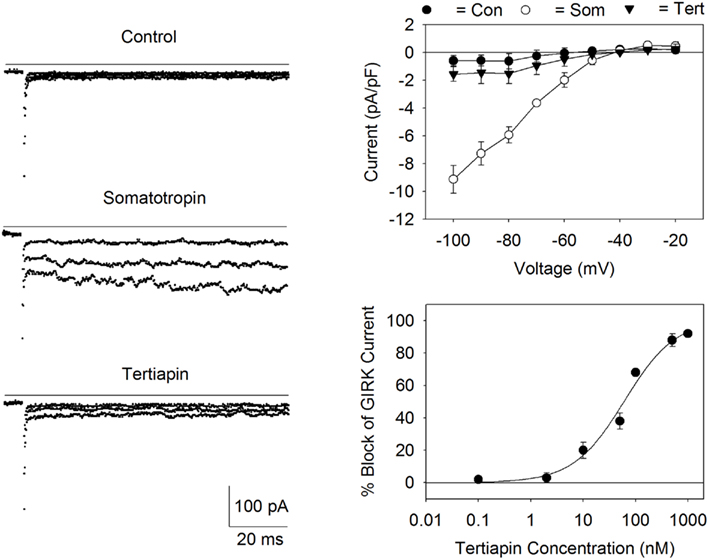
Figure 1. Expression of GIRK channels in AtT20 cells. Left panel: Currents were measured before (control) and after the addition of somatostatin (200 nM) during voltage steps applied from the holding potential of −40 to −60, −80, and −100 mV. Addition of tertiapin (500 nM) inhibited the current (bottom records). The solid lines represent zero current. Top right panel: I/V relationship for the somatostatin activated current (Som) displayed inward rectification and a reversal potential of −40 mV. Current values were obtained by subtracting the currents by records obtained in the presence of 1 mM BaCl2. Each point represents the mean ± SE current (pA/pF) measured in three to five cells. Bottom right panel: dose versus response curve for tertiapin-Q block of the AtT20 cell GIRK current. Each point represents the mean ± SE inhibition obtained from three to six experiments. The calculated IC50 value for tertiapin-Q block was 60 nM.
GIRK Channel Fluorescent Plate Reader Assay
For measuring changes in the membrane potential, cells were incubated for 1 h in normal buffer solution containing the oxonol dye HLB 021-152 (5 μM; AnaSpec) and fluorescent signals recorded using a Synergy2 microplate reader (Biotek) at 28°C. Test compounds were dissolved in DMSO at a stock concentration of 10 mM, diluted to various concentrations and applied to the cells for 5 min prior to the fluorescent measurements. DMSO, up to 1% in normal buffer solution, had no adverse effects on the cell response. Carbachol, somatostatin, or control solution (20 μL) was added to each well (total volume = 220 μL) at time zero using the Synergy2 injector system. Data points were collected at 10 s intervals over a 300-s sampling period at excitation and emission wavelengths of 520 and 560 nm, respectively. Z′-factors (Zhang et al., 1999) were obtained by (1) comparing the fluorescent signal between wells injected with control solution and wells injected with the GPCR ligand and (2) comparing the signal in wells injected with the GPCR ligand in the presence and absence of 2 mM BaCl2. The Z′-factor is defined as: Z′ = 1 − (3σP + 3σN)/| μP − μN |, where μP and μN are the means of the positive control and negative control signals, and σP and σN are the SD of the positive control and negative control signals, respectively (Zhang et al., 1999). Plates used for Z′-factor determination contained two rows each of positive and negative controls. Dose–response curves for selected compounds were obtained and drug potencies determined by fitting the data with the curve: Max/(1 + ([drug]/IC50)k), where the IC50 is the concentration of the compound producing a 50% decrease in the maximal GPCR response (Max) and k is the slope factor. Interference of the test compounds with the HLB 021-152 fluorescence was determined in the absence of cells by adding various concentrations of the compounds to buffer solution containing the dye.
Drugs and Chemicals
Carbachol, somatostatin, and a Na+, K+ channel modulator kit (catalog # LO2220), containing 68 compounds, were purchased from Sigma-Aldrich Chemical Corp. Tertiapin-Q (synthetic formulation) was obtained from Alomone Labs.
Results
Measurement of GIRK Currents in AtT20 Cells
G protein-coupled inward rectifier K+ channels are highly expressed in the central and peripheral nervous system as well as in endocrine tissue. The AtT20 cell line was originally derived from a mouse pituitary tumor and expresses the somatostatin type 2 and 5 receptors (SSTR2 and SSTR5), voltage-gated Ca2+ channels, and GIRK1/GIRK2 channels (Mackie et al., 1995; Kuzhikandathil et al., 1998). Although the AtT20 cells were first thought to be related to pituitary corticotrophs, recent evidence indicates that these cells have functional and structural features more in common with neuropeptide-secreting neurons. Thus, the AtT20 cells provide a good model system for studying neuronal signaling.
Figure 1 displays GIRK currents measured in the AtT20 cells using the whole-cell arrangement of the patch clamp technique. Cells were bathed in the high K+ buffer solution (30 mM KCl) and the internal (pipette) solution contained KCl/K+–glutamate (140 mM K+) with GTP. Application of somatostatin to the recording chamber resulted in the activation of a Ba2+-sensitive, inward rectifying current (Figure 1). The somatostatin–activated current was blocked by addition of the GIRK channel blocker tertiapin-Q (Jin and Lu, 1998; Figure 1). Tertiapin-Q blocked the current with an IC50 of 60 nM (Figure 1). The reversal potential (Erev) for the current (−40 mV) was close to the calculated Nernst equilibrium potential (EK; −39 mV) for a K+-selective channel under the conditions of the experiment. Thus, the inward rectifying current versus voltage relationship, the tertiapin-Q-sensitivity, and the K+ selectivity of the channel are all consistent with the expression of GIRK channels in the AtT20 cells (Mackie et al., 1995; Kuzhikandathil et al., 1998).
Development of a GIRK Channel Screening Assay
Having confirmed the presence of GIRK channels in the AtT20 cell line, it was determined if these cells could be used for drug screening. The experimental design of the screening assay is outlined in Figure 2. AtT20 cells were cultured in 96-well plates and loaded with the fluorescent membrane potential-sensitive dye HLB 021-152 (AnaSpec). Membrane potential-sensitive dyes such as HLB 021-152 (D) distribute across the plasma membrane when cells are in the rested state and reach equilibrium (Figure 2, top panel). The dye molecules inside the cells become strongly fluorescent upon binding to intracellular proteins and other cytoplasmic components. Treatment of the cells with somatostatin (Som), which binds to the somatostatin receptor (SSTR), activates the G inhibitory protein (Gi) stimulating the GIRK channels to open (Figure 2, bottom panel). The resulting efflux of K+ out of the cell (resting potential of −50 to −60 mV for AtT20 cells) causes the resting membrane potential (Rm) to become more negative and the HLB 021-152 molecules to redistribute to the outside of the cell. As a result the cell fluorescent signal is decreased.
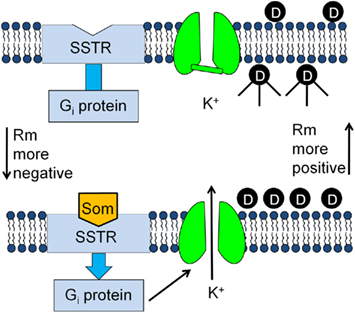
Figure 2. Development of a screening assay for identifying new neuronal GIRK channel blockers.
The applicability of the GIRK channel fluorescent assay for drug discovery was evaluated in the experiments displayed in Figure 3. Each figure plots the HLB 021-152 fluorescence signal measured over time in the 96-well plates using a microplate reader. As predicted by the experimental model, addition of somatostatin (200 nM) to the AtT20 cells caused a rapid, time-dependent decrease in the HLB 021-152 fluorescent signal (Figure 3). In contrast, addition of control solution produced only a small instantaneous rise in the fluorescence (Figure 3). Comparison of peak fluorescent values in wells injected with control and somatostatin solutions gave a Z′-factor of 0.7. The Z′-factor is used to quantify the reliability of the assay (Zhang et al., 1999). Z′-factors in the range of 0.5–1.0 indicate that the quality of the assay is excellent (Zhang et al., 1999). In order to quantify the GIRK channel fluorescent signal, the averaged control measurement, obtained in each 96-well plate, was subtracted from the records measured in the presence of somatostatin (Som – Con; Figure 3). This control-subtracted fluorescent signal (i.e., the somatostatin-sensitive component) was subsequently obtained and analyzed throughout the rest of the study. As shown in the top panel of Figure 3, somatostatin activated the AtT20 cell GIRK channel in a concentration dependent manner with a half-maximal effective concentration (EC50) of 4 nM. At a concentration of 200 nM somatostatin caused a fluorescent change (F/FO) of −0.088 (n = 6 wells). In comparison, addition of 10, 20, and 30 mM KCl, to depolarize the cell membrane, produced fluorescent changes of 0.058, 0.13, and 0.15, respectively (n = 6 wells for each KCl concentration). It was also determined if stimulation of other Gi-coupled GPCRs might activate the AtT20 cell GIRK channels. Application of the muscarinic agonist carbachol resulted in a peak fluorescent change of −0.037 (n = 6 wells; Figure 3, bottom panel). The EC50 for carbachol activation of the channel was 1 μM (Figure 3, bottom panel). In contrast, application of 10 μM adenosine caused no change in the fluorescent signal.
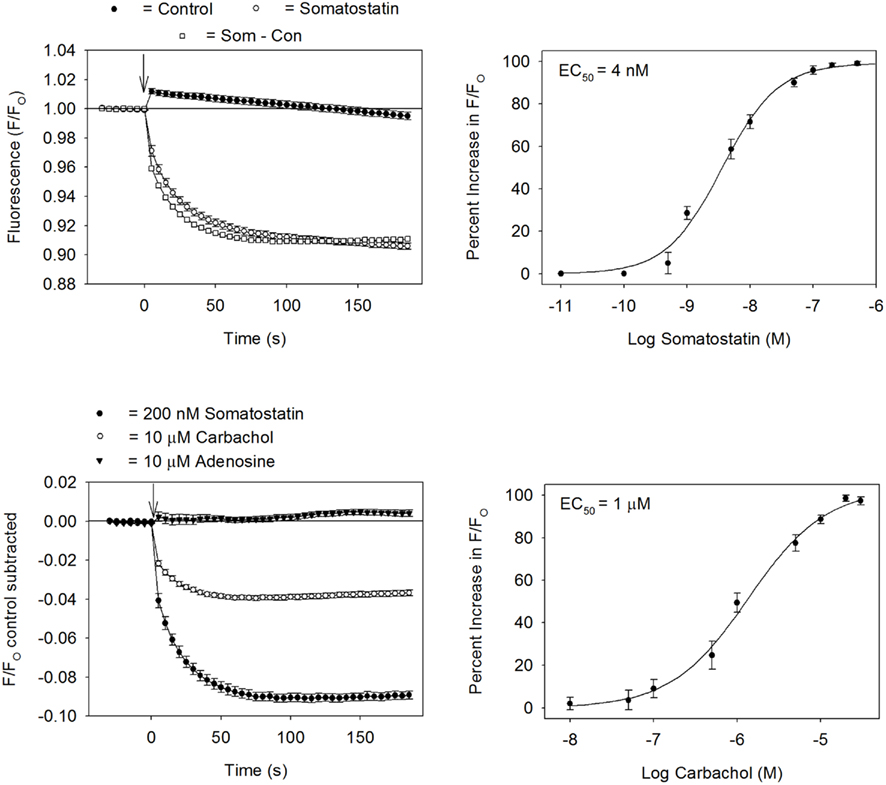
Figure 3. Development of a GIRK channel blocker screening assay. Top left panel: HLB 021-152 fluorescent intensities obtained in AtT20 cells in the presence or absence of somatostatin. The ratio of the fluorescent intensity (F/FO) was calculated by dividing the signal in the presence (F) of somatostatin (or control solution) by the baseline signal measured before (FO) addition of somatostatin (or control solution). Each point represents the mean ± SE obtained in six to eight wells. Somatostatin was added at time zero (↓). Top right panel: dose–response curve for the somatostatin-sensitive fluorescent signal (EC50 = 4 nM). Each point represents the mean ± SE of six measurements. Bottom left panel: changes in fluorescent signal obtained during injection of somatostatin, carbachol, and adenosine. Each point represents the mean ± SE obtained in six wells. Bottom right panel: dose–response curve for the carbachol-sensitive fluorescent signal (EC50 = 1 μM). Each point represents the mean ± SE of six measurements.
Identification of GIRK Channel Blockers in AtT20 Cells and Cardiac HL-1 Cells
It was next determined if the fluorescent assay system could be utilized for screening drugs that block GIRK channels. Inhibition of the GIRK channel should reduce somatostatin-mediated changes in Rm by preventing K+ efflux from the cells. To provide proof of concept, the AtT20 cells were incubated for 5 min with either BaCl2 or tertiapin-Q prior to somatostatin addition. As expected for a signal arising from GIRK channel activation, treatment with BaCl2 (2 mM) or tertiapin (500 nM) produced a complete inhibition of the somatostatin-mediated fluorescent change (Figure 4). BaCl2 at concentrations of 0.5 and 1 mM reduced the fluorescent change by 67 ± 4% and 92 ± 2% (n = 6 wells each), respectively.
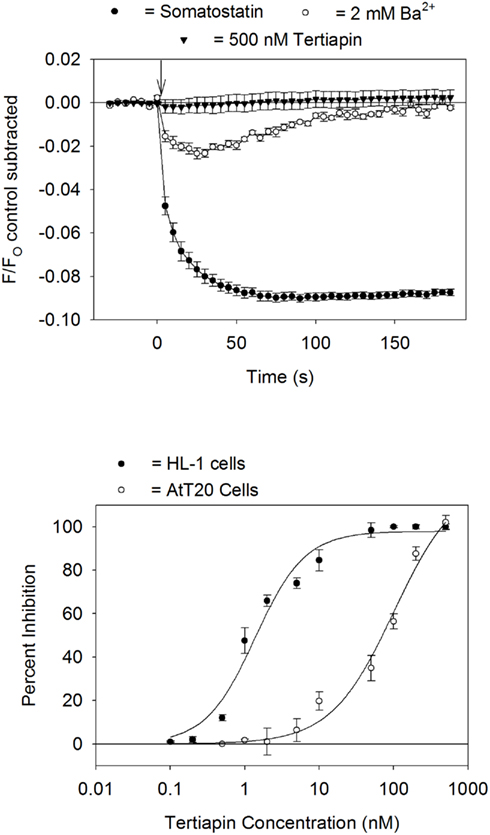
Figure 4. Block of GIRK channels by Ba2+ and tertiapin-Q. Top panel: somatostatin-sensitive fluorescent signal measured in AtT20 cells pretreated for 5 min with either 2 mM BaCl2 or 500 nM tertiapin-Q. Each point represents the mean ± SE value obtained in six wells in one plate. Bottom panel: dose versus response curve for tertiapin-Q block of the HL-1 cell (carbachol stimulation) or AtT20 cell (somatostatin stimulation) GIRK fluorescent signal. Each point represents the mean ± SE inhibition obtained from three to five experiments. Calculated IC50 values for tertiapin-Q block were HL-1 cells = 1.4 nM, AtT20 cells = 102 nM.
We previously demonstrated that the GIRK channel fluorescent assay could be utilized to study the pharmacology of the GIRK1/GIRK4 channel in cardiac HL-1 cells (Walsh, 2010). The HL-1 cells are an immortalized atrial cell line that displays an adult-like cardiac genotype and contracts spontaneously in cell culture (Whit et al., 2004). The cardiac GIRK channel is strongly blocked by tertiapin (Nobles et al., 2010; Walsh, 2010). In Figure 4 the concentration versus inhibition response of tertiapin-Q on the GIRK channel fluorescent signal was compared in the AtT20 and HL-1 cells. Tertiapin-Q was nearly a 100-fold more potent in blocking the GIRK channel in the HL-1 cells (IC50 = 1.4 nM) as compared to the AtT20 cells (IC50 = 102 nM).
G protein-coupled inward rectifier K+ channels are non-specifically blocked by a number of drugs including anti-arrhythmic agents, antipsychotic drugs, and antidepressants (Kobayashi and Ikeda, 2006). As a first step in expanding the limited pharmacology of the channel, the AtT20 and HL-1 cells were screened against a small compound library of Na+ and K+ channel modulators. Dose versus response curves were obtained for compounds producing a 50% or greater inhibition of the GPCR ligand-induced signal when tested at a concentration of 1 μM. Several ion transporter inhibitors were identified as GIRK channel blockers. The Na+/H+ exchange inhibitors 5-(N,N-hexamethylene)-amiloride (HMEA; Figure 5) and 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) blocked the GIRK channel with IC50s of 1 μM in both the AtT20 and HL-1 cells. In contrast, the parent compound amiloride had no blocking effect when tested up to a concentration of 10 μM. The gastric H+/K+ transport inhibitor SCH-28080 (2-methyl-8-(phenylmethoxy)imidazo[1,2-A]pyridine-3-acetonitrile) potently reduced the GPCR-sensitive fluorescent signal (IC50 = 0.2 μM for the AtT20 cells; IC50 = 0.3 μM for the HL-1 cells; Figure 5). However, no change in fluorescence was measured during treatment with the structurally related transport inhibitor omeprazole (Figure 5).
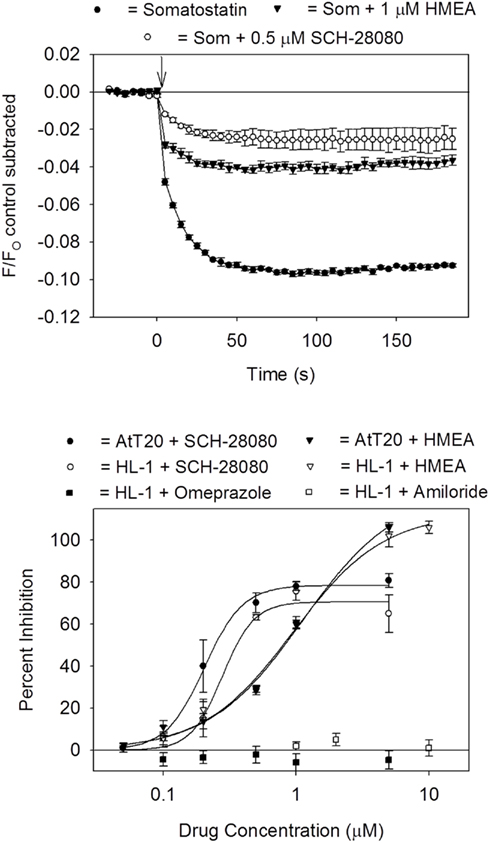
Figure 5. Block of GIRK channels by ion transporter inhibitors. Top panel: somatostatin-sensitive fluorescent signal measured in AtT20 cells pretreated for 5 min with drug vehicle solution (DMSO; Som) and either 1 μM 5-(N,N-hexamethylene)-amiloride (HMEA) or 0.5 μM SCH-28080. Each point represents the mean ± SE value obtained in six wells in one plate. Bottom panel: dose versus response curve for drug block of the somatostatin (AtT20 cells) or carbachol (HL-1 cells) signal. Each point represents the mean ± SE inhibition obtained from three to six experiments. Calculated IC50 values were HMEA = 1 μM (AtT20 and HL-1 cells) and SCH-28020 (200 nM for AtT20 cells and 300 nM for HL-1 cells).
Discussion
GIRK Channels as Targets for Drug Therapy
The goal of this study was to develop a screening assay for identifying modulators of neuronal and cardiac GIRK channels. For this purpose immortalized pituitary AtT20 and cardiac HL-1 cells were cultured in 96-well plates, loaded with an oxonol membrane potential-sensitive dye and analyzed with a fluorescent plate reader. Activation of the endogenous GPCRs in the cells, through application of either somatostatin or carbachol, caused a rapid, time-dependent decrease in the fluorescent signal. This change in the fluorescent signal was blocked by treatment with the GIRK channel blockers Ba2+ and tertiapin-Q. Thus, the GIRK channel fluorescent assay provides a fast, reliable, and inexpensive procedure for identifying drugs that modulate GIRK channels.
G protein-coupled inward rectifier K+ channel dysfunction has been implicated in the patho-physiology of a number of disorders including neuropathic pain, drug addiction, and cardiac arrhythmias. Support for a role of GIRK channels in these disorders has come primarily from studies with GIRK channel knockout mice. For example, GIRK channel knockout mice display a reduced sensitivity to analgesics (Lusscher and Slesinger, 2010). Dose–response curves for morphine-induced analgesia are shifted to higher doses in both GIRK2 and GIRK1 subunit knockout mice (Marker et al., 2004). Anti-nociceptive effects of ethanol, oxotremorine (muscarinic agonist), nicotine, baclofen (GABA agonist), clonidine (α2-adrenergic agonist), and WIN 55,212 (cannabinoid agonist) are also diminished in GIRK2 null mice (Blednov et al., 2003; Mitrovic et al., 2003). Kovoor et al. (2001) demonstrated that mice lacking the GIRK4 subunit are less susceptible to the development of atrial fibrillation. This finding is supported by recent studies indicating that the GIRK channel is constitutively activated in patients with atrial fibrillation (Dobrev et al., 2005). Therefore, although GIRK channels represent desirable targets for pharmacological therapy, drug development has been hampered by the absence of a cell-based screening system.
Development of a GIRK Channel Screening Assay
The traditional approach to ion channel drug discovery has involved the expression of recombinant channels in heterologous cell lines such HEK-293 and CHO cells. However, these cells often lack the relevant “physiological environment” that is present in native tissues, and required for normal pharmacological responses (Numann and Negulescu, 2001; Eglen and Reisine, 2010). Of particular concern, the expression profile of G proteins and ion channels in HEK-293 cells differs from that found in muscle and nerve (Numann and Negulescu, 2001; Eglen and Reisine, 2010). Thus, recent interest has focused on the suitability of primary cells, embryonic stem cells and clonal cell lines for drug discovery (Eglen and Reisine, 2010).
In the present study, GIRK channels were screened using the membrane potential-sensitive dye HLB 021-152. Membrane potential-sensitive dyes have previously been used to study Kir channels (Wolff et al., 2003). More recently, HEK-293 cells overexpressing GIRK channels were successfully utilized to screen for metabotropic glutamate receptor ligands using the thallium flux assay (Niswender et al., 2008). During the course of our experiments we performed preliminary experiments using the thallium flux assay with the fluorescent dye BTC–AM. Application of the thallium containing solution to the HL-1 and AtT20 cells produced large non-linear background increases in the BTC fluorescence. This background signal was larger than the subsequent fluorescent change produced by addition of the GPCR ligand. HL-1 cells express the ether-à-go-go related gene (ERG) channel and other K+ channels (Whit et al., 2004) that may allow thallium influx under basal conditions. Therefore, the thallium influx assay is not currently suitable for the AtT20 and HL-1 cell lines.
The AtT20 and Hl-1 cell lines represent ideal systems for GIRK channel drug discovery. AtT20 cells express the GIRK1 and GIRK2 subunits as well as SSTR2 and SSTR5. HL-1 cells express the GIRK1 and GIRK4 subunits along with the muscarinic type2 (M2) receptor. Whole-cell acetylcholine-activated K+ currents (IK,Ach; GIRK1/GIRK4 channel) are blocked by 100 nM tertiapin-Q in the HL-1 cells (Nobles et al., 2010; Walsh, 2010). In the present study, tertiapin-Q was nearly 100-fold more potent in blocking the HL-1 cell GIRK channel fluorescent signal (Figure 4). Lu and colleagues (Jin and Lu, 1998; Ramu et al., 2004) identified a short sequence of amino acids (approximately 10 amino acid residues), located in the M1–M2 linker of Kir subunits, that determines the binding affinity of tertiapin. Slight variances in this region greatly alter the potency of tertiapin in blocking Kir channels (Ramu et al., 2004). When expressed in Xenopus oocytes, homomeric GIRK1 and GIRK4 channel constructs are blocked by tertiapin-Q with Kd values of 20 μM and 2 nM, respectively (Ramu et al., 2004). Variances in this linker sequence may also account for the different IC50s that were measured with tertiapin in the AtT20 and HL-1 cells.
Despite the different potencies of GIRK channel inhibition produced by tertiapin-Q in the two cell lines, both cell types displayed a similar sensitivity to the ionic transport inhibitors HMEA, EIPA, and SCH-28080. GIRK channels are blocked by a wide range of pharmacological agents including volatile anesthetics (halothane, isoflurane, and enflurane), antipsychotic drug (clozapine, pimozide, and haloperidol), and antidepressants (imipramine, amitriptyline, and clomipramine; Kobayashi and Ikeda, 2006; Lusscher and Slesinger, 2010). Furthermore, a number of anti-arrhythmic agents including amiodarone, flecainide, quinidine, and propafenone block GIRK channels in primary cultures of atrial myocytes (Inomata et al., 1993; Watanabe et al., 1996; Hashimoto et al., 2006) and in HL-1 cells (Walsh, 2010). However, GIRK channel inhibition by these agents typically requires drug concentrations in the micromolar range. Recently, two benzopyran derivatives, NIP-151 and NTC-801, were demonstrated to block GIRK1/GIRK4 channels at nanomolar concentrations (Hashimoto et al., 2008; Machida et al., 2011). When tested in a canine model of atrial fibrillation, both compounds decreased atrial excitability and converted AF to sinus rhythm (Hashimoto et al., 2008; Machida et al., 2011). While the clinical efficacy of these agents is yet to be established, the results of these studies support the further development of new and selective GIRK channel modulators.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Ms. Charity Fix and Mr. David Stepp for their excellent technical assistance. This work was supported by US Public Health Service award NS-071530 and a grant from the University of South Carolina.
References
Blednov, Y. A., Stoffel, M., Alva, H., and Harris, R. A. (2003). A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc. Natl. Acad. Sci. U.S.A. 100, 277–282.
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., Knaut, M., and Ravens, U. (2005). The G protein-gated potassium current IK, Ach is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706.
Eglen, R., and Reisine, T. (2010). Primary cells and stem cells in drug discovery: emerging tools for high-throughput screening. Assay Drug Dev. Technol. 9, 108–124.
Hamill, O. P., Marty, A., Neher, E., Sakmann, B., and Sigworth, J. (1981). Improved patch-clamp techniques for high resolution current recordings from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100.
Hashimoto, N., Yamashita, T., and Nobutomo, T. (2008). Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IK, ACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J. Cardiovasc. Pharmacol. 51, 162–169.
Hashimoto, N., Yamashita, T., and Tsuruzoe, N. (2006). Tertiapin, a selective IK, Ach blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol. Res. 54, 136–141.
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inward rectifying potassium channels: their structure, function and physiological roles. Physiol. Rev. 90, 291–366.
Inomata, N., Ohno, T., Ishihara, K., and Akaike, N. (1993). Antiarrhythmic agents act differently on the activation phase of the Ach-response in guinea pig atrial myocytes. Br. J. Pharmacol. 108, 111–115.
Jin, W., and Lu, Z. (1998). A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 37, 13291–13299.
Kobayashi, T., and Ikeda, K. (2006). G protein-activated inwardly rectifying potassium channels as potential therapeutic targets. Curr. Pharm. Des. 12, 4513–4523.
Kovoor, P., Wickman, K., Maguire, C., Pu, W., Gehrmann, J., Berul, C., and Clapham, D. E. (2001). Evaluation of the role of IK, Ach in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143.
Kuzhikandathil, E. V., Yu, W., and Oxford, G. S. (1998). Human dopamine D3 and D2L receptors couple to inward rectifier potassium channels in mammalian cell lines. Mol. Cell. Neurosci. 12, 390–402.
Lusscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315.
Machida, T., Hashimoto, N., Kuwahara, I., Ogino, Y., Matsuura, J., Yamamoto, W., Itano, Y., Zamma, A., Matsumoto, R., Kamon, J., Kobayashi, T., Ishiwata, N., Yamashita, T., Ogura, T., and Nakaya, H. (2011). Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ. Arrhythm. Electrophysiol. 4, 94–102.
Mackie, K., Lai, Y., Westenbroek, R., and Mitchell, R. (1995). Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J. Neurosci. 15, 6552–6561.
Marker, C. L., Stoffel, M., and Wickman, K. (2004). Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J. Neurosci. 24, 2806–2812.
Mitrovic, I., Margeta-Mitrovic, M., Bader, S., Stoffel, M., Jan, L. Y., and Basbaum, A. I. (2003). Contribution of GIRK2-mediated postsynaptic signaling to opiate and α2-adrenergic analgesia and analgesic sex differences. Proc. Natl. Acad. Sci. U.S.A. 100, 271–276.
Niswender, C. M., Johnson, K. A., Luo, Q., Ayala, J. E., Kim, C., Conn, P. J., and Weaver, C. D. (2008). A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 73, 1213–1224.
Nobles, M., Sebastian, S., and Tinker, A. (2010). HL-1 cells express an inwardly rectifying K+ current activated via muscarinic receptors comparable to that in mouse atrial myocytes. Pflugers Arch. 460, 99–108.
Numann, R., and Negulescu, P. (2001). High throughput screening strategies for cardiac ion channels. Trends Cardiovasc. Med. 11, 54–59.
Ramu, Y., Klem, A. M., and Lu, Z. (2004). Short variable sequence acquired in evolution enables selective inhibition of various inward-rectifier K+ channels. Biochemistry 43, 10701–10709.
Walsh, K. B. (2010). A real-time screening assay for GIRK1/4 channel blockers. J. Biomol. Screen. 15, 1229–1237.
Walsh, K. B., and Zhang, J. (2008). Neonatal rat cardiac fibroblasts express three types of voltage-gated K+ channels: regulation of a transient outward current by protein kinase C. Am. J. Physiol. Cell Physiol. 294, H1010–H1017.
Watanabe, Y., Hara, Y., Tamagawa, M., and Nakaya, H. (1996). Inhibitory effect of amiodarone on the muscarinic acetylcholine receptor-operated potassium current in guinea pig atrial cells. J. Pharmacol. Exp. Ther. 279, 617–624.
Whit, S. M., Constantin, P. E., and Claycomb, W. C. (2004). Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am. J. Physiol. Heart Circ. Physiol. 286, H823–H829.
Wolff, C., Fuks, B., and Chatelain, P. (2003). Comparative study of membrane potential-sensitive fluorescent probes and their use in ion channel screening assays. J. Biomol. Screen. 8, 533–543.
Keywords: GIRK channel, clonal cell lines, drug screening, neuropathic pain, atrial fibrillation
Citation: Walsh KB (2011) Targeting GIRK channels for the development of new therapeutic agents. Front. Pharmacol. 2:64. doi: 10.3389/fphar.2011.00064
Received: 29 June 2011;
Accepted: 04 October 2011;
Published online: 31 October 2011.
Edited by:
Ralf Franz Kettenhofen, Axiogenesis AG, GermanyReviewed by:
Maurizio Taglialatela, University of Molise, ItalyOwen McManus, Johns Hopkins University, USA
Gregory John Kaczorowski, Kanalis Consulting, L.L.C., USA
Copyright: © 2011 Walsh. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Kenneth B. Walsh, Department of Pharmacology, Physiology and Neuroscience, School of Medicine, University of South Carolina, Columbia, SC 29208, USA. e-mail:d2Fsc2hAdXNjbWVkLnNjLmVkdQ==