Abstract
Inflammation is a protective response to a variety of infectious agents. To develop a new anti-inflammatory drug, we explored a pharmacologically important thiazole scaffold in this study. In a multi-step synthetic approach, we synthesized seven new thiazole derivatives (5a–5g). Initially, we examined the in vitro anti-inflammatory potentials of our compounds using COX-1, COX-2, and 5-LOX enzyme assays. After in vitro confirmation, the potential compounds were subjected to in vivo analgesic and anti-inflammatory studies. The hot plate method was used for analgesia, and carrageenan-induced inflammation was also assayed. Overall, all our compounds proved to be potent inhibitors of COX-2 compared to celecoxib (IC50 0.05 μM), exhibiting IC50 values in the range of 0.76–9.01 μM .Compounds 5b, 5d, and 5e were dominant and selective COX-2 inhibitors with the lowest IC50 values and selectivity index (SI) values of 42, 112, and 124, respectively. Similarly, in the COX-1 assay, our compounds were relatively less potent but still encouraging. Standard aspirin exhibited an IC50 value of 15.32 μM. In the 5-LOX results, once again, compounds 5d and 5e were dominant with IC50 values of 23.08 and 38.46 μM, respectively. Standard zileuton exhibited an IC50 value of 11.00 μM. Based on the COX/LOX and SI potencies, the compounds 5d and 5e were subjected to in vivo analgesic and anti-inflammatory studies. Compounds 5d and 5e at concentrations of 5, 10, and 20 mg/kg body weight were significant in animal models. Furthermore, we explored the potential role of compounds 5d and 5e in various phlogistic agents. Similarly, both compounds 5d and 5e were also significantly potent in the anti-nociceptive assay. The molecular docking interactions of these two compounds with the target proteins of COX and LOX further strengthened their potential for use in COX/LOX pathway inhibitions.
Introduction
Inflammation is a physiological response that occurs naturally in response to a variety of infectious agents, trauma, autoimmune diseases, injury, tissue ischemia, and an imbalance. It is a process that is required for the body to heal from infection and damage. Inflammation causes biochemical changes that result in swelling, redness, pain, itching, and an increase in body temperature. However, if inflammation is not appropriately treated, leukocytes, lymphocytes, or damaged collagen can infiltrate the tissue and cause long-lasting tissue damage (Nathan, 2002; Mahmood et al., 2022). A significant current public health concern is chronic inflammatory diseases (Killeen et al., 2014; Shin et al., 2014). Specific illnesses such as psoriasis, rheumatoid arthritis, periodontal disease, asthma, and atherosclerosis are caused by the dysregulation of inflammatory processes. Additionally, it plays a significant role in the development of several degenerative ailments, including diabetes, Alzheimer’s disease, immunological disorders, and periodontal disease (Libby, 2002; Hansson and Peter, 2006; Sadiq et al., 2018). Additionally, the primary contributor to the harm caused by autoimmune illnesses is the inflammatory response (Jan et al., 2020). As a result, controlling inflammatory processes is a crucial strategy for treating a variety of illnesses. The quest for novel anti-inflammatory substances is still a crucial topic of research, despite the fact that there have been several previous attempts in this direction. This is because conventional medicines employing steroidal or nonsteroidal drugs are frequently associated with ineffectiveness and unfavorable side effects. In the traditional medicine of many cultures, plants have been utilized for generations to treat inflammation-related pain (Alam et al., 2020; Mahnashi et al., 2021). Chronic inflammation causes an overproduction of inflammatory mediators by activating Toll-like receptors found in macrophages. These mediators activate transcription factors that are either directly or indirectly controlled by mitogen-activated protein kinases (MAPKs), including nuclear factor kappa B (NF-kB) and nuclear factor erythroid 2-related factor 2 (Nrf2) (He et al., 2020). Additionally, a number of enzymes are activated, including COX-2 (cyclooxygenase), I kappa B kinase (IKK), inducible nitric oxide synthase (iNOS), and 5-LOX (lipoxygenase) (Ko et al., 2013; Tambuwala, 2016).
An endogenous enzyme called cyclooxygenase (COX) is primarily responsible for turning arachidonic acid into prostaglandins. COX-1 and COX-2 are two prevalent isoforms of this enzyme. COX-2 is an inducible enzyme that is expressed in response to an inflammatory stimulus and promotes inflammation, whereas COX-1 is a constitutive enzyme (Munir et al., 2020; Sadiq et al., 2021). Inflammation can be treated effectively by blocking the enzyme COX-2 (Osama et al., 2024). The untreated inflammation causes an increase in free radicals around the swelling area. Furthermore, the increase in temperature of the inflammatory site and even the whole body is due to the inflammatory response. Therefore, the analgesic and anti-inflammatory drugs work side by side due to their common enzymatic targets (Javed et al., 2022). Due to the uncoupling effects on oxidative phosphorylation, anti-inflammatory, analgesic, and antipyretic effects have been related to the suppression of prostaglandin formation. The pro-inflammatory cytokine tumor necrosis factor (TNF), which is released by a variety of cells and has a wide range of cellular effects, is significant (Montgomery and Bowers, 2012; Zelová and Jan 2013). TNF-α has been linked to a variety of disease states in people, including cancer, psychological disorders, and immunological and inflammatory diseases. IL-1 is a different cytokine that primarily promotes inflammation (Fenton, 1992; Alshehri et al., 2024).
Inflammation has typically been treated with steroids, although the use of these medications has increasingly decreased because of their negative effects (Poetker and Reh, 2010). One of the main classes of medications used to treat inflammation is nonsteroidal NSAIDs, which operate on the afflicted tissues by blocking COX, an enzyme involved in the manufacture of prostaglandins (Javed et al., 2021). Celecoxib (1), rofecoxib (2), and etoricoxib (3) are/were some of the very efficient COX-2 inhibitors, and they represent the COX-2 category of medications (Alqahtani et al., 2022). Rofecoxib and etoricoxib had to be taken off the market due to their damage to the gastrointestinal tract and increased risk of cardiotoxicity and hepatotoxicity (Dvorakova and Landa, 2017). Most of the currently used anti-inflammatory drugs, particularly those with established clinical efficacy, such as aspirin and ibuprofen, have an acidic character. It is, therefore, necessary to develop new anti-inflammatory drugs with great therapeutic efficacy and minimal adverse effects.
The researchers working on organo–medicinal compounds provide effective methods for the development of new drugs, natural products, or building blocks of bioactive compounds (Ahmad et al., 2019; Qayyum et al., 2024). Various synthetic reactions provide baseline structural units for pharmacologically important molecules. Previously, we reported the synthetic approaches for the synthesis of various medicinally important compounds (Bibi et al., 2020; Qayyum et al., 2022). The medicinal compounds, particularly the nitrogenous moieties, have been explored for various medicinal purposes (Nugent et al., 2012; Sadiq et al., 2015; Sadiq and Nugent, 2020; Sadiq et al., 2022). The thiazole nucleus has been regarded as one of the prospective bioactive scaffolds in a variety of biological systems based on the literature that is currently accessible (Shah et al., 2023). Thiazole derivatives have a wide range of medicinal uses, including anti-Alzheimer, anticancer, antibacterial, and anti-diuretic properties. Certain medicines have thiazole rings (Tacconelli et al., 2002; Kovala-Demertzi, 2006). For many years, various natural and synthesized compounds containing the thiazole core moiety have been investigated for their anticancer, antibacterial, anti-HIV, anticoagulant, anti-inflammatory, and antioxidant effects. The bioactivities of thiazole scaffolds are further supported by the literature (Kamat et al., 2020). Anti-inflammatory medications lessen pain by completely or partially halting prostaglandin synthesis (Kawabata, 2011). Well-known anti-inflammatory medications (Figure 1), like meloxicam (5), are hybrid substances containing a thiazole core (Kamat et al., 2020). This study has been designed to synthesize new derivatives of thiazole for the treatment of analgesia and inflammation.
FIGURE 1
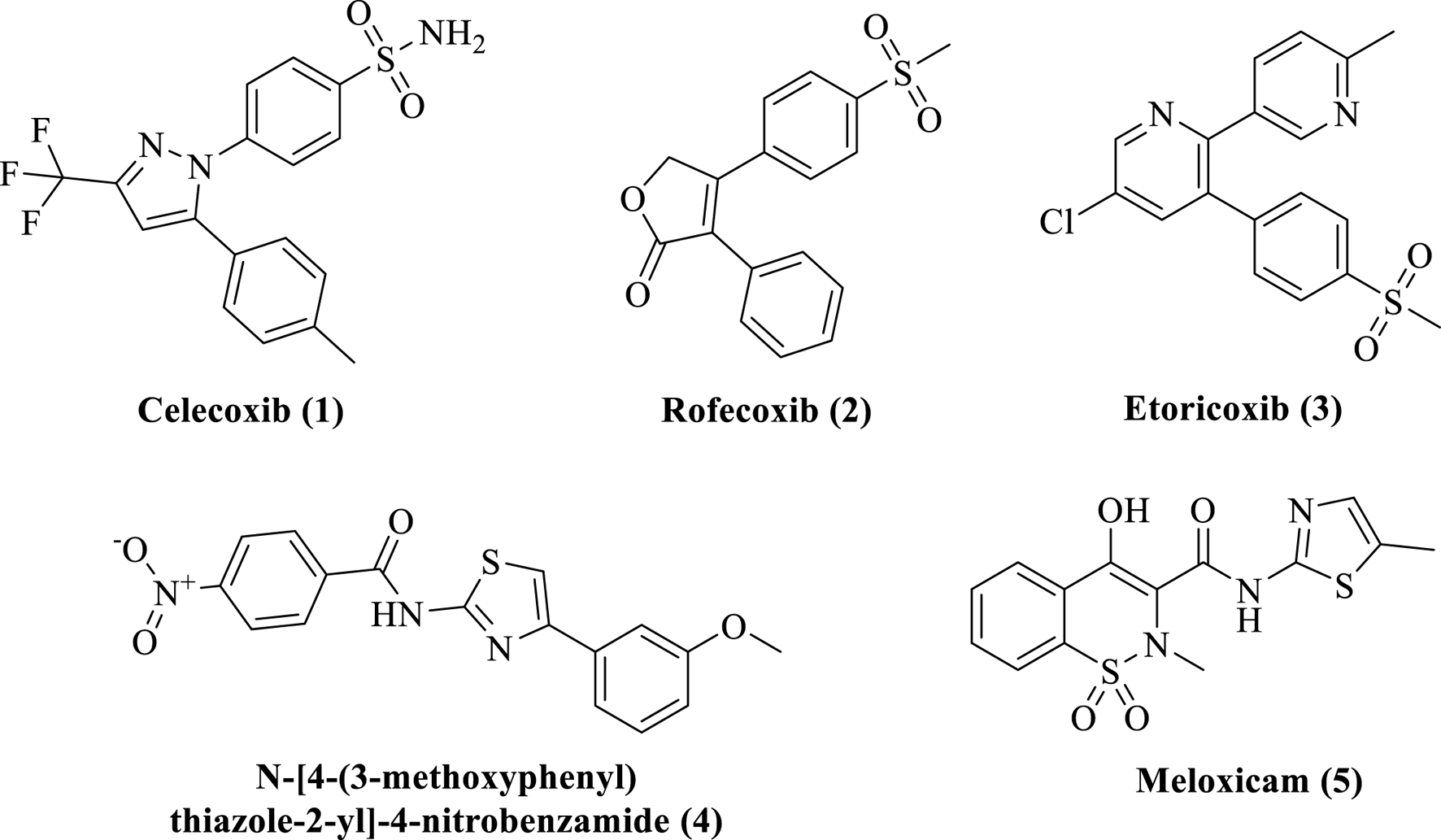
Marketed and reported anti-inflammatory drugs and agents.
Methodology
Chemical synthesis
The chemical synthesis of the target compound is a multi-step procedure. The synthesis was initiated by the bromination of 1-(4-chlorothiophen-2-yl)ethan-1-one (1, 1.0 equivalent) in the presence of diethyl ether (1.0 M) at room temperature. The brominated product (2, 1.0 equivalent) of 1-(4-chlorothiophen-2-yl)ethan-1-one was then treated with thiourea (1.2 equivalent) at a high temperature of 80 C to obtain compound 4-(4-chlorothiophen-2-yl)thiazol-2-amine (3). Compound 3, i.e., 4-(4-chlorothiophen-2-yl)thiazol-2-amine, was then treated with N-bromosuccinimide (NBS) (1.0 equivalent) at low temperature (0 C), followed by treatment with primary or secondary amines (1.5 equivalent) to obtain compound 4. Compound 4 was finally treated with potassium carbonate (0.5 equivalent) in the presence of dimethylformamide (1.0 M) at 70 C to obtain compound 5.
COX-1 activity
The anti-cyclooxygenase-1 activities of the compounds (5a–5g) were assessed using a modified protocol of the Glassman and White experiment. In brief, the COX-1 enzyme was added to a co-factor solution containing glutathione, hematin, and N, N, N-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) prepared in a Tris-HCl buffer (pH 8.0). Then, test samples were added to the COX-1 enzymes and co-factor solution mixture. After 5 min, arachidonic acid was added and the solution was incubated. After incubation, the absorbance of the final reaction was measured at 570 nm using a UV spectrophotometer. Aspirin was used as a positive control for cyclooxygenase-1 enzyme inhibition (Ahmad et al., 2021).
COX-2 activity
The anti-cyclooxygenase-2 activities of the compounds (5a–5g) were assessed using a slightly modified protocol of the Glassman and White experiment. In short, COX-2 enzymes (300 units per ml) were added to a co-factor solution containing glutathione, hematin, and N, N, N-TMPD prepared in a Tris-HCl buffer (pH 8.0). Then, test samples were added to the COX-2 enzymes and co-factor solution mixture. After 5 min, arachidonic acid was added and incubated. After incubation, the absorbance of the final reaction was measured at 570 nm using a UV spectrophotometer. Celecoxib was used as a positive control for cyclooxygenase-2 enzyme inhibition (Jan et al., 2020). The selective COX-2 inhibitors are normally determined using the selectivity index (SI) of the compounds in both COX-1 and COX-2 assays. A greater SI value shows that the compound is a more selective COX-2 inhibitor. The SI of the compounds can be calculated from the IC50 values of their respective COX-1 and COX-2 inhibitors as per the procedure.
5-LOX activity
The 5-lipoxygenase inhibitory activities of the compounds (5a–5g) were evaluated using different concentrations of the compounds against the standard drug zileuton. In brief, a concentration of 10,000 U per ml of the 5-LOX enzyme was used in the experiment. Moreover, linoleic acid was added as a substrate. Afterward, we dissolved different concentrations of test samples in a phosphate buffer (with pH of 6.3), followed by the addition of the 5-LOX enzyme solution. The mixture was then incubated for 5 min at 25°C. After incubation, we added and mixed the linoleic acid solution with it and kept it for 5 min. Finally, the absorbance was measured at 234 nm using a UV spectrophotometer, and the experiment was repeated three times for optimal results (Jan et al., 2020).
Experimental animals and ethical approval
The experimental animals used in this study were obtained from the breeding house and were used as per the Animal By-Laws of the University of Malakand. The animals were maintained at a controlled temperature, humidity, and a 12-h dark/light cycle. The animals were provided food and water as per the approval of the ethical committee. The experimental animals used in this study were reviewed and approved by the Research Ethics Committee of the Department of Pharmacy, University of Malakand, Pakistan (Ref No. /055A).
Carrageenan-induced in vivo inflammation
For the in vivo anti-inflammatory test, the potential compounds (5d and 5e) were put to the test with carrageenan in albino mice that weighed 25–30 g. In total, there were 5 groups of 40 mice. Each group had eight animals. For group I, the negative control was 10 mL of 1% DMSO per kg of body weight. Group II was the positive control and received 100 mg of aspirin per kg of body weight. Compounds 5d and 5e were administered to groups III, IV, and V at amounts of 5, 10, and 20 mg/kg, respectively. Every 30 min, a newly prepared saline solution (0.05 mL) with 1% carrageenan was applied to the sub-planter surface of each mouse. A plethysmometer (LE 7500 Plan Lab SL) was used to measure the paw edema volume every 1–5 h after carrageenan was given. We measured the paw edema of the tested samples and the usual medicine at different times and compared them with the negative control group (Jan et al., 2020). The following method was used to obtain the percentage inhibition of inflammation:
where “EC” is the average edema of the control group and “ET” is the edema of the group that was measured.
Mechanism of inflammation of the potent compounds
The potent synthesized compounds (5d and 5e) might help reduce inflammation by using paw edema tests that were triggered by prostaglandin E2, bradykinin, leukotriene, and histamine. It was done by injecting 10% DMSO, a Bradykinin inhibitor (HOE 140) at a dose of 1 mg/kg, a lipoxygenase inhibitor (montelukast) at a dose of 25 mg/kg, an antihistamine at a dose of 100 mg/kg, the tested compound at a dose of 100 mg/kg, or a cyclooxygenase inhibitor (celecoxib) at a dose of 50 mg/kg. After 1 h, doses of prostaglandin E2 (0.01 mg/mL), leukotriene (10 mg/mL), bradykinin (20 mg/mL), or histamine (0.1 mg/mL) under the planter caused paw swelling. The volume of the paw of each mouse was measured immediately before and after irritants (inflammatory agents) were injected under the planter at 1–5 h (Jan et al., 2020).
Hot plate test
All of the animals were used to being in the laboratory for at least 2 h before the experiments began. An analgesiometer was used to test how well the painkillers worked on a hot plate (Shah et al., 2012). It was kept at 54.0 ± 0.1°C on the hot plates. The animals were given a pre-test, and those with a delay time of less than 30 s were chosen. After the animals were chosen, they were divided into different groups, such as the normal saline group, the standard group, and other groups that would be tried on more powerful samples. Normal water, a standard drug, and different doses of the test sample were all given via abdominal administration. It was possible to determine the duration for which the animals remained on the hot plates at various time intervals.
Docking study
To investigate the anti-inflammatory inhibition potential of our newly synthesized 4-(4-chlorothiophen-2-yl) thiazole-based compounds, all the compounds were docked deep inside the receptors of the target proteins using Molecular Operating Environment (MOE) software. The crystal structures of the target proteins COX-2 and 5-LOX were retrieved from the Protein Data Bank (PDB) using the four-letter alphanumeric access codes 1CX2 and 6N2W, respectively (Sadiq et al., 2021). All compounds and target proteins were protonated, minimized, and prepared according to the previously reported standard procedure. The docking protocol was verified by the redocking method with RMSD >2.0. After docking calculations, all the orientations were evaluated based on the binding energy of the protein–ligand complex (ΔE = S-score) and the non-covalent interaction deep inside the pocket of target proteins.
Results and discussion
Chemistry portion
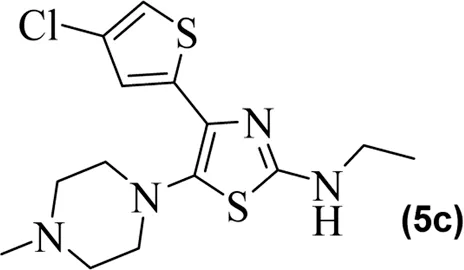
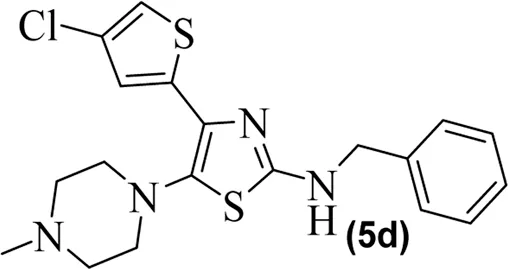
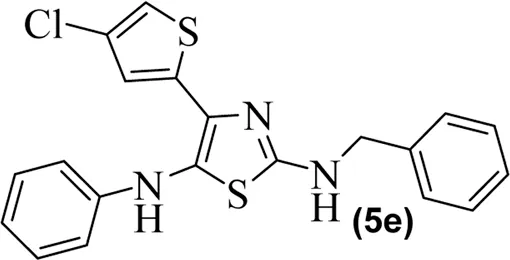
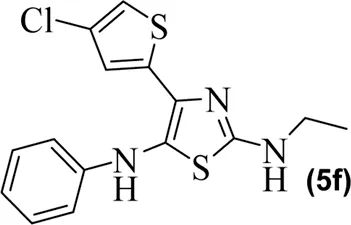
The synthetic steps for approaching the target compounds (5a–5g) are summarized in Scheme 1. Initially, we brominated 1-(4-chlorothiophen-2-yl)ethanone (1) by adding bromine (Br2) in the presence of ether for 2 h to produce the brominated product 2-bromo-1-(4-chlorothiophen-2-yl)ethan-1-one (2). The condensation of 2-bromo-1-(4-chlorothiophen-2-yl)ethan-1-one (2) with thiourea produced 4-(4-chlorothiophen-2-yl)thiazol-2-amine (3) in 5 h. Compound 3 was then brominated by reacting with NBS, and then, by nucleophilic reaction, the intermediate product was treated with amines to obtain compound 4. The combined reaction was completed within 24 h. The intermediates produced in the previous step were treated with K2CO3 to obtain the target products 5a–5g in 24 h, depending on the product. The combined yields for the synthetic steps were in the range of 45%–59%. The structures of the synthesized compounds (5a–5g) are shown in Figure 2.
SCHEME 1

Chemical synthesis of 4-(4-chlorothiophen-2-yl)thiazol-2-amine (5).
FIGURE 2
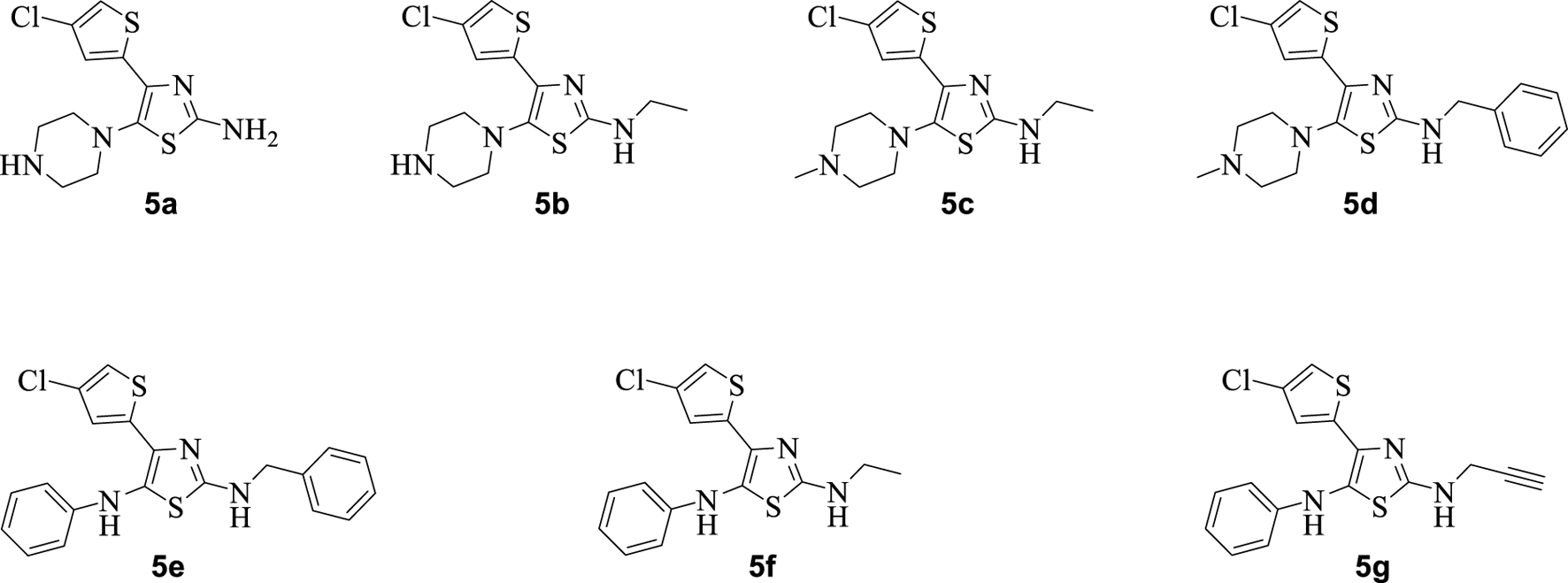
Structures of the synthesized compounds 5a–5g.
In vitro COX/LOX results
The in vitro results of cyclooxygenases (COX-1 and COX-2) and lipoxygenase (5-LOX) of the compounds (5a–5g) are shown in Table 1. Compared to standard zileuton, our compounds (5a–5g) were less potent as inhibitors of 5-lipoxygenase.
TABLE 1
| Compound/std. | IC50 (μM) ± SEM | Selectivity index (SI) | ||
|---|---|---|---|---|
| 5-LOX | COX-1 | COX-2 | ||
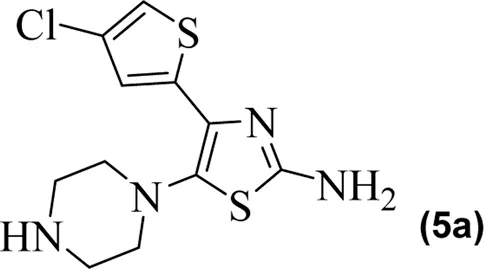
|
93.15 ± 0.07 | 52.38 ± 0.04 | 4.21 ± 0.09 | 52 |
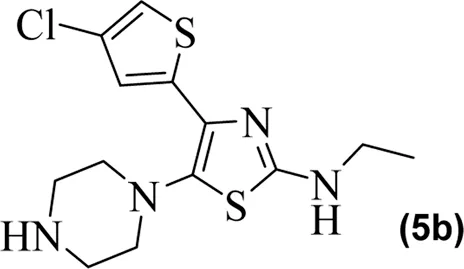
|
98.26 ± 0.08 | 39.64 ± 0.10 | 0.93 ± 0.11 | 42 |
|
72.32 ± 0.11 | 66.18 ± 0.18 | 1.98 ± 0.18 | 33 |
|
23.08 ± 0.18 | 93.41 ± 0.06 | 0.83 ± 0.03 | 112 |
|
38.45 ± 0.20 | 94.33 ± 0.14 | 0.76 ± 0.17 | 124 |
|
56.21 ± 0.15 | 34.09 ± 0.13 | 9.01 ± 0.10 | 4 |
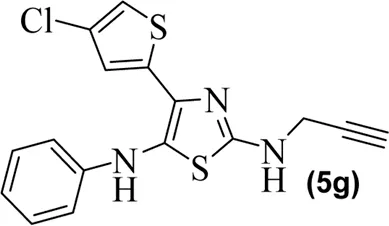
|
83.11 ± 0.16 | 25.81 ± 0.12 | 6.37 ± 0.07 | 4 |
| Zileuton | 11.00 ± 0.12 | — | — | — |
| Aspirin | — | 15.32 ± 0.17 | — | — |
| Celecoxib | — | — | 0.05 ± 0.01 | - |
In vitro cyclooxygenase inhibition results.
The observed IC50 value of zileuton in 5-LOX inhibition was 11.00 μM. Among the compounds, 5d had the highest activity, showing IC50 of 23.08 μM. The 5-LOX inhibition potency of compound 5d was almost equal to half of that of standard drug zileuton. All the other compounds exhibited IC50 values in the range of 38.45–98.26 μM for 5-LOX inhibition. Compared to 5-LOX, our compounds (5a–5g) were much more potent inhibitors of COX pathways. The leading COX-1 inhibitors were 5b, 5f, and 5g with IC50 values of 39.64, 34.09, and 25.81 μM, respectively. All our compounds were very dominant and significant inhibitors of COX-2. The observed range of IC50 values was from 0.76 to 9.01 μM. Among the compounds, 5d and 5e were the most potent inhibitors of COX-2, with IC50 values of 0.83 and 0.76 μM, respectively. Similarly, compound 5b was also dominant with an IC50 value of 0.93 μM. The selectivity index is one of the major parameters for cyclooxygenase inhibitors. A greater value of SI shows that the compound is a more selective inhibitor of COX-2. Among our compounds, 5d and 5e had the highest SI values of 112 and 124, respectively. Based on the potencies of these compounds (5d and 5e) and their selectivity indexes, we further used these two compounds for in vivo experiments.
In vivo anti-inflammatory activity
Carrageenan-induced inflammation results
In carrageenan-induced inflammation, the anti-inflammatory action of the synthesized compounds 5d and 5e produced was remarkable. Compound 5d at the doses of 5, 10, and 20 mg/kg body weight exhibited good activity with percentage inhibition of 45.47 ± 1.11, 49.68 ± 1.44, and 53.44 ± 2.22 at 1 h and was still significant (***p < 0.001) up to 5 h with percentage inhibition of 55.34 ± 1.12, 58.56 ± 1.52, and 61.64 ± 1.10, respectively. Likewise, compound 5e demonstrated the greatest anti-inflammatory activity at doses of 5, 10, and 20 mg/kg. Compound 5e has outstanding anti-inflammatory activity (55.29% ± 1.04%) at a maximum dose (20 mg/kg) at 1 h, which peaked at 5 h (64.59% ± 1.49%) after carrageenan insertion and remained significant (***p < 0.001) throughout investigated drug administration, as shown in Table 2. At a dose of 50 mg/kg for 5 h, aspirin standard medication had a significant effect (59.60 1.52%, ***p < 0.001), which was roughly half of the effects caused by compound 5e at a dose of 20 mg/kg.
TABLE 2
| Treatment | Dose (mg/kg) | Percent inhibition of paw edema | ||||
|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | 5 h | ||
| Vehicle | -------- | 7.720 ± 2.70 | 7.690 ± 1.42 | 9.40 ± 1.20 | 10.42 ± 1.52 | 7.90 ± 2.20 |
| Aspirin | 50 | 48.52 ± 1.12*** | 54.63 ± 1.47*** | 55.22 ± 1.38*** | 58.92 ± 1.95*** | 59.60 ± 1.52*** |
| 5d | 5 | 45.47 ± 1.11*** | 48.10 ± 1.42*** | 51.78 ± 2.10*** | 52.11 ± 2.67*** | 55.34 ± 1.12*** |
| 10 | 49.68 ± 1.44*** | 52.23 ± 1.65*** | 54.45 ± 1.47*** | 55.56 ± 1.52*** | 58.56 ± 1.52*** | |
| 20 | 53.44 ± 2.22*** | 55.65 ± 2.21*** | 57.33 ± 1.31*** | 56.89 ± 1.29*** | 61.64 ± 1.10*** | |
| 5e | 5 | 47.70 ± 1.68*** | 51.47 ± 1.50*** | 54.56 ± 1.34*** | 53.35 ± 1.47*** | 56.25 ± 1.65*** |
| 10 | 52.36 ± 2.26*** | 54.58 ± 2.58*** | 57.59 ± 1.59*** | 56.78 ± 2.66*** | 62.65 ± 2.79*** | |
| 20 | 55.29 ± 1.04*** | 57.39 ± 1.69*** | 60.76 ± 2.68*** | 59.08 ± 1.98*** | 64.59 ± 1.49*** | |
Percentage inhibitions of the synthesized compounds in carrageenan-induced paw edema.
The percentage of carrageenan-induced paw edema model in mice was blocked by tested molecules at doses of 5, 10, and 20 mg/kg. The mean (±SEM) for a group of eight mice is shown by each percentage point. Two-way repeated measures ANOVA and Bonferroni’s post hoc test were used to analyze the data. Asterisks indicate numbers that are important from the control (car). *p < 0.05, **p < 0.01, and ***p < 0.001; n.s. stands for “not significant.” There are eight mice in each group.
Anti-inflammatory mechanism of the potent synthesized compounds 5d and 5e
After observing potential anti-inflammatory effects in vitro and in vivo, we further investigated the synthesized compounds 5d and 5e to understand how they work to fight inflammation. We used different phlogistic agents, such as histamine, bradykinin, prostaglandin, and leukotriene, to test the process. It was found that compound 5d had a slight antihistaminic effect (27.12%) at the highest dose (20 mg/kg) at 3 h. This may be because it stopped mast cells from releasing mediators. In the same way, compound 5e showed 35% suppression at 3 h. In addition, the standard drug chlorpheniramine maleate greatly reduced swelling observed from the first to the fifth hour, resulting in a reduction of 71.62%–64.02% caused by histamine (Figure 3A). In the second part, swelling in the mouse paw reached its peak 60 min after the injection of bradykinin. The swelling of the mouse paw was not alleviated by compound 5d. At the highest doses (20 mg/kg), compound 5d stopped paw swelling by 9.20% in the first hour. It then decreased but not as much as the normal drug (HOE 140). Similarly, compound 5e did not yield excellent results either. Its effect peaked at 21.30% in the first hour and then decreased from the second to the fifth hour, as shown in Figure 3B. Figure 3C shows a notable increase in the amount of prostaglandin E2 in the paw tissue after the injection of the prostaglandin E2 mediator. When mice were given compound 5d (20 mg/kg) or celecoxib (50 mg/kg), the level of increased PGE2 significantly decreased. At the fourth hour, compound 5d had the strongest effect (76.56 ± 0.58), about the same as a normal drug. It was still significant (***p < 0.001) at the fifth hour. In the same way, compound 5e was stronger than compound 5d and showed a suppression of 66.50% ± to 88.0% ± 0.66% from the first to the fourth hour. At the 4th hour after inflammogen, the usual drug celecoxib showed an 88% reduction. Furthermore, leukotriene caused inflammation, and the mouse paws swelled up after it was applied. The swelling reached its highest point 30 min after it was applied. It was found that compound 5d was very effective at treating mouse paw edema. Compound 5d showed good results, causing a percentage inhibition of 51.80 ± 0.58, 55.90 ± 0.66, 59.46 ± 0.52, 64.56 ± 0.78, and 59.90 ± 0.68. The compound 5d result was significant (**p < 0.01) at the first hour and reached its highest level at the fourth hour after the leukotriene mediator was given. It remained significant (***p < 0.001) until the fifth hour. Similarly, compound 5e showed excellent results, showing 69.84% inhibition at the fourth hour. At the fourth hour (Figure 3D), the usual drug montelukast showed a percentage reduction of 72.79 ± 1.10.
FIGURE 3
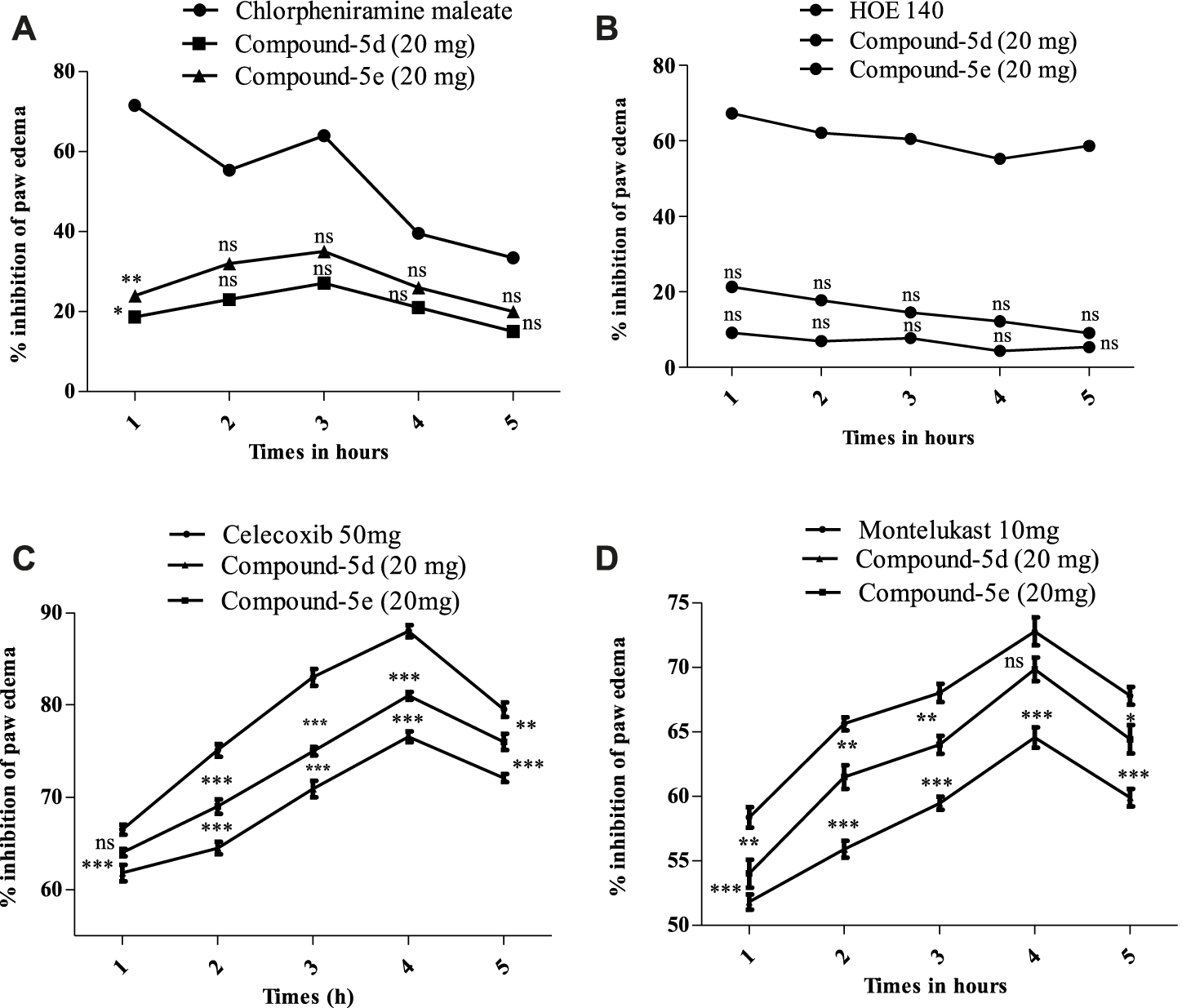
(A) Percentage of blockage caused by histamine and compounds 5d and 5e (20 mg/kg) in a mouse model of histamine-induced paw edema. (B) Amount of blockage caused by bradykinin and compounds 5d and 5e (20 mg/kg) in a mouse model of bradykinin-induced paw edema. (C) Amount of inhibition that the studied compounds 5d and 5e (20 mg/kg) created in a mouse model of prostaglandin E2-induced paw edema. (D) Amount of reduction that tested compounds 5d and 5e (20 mg/kg) caused in a mouse model of leukotriene-induced paw edema. The mean (±SEM) for a group of eight mice is shown by each percentage point. ANOVA and Dunnett’s test were used to analyze the data. *p < 0.05, **p < 0.01, and ***p < 0.001 are all significant, and n.s. means that there was no difference between the experimental and normal drugs.
Evaluation of anti-nociceptive activity on the hot plate model
The results of the hot plate method of analgesia are presented in Table 3. By the administration of the positive control group after 30 min, the delay time was 17.23, and the painkilling effect was 47.59% ± 0.71% at 100 mg/kg body weight. Synthesized compound 5e changed the delay time by 14.33 ± 0.44 (p < 0.01) and stopped thermal shocks 36.99% ± 0.94% of the time at a dose of 20 mg/kg I/p. Furthermore, compound 5d greatly reversed the effect, stopping 29.62% ± 0.84% (p < 0.05) of the activity at a dose of 20 mg/kg (Table 3). Similarly, after 60 min, the delay time was significantly longer in animals that had been given the usual drug (5 mg/kg tramadol I/P) than in those that had been given the vehicle control group (p < 0.001). At a dose of 20 mg/kg I/p, compound 5e significantly changed the pain-relieving effect, stopping 44.16% ± 0.70% of it (p < 0.01). Synthesized compound 5d that was tested had a delay time of 14 s and blocked the good effects of heat stimuli with a significance level of 35.50% ± 0.50% (p < 0.05) at a dose of 20 mg/kg (Table 3). In addition, the latency time was significantly longer in animals that were given the usual drug (5 mg/kg tramadol I/P) than in those given the vehicle control group (latency time 13.27 s; p < 0.001). The pain effect was greatly offset by compound 5e, which had a delay time of 11.25 (p < 0.01) and caused percentage analgesic inhibitions of 22.93 ± 0.60 at a dose of 20 mg/kg I/p. Compared to the negative control at the same amount, compound 5d had a delay time of 8.8 and had a lesser effect than compound 5e.
TABLE 3
| Treatment | Dose (mg/kg) | Latency time in seconds (mean ± SEM) | ||
|---|---|---|---|---|
| After 30 min | After 60 min | After 90 min | ||
| Normal saline | 10 mL/kg | 8.01 ± 0.22 | 9.03 ± 0.28 | 7.55 ± 0.59 |
| Standard | 5 mg/kg | 16.31 ± 0.47*** | 18.50 ± 0.58*** | 12.25 ± 0.31*** |
| 5d | 20 mg/kg | 12.83 ± 0.52* | 14.00 ± 0.10*** | 8.83 ± 0.80* |
| 5e | 20 mg/kg | 14.33 ± 0.44** | 16.17 ± 0.42** | 11.25 ± 0.52** |
Analgesic activity of the synthesized compounds assessed using the hot plate method.
The information shows pain-relieving effects using a hot plate test on mice. The results are shown as the mean ± SEM (n = 6) and were analyzed using a two-way ANOVA, followed by Bonferroni’s post hoc test. ∗M < 0.05; ∗∗p < 0.01, and ∗∗∗p < 0.001; ns; not important.
Docking studies
Docking studies on COX-2
The structure of COX-2 was retrieved from the protein database with PDB ID 1CX2. It contains the COX-2 enzyme complexed with the native ligand (4-[3-(trifluoromethyl)-5-(4-bromophenyl)-1H-pyrazol-1-yl]benzenesulfonamide). The native ligand displayed important interactions with amino acid residues, including His90, Leu352, Ser353, Arg513, Tyr355, Arg120, Val349, Val523, Tyr385, Trp387, Phe381, Leu384, and Ala529.
Compound 5d displayed important binding affinities similar to those of native ligands, including vital hydrophilic hydrogen bond interactions with His90 and Ser353 through the hydrogen of the NH group. It also displayed pi–sulfur interaction (hydrophobic) with Tyr355 and Phe518 by the sulfur of the thiazole and thiophene regions. Other hydrophobic interactions include the π–π stacked interaction with Asp515 and the π–alkyl interaction with Val523.
Compound 5e also displayed binding affinities with important residues similar to the native ligand. It includes pi–sulfur hydrophobic interaction with His90 through the sulfur of the thiophene region. The aromatic region in the benzyl region showed pi–sulfur interaction with Met522 and Met113. Pi–sigma interaction was also displayed between the Ser353 amino acid residue and the aromatic region of thiophene. The halogen attached to the thiophene group also displayed interaction with Gln192. The aromatic region of one terminal side of the molecule displayed binding affinity (hydrophobic interaction) with Trp387 (Figure 4).
FIGURE 4
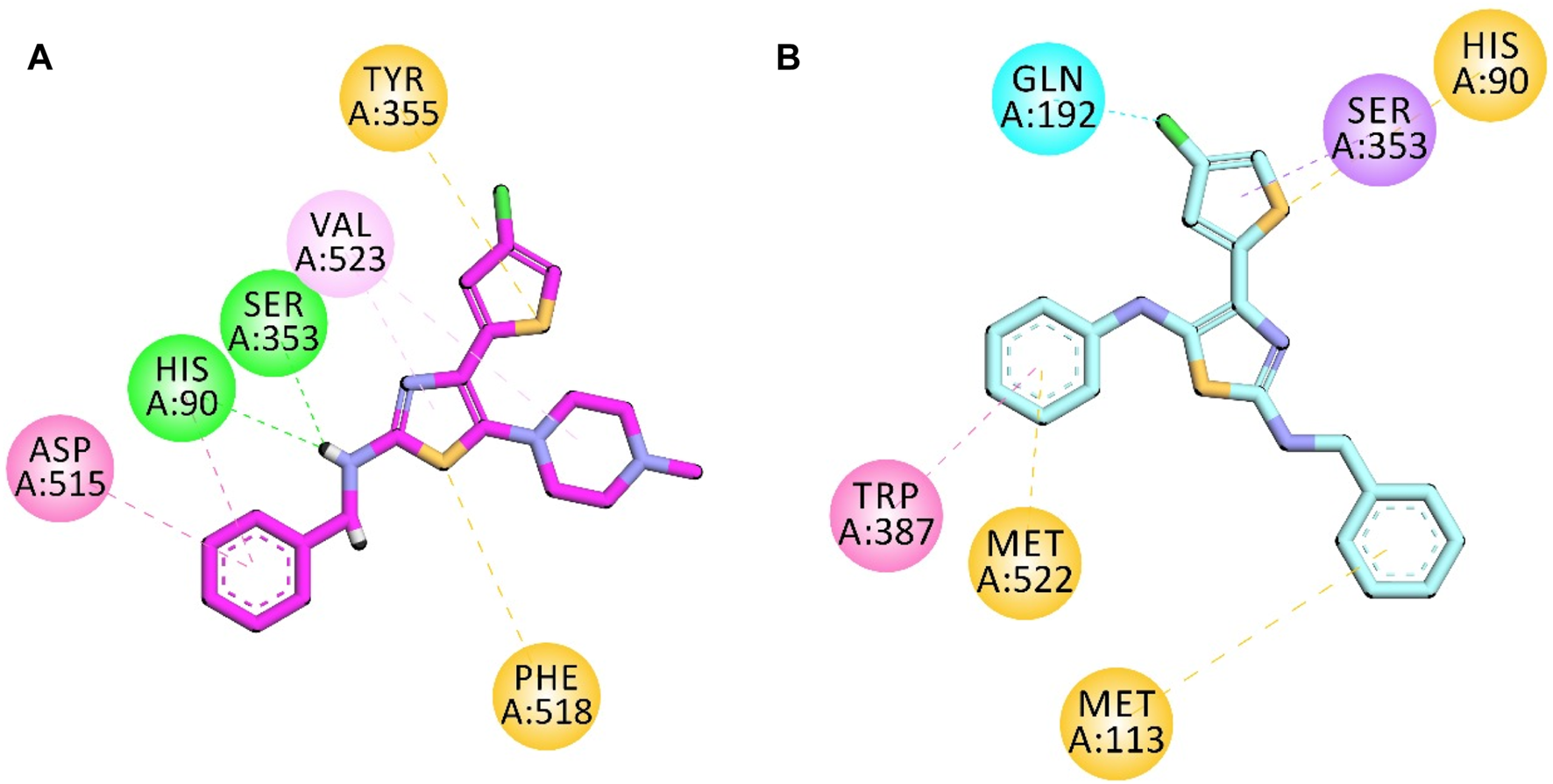
2D interaction diagram of synthesized compounds (A) 5d and (B) 5e in the binding site of COX-2 (PDB ID = COX2).
Docking studies on 5-LOX
The co-crystalized structure of enzyme 5-LOX having native ligand nordihydroguaiaretic acid (NDGA) was obtained from the PDB having a code 6N2W. The native ligand displayed binding affinities with the following amino acid residues: His600, Leu607, Arg596, Asn407, Phe359, Trp599, His372, His367, and Ala410.
Compound 5d displayed binding affinities with amino acid residues similar to those of native ligands, including hydrophobic interactions with His367, Trp599, and Phe359 (pi–pi T-shaped) through the thiophene aromatic region and aromatic region present at the one terminal end. The halogen group attached to the thiophene ring displayed halogen interactions. Pi–sulfur hydrophobic binding affinity was displayed with the His372 residue through the sulfur of the thiophene group. Other hydrophobic binding affinities include Leu607 and Ala410.
Compound 5e also displayed binding affinities similar to those of native ligands, including hydrophobic interactions with His367 (pi–pi T-shaped), His 432, and Trp599 (pi–pi stacked) with an aromatic region present on both terminals of the synthesized molecule and thiophene aromatic region. It also displayed pi–alkyl hydrophobic interactions with Leu268 and Ala603 (Figure 5).
FIGURE 5
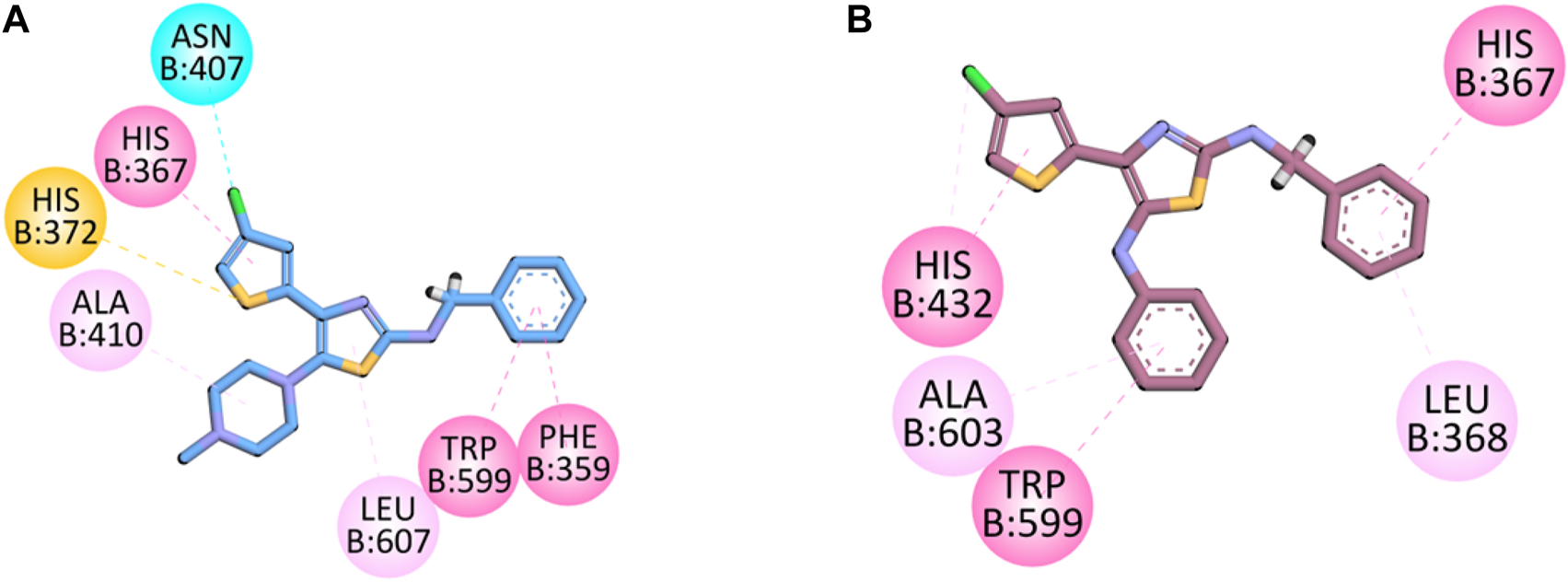
2D interaction diagram of synthesized compounds (A) 5d and (B) 5e in the binding pocket of 5-LOX (PDB ID: 6N2W).
Analgesia and its major cause, inflammation, are medical complications that go side by side in many ailments (Zhang and An, 2007; Mahnashi et al., 2021). Both of these ailments are protective responses to injuries and external stimuli (Antonelli and Kushner, 2017). Hotness, or analgesia, is among the major symptoms of inflammation. The current marketed analgesic and anti-inflammatory drugs are associated with severe unwanted effects (Carter et al., 2014). To minimize toxicity and improve the activity profile, medicinal chemists synthesize various potent molecules for the treatment of analgesia and inflammation (Zayed, 2022). Previously, we synthesized simple pyrrolidinedione and other Michael products as potential analgesic and anti-inflammatory agents (Ahmad et al., 2021; Sadiq et al., 2021). In this research, we found that bis-succinimide efficiently inhibits the COX/LOX pathway for the management of analgesia and inflammation.
Conclusion
In this study, we synthesized 7 new 4-(4-chlorothiophen-2-yl)thiazol-2-amine derivatives (5a–5g) as potential anti-inflammatory and analgesic agents. The compounds were synthesized efficiently through multistep chemical reactions. The pharmacologically important building blocks of thiazoles, specifically, compounds 5d and 5e, have been proven to be potential inhibitors of COX/LOX pathways. Compared to the respective standard drugs (zileuton for 5-LOX, aspirin for COX-1, and celecoxib for COX-2), compounds 5d and 5e were potent anti-COX/LOX agents. Moreover, our compounds also proved to be safe in experimental animals. The compounds proved to be very active analgesic and anti-inflammatory agents in in vivo models. The molecular docking of the selected compounds in the enzymes COX-1, COX-2, and 5-LOX further supported our results and confirmed that our synthesized compounds effectively bind to the amino acid residues of the target site. In this study, we explored the thiazole scaffold as a potential analgesic and anti-inflammatory building block.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary materials, further inquiries can be directed to the corresponding authors.
Ethics statement
The experimental animals used in this study were obtained from the breeding house and were used as per the Animal By-Laws of the University of Malakand. The experimental animals used in this study were reviewed and approved by the Research Ethics Committee of the Department of Pharmacy, University of Malakand, Pakistan (Ref No. /055A). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MM: funding acquisition, investigation, methodology, project administration, resources, visualization, and writing–original draft. UR: conceptualization, formal analysis, investigation, methodology, software, and writing–original draft. HA: data curation, formal analysis, funding acquisition, project administration, resources, validation, and writing–original draft. MN: data curation, formal analysis, funding acquisition, investigation, methodology, validation, and writing–original draft. AB: data curation, formal analysis, funding acquisition, project administration, resources, software, and writing–original draft. OA: data curation, formal analysis, funding acquisition, investigation, methodology, software, and writing–original draft. MA: data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, and writing–original draft. MJ: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, software, validation, and writing–original draft. AS: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, software, supervision, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Authors would like to acknowledge the support of the Deputy for Research and Innovation, Ministry of Education, Kingdom of Saudi Arabia for this research through a grant (NU/IFC/02/MRC/-/010) under the Institutional Funding Committee at Najran University, Kingdom of Saudi Arabia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Ahmad G. Rasool N. Rizwan K. Imran I. Ameer F. Z. Zubair M. et al (2019). Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl) thiophene-2-carboxamide analogs. Bioorg. Chem.92, 103216. 10.1016/j.bioorg.2019.103216
2
Ahmad S. Mahnashi M. H. Alyami B. A. Alqahtani Y. S. Ullah F. Ayaz M. et al (2021). “Synthesis of michael adducts as key building blocks for potential analgesic drugs: in vitro, in vivo and in silico explorations,” in Drug design (Development and Therapy)15, 1299–1313. 10.2147/DDDT.S292826
3
Alam F. Kinza M. D. Rasheed R. Abdul S. Jan M. S. Amber M. M. et al (2020). Phytochemical investigation, anti-inflammatory, antipyretic and antinociceptive activities of Zanthoxylum armatum DC extracts-in vivo and in vitro experiments. Heliyon6 (11), e05571. 10.1016/j.heliyon.2020.e05571
4
Alqahtani Y. S. Muhammad S. J. Mahnashi M. H. Alyami B. A. Alqarni A. O. Rashid U. et al (2022). Anti-inflammatory potentials of β-ketoester derivatives of N-ary succinimides: in vitro, in vivo, and molecular docking studies. J. Chem.2022, 1–11. 804032210.1155/2022/8040322
5
Alshehri O. M. Zeb A. Shah S. M. M. Mahnashi M. H. Asiri S. A. Alqahtani O. (2024). Investigation of anti-nociceptive, anti-inflammatory potential and ADMET studies of pure compounds isolated from Isodon rugosus Wall. ex Benth. Front. Pharmacol.15. 10.3389/fphar.2024.1328128
6
Antonelli M. Kushner I. (2017). It's time to redefine inflammation. FASEB J.31 (5), 1787–1791. 10.1096/fj.201601326R
7
Bibi M. Qureshi N. A. Sadiq A. Farooq U. Hassan A. Shaheen N. et al (2020). Exploring the ability of dihydropyrimidine-5-carboxamide and 5-benzyl-2, 4-diaminopyrimidine-based analogues for the selective inhibition of L. major Dihydrofolate reductase. Eur. J. Med. Chem.210 (2021), 112986. 10.1016/j.ejmech.2020.112986
8
Carter G. T. Duong V. Ho S. Kathryn C. N. Christopher L. G. Weeks D. L. (2014). Side effects of commonly prescribed analgesic medications. Phys. Med. Rehabilitation Clin.25 (2), 457–470. 10.1016/j.pmr.2014.01.007
9
Dvorakova M. Landa P. (2017). Anti-inflammatory activity of natural stilbenoids: a review. Pharmacol. Res.124, 126–145. 10.1016/j.phrs.2017.08.002
10
Fenton M. J. (1992). Review: transcriptional and post-transcriptional regulation of interleukin 1 gene expression. Int. J. Immunopharmacol.14 (3), 401–411. 10.1016/0192-0561(92)90170-p
11
Hansson G. K. Peter L. (2006). The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol.6 (7), 508–519. 10.1038/nri1882
12
He F. Antonucci L. Karin M. (2020). NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis41 (4), 405–416. 10.1093/carcin/bgaa039
13
Jan M. S. Ahmad S. Hussain F. Ahmad A. Mahmood F. Rashid U. et al (2020). Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2, 5-dione derivatives as multitarget anti-inflammatory agents. Eur. J. Med. Chem.186, 111863. 10.1016/j.ejmech.2019.111863
14
Javed M. A. Ashraf N. Jan M. S. Mahnashi M. H. Alqahtani Y. S. Alyami B. A. et al (2021). Structural modification, in vitro, in vivo, ex vivo, and in silico exploration of pyrimidine and pyrrolidine cores for targeting enzymes associated with neuroinflammation and cholinergic deficit in Alzheimer’s disease. ACS Chem. Neurosci.12 (21), 4123–4143. 10.1021/acschemneuro.1c00507
15
Javed M. A. Bibi S. Jan M. S. Ikram M. Zaidi A. Farooq U. et al (2022). Diclofenac derivatives as concomitant inhibitors of cholinesterase, monoamine oxidase, cyclooxygenase-2 and 5-lipoxygenase for the treatment of Alzheimer's disease: synthesis, pharmacology, toxicity and docking studies. RSC Adv.12 (35), 22503–22517. 10.1039/d2ra04183a
16
Kamat V. Santosh R. Poojary B. SureshNayak P. Karan Kumar B. Sankaranarayanan M. et al (2020). Pyridine-and thiazole-based hydrazides with promising anti-inflammatory and antimicrobial activities along with their in silico studies. ACS omega5 (39), 25228–25239. 10.1021/acsomega.0c03386
17
Kawabata A. (2011). Prostaglandin E2 and pain—an update. Biol. Pharm. Bull.34 (8), 1170–1173. 10.1248/bpb.34.1170
18
Killeen M. J. Linder M. Pontoniere P. Crea R. (2014). NF-κβ signaling and chronic inflammatory diseases: exploring the potential of natural products to drive new therapeutic opportunities. Drug Discov. today19 (4), 373–378. 10.1016/j.drudis.2013.11.002
19
Ko Y.-J. Kim H.-H. Kim E.-J. Katakura Y. Lee W.-S. Kim G.-S. et al (2013). Piceatannol inhibits mast cell-mediated allergic inflammation. Int. J. Mol. Med.31 (4), 951–958. 10.3892/ijmm.2013.1283
20
Kovala-Demertzi D. (2006). Recent advances on non-steroidal anti-inflammatory drugs, NSAIDs: organotin complexes of NSAIDs. J. Organomet. Chem.691 (8), 1767–1774. 10.1016/j.jorganchem.2005.11.058
21
Libby P. (2002). Atherosclerosis: the new view. Sci. Am.286 (5), 46–55. 10.1038/scientificamerican0502-46
22
Mahmood F. Khan J. A. Mahnashi M. H. Jan M. S. Javed M. A. Rashid U. et al (2022). Anti-inflammatory, analgesic and antioxidant potential of new (2S,3S)-2-(4-isopropylbenzyl)-2-methyl-4-nitro-3-phenylbutanals and their corresponding carboxylic acids through in vitro, in silico and in vivo studies. Molecules27 (13), 4068. 10.3390/molecules27134068
23
Mahnashi M. H. Alyami B. A. Alqahtani Y. S. Muhammad S. J. Rashid U. Abdul S. et al (2021). Phytochemical profiling of bioactive compounds, anti-inflammatory and analgesic potentials of Habenaria digitata Lindl.: molecular docking based synergistic effect of the identified compounds. J. Ethnopharmacol.273 (2021), 113976. 10.1016/j.jep.2021.113976
24
Montgomery S. L. Bowers W. J. (2012). Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. neuroimmune Pharmacol.7 (1), 42–59. 10.1007/s11481-011-9287-2
25
Munir A. Adil K. Saeed K. Abdul S. Ullah R. Ali G. et al (2020). Synthesis, in-vitro, in-vivo anti-inflammatory activities and molecular docking studies of acyl and salicylic acid hydrazide derivatives. Bioorg. Chem.104, 104168. 10.1016/j.bioorg.2020.104168
26
Nathan C. (2002). Points of control in inflammation. Nature420 (6917), 846–852. 10.1038/nature01320
27
Nugent T. C. Bibi A. Abdul S. Shoaib M. Umar M. N. Tehrani F. N. (2012). Chiral picolylamines for Michael and aldol reactions: probing substrate boundaries. Org. Biomol. Chem.10 (46), 9287–9294. 10.1039/c2ob26382c
28
Osama M. Alshehri A. Z. Syed M. M. S. Mahnashi M. H. Saeed A. A. Omaish A. et al (2024) Investigation of anti-nociceptive and anti-inflammatory potential and ADMET studies of pure compounds isolated from Isodon rugosus Wall. ex Benth. Front. Pharmacol.,15132812810.3389/fphar.2024.1328128
29
Poetker D. M. Reh D. D. (2010). A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryngologic Clin. N. Am.43 (4), 753–768. 10.1016/j.otc.2010.04.003
30
Qayyum M. I. Ullah S. Rashid U. Abdul S. Mahnashi M. H. Khalil S. U. K. et al (2024). N-phenyl and N-benzyl substituted succinimides: preclinical evaluation for their antihypertensive effect and underlying mechanism. Eur. J. Pharmacol.964 (2024), 176195. 10.1016/j.ejphar.2023.176195
31
Qayyum M. I. Ullah S. Rashid U. Abdul S. Obaidullah M. H. M. Alshehri O. M. et al (2022). Synthesis, molecular docking, and preclinical evaluation of a new succinimide derivative for cardioprotective, hepatoprotective and lipid-lowering effects. Molecules27 (19), 6199. 10.3390/molecules27196199
32
Sadiq A. Mahmood F. Ullah F. Ayaz M. Ahmad S. Ul Haq F. et al (2015). Synthesis, anticholinesterase and antioxidant potentials of ketoesters derivatives of succinimides: a possible role in the management of Alzheimer’s. Chem. Central J.9, 31–39. 10.1186/s13065-015-0107-2
33
Sadiq A. Mahnashi M. H. Alyami B. A. Alqahtani Y. S. Alqarni A. O. Rashid U. (2021). Tailoring the substitution pattern of Pyrrolidine-2, 5-dione for discovery of new structural template for dual COX/LOX inhibition. Bioorg. Chem.112, 104969. 10.1016/j.bioorg.2021.104969
34
Sadiq A. Mater H. M. Umer R. Muhammad S. J. Mohammed A. A. Mohammed A. H. (2022). 3-(((1 S, 3 S)-3-((R)-Hydroxy (4-(trifluoromethyl) phenyl) methyl)-4-oxocyclohexyl) methyl) pentane-2, 4-dione: design and synthesis of new stereopure multi-target antidiabetic agent. Molecules27 (10), 3265. 10.3390/molecules27103265
35
Sadiq A. Nugent T. C. (2020). Catalytic access to succinimide products containing stereogenic quaternary carbons. ChemistrySelect5 (38), 11934–11938. 10.1002/slct.202003664
36
Sadiq A. Zeb A. Ullah F. Ahmad S. Ayaz M. Rashid U. et al (2018). Chemical characterization, analgesic, antioxidant, and anticholinesterase potentials of essential oils from Isodon rugosus Wall. ex. Benth. Front. Pharmacol.9, 623. 10.3389/fphar.2018.00623
37
Shah M. Jan M. S. Abdul S. Khan S. Rashid U. (2023). SAR and lead optimization of (Z)-5-(4-hydroxy-3-methoxybenzylidene)-3-(2-morpholinoacetyl) thiazolidine-2, 4-dione as a potential multi-target antidiabetic agent. Eur. J. Med. Chem.258 (2023), 115591. 10.1016/j.ejmech.2023.115591
38
Shah S. M. M. Ullah F. Syed M. H. S. Zahoor M. Abdul S. (2012). Analysis of chemical constituents and antinociceptive potential of essential oil of Teucrium Stocksianum bioss collected from the North West of Pakistan. BMC complementary Altern. Med.12, 244–246. 10.1186/1472-6882-12-244
39
Shin I.-S. Park J.-W. Shin N.-R. Jeon C.-Mi Kwon O.-K. Kim J.-S. et al (2014). Melatonin reduces airway inflammation in ovalbumin-induced asthma. Immunobiology219 (12), 901–908. 10.1016/j.imbio.2014.08.004
40
Tacconelli S. Capone M. L. Sciulli M. G. Patrignani P. (2002). The biochemical selectivity of novel COX-2 inhibitors in whole blood assays of COX-isozyme activity. Curr. Med. Res. Opin.18 (8), 503–511. 10.1185/030079902125001335
41
Tambuwala M. M. (2016). Natural nuclear factor kappa beta inhibitors: safe therapeutic options for inflammatory bowel disease. Inflamm. bowel Dis.22 (3), 719–723. 10.1097/MIB.0000000000000655
42
Zayed M. F. (2022). Medicinal chemistry of quinazolines as analgesic and anti-inflammatory agents. ChemEngineering6 (6), 94. 10.3390/chemengineering6060094
43
Zelová H. Jan H. (2013). TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm. Res.62, 641–651. 10.1007/s00011-013-0633-0
44
Zhang J.-M. An J. (2007). Cytokines, inflammation and pain. Int. Anesthesiol. Clin.45 (2), 27–37. 10.1097/AIA.0b013e318034194e
Summary
Keywords
analgesic, inflammation, thiazole, cyclooxygenase, LOX, in vivo mechanism
Citation
Mahnashi MH, Rashid U, Almasoudi HH, Nahari MH, Ahmad I, Binshaya AS, Abdulaziz O, Alsuwat MA, Jan MS and Sadiq A (2024) Modification of 4-(4-chlorothiophen-2-yl)thiazol-2-amine derivatives for the treatment of analgesia and inflammation: synthesis and in vitro, in vivo, and in silico studies . Front. Pharmacol. 15:1366695. doi: 10.3389/fphar.2024.1366695
Received
07 January 2024
Accepted
16 February 2024
Published
29 February 2024
Volume
15 - 2024
Edited by
Hafiz Muhammad Asif, Islamia University of Bahawalpur, Pakistan
Reviewed by
Qian Bai, Second Affiliated Hospital of Zhengzhou University, China
Abdul Bari Shah, Seoul National University, Republic of Korea
Updates
Copyright
© 2024 Mahnashi, Rashid, Almasoudi, Nahari, Ahmad, Binshaya, Abdulaziz, Alsuwat, Jan and Sadiq.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umer Rashid, umerrashid@cuiatd.edu.pk; Abdul Sadiq, sadiquom@yahoo.com; Hassan Hussain Almasoudi, hhalmasoudi@nu.edu.sa
ORCID ID: Meshari A. Alsuwat, orcid.org/0000-0003-0953-0692
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.