 Timothy M. Thomson1,2,3*
Timothy M. Thomson1,2,3*- 1Institute for Molecular Biology (IBMB-CSIC), Barcelona, Spain
- 2CIBER de Enfermedades Hepáticas y Digestivas (CIBERehd), Madrid, Spain
- 3Universidad Peruana Cayetano Heredia, Lima, Peru
Introduction
Drug development is a complex, risky, expensive and time-consuming process that requires the accurate execution of multiple stages, from identification and selection of collections of potentially druggable molecules to proof-of-concept validation. Prior to embarking on a drug discovery project, a detailed strategic plan must be designed that includes hundreds of critical considerations such as source of compounds for screening, feasibility of their synthetic pathways, target selection (molecules, cells, organisms), type of output (binding, function, phenotype), throughput, nature, layers and iterations of the screening process, scoring systems, lead optimization approaches or model systems for validation (biochemical activity, cellular function, organismal properties) and, crucially, good contingency plans.
The need to discover new drugs rests on practical matters of human progress, rather than mere market considerations. For example, the global emergence of multidrug resistant pathogens makes it a research target to find alternative antibiotics; current cancer drugs, including advanced biologicals, face drug resistance; many parasitic diseases lack effective drug treatments; highly prevalent neurodegenerative diseases and rare diseases, which collectively affect significant numbers of people, are essentially drug orphan; vital agricultural crops are plagued by fungal and parasitic diseases that have evolved to become increasingly resistant to currently available chemicals. The current COVID-19 pandemic illustrates how pharmaceutically unprepared humanity is (vaccines aside) to confront the sudden emergence of a novel, deadly and highly transmissible pathogen. To date, only a handful of repurposed drugs display demonstrable therapeutic efficacy against infection and disease by the causing agent, SARS-CoV-2 (Lui and Guaraldi, 2023; Sandulescu et al., 2023). Despite an unprecedented parallel effort by hundreds of thousands of industrial and academic scientists worldwide, employing leading-edge technologies, no new drugs have been discovered over the past 3 years to effectively treat this disease.
Natural products as sources of chemical diversity
That some of these targets and diseases may be truly undruggable remains a possibility. However, the general working hypothesis is that small molecules or biologicals will be eventually found to match the majority of designated new targets and to significantly improve upon existing drugs that act on more conventional targets. Although this tenet may seem like wishful thinking, it is at least partly based on sound estimates of ligand structural diversity and druggable chemical space, as well as on the evidence that combinatorial approaches remain largely untested. As such, while the largest currently available compound and fragment libraries, used in ultra-large virtual screenings, contain up to 2 × 109 unique structures (Lyu et al., 2019; Grygorenko et al., 2020; Crunkhorn, 2022), the total chemical space of small organic compounds suitable for drug discovery is estimated as more than 1060 molecules (Bohacek et al., 1996). The underlying concept is that the more compounds are screened, the higher the likelihood of finding true positives (Lyu et al., 2019; Gorgulla et al., 2020).
The above considerations suggest that there is still ample margin to finding new structures to be used as ligands for drug discovery screening efforts, which begs the question: Where will new chemical entities (NCEs) likely come from? Because of their special features as compared to currently available synthetic molecules, natural products (NPs) offer advantages as sources of future NCEs. As such, NPs provide large scaffold diversity and structural complexity accompanied with generally higher molecular rigidity, more chiral centers, higher fraction of sp3 atoms, more oxygen atoms and hydrogen bond acceptors and donors, lower octanol–water partition coefficients (cLog) indicating higher hydrophilicity, low ratio of aromatic ring atoms or diversity of ring systems (Koehn and Carter, 2005; Atanasov et al., 2021; Najmi et al., 2022).
A major limitation for expanding the ligand chemical space with NPs is the laborious nature of NP isolation and structural characterization towards drug discovery. Traditionally, this is done through producing crude extracts with a variety of solvents, screened and fractionated guided by biological activity, and hit compounds purified and structurally characterized. Given the availability of large compound structural databases, a virtual screening-centric strategy may afford to reverse conventional drug discovery schemes. As such, compounds can be structurally characterized after minimal purification or fractionation from crude extracts, by means of NMR spectroscopy, high-resolution mass spectrometry (HRMS), liquid chromatography HRMS (LC–HRMS) (Giavalisco et al., 2008; Wolfender et al., 2019; Garcia-Perez et al., 2020; Stavrianidi, 2020). These methods enable routine acquisition of accurate molecular mass information and unambiguous assignment of formulae for hundreds to thousands of metabolites in a single extract over a broad dynamic range (Fontana et al., 2020), thus facilitating chemical entity dereplication (Arora and Banerjee, 2019). In turn, dereplication is aided by accessing databases such as the Dictionary of Natural Products (https://dnp.chemnetbase.com/), which encompasses all NP structures reported with links to their biological sources, the Global Natural Products Social (GNPS) molecular networking platform (https://gnps.ucsd.edu/) (Wang M. et al., 2016), in which thousands of sets of MS/MS data are recorded from a given set of extracts, clustering compounds by their structural relationships (Allard et al., 2016; Zhou et al., 2017; da Silva et al., 2018), Compound Structure Identification (CSI) (Aksenov et al., 2017) or METLIN (Guijas et al., 2018), containing fragment ion spectra that can be used for the identification of unknown compounds. In summary, quantitative NMR and LC–MS approaches can yield novel structures to populate screening-ready databases at early stages in virtual screening drug discovery strategies, thus avoiding futile downstream development efforts (Wohlgemuth et al., 2016).
In spite of these technological advances that facilitate the expansion of the known chemical space, NPs may contain only a fraction of the theoretical space and scaffold diversity (Pye et al., 2017). In order to further expand chemical space and structural diversity, several strategies have been used to create new biologically active compounds by adding appendages on NP core scaffolds (Grigalunas et al., 2022), such as diversity-oriented synthesis (DOS) (Schreiber, 2009), DNA encoded libraries (DEL) (Franzini and Randolph, 2016) or biology-oriented synthesis (BOS) (van Hattum and Waldmann, 2014). Other strategies go beyond the available NP scaffolds by resorting to ring distortion reactions (Motika and Hergenrother, 2020), albeit still relying on the original scaffolds. The pseudo-NP strategy deconstructs NPs into fragments and recombines them into novel scaffolds that are not possible to attain through known biosynthetic pathways but retain the chemical and biological relevance of NPs (Grigalunas et al., 2020; Karageorgis et al., 2020).
Additional efforts to expand the NP chemical space include engineering biosynthetic pathways aimed at yielding new NP analogues with potentially improved pharmacological properties (Atanasov et al., 2021). Such strategies include the activation of cryptic or occult biosynthetic gene clusters that remain otherwise silent (Macheleidt et al., 2016), which can be achieved through the manipulation of culture conditions (Pan et al., 2019), micro-organism co-cultures (Bertrand et al., 2014) or exposure to small molecule epigenetic modulators (Pillay et al., 2022), among other approaches. The expansion of chemical space through various strategies entails the parallel development of new chemical methods capable of solving previously untested synthetic paths, so as to produce compounds corresponding to the newly designed structures and in cost-effective yields (Cai et al., 2023).
Further to approaching theoretical ligand chemical space limits and structural characterization of NP and NP-like molecules, a major challenge is to make them available as large screening-ready libraries. Several databases provide information on NPs and their structures (Table 1). Compared to these databases, the currently accessible databases of chemical entities with focus on NPs of Latin American origin contain information on relatively few compounds (reviewed in (Medina-Franco, 2020; Nunez et al., 2021; Gomez-Garcia and Medina-Franco, 2022)) (Table 2). As such, given the estimated share of Latin American biodiversity in global biodiversity (Raven et al., 2020), it is apparent that NPs of Latin American origin are heavily underrepresented in databases of NP physicochemical properties and structures.
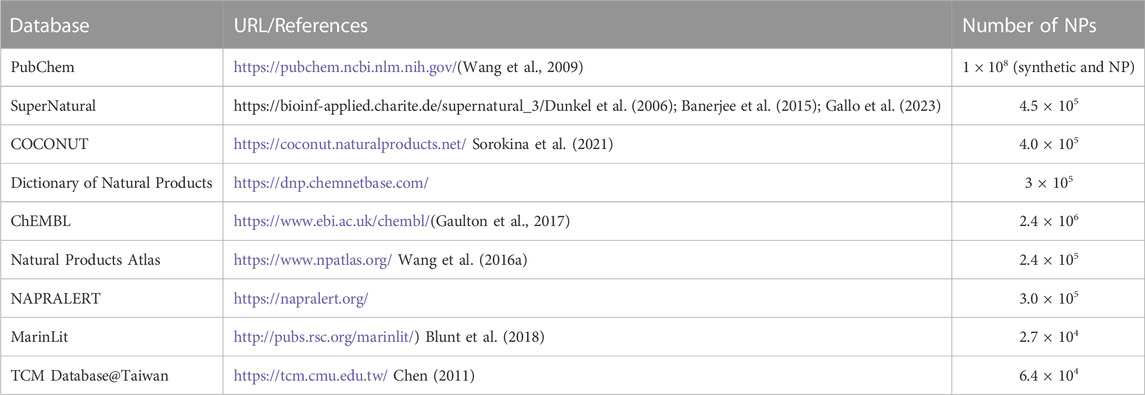
TABLE 1. Natural Product databases containing NP structural information.
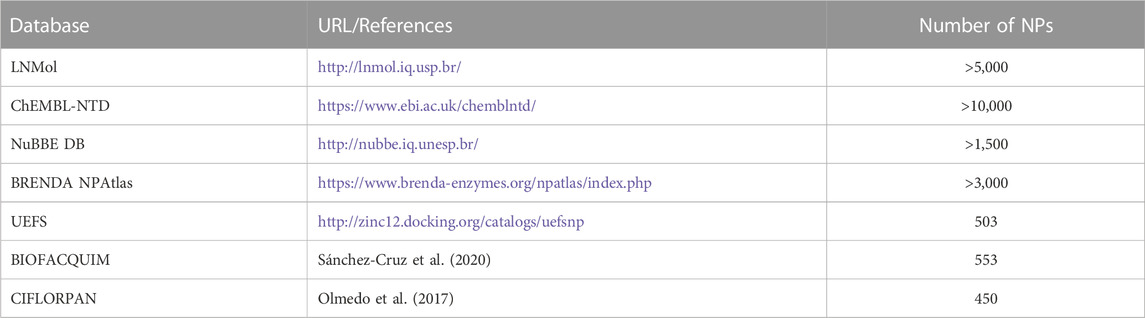
TABLE 2. Databases of chemical entities with focus on NPs of Latin American origin.
Large chemical structure databases are necessary for next-generation virtual drug discovery efforts, but they are not sufficient. Open-source, robust platforms are also needed that can integrate tasks in virtual screening and provide smooth connectivity to docking tools, such as VirtualFlow (Gorgulla et al., 2020), which can dock 1 billion compounds in about 2 weeks when run on 10,000 CPU cores, or V-SYNTHES (Sadybekov et al., 2022), which performs iterative steps of library preparation, enumeration, docking and hit selection, handling fragment-like libraries representing all possible scaffold–synthon combinations for all reactions in the 11 billion compound REAL Space library.
Novel approaches to unbiased high-throughput target identification
Experimental approaches for target identification require molecular and biochemical studies of disease pathophysiology (McFedries et al., 2013; Shaker et al., 2021; Zecha et al., 2023), which can be costly, labor-intensive and time-consuming. Conventional virtual screening approaches for target selection focus on a preferred molecular target to conduct structure-directed screenings. For better outcomes, such targets need to be structurally resolved at the highest possible atomic resolution. Until recently, that entailed “one target at a time” strategies. For decades, conventional approaches to the resolution of macromolecular structures have relied on techniques such X-ray crystallography or NMR, which are labor-intensive and low-throughput. The advent to fruition of cryoelectron microscopy (Baumeister, 2022) has enormously speeded up this process. As a result of these collective efforts, there are currently over 200,000 experimentally resolved structures deposited in Protein Data Bank (https://www.rcsb.org/), as unique entries corresponding to full-length proteins, fragments and complexes (protein-protein, protein-DNA, protein-ligand). This volume of structural information has laid the foundation for, and enabled, the use of machine learning tools, such as AlphaFold2 (Jumper et al., 2021) or RoseTTA fold (Baek et al., 2021), to accurately predict the structures of millions of proteins. As such, the AlphaFold protein structure database (https://alphafold.ebi.ac.uk) currently contains 214,683,829 predicted structures, including 48 complete proteomes. An added bonus to these predictive tools is that targets for which the experimentally determined structures are incomplete or ambiguous at specific regions can be completed or “polished” for subsequent use in virtual screening. For virtual drug discovery, potential binding sites must be defined on target proteins. To this end, a number of tools have been developed to predict pockets amenable to blocking by small drug-like molecules on proteins with known (Bhagavat et al., 2018) or predicted (Wang et al., 2022; Sim et al., 2023) structures.
While the availability of large ligand structural libraries improves hit rates on pre-determined targets, the availability of large target structural libraries covering complete proteomes allows to perform near-complete screenings of hit and lead compounds for target selectivity. A major reason for candidate compound failure in drug discovery schemes is undesired or adverse effects, which is why characterization of absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of candidate molecules at the earliest possible stage is relevant (Selick et al., 2002; Caldwell et al., 2009; Wu et al., 2020). Traditional ADMET prediction methods, such as quantitative structure activity relationship (QSAR) models, require costly and time-consuming data generation and are generally used relatively late in drug discovery programs. The increasing availability of data and resources enables ADMET predictions earlier in the process, with the use of machine learning tools to predict drug-target interactions, the blood-brain-barrier permeability of compounds, or toxic properties of drug candidates (reviewed in (Shaker et al., 2021)). The availability of predicted structures for complete proteomes should represent a paradigm shift in ADMET predictions, by affording approaches such as large-scale reverse docking, by which small molecules are simultaneously docked on many protein and cavity targets. This provides information on selectivity and thus potential off-target effects of the small molecules. For example, Wong et al. (2022) docked 319 compounds, of which 218 had antibacterial activity, on 296 essential E. coli proteins with structures predicted with AlphaFold2, finding unexpectedly promiscuous interactions and demonstrating the feasibility of the approach, albeit also highlighting the need to improve the performance of machine learning-based protein-ligand modeling methods.
Target-agnostic approaches have been applied as exploratory efforts to identify activities of interest prior to targeted drug discovery (Wang Y. et al., 2016). As such, metabolomics data can be integrated with data obtained by other omics techniques such as transcriptomics, proteomics or functional genomics with imaging-based or phenotypic screens (Kasap et al., 2014; Kurita et al., 2015; Bray et al., 2016; Subramanian et al., 2017; Earl et al., 2018; Setten et al., 2019; Ziegler et al., 2021). Eventually, as current criteria followed by drug approval agencies require the identification of molecular mechanisms, these exploratory approaches need to be followed up by biochemical, molecular and structural studies for a precise mechanistic characterizations of candidate drug activities.
Perspectives and proposal
Significant constraints for the implementation of effective drug discovery programs in Latin America include relatively limited funding and failure to assemble coordinated transnational efforts. To date, scarce numbers of virtual screening projects in Latin America have led to the discovery of NP or NP-inspired compounds from isolation to proof-of-concept experimental activities of identified compounds (Fernandes et al., 2019; Rodrigues et al., 2019; Belgamo et al., 2020; Fernandez et al., 2020; Battini et al., 2021; Ferreira et al., 2021; Vargas et al., 2021; Valera-Vera et al., 2022; Adessi et al., 2023; Almeida et al., 2023; Araujo et al., 2023; Llanos et al., 2023; Peralta-Moreno et al., 2023). With the increasing availability and accessibility of advanced virtual screening tools that facilitate many stages in drug discovery pipelines (Daina and Zoete, 2019; Gentile et al., 2020; Ghislat et al., 2021; Singh et al., 2021; Arul Murugan et al., 2022; Blanes-Mira et al., 2022; Gorgulla et al., 2022; Muller et al., 2022; Sarkar et al., 2023; Thomas et al., 2023), which under conventional schemes are costly, labor intensive and time-consuming, a window of opportunity opens to change the tide towards NP-inspired drug discovery in less affluent economies.
Although less costly than conventional approaches, large next-generation virtual screening-centric drug discovery efforts based on NPs still require expertise and equipment for modern compound isolation and structural characterization, chemical synthetic and biosynthetic capabilities and, most importantly, ample computing power and connectivity. A further practical issue is the availability and cost of NPs and NP-inspired compounds for experimental validation of candidate molecules identified by virtual screening. The cost of NPs through conventional commercial channels can be relatively high, particularly for those with low yields in standard isolation procedures. As argued above, current technology enables early structural characterization of individual compounds, even as part of relatively complex mixtures, thus affording to bypass purification prior to structural characterization. In this scheme, NPs provide structures of interest, while experimental activity validation is performed with synthetic compounds that recapitulate NP structural features of pharmacological interest. This approach reduces the problem of yield, but does not totally solve the issue of cost per compound to be tested, particularly for those that may require difficult synthetic paths. New, more cost-effective synthetic strategies are expected to mitigate cost issues, as will entrusting non-profit institutions with on-demand synthesis of NP-inspired compounds for drug discovery. Together with building strong computational capabilities, this requires a concerted effort by individual teams, academic and industry organizations, transnational societies, institutional instances and public and private funding agencies, to design long-term, outcome-oriented plans coupled to commensurate multi-year funding schemes.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The author’s work is supported by the Spanish Ministry of Science and Technology (PID 2019-107139RB-C21), the Interdisciplinary Platform-Global Health (Plataforma Temática Interdisciplinar-Salud Global, PTI-SG) (SGL2103019) and the Networked Researched Center on Liver and Digestive Diseases (Centro de Investigación en Red en Enfermedades Hepáticas y Digestivas, CIBER-EHD).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adessi, T. G., Cantero, J., Ballesteros-Casallas, A., Garcia, M. E., Nicotra, V. E., and Paulino, M. (2023). Identification of potential biological target for trypanocidal sesquiterpene lactones derivatives. J. Biomol. Struct. Dyn. 2023, 1–14. doi:10.1080/07391102.2023.2183031
Aksenov, A. A., Da Silva, R., Knight, R., Lopes, N. P., and Dorrestein, P. C. (2017). Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 1, 0054. doi:10.1038/s41570-017-0054
Allard, P. M., Peresse, T., Bisson, J., Gindro, K., Marcourt, L., Pham, V. C., et al. (2016). Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 88, 3317–3323. doi:10.1021/acs.analchem.5b04804
Almeida, E. S. F. H., Silva, A. R. N., De Oliveira, T. J. S., Guimaraes, A. L., De Azevedo, F. R., Brito Dos Santos, M., et al. (2023). A chalcone identified by in silico and in vitro assays possesses high larvicidal activity against Aedes aegypti. Acta Trop. 238, 106791. doi:10.1016/j.actatropica.2022.106791
Araujo, S. C., De Angelo, R. M., Barbosa, H., Costa-Silva, T. A., Tempone, A. G., Lago, J. H. G., et al. (2023). Identification of inhibitors as drug candidates against Chagas disease. Eur. J. Med. Chem. 248, 115074. doi:10.1016/j.ejmech.2022.115074
Arora, N., and Banerjee, A. K. (2019). Dereplication in natural product discovery. Curr. Top. Med. Chem. 19, 101–102. doi:10.2174/156802661902190328145951
Arul Murugan, N., Ruba Priya, G., Narahari Sastry, G., and Markidis, S. (2022). Artificial intelligence in virtual screening: Models versus experiments. Drug Discov. Today 27, 1913–1923. doi:10.1016/j.drudis.2022.05.013
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., International Natural Product Sciences, T., and Supuran, C. T. (2021). Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 20, 200–216. doi:10.1038/s41573-020-00114-z
Baek, M., Dimaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. doi:10.1126/science.abj8754
Banerjee, P., Erehman, J., Gohlke, B. O., Wilhelm, T., Preissner, R., and Dunkel, M. (2015). Super Natural II--a database of natural products. Nucleic Acids Res. 43, D935–D939. doi:10.1093/nar/gku886
Battini, L., Fidalgo, D. M., Alvarez, D. E., and Bollini, M. (2021). Discovery of a potent and selective chikungunya virus envelope protein inhibitor through computer-aided drug design. ACS Infect. Dis. 7, 1503–1518. doi:10.1021/acsinfecdis.0c00915
Baumeister, W. (2022). Cryo-electron tomography: A long journey to the inner space of cells. Cell 185, 2649–2652. doi:10.1016/j.cell.2022.06.034
Belgamo, J. A., Alberca, L. N., Porfido, J. L., Romero, F. N. C., Rodriguez, S., Talevi, A., et al. (2020). Application of target repositioning and in silico screening to exploit fatty acid binding proteins (FABPs) from Echinococcus multilocularis as possible drug targets. J. Comput. Aided Mol. Des. 34, 1275–1288. doi:10.1007/s10822-020-00352-8
Bertrand, S., Bohni, N., Schnee, S., Schumpp, O., Gindro, K., and Wolfender, J. L. (2014). Metabolite induction via microorganism co-culture: A potential way to enhance chemical diversity for drug discovery. Biotechnol. Adv. 32, 1180–1204. doi:10.1016/j.biotechadv.2014.03.001
Bhagavat, R., Sankar, S., Srinivasan, N., and Chandra, N. (2018). An augmented pocketome: Detection and analysis of small-molecule binding pockets in proteins of known 3D structure. Structure 26, 499–512. doi:10.1016/j.str.2018.02.001
Blanes-Mira, C., Fernandez-Aguado, P., De Andres-Lopez, J., Fernandez-Carvajal, A., Ferrer-Montiel, A., and Fernandez-Ballester, G. (2022). Comprehensive survey of consensus docking for high-throughput virtual screening. Molecules 28, 175. doi:10.3390/molecules28010175
Blunt, J. W., Carroll, A. R., Copp, B. R., Davis, R. A., Keyzers, R. A., and Prinsep, M. R. (2018). Marine natural products. Nat. Prod. Rep. 35, 8–53. doi:10.1039/c7np00052a
Bohacek, R. S., Mcmartin, C., and Guida, W. C. (1996). The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 16, 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6
Bray, M. A., Singh, S., Han, H., Davis, C. T., Borgeson, B., Hartland, C., et al. (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 11, 1757–1774. doi:10.1038/nprot.2016.105
Cai, J. H., Zhu, X. Z., Guo, P. Y., Rose, P., Liu, X. T., Liu, X., et al. (2023). Recent updates in click and computational chemistry for drug discovery and development. Front. Chem. 11, 1114970. doi:10.3389/fchem.2023.1114970
Caldwell, G. W., Yan, Z., Tang, W., Dasgupta, M., and Hasting, B. (2009). ADME optimization and toxicity assessment in early- and late-phase drug discovery. Curr. Top. Med. Chem. 9, 965–980. doi:10.2174/156802609789630929
Chen, C. Y. (2011). TCM Database@Taiwan: The world's largest traditional Chinese medicine database for drug screening in silico. PLoS One 6, e15939. doi:10.1371/journal.pone.0015939
Crunkhorn, S. (2022). Screening ultra-large virtual libraries. Nat. Rev. Drug Discov. 21, 95. doi:10.1038/d41573-022-00002-8
Da Silva, R. R., Wang, M., Nothias, L. F., Van Der Hooft, J. J. J., Caraballo-Rodriguez, A. M., Fox, E., et al. (2018). Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 14, e1006089. doi:10.1371/journal.pcbi.1006089
Daina, A., and Zoete, V. (2019). Application of the SwissDrugDesign online resources in virtual screening. Int. J. Mol. Sci. 20, 4612. doi:10.3390/ijms20184612
Dunkel, M., Fullbeck, M., Neumann, S., and Preissner, R. (2006). SuperNatural: A searchable database of available natural compounds. Nucleic Acids Res. 34, D678–D683. doi:10.1093/nar/gkj132
Earl, D. C., Ferrell, P. B., Leelatian, N., Froese, J. T., Reisman, B. J., Irish, J. M., et al. (2018). Discovery of human cell selective effector molecules using single cell multiplexed activity metabolomics. Nat. Commun. 9, 39. doi:10.1038/s41467-017-02470-8
Fernandes, D. A., Barros, R. P. C., Teles, Y. C. F., Oliveira, L. H. G., Lima, J. B., Scotti, M. T., et al. (2019). Larvicidal compounds extracted from helicteres velutina K. Schum (sterculiaceae) evaluated against Aedes aegypti L. Molecules 24, 2315. doi:10.3390/molecules24122315
Fernandez, G. A., Castro, E. F., Rosas, R. A., Fidalgo, D. M., Adler, N. S., Battini, L., et al. (2020). Design and optimization of quinazoline derivatives: New non-nucleoside inhibitors of bovine viral diarrhea virus. Front. Chem. 8, 590235. doi:10.3389/fchem.2020.590235
Ferreira, L. T., Borba, J. V. B., Moreira-Filho, J. T., Rimoldi, A., Andrade, C. H., and Costa, F. T. M. (2021). QSAR-based virtual screening of natural products database for identification of potent antimalarial hits. Biomolecules 11, 459. doi:10.3390/biom11030459
Fontana, A., Iturrino, L., Corens, D., and Crego, A. L. (2020). Automated open-access liquid chromatography high resolution mass spectrometry to support drug discovery projects. J. Pharm. Biomed. Anal. 178, 112908. doi:10.1016/j.jpba.2019.112908
Franzini, R. M., and Randolph, C. (2016). Chemical space of DNA-encoded libraries. J. Med. Chem. 59, 6629–6644. doi:10.1021/acs.jmedchem.5b01874
Gallo, K., Kemmler, E., Goede, A., Becker, F., Dunkel, M., Preissner, R., et al. (2023). SuperNatural 3.0-a database of natural products and natural product-based derivatives. Nucleic Acids Res. 51, D654–D659. doi:10.1093/nar/gkac1008
Garcia-Perez, I., Posma, J. M., Serrano-Contreras, J. I., Boulange, C. L., Chan, Q., Frost, G., et al. (2020). Identifying unknown metabolites using NMR-based metabolic profiling techniques. Nat. Protoc. 15, 2538–2567. doi:10.1038/s41596-020-0343-3
Gaulton, A., Hersey, A., Nowotka, M., Bento, A. P., Chambers, J., and Mendez, D. (2017). The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954.
Gentile, F., Agrawal, V., Hsing, M., Ton, A. T., Ban, F., Norinder, U., et al. (2020). Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 6, 939–949. doi:10.1021/acscentsci.0c00229
Ghislat, G., Rahman, T., and Ballester, P. J. (2021). Recent progress on the prospective application of machine learning to structure-based virtual screening. Curr. Opin. Chem. Biol. 65, 28–34. doi:10.1016/j.cbpa.2021.04.009
Giavalisco, P., Hummel, J., Lisec, J., Inostroza, A. C., Catchpole, G., and Willmitzer, L. (2008). High-resolution direct infusion-based mass spectrometry in combination with whole 13C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Anal. Chem. 80, 9417–9425. doi:10.1021/ac8014627
Gomez-Garcia, A., and Medina-Franco, J. L. (2022). Progress and impact of Latin American natural product databases. Biomolecules 12, 1202. doi:10.3390/biom12091202
Gorgulla, C., Boeszoermenyi, A., Wang, Z. F., Fischer, P. D., Coote, P. W., Padmanabha Das, K. M., et al. (2020). An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663–668. doi:10.1038/s41586-020-2117-z
Gorgulla, C., Jayaraj, A., Fackeldey, K., and Arthanari, H. (2022). Emerging frontiers in virtual drug discovery: From quantum mechanical methods to deep learning approaches. Curr. Opin. Chem. Biol. 69, 102156. doi:10.1016/j.cbpa.2022.102156
Grigalunas, M., Brakmann, S., and Waldmann, H. (2022). Chemical evolution of natural product structure. J. Am. Chem. Soc. 144, 3314–3329. doi:10.1021/jacs.1c11270
Grigalunas, M., Burhop, A., Christoforow, A., and Waldmann, H. (2020). Pseudo-natural products and natural product-inspired methods in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 56, 111–118. doi:10.1016/j.cbpa.2019.10.005
Grygorenko, O. O., Radchenko, D. S., Dziuba, I., Chuprina, A., Gubina, K. E., and Moroz, Y. S. (2020). Generating multibillion chemical space of readily accessible screening compounds. iScience 23, 101681. doi:10.1016/j.isci.2020.101681
Guijas, C., Montenegro-Burke, J. R., Domingo-Almenara, X., Palermo, A., Warth, B., Hermann, G., et al. (2018). Metlin: A technology platform for identifying knowns and unknowns. Anal. Chem. 90, 3156–3164. doi:10.1021/acs.analchem.7b04424
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Karageorgis, G., Foley, D. J., Laraia, L., and Waldmann, H. (2020). Principle and design of pseudo-natural products. Nat. Chem. 12, 227–235. doi:10.1038/s41557-019-0411-x
Kasap, C., Elemento, O., and Kapoor, T. M. (2014). DrugTargetSeqR: A genomics- and CRISPR-cas9-based method to analyze drug targets. Nat. Chem. Biol. 10, 626–628. doi:10.1038/nchembio.1551
Koehn, F. E., and Carter, G. T. (2005). The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 4, 206–220. doi:10.1038/nrd1657
Kurita, K. L., Glassey, E., and Linington, R. G. (2015). Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc. Natl. Acad. Sci. U. S. A. 112, 11999–12004. doi:10.1073/pnas.1507743112
Llanos, M. A., Alberca, L. N., Ruiz, M. D., Sbaraglini, M. L., Miranda, C., Pino-Martinez, A., et al. (2023). A combined ligand and target-based virtual screening strategy to repurpose drugs as putrescine uptake inhibitors with trypanocidal activity. J. Comput. Aided Mol. Des. 37, 75–90. doi:10.1007/s10822-022-00491-0
Lui, G., and Guaraldi, G. (2023). Drug treatment of COVID-19 infection. Curr. Opin. Pulm. Med. 29, 174–183. doi:10.1097/MCP.0000000000000953
Lyu, J., Wang, S., Balius, T. E., Singh, I., Levit, A., Moroz, Y. S., et al. (2019). Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229. doi:10.1038/s41586-019-0917-9
Macheleidt, J., Mattern, D. J., Fischer, J., Netzker, T., Weber, J., Schroeckh, V., et al. (2016). Regulation and role of fungal secondary metabolites. Annu. Rev. Genet. 50, 371–392. doi:10.1146/annurev-genet-120215-035203
Mcfedries, A., Schwaid, A., and Saghatelian, A. (2013). Methods for the elucidation of protein-small molecule interactions. Chem. Biol. 20, 667–673. doi:10.1016/j.chembiol.2013.04.008
Medina-Franco, J. L. (2020). Towards a unified Latin American natural products database: LANaPD. Future Sci. OA 6, FSO468. doi:10.2144/fsoa-2020-0068
Motika, S. E., and Hergenrother, P. J. (2020). Re-engineering natural products to engage new biological targets. Nat. Prod. Rep. 37, 1395–1403. doi:10.1039/d0np00059k
Muller, C., Rabal, O., and Diaz Gonzalez, C. (2022). Artificial intelligence, machine learning, and deep learning in real-life drug design cases. Methods Mol. Biol. 2390, 383–407. doi:10.1007/978-1-0716-1787-8_16
Najmi, A., Javed, S. A., Al Bratty, M., and Alhazmi, H. A. (2022). Modern approaches in the discovery and development of plant-based natural products and their analogues as potential therapeutic agents. Molecules 27, 349. doi:10.3390/molecules27020349
Nunez, M. J., Diaz-Eufracio, B. I., Medina-Franco, J. L., and Olmedo, D. A. (2021). Latin American databases of natural products: Biodiversity and drug discovery against SARS-CoV-2. RSC Adv. 11, 16051–16064. doi:10.1039/d1ra01507a
Olmedo, D. A., Gonzalez-Medina, M., Gupta, M. P., and Medina-Franco, J. L. (2017). Cheminformatic characterization of natural products from Panama. Mol. Divers 21, 779–789. doi:10.1007/s11030-017-9781-4
Pan, R., Bai, X., Chen, J., Zhang, H., and Wang, H. (2019). Exploring structural diversity of microbe secondary metabolites using osmac strategy: A literature review. Front. Microbiol. 10, 294. doi:10.3389/fmicb.2019.00294
Peralta-Moreno, M. N., Anton-Muñoz, V., Ortega-Alarcon, D., Jimenez-Alesanco, A., Vega, S., Abian, O., et al. (2023). Autochthonous Peruvian natural plants as potential SARS-CoV-2 mpro main protease inhibitors. Pharm. (Basel) 16, 585. doi:10.3390/ph16040585
Pillay, L. C., Nekati, L., Makhwitine, P. J., and Ndlovu, S. I. (2022). Epigenetic activation of silent biosynthetic gene clusters in endophytic fungi using small molecular modifiers. Front. Microbiol. 13, 815008. doi:10.3389/fmicb.2022.815008
Pye, C. R., Bertin, M. J., Lokey, R. S., Gerwick, W. H., and Linington, R. G. (2017). Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. U. S. A. 114, 5601–5606. doi:10.1073/pnas.1614680114
Raven, P. H., Gereau, R. E., Phillipson, P. B., Chatelain, C., Jenkins, C. N., and Ulloa Ulloa, C. (2020). The distribution of biodiversity richness in the tropics. Sci. Adv. 6, eabc6228. doi:10.1126/sciadv.abc6228
Rodrigues, R. P., Ardisson, J. S., Ribeiro Goncalves, R. C., Oliveira, T. B., Barreto Da Silva, V., Kawano, D. F., et al. (2019). Search for potential inducible nitric oxide synthase inhibitors with favorable ADMET profiles for the therapy of Helicobacter pylori infections. Curr. Top. Med. Chem. 19, 2795–2804. doi:10.2174/1568026619666191112105650
Sadybekov, A. A., Sadybekov, A. V., Liu, Y., Iliopoulos-Tsoutsouvas, C., Huang, X. P., Pickett, J., et al. (2022). Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459. doi:10.1038/s41586-021-04220-9
Sánchez-Cruz, N., Pilón-Jiméne, B. A., and Medina-Franc, J. L. (2020). Functional group and diversity analysis of biofacquim: A Mexican natural product database. F1000Research 8, 2071. doi:10.12688/f1000research.21540.2
Sandulescu, O., Apostolescu, C. G., Preotescu, L. L., Streinu-Cercel, A., and Sandulescu, M. (2023). Therapeutic developments for SARS-CoV-2 infection-Molecular mechanisms of action of antivirals and strategies for mitigating resistance in emerging variants in clinical practice. Front. Microbiol. 14, 1132501. doi:10.3389/fmicb.2023.1132501
Sarkar, C., Das, B., Rawat, V. S., Wahlang, J. B., Nongpiur, A., Tiewsoh, I., et al. (2023). Artificial intelligence and machine learning technology driven modern drug discovery and development. Int. J. Mol. Sci. 24, 2026. doi:10.3390/ijms24032026
Schreiber, S. L. (2009). Organic chemistry: Molecular diversity by design. Nature 457, 153–154. doi:10.1038/457153a
Selick, H. E., Beresford, A. P., and Tarbit, M. H. (2002). The emerging importance of predictive ADME simulation in drug discovery. Drug Discov. Today 7, 109–116. doi:10.1016/s1359-6446(01)02100-6
Setten, R. L., Rossi, J. J., and Han, S. P. (2019). The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 18, 421–446. doi:10.1038/s41573-019-0017-4
Shaker, B., Ahmad, S., Lee, J., Jung, C., and Na, D. (2021). In silico methods and tools for drug discovery. Comput. Biol. Med. 137, 104851. doi:10.1016/j.compbiomed.2021.104851
Sim, J., Kwon, S., and Seok, C. (2023). HProteome-BSite: Predicted binding sites and ligands in human 3D proteome. Nucleic Acids Res. 51, D403–D408. doi:10.1093/nar/gkac873
Singh, N., Chaput, L., and Villoutreix, B. O. (2021). Virtual screening web servers: Designing chemical probes and drug candidates in the cyberspace. Brief. Bioinform 22, 1790–1818. doi:10.1093/bib/bbaa034
Sorokina, M., Merseburger, P., Rajan, K., Yirik, M. A., and Steinbeck, C. (2021). COCONUT online: Collection of open natural products database. J. Cheminform 13, 2. doi:10.1186/s13321-020-00478-9
Stavrianidi, A. (2020). A classification of liquid chromatography mass spectrometry techniques for evaluation of chemical composition and quality control of traditional medicines. J. Chromatogr. A 1609, 460501. doi:10.1016/j.chroma.2019.460501
Subramanian, A., Narayan, R., Corsello, S. M., Peck, D. D., Natoli, T. E., Lu, X., et al. (2017). A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452. doi:10.1016/j.cell.2017.10.049
Thomas, M., Bender, A., and De Graaf, C. (2023). Integrating structure-based approaches in generative molecular design. Curr. Opin. Struct. Biol. 79, 102559. doi:10.1016/j.sbi.2023.102559
Valera-Vera, E., Reigada, C., Saye, M., Digirolamo, F. A., Galceran, F., Miranda, M. R., et al. (2022). Trypanocidal activity of the anthocyanidin delphinidin, a non-competitive inhibitor of arginine kinase. Nat. Prod. Res. 36, 3153–3157. doi:10.1080/14786419.2021.1947270
Van Hattum, H., and Waldmann, H. (2014). Biology-oriented synthesis: Harnessing the power of evolution. J. Am. Chem. Soc. 136, 11853–11859. doi:10.1021/ja505861d
Vargas, E. L. G., De Almeida, F. A., De Freitas, L. L., Pinto, U. M., and Vanetti, M. C. D. (2021). Plant compounds and nonsteroidal anti-inflammatory drugs interfere with quorum sensing in Chromobacterium violaceum. Arch. Microbiol. 203, 5491–5507. doi:10.1007/s00203-021-02518-w
Wang, M., Carver, J. J., Phelan, V. V., Sanchez, L. M., Garg, N., Peng, Y., et al. (2016a). Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34, 828–837. doi:10.1038/nbt.3597
Wang, S., Lin, H., Huang, Z., He, Y., Deng, X., Xu, Y., et al. (2022). CavitySpace: A database of potential ligand binding sites in the human proteome. Biomolecules 12, 967. doi:10.3390/biom12070967
Wang, Y., Cornett, A., King, F. J., Mao, Y., Nigsch, F., Paris, C. G., et al. (2016b). Evidence-based and quantitative prioritization of tool compounds in phenotypic drug discovery. Cell Chem. Biol. 23, 862–874. doi:10.1016/j.chembiol.2016.05.016
Wang, Y., Xiao, J., Suzek, T. O., Zhang, J., Wang, J., and Bryant, S. H. (2009). PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 37, W623–W633.
Wohlgemuth, G., Mehta, S. S., Mejia, R. F., Neumann, S., Pedrosa, D., Pluskal, T., et al. (2016). SPLASH, a hashed identifier for mass spectra. Nat. Biotechnol. 34, 1099–1101. doi:10.1038/nbt.3689
Wolfender, J. L., Nuzillard, J. M., Van Der Hooft, J. J. J., Renault, J. H., and Bertrand, S. (2019). Accelerating metabolite identification in natural product research: Toward an ideal combination of liquid chromatography-high-resolution tandem mass spectrometry and NMR profiling, in silico databases, and chemometrics. Anal. Chem. 91, 704–742. doi:10.1021/acs.analchem.8b05112
Wong, F., Krishnan, A., Zheng, E. J., Stark, H., Manson, A. L., Earl, A. M., et al. (2022). Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 18, e11081. doi:10.15252/msb.202211081
Wu, F., Zhou, Y., Li, L., Shen, X., Chen, G., Wang, X., et al. (2020). Computational approaches in preclinical studies on drug discovery and development. Front. Chem. 8, 726. doi:10.3389/fchem.2020.00726
Zecha, J., Bayer, F. P., Wiechmann, S., Woortman, J., Berner, N., Muller, J., et al. (2023). Decrypting drug actions and protein modifications by dose- and time-resolved proteomics. Science 380, 93–101. doi:10.1126/science.ade3925
Zhou, Z., Xiong, X., and Zhu, Z. J. (2017). MetCCS predictor: A web server for predicting collision cross-section values of metabolites in ion mobility-mass spectrometry based metabolomics. Bioinformatics 33, 2235–2237. doi:10.1093/bioinformatics/btx140
Keywords: natural products, virtual screening, Latin America, databases, computation
Citation: Thomson TM (2023) On the importance for drug discovery of a transnational Latin American database of natural compound structures. Front. Pharmacol. 14:1207559. doi: 10.3389/fphar.2023.1207559
Received: 17 April 2023; Accepted: 15 June 2023;
Published: 22 June 2023.
Edited by:
Carmenza Spadafora, Instituto de Investigaciones Científicas y Servicios de Alta Tecnología, PanamaReviewed by:
Alan Hesketh, Independent Researcher, Gerrards Cross, United KingdomAbraham Madariaga-Mazon, National Autonomous University of Mexico, Mexico
Copyright © 2023 Thomson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy M. Thomson, dGl0Ym1jQGlibWIuY3NpYy5lcw==