 Teja Celhar
Teja Celhar- Singapore Immunology Network, Agency for Science, Technology and Research (A*STAR), Singapore, Singapore
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by the loss of tolerance to self-nuclear antigens. The symptoms of SLE, progression of pathology and the array of autoantibodies present in the serum differ significantly from patient to patient, which calls for a personalized approach to treatment. SLE is polygenic and strongly influenced by gender, ethnicity, and environmental factors. Data from genome-wide association studies suggests that polymorphisms in as many as 100 genes contribute to SLE susceptibility. Recent research has focused on genes associated with Toll-like receptors (TLRs), type I interferons, immune regulation pathways, and immune-complex clearance. TLR7 and TLR9 have been extensively studied using lupus-prone mouse models. In multiple systems overexpression of TLR7 drives disease progression but interestingly, a loss of TLR9 results in an almost identical phenotype. While TLR7 overexpression has been linked to human SLE, the possible role of TLR9 in human disease remains elusive. In the present review, we focus on TLR polymorphisms and TLR expression in SLE patients and discuss their potential as biomarkers for individualized treatment.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with a variety of clinical manifestations that differ from patient to patient. The heterogeneity of the symptoms represents a challenge for both diagnosis and treatment. While the diagnostic criteria for SLE have evolved over the past decades (reviewed in Yu et al., 2014), the treatment has remained largely symptomatic using non-specific conventional therapies which include non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, hydroxychloroquine (HCQ), and immunosuppressants (Murphy et al., 2013). In addition to their modest therapeutic effect, these drugs have severe side effects leading to substantial morbidity and mortality, resulting in a substantial economic burden to many societies (Lau and Mak, 2009). In this short review, we briefly discuss novel therapeutic agents and emerging immunological targets. We will focus on the burgeoning data on Toll-like receptor (TLR) 7 and TLR9 in SLE and on the factors that could influence their expression. We propose altered expression of TLR7/9 as a biomarker for identification of a subset of SLE patients that might benefit from a targeted therapeutic approach.
Recent Developments in B Cell Therapeutics
Recently, therapeutic avenues in SLE have pursued biologic drugs which either deplete B cells or reduce their activity. Belimumab (Benlysta®) is the only biological drug and the first therapeutic in 50 years to be approved by the Federal Drug Administration (FDA) for the treatment of lupus. Belimumab is a human monoclonal antibody (mAb) specific for B lymphocyte stimulator (BLyS) protein/B cell activating factor (BAFF; Hahn, 2011). Multicenter randomized controlled trials demonstrated a significant reduction in the Safety of Estrogen in Lupus Erythematosus National Assessment–SLE Disease Activity Index (SELENA–SLEDAI) score and risk of severe flares (Furie et al., 2011; Navarra et al., 2011). Recent results also suggest that belimumab may improve renal disease; however, further studies are needed to demonstrate a benefit in patients with severe active renal nephritis (Dooley et al., 2013). Based on the success of belimumab, other B cell targeting therapies are being assessed in clinical trials, including rituximab (anti-CD20), epratuzumab (anti-CD22), blisibimod and tabalumab (anti-BLyS), and atacicept [anti-BLyS/APRIL (A proliferation-inducing ligand); Kamal, 2014]. So far, the outcomes of B cell depletion with rituximab in clinical trials have been disappointing, partially due to poor trial design, problems with outcome measures and lack of long-term follow-up (Looney et al., 2010; Murphy et al., 2013; Kamal, 2014). In a randomized, double-blind, placebo-controlled Phase III trial rituximab successfully depleted B cells in lupus nephritis patients and decreased anti-dsDNA antibody levels, but failed to improve renal disease (Rovin et al., 2012). Since human prospective studies and mouse models have suggested a multi-step hypothesis to the development of SLE, it is perhaps unsurprising that reducing B cells has only moderate effects in end organ disease such as nephritis (Arbuckle et al., 2003; Fairhurst et al., 2006; Kanta and Mohan, 2009). Based on this hypothesis, the B cells have a critical role in autoantibody production due to initial loss of tolerance. However, an additional amplification step within the innate immune system is needed for the development of end organ disease.
Accordingly, new therapeutic approaches which focus on targeting innate immune cells are being developed (Figure 1).
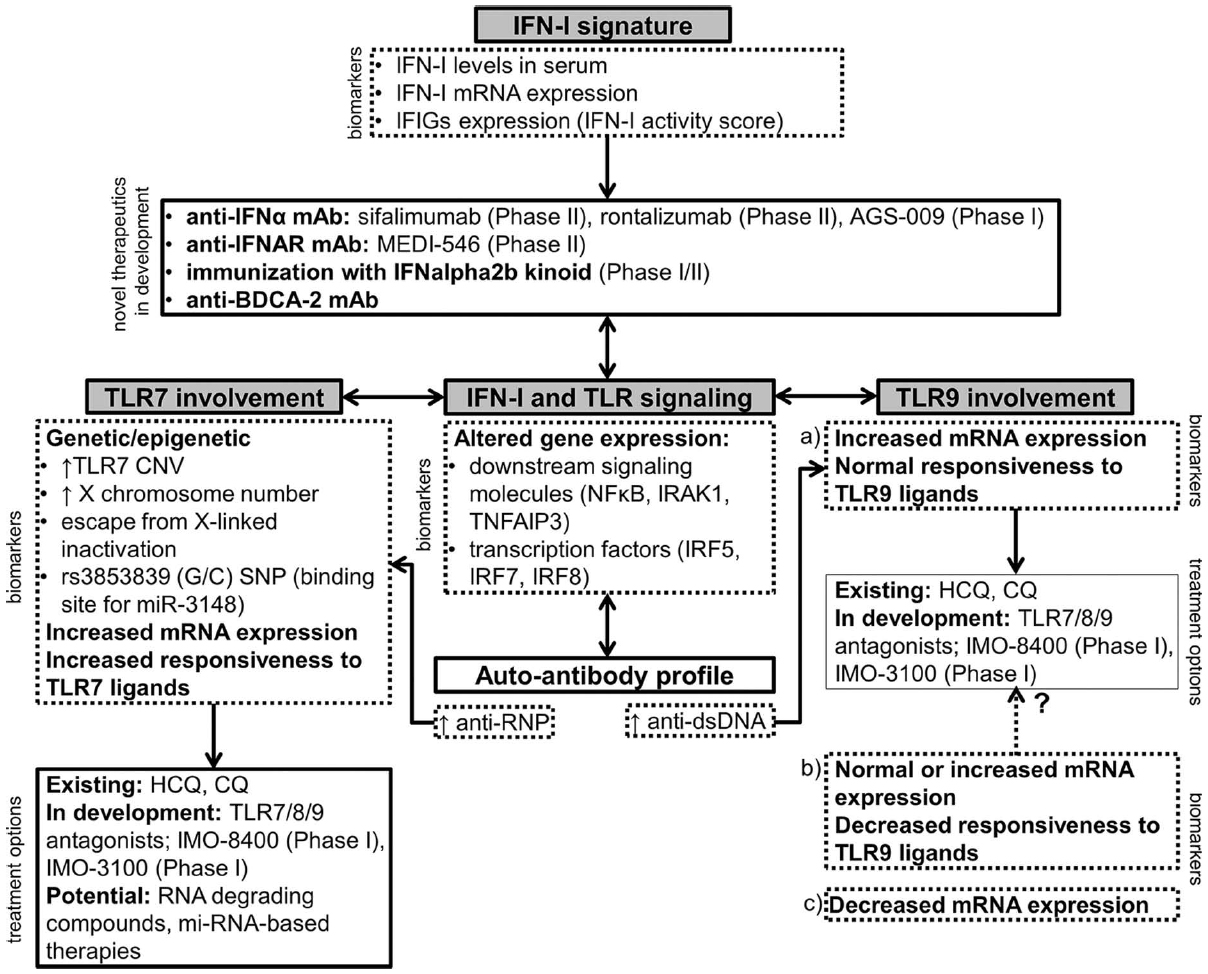
Figure 1. A schematic representation of novel therapeutic approaches in SLE involving the IFN-I signature, TLR7 and TLR9. The choice of the appropriate treatment option should ideally be based on the expression of relevant biomarkers in an individual patient. Drugs that are currently in clinical development are listed with the corresponding phase of the clinical trial (Clinicaltrials.gov, 2014). IFIGs, interferon-inducible genes; HCQ, hydroxychloroquine; CQ, chloroquine.
TLR7 as a Target for SLE Therapy
Immunological Evidence for TLRs in SLE
The type I-interferon (IFN-I) gene signature is one of the main immunological characteristics which was identified just over a decade ago by multiple groups (Baechler et al., 2003; Bennett et al., 2003; Crow, 2014). The increase in IFN-I-related genes was identified in the peripheral blood monocytes (PBMCs) from patients with SLE using gene expression profiling and has been identified in the majority of pediatric SLE patients and in the majority of adult SLE patients with active disease (Baechler et al., 2003; Bennett et al., 2003). Interestingly, the gene expression signature does not correlate with either elevated IFN-I levels in SLE serum, which is detectable only in a fraction of patients, or with elevated IFN-I mRNA (Blanco et al., 2001; Baechler et al., 2003). The reason for this discrepancy might be the low sensitivity of enzyme-linked immunosorbent assays (ELISAs) to detect serum IFN-I and the migration of IFN-I-producing cells to the tissues. However, there is also the possibility that the IFN-I signature is caused by another stimulus aside from IFN-I itself (Baechler et al., 2003; Bennett et al., 2003).
To overcome the limitations of serum IFN-I measurements “IFN-I activity score” assays were developed by several groups (Hua et al., 2006; Burgi Mde et al., 2012). These assays use serum or plasma of patients and the activity score strongly correlates with the titer of antinuclear autoantibodies (ANAs; Kirou et al., 2005; Hua et al., 2006; Niewold et al., 2007). High IFN-I activity score and the tendency to develop anti-ribonucleoprotein (RNP) and anti-dsDNA antibodies appear to be independent risk factors for SLE, since healthy first-degree relatives of SLE patients frequently display elevated IFN-I activity but no detectable ANAs (Niewold et al., 2007). The crucial connection between both traits is nucleic acid-sensing by endosomal TLRs, which evolved as sensors of foreign RNA and DNA (Blasius and Beutler, 2010; Kawai and Akira, 2010). In SLE, where self-nucleic acids are associated with autoantibodies in immune complexes (ICs), endosomal TLRs might become aberrantly activated in the absence of foreign molecules (reviewed in Celhar et al., 2012). Multiple in vitro studies in both mouse and human cells proved that RNA/DNA-containing ICs activate TLR9 and TLR7 through B cell receptor (BCR)-mediated internalization in B cells (Leadbetter et al., 2002; Lau et al., 2005) and through Fc gamma receptor (FcγR)-mediated internalization in dendritic cells (DCs; Boule et al., 2004), plasmacytoid DCs (pDCs; Bave et al., 2003; Barrat et al., 2005; Means et al., 2005; Vollmer et al., 2005; Lovgren et al., 2006), macrophages (Henault et al., 2012), and neutrophils (Garcia-Romo et al., 2011). Upon such activation, pDCs produce IFNα (Bave et al., 2003; Barrat et al., 2005; Means et al., 2005; Vollmer et al., 2005; Lovgren et al., 2006), conventional DCs produce cytokines (Boule et al., 2004) and neutrophils release neutrophil extracellular traps (NETs; Garcia-Romo et al., 2011). These findings highlight the central role of TLR7 and TLR9 in the induction and modulation of immune responses in SLE, and their association with the IFN-I signature (recently reviewed in Kono et al., 2013; Shrivastav and Niewold, 2013; Crow, 2014). Genome-wide association studies (GWAS) have provided supporting evidence by identifying SLE susceptibility variants in both TLR and IFN-I pathways (Rullo and Tsao, 2013).
Genetic Evidence: Polymorphisms in TLR7-Associated Pathways
Overexpression of TLR7 causes severe lupus in multiple mouse models of SLE (reviewed in Celhar et al., 2012). Male BXSB lupus mice develop severe disease due to a translocation of a segment near the pseudoautosomal region of the X chromosome onto the Y chromosome, identified as the y-linked autoimmune accelerating (yaa) locus (Pisitkun et al., 2006; Subramanian et al., 2006). Among the duplicated genes, TLR7 was identified as the major gene responsible for the development of severe disease (Deane et al., 2007; Fairhurst et al., 2008; Santiago-Raber et al., 2008). Genetically modified mouse models have proven to be an indispensable tool for the study of single gene modifications. However, correlations with the highly genetically variable human population are rarely straightforward. This is particularly true for polygenic disorders such as SLE, where multiple genes are associated with disease susceptibility (Rullo and Tsao, 2013; Armstrong et al., 2014). Genomic data suggests that common copy number variations (CNVs) similar to yaa are very rare in human SLE (Conrad et al., 2010; Park et al., 2010; Shen et al., 2010). Nevertheless, they may be an important risk factor childhood-onset SLE, as recently shown in the Mexican population (Garcia-Ortiz et al., 2010). Male patients with more than one copy and female patients with greater than two copies of TLR7 had a higher disease susceptibility and the TLR7 copy number correlated with TLR7 mRNA expression levels (Garcia-Ortiz et al., 2010). In addition to altered gene copy number, the gene dosage of TLR7 can be altered in individuals affected by aneuploidy. Indeed, SLE is more common in men affected by conditions with additional X chromosomes, such as Klinefelter’s syndrome (47,XXY; Ortiz-Neu and LeRoy, 1969; Dillon et al., 2012).
Overall, however, it is unlikely that a single gene, in this case TLR7, would have such a large impact. Consistent with this theory, recent investigations have identified polymorphisms in genes that are shared among TLR and IFN-I signaling pathways, including the transcription factors interferon regulatory factor (IRF) 5, IRF7, and IRF8 and components of the downstream NFκB pathway, such as interleukin-1 receptor-associated kinase 1 (IRAK1) and tumor necrosis factor, alpha-induced protein 3 (TNFAIP3; Rullo and Tsao, 2013). Therefore, polymorphisms in several genes could ultimately together lead to an excessive response to RNA sensing by TLR7. Additionally, variants in regulatory single nucleotide polymorphisms (SNPs) which bind microRNA (miRNA), X-chromosome linked modifications and induction of TLR7 expression by various infectious agents may be involved in subgroups of SLE patients.
X-Chromosome Linked Alterations of TLR7 Expression
Recent findings suggest that approximately 15% of genes on the X chromosome escape X-linked inactivation and are thus bi-allelicially expressed, ultimately increasing gene dosage (reviewed in Berletch et al., 2011). X-linked genes are particularly interesting in dissecting out causes of conditions that predominately affect women, including autoimmune diseases and some viral infections such as HIV and herpes simplex virus (HSV; Fish, 2008). Several studies have attributed these sexual dimorphisms to differential RNA-sensing by TLR7, since female immune cells produce higher levels of IFNα upon stimulation with TLR7 ligands or virus-derived RNA compared to male counterparts (Berghofer et al., 2006; Meier et al., 2009; Torcia et al., 2012). The underling mechanism for this sex bias is currently not clear. Berghofer et al. (2006) found equivalent TLR7 mRNA expression in pDCs and B cells from healthy males and females with no evidence of escaping X-linked inactivation or involvement of estrogen receptor signaling. However, these findings do not exclude escape from X-linked inactivation in SLE patients.
Epigenetic Factors and microRNA Binding
It has now been established that miRNAs can act to fine tune TLR signaling by targeting expression itself or by modulating adaptor molecules, downstream regulators and cytokines (reviewed in O’Neill et al., 2011). Variants in the 3′ untranslated region (UTR) regions may alter miRNAs binding and ultimately TLR7 expression and/or responsiveness. A large multi-centered and multi-ethnic study identified such SNP in the 3′ UTR region of TLR7 (rs3853839 (G/C)) as a risk factor for SLE (Shen et al., 2010; Deng et al., 2013). The G-allele carriers have increased TLR7 transcripts and are more likely to have anti-RNA associated autoantibodies than C-allele carriers (Shen et al., 2010). The non-risk C allele bears a binding site of microRNA-3148 (miR-3148), which confers faster degradation of the transcript and thus lower levels of TLR7 gene product (Deng et al., 2013).
Induction of TLR7 Expression
Several studies have reported the induction of TLR7 transcription in immune cells (B cells, eosinophils, monocytes, macrophages, pDCs) following stimulation with various inflammatory agents, including bacteria (Zarember and Godowski, 2002; Bourke et al., 2003; Miettinen et al., 2008), viruses (Miettinen et al., 2001) CpG (Hornung et al., 2002; Bourke et al., 2003), IFNα (Miettinen et al., 2001; Sirén et al., 2005), and IFNγ (Miettinen et al., 2001; Nagase et al., 2003). This increase in TLR7 during infection may be part of a positive feedback mechanism to increase IFNα, which is essential for a fast and robust anti-microbial response (Rönnblom et al., 2006). It is possible that this pathway is deregulated in SLE patients and explains why many features of SLE resemble a chronic viral infection in the absence of a detectable virus (Crow, 2014). Indeed, upregulated TLR7 and TLR9 mRNA expression have been reported in PBMCs from SLE patients and levels correlate with the expression of IFNα (Komatsuda et al., 2008; Lyn-Cook et al., 2014). Upregulation of TLR7, but not other TLRs, has also been observed when healthy neutrophils were cultured with sera from SLE patients with active disease (Garcia-Romo et al., 2011). This may be due to IFNα present in patient’s sera, since pre-treatment with purified IFNα resulted in a similar response of increased TLR7. Furthermore, IFNα increased susceptibility to anti-RNP antibody-induced NETosis (Garcia-Romo et al., 2011). Consistent with these findings, monocyte-derived macrophages produce IFNα upon TLR7/8 ligand stimulation only after pre-treatment with IFNα and subsequent induction of TLR7 (Sirén et al., 2005). TLR7 may also be induced by serum-derived ICs containing TLR7 ligands. A recent study by Chauhan et al. (2013) showed that TLR7 was preferentially increased in SLE patients with antibodies against RNA-associated antigens, while TLR9 induction correlated with anti-dsDNA antibody titers. Moreover, flu and synthetic TLR7 ligands have the capacity to induce early IFN-inducible genes in pDCs independently of IFN-α production (Di Domizio et al., 2009). Overall, accumulating data suggest that persistently higher levels of TLR7 may lead to an acquired responsiveness in cells which are normally unresponsive to TLR7 ligands. This is supported by the observation that pDCs are responsible for the majority IFNα produced by healthy PBMCs, but account only for 57% of IFNα produced by PBMCs from SLE patients (Blanco et al., 2001).
TLR9 as a Target for Therapy for SLE
While TLR7 hyper-responsiveness in human SLE is consistent with the TLR7-associated nephritis shown by mouse models, the role of TLR9 remains controversial. Multiple mouse studies have shown the importance of TLR9 expression in B cells for the generation of anti-dsDNA, anti-chromatin, and anti-nucleosome autoantibodies (Christensen et al., 2005, 2006; Lartigue et al., 2006; Yu et al., 2006; Nickerson et al., 2010). However, the deletion of TLR9 in these lupus-prone models did not lead to amelioration, but rather to exacerbation of disease, suggesting a protective/regulatory role of TLR9 in cells other than B cells. Nonetheless, the manifestation of the disease in TLR9-deficient mice was dependent on TLR7 expression (Nickerson et al., 2010).
Increases in TLR9 expression have been shown in PBMCs from SLE patients and levels usually correlate with IFNα expression and anti-dsDNA antibodies (Komatsuda et al., 2008; Mu et al., 2012; Chauhan et al., 2013; Lyn-Cook et al., 2014). Additionally, B cells and monocytes from patients with active disease express higher TLR9 levels compared to patients with inactive disease (Papadimitraki et al., 2006; Nakano et al., 2008). An increase in the frequency of TLR9-expressing B cells, but not monocytes, correlated with anti-dsDNA antibodies (Papadimitraki et al., 2006). These results suggest that, similarly to murine studies, increased TLR9 expression in B cells may be involved in autoantibody production, especially anti-dsDNA
However, paradoxically, the increased expression of TLR9 does not lead to increased responsiveness to TLR9 ligands. Despite an increase in TLR9 on B cells and BDCA3+ DCs from patients with severe disease, cells were less activated and hyporesponsive to TLR9 stimulation (Zorro et al., 2009). Similarly, despite an increased TLR9 expression in SLE PBMCs, TLR9-induced IFN-α production was markedly reduced (Kwok et al., 2008). This desensitization has been directly attributed to HCQ treatment (Kwok et al., 2008; Sacre et al., 2012). However, in patients not taking HCQ, alterations in downstream TLR9-signaling or induction of negative regulators may be involved (Kwok et al., 2008; Zorro et al., 2009).
Limited data is available on the effect of decreased TLR9 expression in human SLE. A recent study conducted in a cohort of Danish SLE patients found significantly decreased TLR9 mRNA expression in lupus PBMCs, which did not correlate with dsDNA antibody titers (Laska et al., 2014). It would be interesting to further identify the main cell type within the PBMCs responsible for the downregulation and assess the responsiveness to TLR9 ligands in this cohort.
Overall, further research is required to establish the role of TLR9 in mediating the progression of benign autoimmunity to severe disease.
TLR9 Polymorphisms
Several TLR9 polymorphisms have been associated with SLE, particularly in patients of Asian or European ancestry (Tao et al., 2007; Xu et al., 2009; Lu et al., 2011; dos Santos et al., 2012; Huang et al., 2012; Piotrowski et al., 2013; Laska et al., 2014). However, these results were not confirmed by other studies or by a recent meta-analysis (Hur et al., 2005; Ng et al., 2005; De Jager et al., 2006; Yang et al., 2012). Similarly, as mentioned earlier for TLR7, polymorphisms in a single gene are not very likely to recapitulate a polygenic disease like SLE. Additional genetic variants in TLR9-downstream signaling pathways, regulators of TLR signaling and epigenetic modifications should also be considered and need further examination.
Development of Novel Therapeutics for SLE
Novel Therapies Directed Toward IFNα
IFNα signaling may be suppressed by several strategies: direct neutralization by an anti-IFNα mAb, suppression of the IFN-I signature using an anti-IFNα receptor (IFNAR) antibody, or immunization to achieve endogenous anti-IFNα production (reviewed in Crow, 2014). Three anti-IFN-α mAbs, sifalimumab, AGS-009, and rontalizumab, have achieved safety and dose-dependent reduction of IFN-I signature in Phase I SLE trials (Yao et al., 2009; Merrill et al., 2011; Petri et al., 2013; Stohl, 2013). Furthermore, a Phase II trial has shown that rontalizumab decreased the incidence of flares and reduced prednisone dosage in a subgroup of patients who had a low IFN-I signature at baseline (Stohl, 2013).
Targeting the IFNAR with MEDI-546, a fully human mAb against huIFNAR, achieved success in Phase I trials for systemic sclerosis (SSc; Wang et al., 2013). Pharmacogenomics and translational simulations were used to build a model to predict responses to MEDI-546 in SLE patients and facilitated the progression of this candidate molecule to a Phase II trial (Wang et al., 2013).
Immunization with human (hu) IFNalpha2b kinoid (IFN-K) generates endogenous polyclonal antibodies directed against all 13 subtypes of huIFNα (Mathian et al., 2011). The results from the first clinical study show that IFN-K is well tolerated and significantly reduces the IFN-I signature in IFN signature-positive patients (Lauwerys et al., 2013).
An additional approach is the blockage of BDCA-2 expressed on the surface of pDCs. Anti-BDCA-2 mAb successfully inhibited IFN-α production from healthy donors and SLE patients (Dzionek et al., 2001; Blomberg et al., 2003). A patent application has recently been submitted for an anti-BDCA-2 antibody in the context of preventing or treating pathological conditions involving pDC activation (De and Fournier, 2012).
TLR Inhibition in SLE
Several compounds that bind endosomal TLR7 and/or TLR9 activation have been developed with the goal to inhibit IFNα production and activation of autoimmune B cells (Barrat and Coffman, 2008; Kandimalla et al., 2013). This approach successfully ameliorated disease in lupus-prone mice but it needs further clinical evaluation (Barrat et al., 2007). IMO-3100, a TLR7/9 antagonist, and IMO-8400, a TLR7/8/9 antagonist, suppressed inflammation in a mouse model of psoriasis (Suarez-Farinas et al., 2013). Both compounds were well tolerated by healthy adults and are now undergoing Phase 2 trials in psoriasis patients (Clinicaltrials.gov, 2014). It is important to note that the therapeutic value of the standard SLE care drugs HCQ and chloroquine have been attributed to indirect inhibition of TLR7 and TLR9, through their inhibitory action on endosomal acidification which is necessary for TLR7/9 activation (Sacre et al., 2012).
Finally, miRNA-based therapies and therapeutics design to degrade RNA may also represent potential treatment options in specific groups of SLE patients (Deng et al., 2013; Sun et al., 2013).
Conclusion
Advances in the understanding of the innate immune system have revolutionized the development of novel SLE therapies. Owing to the heterogeneous nature of the clinical manifestations, much thought is being given to a stratification of lupus patients based on clinical symptoms and immunological dysfunctions, targeting disease development in a personalized medicine approach. This is particularly important for the design of clinical trials, where the correct recruitment and sub-grouping of patients may directly impact the outcome measures. Pharmacogenomic and pharmacogenetic studies will ultimately be essential to identify patients that will most likely benefit from a specific treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arbuckle, M. R., Mcclain, M. T., Rubertone, M. V., Scofield, R. H., Dennis, G. J., James, J. A., et al. (2003). Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 349, 1526–1533. doi: 10.1056/NEJMoa021933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Armstrong, D. L., Zidovetzki, R., Alarcon-Riquelme, M. E., Tsao, B. P., Criswell, L. A., Kimberly, R. P., et al. (2014). GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 15, 347–354. doi: 10.1038/gene.2014.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baechler, E. C., Batliwalla, F. M., Karypis, G., Gaffney, P. M., Ortmann, W. A., Espe, K. J., et al. (2003). Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U.S.A. 100, 2610–2615. doi: 10.1073/pnas.0337679100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrat, F. J., and Coffman, R. L. (2008). Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol. Rev. 223, 271–283. doi: 10.1111/j.1600-065X.2008.00630.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrat, F. J., Meeker, T., Chan, J. H., Guiducci, C., and Coffman, R. L. (2007). Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol. 37, 3582–3586. doi: 10.1002/eji.200737815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrat, F. J., Meeker, T., Gregorio, J., Chan, J. H., Uematsu, S., Akira, S., et al. (2005). Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202, 1131–1139. doi: 10.1084/jem.20050914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bave, U., Magnusson, M., Eloranta, M. L., Perers, A., Alm, G. V., and Ronnblom, L. (2003). Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 171, 3296–3302. doi: 10.4049/jimmunol.171.6.3296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennett, L., Palucka, A. K., Arce, E., Cantrell, V., Borvak, J., Banchereau, J., et al. (2003). Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723. doi: 10.1084/jem.20021553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berghofer, B., Frommer, T., Haley, G., Fink, L., Bein, G., and Hackstein, H. (2006). TLR7 ligands induce higher IFN-alpha production in females. J. Immunol. 177, 2088–2096. doi: 10.4049/jimmunol.177.4.2088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berletch, J. B., Yang, F., Xu, J., Carrel, L., and Disteche, C. M. (2011). Genes that escape from X inactivation. Hum. Genet. 130, 237–245. doi: 10.1007/s00439-011-1011-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blanco, P., Palucka, A. K., Gill, M., Pascual, V., and Banchereau, J. (2001). Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294, 1540–1543. doi: 10.1126/science.1064890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blasius, A. L., and Beutler, B. (2010). Intracellular Toll-like receptors. Immunity 32, 305–315. doi: 10.1016/j.immuni.2010.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blomberg, S., Eloranta, M. L., Magnusson, M., Alm, G. V., and Ronnblom, L. (2003). Expression of the markers BDCA-2 and BDCA-4 and production of interferon-alpha by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. 48, 2524–2532. doi: 10.1002/art.11225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boule, M. W., Broughton, C., Mackay, F., Akira, S., Marshak-Rothstein, A., and Rifkin, I. R. (2004). Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J. Exp. Med. 199, 1631–1640. doi: 10.1084/jem.20031942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bourke, E., Bosisio, D., Golay, J., Polentarutti, N., and Mantovani, A. (2003). The Toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood 102, 956–963.
Burgi Mde, L., Prieto, C., Etcheverrigaray, M., Kratje, R., Oggero, M., and Bollati-Fogolin, M. (2012). WISH cell line: from the antiviral system to a novel reporter gene assay to test the potency of human IFN-alpha and IFN-beta. J. Immunol. Methods 381, 70–74. doi: 10.1016/j.jim.2012.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Celhar, T., Magalhaes, R., and Fairhurst, A. M. (2012). TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol. Res. 53, 58–77. doi: 10.1007/s12026-012-8270-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chauhan, S. K., Singh, V. V., Rai, R., Rai, M., and Rai, G. (2013). Distinct Autoantibody Profiles in Systemic Lupus Erythematosus Patients are Selectively Associated with TLR7 and TLR9 Upregulation. J. Clin. Immunol. 33, 954–964. doi: 10.1007/s10875-013-9887-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christensen, S. R., Kashgarian, M., Alexopoulou, L., Flavell, R. A., Akira, S., and Shlomchik, M. J. (2005). Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med. 202, 321–331. doi: 10.1084/jem.20050338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christensen, S. R., Shupe, J., Nickerson, K., Kashgarian, M., Flavell, R. A., and Shlomchik, M. J. (2006). Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428. doi: 10.1016/j.immuni.2006.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clinicaltrials.gov (2014). Available at: https://clinicaltrials.gov [accessed November 21, 2014].
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y., et al. (2010). Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712. doi: 10.1038/nature08516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crow, M. K. (2014). Type I interferon in the pathogenesis of lupus. J. Immunol. 192, 5459–5468. doi: 10.4049/jimmunol.1002795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, J. A., Pisitkun, P., Barrett, R. S., Feigenbaum, L., Town, T., Ward, J. M., et al. (2007). Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27, 801–810. doi: 10.1016/j.immuni.2007.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Jager, P. L., Richardson, A., Vyse, T. J., and Rioux, J. D. (2006). Genetic variation in Toll-like receptor 9 and susceptibility to systemic lupus erythematosus. Arthritis Rheum. 54, 1279–1282. doi: 10.1002/art.21755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De, R. C., and Fournier, N. (2012). Use of an Antibody Directed Against a Membrane Protein. Google Patents.
Deng, Y., Zhao, J., Sakurai, D., Kaufman, K. M., Edberg, J. C., Kimberly, R. P., et al. (2013). MicroRNA-3148 modulates allelic expression of Toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 9:e1003336. doi: 10.1371/journal.pgen.1003336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Domizio, J., Blum, A., Gallagher-Gambarelli, M., Molens, J. P., Chaperot, L., Plumas, J. (2009). TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood 114, 1794–1802. doi: 10.1182/blood-2009-04-216770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dillon, S. P., Kurien, B. T., Li, S., Bruner, G. R., Kaufman, K. M., Harley, J. B., et al. (2012). Sex chromosome aneuploidies among men with systemic lupus erythematosus. J. Autoimmun. 38, J129–J134. doi: 10.1016/j.jaut.2011.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dooley, M. A., Houssiau, F., Aranow, C., D’Cruz, D. P., Askanase, A., Roth, D. A., et al. (2013). Effect of belimumab treatment on renal outcomes: results from the phase 3 belimumab clinical trials in patients with SLE. Lupus 22, 63–72. doi: 10.1177/0961203312465781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
dos Santos, B. P., Valverde, J. V., Rohr, P., Monticielo, O. A., Brenol, J. C., Xavier, R. M., et al. (2012). TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 21, 302–309. doi: 10.1177/0961203311425522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dzionek, A., Sohma, Y., Nagafune, J., Cella, M., Colonna, M., Facchetti, F., et al. (2001). BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 194, 1823–1834. doi: 10.1084/jem.194.12.1823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fairhurst, A. M., Hwang, S. H., Wang, A., Tian, X. H., Boudreaux, C., Zhou, X. J., et al. (2008). Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur. J. Immunol. 38, 1971–1978. doi: 10.1002/eji.200838138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fairhurst, A. M., Wandstrat, A. E., and Wakeland, E. K. (2006). Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv. Immunol. 92, 1–69. doi: 10.1016/S0065-2776(06)92001-X
Fish, E. N. (2008). The X-files in immunity: sex-based differences predispose immune responses. Nat. Rev. Immunol. 8, 737–744. doi: 10.1038/nri2394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Furie, R., Petri, M., Zamani, O., Cervera, R., Wallace, D. J., Tegzova, D., et al. (2011). A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 63, 3918–3930. doi: 10.1002/art.30613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia-Ortiz, H., Velazquez-Cruz, R., Espinosa-Rosales, F., Jimenez-Morales, S., Baca, V., and Orozco, L. (2010). Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 69, 1861–1865. doi: 10.1136/ard.2009.124313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia-Romo, G. S., Caielli, S., Vega, B., Connolly, J., Allantaz, F., Xu, Z., et al. (2011). Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3:73ra20. doi: 10.1126/scitranslmed.3001201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hahn, B. H. (2011). Targeted therapies in systemic lupus erythematosus: successes, failures and future. Ann. Rheum. Dis. 70(Suppl. 1), i64–i66. doi: 10.1136/ard.2010.142208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henault, J., Martinez, J., Riggs, J. M., Tian, J., Mehta, P., Clarke, L., et al. (2012). Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37, 986–997. doi: 10.1016/j.immuni.2012.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hornung, V., Rothenfusser, S., Britsch, S., Krug, A., Jahrsdörfer, B., Giese, T., et al. (2002). Quantitative expression of Toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168, 4531–4537.
Hua, J., Kirou, K., Lee, C., and Crow, M. K. (2006). Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 54, 1906–1916. doi: 10.1002/art.21890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, C. M., Huang, P. H., Chen, C. L., Lin, Y. J., Tsai, C. H., Huang, W. L., et al. (2012). Association of Toll-like receptor 9 gene polymorphism in Chinese patients with systemic lupus erythematosus in Taiwan. Rheumatol. Int. 32, 2105–2109. doi: 10.1007/s00296-011-1925-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hur, J. W., Shin, H. D., Park, B. L., Kim, L. H., Kim, S. Y., and Bae, S. C. (2005). Association study of Toll-like receptor 9 gene polymorphism in Korean patients with systemic lupus erythematosus. Tissue Antigens 65, 266–270. doi: 10.1111/j.1399-0039.2005.00374.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kamal, A. (2014). The efficacy of novel B cell biologics as the future of SLE treatment: a review. Autoimmun. Rev. doi: 10.1016/j.autrev.2014.08.020 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kandimalla, E. R., Bhagat, L., Wang, D., Yu, D., Sullivan, T., La Monica, N., et al. (2013). Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 41, 3947–3961. doi: 10.1093/nar/gkt078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanta, H., and Mohan, C. (2009). Three checkpoints in lupus development: central tolerance in adaptive immunity, peripheral amplification by innate immunity and end-organ inflammation. Genes Immun. 10, 390–396. doi: 10.1038/gene.2009.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirou, K. A., Lee, C., George, S., Louca, K., Peterson, M. G., and Crow, M. K. (2005). Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 52, 1491–1503. doi: 10.1002/art.21031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Komatsuda, A., Wakui, H., Iwamoto, K., Ozawa, M., Togashi, M., Masai, R., et al. (2008). Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 152, 482–487. doi: 10.1111/j.1365-2249.2008.03646.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kono, D. H., Baccala, R., and Theofilopoulos, A. N. (2013). TLRs and interferons: a central paradigm in autoimmunity. Curr. Opin. Immunol. 25, 720–727. doi: 10.1016/j.coi.2013.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kwok, S. K., Lee, J. Y., Park, S. H., Cho, M. L., Min, S. Y., Kim, H. Y., et al. (2008). Dysfunctional interferon-alpha production by peripheral plasmacytoid dendritic cells upon Toll-like receptor-9 stimulation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 10, R29. doi: 10.1186/ar2382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lartigue, A., Courville, P., Auquit, I., Francois, A., Arnoult, C., Tron, F., et al. (2006). Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J. Immunol. 177, 1349–1354. doi: 10.4049/jimmunol.177.2.1349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laska, M. J., Troldborg, A., Hansen, B., Stengaard-Pedersen, K., Junker, P., Nexo, B. A., et al. (2014). Polymorphisms within Toll-like receptors are associated with systemic lupus erythematosus in a cohort of Danish females. Rheumatology (Oxford) 53, 48–55. doi: 10.1093/rheumatology/ket316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lau, C. M., Broughton, C., Tabor, A. S., Akira, S., Flavell, R. A., Mamula, M. J., et al. (2005). RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 202, 1171–1177. doi: 10.1084/jem.20050630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lau, C. S., and Mak, A. (2009). The socioeconomic burden of SLE. Nat. Rev. Rheumatol. 5, 400–404. doi: 10.1038/nrrheum.2009.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauwerys, B. R., Hachulla, E., Spertini, F., Lazaro, E., Jorgensen, C., Mariette, X., et al. (2013). Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis Rheum. 65, 447–456. doi: 10.1002/art.37785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. M., Beaudette, B. C., Shlomchik, M. J., and Marshak-Rothstein, A. (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. doi: 10.1038/416603a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Looney, R. J., Anolik, J., and Sanz, I. (2010). A perspective on B-cell-targeting therapy for SLE. Mod. Rheumatol. 20, 1–10. doi: 10.1007/s10165-009-0213-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lovgren, T., Eloranta, M. L., Kastner, B., Wahren-Herlenius, M., Alm, G. V., and Ronnblom, L. (2006). Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjögren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 54, 1917–1927. doi: 10.1002/art.21893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, K. C., Yang, H. Y., Lin, Y. F., Kao, S. Y., Lai, C. H., Chu, C. M., et al. (2011). The T-1237C polymorphism of the Toll-like receptor-9 gene is associated with chronic kidney disease in a Han Chinese population. Tohoku J. Exp. Med. 225, 109–116. doi: 10.1620/tjem.225.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lyn-Cook, B. D., Xie, C., Oates, J., Treadwell, E., Word, B., Hammons, G., et al. (2014). Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol. Immunol. 61, 38–43. doi: 10.1016/j.molimm.2014.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mathian, A., Amoura, Z., Adam, E., Colaone, F., Hoekman, M. F., Dhellin, O., et al. (2011). Active immunisation of human interferon alpha transgenic mice with a human interferon alpha Kinoid induces antibodies that neutralise interferon alpha in sera from patients with systemic lupus erythematosus. Ann. Rheum. Dis. 70, 1138–1143. doi: 10.1136/ard.2010.141101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Means, T. K., Latz, E., Hayashi, F., Murali, M. R., Golenbock, D. T., and Luster, A. D. (2005). Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115, 407–417. doi: 10.1172/JCI23025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meier, A., Chang, J. J., Chan, E. S., Pollard, R. B., Sidhu, H. K., Kulkarni, S., et al. (2009). Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 15, 955–959. doi: 10.1038/nm.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merrill, J. T., Wallace, D. J., Petri, M., Kirou, K. A., Yao, Y., White, W. I., et al. (2011). Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 70, 1905–1913. doi: 10.1136/ard.2010.144485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miettinen, M., Sareneva, T., Julkunen, I., and Matikainen, S. (2001). IFNs activate Toll-like receptor gene expression in viral infections. Genes Immun. 2, 349–355.
Miettinen, M., Veckman, V., Latvala, S., Sareneva, T., Matikainen, S., and Julkunen, I. (2008). Live Lactobacillus rhamnosus and Streptococcus pyogenes differentially regulate Toll-like receptor (TLR) gene expression in human primary macrophages. J. Leukoc. Biol. 84, 1092–1100. doi: 10.1189/jlb.1206737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mu, R., Sun, X. Y., Lim, L. T., Xu, C. H., Dai, C. X., Su, Y., et al. (2012). Toll-like receptor 9 is correlated to disease activity in Chinese systemic lupus erythematosus population. Chin. Med. J. (Engl.) 125, 2873–2877. doi: 10.3760/cma.j.issn.0366-6999.2012.16.014
Murphy, G., Lisnevskaia, L., and Isenberg, D. (2013). Systemic lupus erythematosus and other autoimmune rheumatic diseases: challenges to treatment. Lancet 382, 809–818. doi: 10.1016/S0140-6736(13)60889-2
Nagase, H., Okugawa, S., Ota, Y., Yamaguchi, M., Tomizawa, H., Matsushima, K., et al. (2003). Expression and function of Toll-like receptors in eosinophils: activation by Toll-like receptor 7 ligand. J. Immunol. 171, 3977–3982.
Nakano, S., Morimoto, S., Suzuki, J., Nozawa, K., Amano, H., Tokano, Y., et al. (2008). Role of pathogenic auto-antibody production by Toll-like receptor 9 of B cells in active systemic lupus erythematosus. Rheumatology (Oxford) 47, 145–149. doi: 10.1093/rheumatology/kem327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Navarra, S. V., Guzman, R. M., Gallacher, A. E., Hall, S., Levy, R. A., Jimenez, R. E., et al. (2011). Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 377, 721–731. doi: 10.1016/S0140-6736(10)61354-2
Ng, M. W., Lau, C. S., Chan, T. M., Wong, W. H., and Lau, Y. L. (2005). Polymorphisms of the Toll-like receptor 9 (TLR9) gene with systemic lupus erythematosus in Chinese. Rheumatology (Oxford) 44, 1456–1457. doi: 10.1093/rheumatology/kei120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nickerson, K. M., Christensen, S. R., Shupe, J., Kashgarian, M., Kim, D., Elkon, K., et al. (2010). TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol. 184, 1840–1848. doi: 10.4049/jimmunol.0902592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Hua, J., Lehman, T. J., Harley, J. B., and Crow, M. K. (2007). High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 8, 492–502. doi: 10.1038/sj.gene.6364408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Neill, L. A., Sheedy, F. J., and Mccoy, C. E. (2011). MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175. doi: 10.1038/nri2957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortiz-Neu, C., and LeRoy, E. C. (1969). The coincidence of Klinefelter’s syndrome and systemic lupus erythematosus. Arthritis Rheum. 12, 241–246. doi: 10.1002/art.1780120312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papadimitraki, E. D., Choulaki, C., Koutala, E., Bertsias, G., Tsatsanis, C., Gergianaki, I., et al. (2006). Expansion of Toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum. 54, 3601–3611. doi: 10.1002/art.22197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, H., Kim, J. I., Ju, Y. S., Gokcumen, O., Mills, R. E., Kim, S., et al. (2010). Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat. Genet. 42, 400–405. doi: 10.1038/ng.555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petri, M., Wallace, D. J., Spindler, A., Chindalore, V., Kalunian, K., Mysler, E., et al. (2013). Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 65, 1011–1021. doi: 10.1002/art.37824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piotrowski, P., Lianeri, M., Wudarski, M., Olesinska, M., and Jagodzinski, P. P. (2013). Contribution of Toll-like receptor 9 gene single-nucleotide polymorphism to systemic lupus erythematosus. Rheumatol. Int. 33, 1121–1125. doi: 10.1007/s00296-012-2509-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pisitkun, P., Deane, J. A., Difilippantonio, M. J., Tarasenko, T., Satterthwaite, A. B., and Bolland, S. (2006). Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672. doi: 10.1126/science.1124978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rönnblom, L., Eloranta, M. L., and Alm, G. V. (2006). The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 54, 408–420.
Rovin, B. H., Furie, R., Latinis, K., Looney, R. J., Fervenza, F. C., Sanchez-Guerrero, J., et al. (2012). Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 64, 1215–1226. doi: 10.1002/art.34359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rullo, O. J., and Tsao, B. P. (2013). Recent insights into the genetic basis of systemic lupus erythematosus. Ann. Rheum. Dis. 72(Suppl. 2), ii56–ii61. doi: 10.1136/annrheumdis-2012-202351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sacre, K., Criswell, L. A., and Mccune, J. M. (2012). Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 14, R155. doi: 10.1186/ar3895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santiago-Raber, M. L., Kikuchi, S., Borel, P., Uematsu, S., Akira, S., Kotzin, B. L., et al. (2008). Evidence for genes in addition to Tlr7 in the Yaa translocation linked with acceleration of systemic lupus erythematosus. J. Immunol. 181, 1556–1562. doi: 10.4049/jimmunol.181.2.1556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sirén, J., Pirhonen, J., Julkunen, I., and Matikainen, S. (2005). IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J. Immunol. 174, 1932–1937.
Shen, N., Fu, Q., Deng, Y., Qian, X., Zhao, J., Kaufman, K. M., et al. (2010). Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc. Natl. Acad. Sci. U.S.A. 107, 15838–15843. doi: 10.1073/pnas.1001337107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shrivastav, M., and Niewold, T. B. (2013). Nucleic Acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol. 4:319. doi: 10.3389/fimmu.2013.00319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stohl, W. (2013). Future prospects in biologic therapy for systemic lupus erythematosus. Nat. Rev. Rheumatol. 9, 705–720. doi: 10.1038/nrrheum.2013.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suarez-Farinas, M., Arbeit, R., Jiang, W., Ortenzio, F. S., Sullivan, T., and Krueger, J. G. (2013). Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE 8:e84634. doi: 10.1371/journal.pone.0084634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subramanian, S., Tus, K., Li, Q. Z., Wang, A., Tian, X. H., Zhou, J., et al. (2006). A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U.S.A. 103, 9970–9975. doi: 10.1073/pnas.0603912103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, X., Wiedeman, A., Agrawal, N., Teal, T. H., Tanaka, L., Hudkins, K. L., et al. (2013). Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. J. Immunol. 190, 2536–2543. doi: 10.4049/jimmunol.1202689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tao, K., Fujii, M., Tsukumo, S., Maekawa, Y., Kishihara, K., Kimoto, Y., et al. (2007). Genetic variations of Toll-like receptor 9 predispose to systemic lupus erythematosus in Japanese population. Ann. Rheum. Dis. 66, 905–909. doi: 10.1136/ard.2006.065961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torcia, M. G., Nencioni, L., Clemente, A. M., Civitelli, L., Celestino, I., Limongi, D., et al. (2012). Sex differences in the response to viral infections: TLR8 and TLR9 ligand stimulation induce higher IL10 production in males. PLoS ONE 7:e39853. doi: 10.1371/journal.pone.0039853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vollmer, J., Tluk, S., Schmitz, C., Hamm, S., Jurk, M., Forsbach, A., et al. (2005). Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J. Exp. Med. 202, 1575–1585. doi: 10.1084/jem.20051696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, B., Higgs, B. W., Chang, L., Vainshtein, I., Liu, Z., Streicher, K., et al. (2013). Pharmacogenomics and translational simulations to bridge indications for an anti-interferon-alpha receptor antibody. Clin. Pharmacol. Ther. 93, 483–492. doi: 10.1038/clpt.2013.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, C. J., Zhang, W. H., Pan, H. F., Li, X. P., Xu, J. H., and Ye, D. Q. (2009). Association study of a single nucleotide polymorphism in the exon 2 region of Toll-like receptor 9 (TLR9) gene with susceptibility to systemic lupus erythematosus among Chinese. Mol. Biol. Rep. 36, 2245–2248. doi: 10.1007/s11033-008-9440-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Liang, Y., Qin, B., Li, C., and Zhong, R. (2012). TLR9 polymorphisms and systemic lupus erythematosus risk in Asians: a meta-analysis study. Cytokine 57, 282–289. doi: 10.1016/j.cyto.2011.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yao, Y., Richman, L., Higgs, B. W., Morehouse, C. A., De Los Reyes, M., Brohawn, P., et al. (2009). Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 60, 1785–1796. doi: 10.1002/art.24557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, C., Gershwin, M. E., and Chang, C. (2014). Diagnostic criteria for systemic lupus erythematosus: a critical review. J. Autoimmun. 48–49, 10–13. doi: 10.1016/j.jaut.2014.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, P., Wellmann, U., Kunder, S., Quintanilla-Martinez, L., Jennen, L., Dear, N., et al. (2006). Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int. Immunol. 18, 1211–1219. doi: 10.1093/intimm/dxl067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zarember, K. A., and Godowski, P. J. (2002). Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561.
Zorro, S., Arias, M., Riano, F., Paris, S., Ramirez, L. A., Uribe, O., et al. (2009). Response to ODN-CpG by B Cells from patients with systemic lupus erythematosus correlates with disease activity. Lupus 18, 718–726. doi: 10.1177/0961203309103098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: systemic lupus erythematosus, TLR7, TLR9, type I interferons, pharmacogenomics
Citation: Celhar T and Fairhurst A-M (2014) Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front. Pharmacol. 5:265. doi: 10.3389/fphar.2014.00265
Received: 15 September 2014; Paper pending published: 13 October 2014;
Accepted: 14 November 2014; Published online: 08 December 2014.
Edited by:
Vita Dolzan, University of Ljubljana, SloveniaReviewed by:
Juergen Reichardt, James Cook University, AustraliaSonja Pavlovic, Institute of Molecular Genetics and Genetic Engineering, Serbia
Copyright © 2014 Celhar and Fairhurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna-Marie Fairhurst, Singapore Immunology Network, Agency for Science, Technology and Research (A*STAR), 8A Biomedical Grove, #03-06 Immunos, Singapore 138648, Singapore e-mail:YW5uYW1hcmllX2ZhaXJodXJzdEBpbW11bm9sLmFzdGFyLmVkdS5zZw==