 Chih-Hsiang Lin
Chih-Hsiang Lin Chen-Jui Ho1
Chen-Jui Ho1 Meng-Han Tsai
Meng-Han Tsai- 1Department of Neurology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan
- 2School of Medicine, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 3Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, College of Medicine, Chang Gung University, Kaohsiung, Taiwan
Antiseizure medications (ASMs) play a central role in seizure management, however, unpredictability in the response to treatment persists, even among patients with similar seizure manifestations and clinical backgrounds. An objective biomarker capable of reliably predicting the response to ASMs would profoundly impact epilepsy treatment. Presently, clinicians rely on a trial-and-error approach when selecting ASMs, a time-consuming process that can result in delays in receiving alternative non-pharmacological therapies such as a ketogenetic diet, epilepsy surgery, and neuromodulation therapies. Pharmacogenetic studies investigating the correlation between ASMs and genetic variants regarding their mechanistic targets offer promise in predicting the response to treatment. Sodium channel subunit genes have been extensively studied along with other ion channels and receptors as targets, however, the results have been conflicting, possibly due to methodological disparities including inconsistent definitions of drug response, variations in ASM combinations, and diversity of genetic variants/genes studied. Nonetheless, these studies underscore the potential effect of genetic variants on the mechanism of ASMs and consequently the prediction of treatment response. Recent advances in sequencing technology have led to the generation of large genetic datasets, which may be able to enhance the predictive accuracy of the response to ASMs.
Introduction
Antiseizure medications (ASMs), previously called antiepileptic drugs (AEDs), are commonly used to manage seizures in individuals with epilepsy (McCorry et al., 2004). The ultimate goal of seizure treatment is to achieve seizure freedom, defined by the International League Against Epilepsy (ILAE) consensus as being free from seizures for a minimum of three times the longest preintervention inter-seizure interval or 12 months, whichever is longer (Kwan et al., 2010). To achieve this goal, physicians select ASMs based on factors such as age, gender, epilepsy syndrome, co-medications, and comorbidities. However, even an ASM carefully chosen by an experienced epileptologist can have a different response in different patients with similar epilepsy syndrome and/or underlying clinical status. Although more than 30 ASMs are currently available, approximately one-third of patients still have seizures after being treated with more than two ASMs (Kwan and Sander, 2004). Such patients are considered to have drug-resistant epilepsy (DRE), defined by the ILAE consensus as the failure of adequate trials of two tolerated and appropriately chosen ASMs to achieve sustained seizure freedom (Kwan et al., 2010). The effectiveness of controlling seizures or developing adverse drug reactions (ADRs) is unpredictable. Therefore, patients have to endure the consequences of inadequately controlled seizures or ADRs before DRE is confirmed, delaying the initiation of other management strategies to alleviate the seizures, such as a ketogenetic diet (Martin et al., 2016), epilepsy surgery (Rugg-Gunn et al., 2020), vagus nerve stimulation (Elliott et al., 2011), deep brain stimulation (Vetkas et al., 2022), or the use of cannabidiol (Gaston and Szaflarski, 2018). The use of ASMs relies on a trial-and-error approach, with careful adjustments of the dosage to achieve a balance between response and ADRs. This approach relies heavily on the clinicians’ expertise, and only a few objective biomarkers currently exist to aid in ASM selection. One notable success in this regard is the prediction of Stevens-Johnson syndrome/toxic epidermal necrolysis in Taiwanese and Southeast Asian populations harboring the human leucocyte antigen HLA-B*1502 allele (Hung et al., 2010). This discovery has influenced clinical practice, prompting genetic tests before prescribing relevant ASMs to prevent deadly skin reactions (Franciotta et al., 2009).
ASMs exert their therapeutic effects by interacting with various targets in the central nervous system (CNS) (Das et al., 2012). Variations in genes encoding these targets theoretically have the potential to alter the response to ASMs. This can occur through changes in the coding region, resulting in amino acid sequence alterations that modify protein structures and their interaction with ASMs (Kimchi-Sarfaty et al., 2007) (Figure 1). Alternatively, variants in the non-coding region may interfere with protein synthesis, thereby affecting protein production and leading to varied responses to ASMs (Tate et al., 2005) (Figure 2). Pharmacogenetic studies offer a viable approach to predict the outcome of ASM treatment. However, despite extensive investigations, no biomarkers have yet demonstrated a predictive power comparable to that for dermatological side effects. This review focuses on pharmacogenetic studies of ASMs and their relevant mechanistic targets, and in particular on predicting the response to ASM treatment.
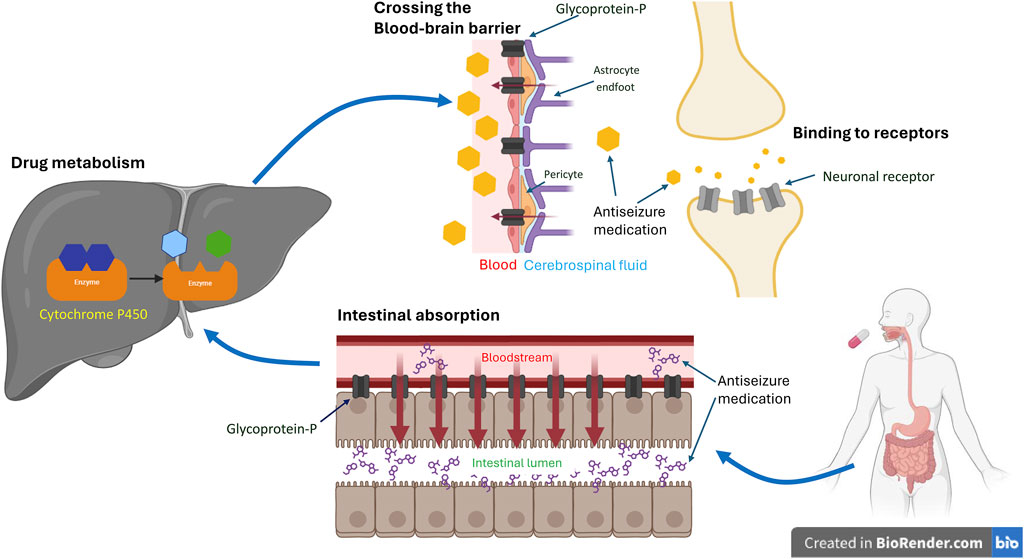
Figure 1. Coding Variants’ Impact on Antiseizure Medication. The schematic illustrates the impact of coding variants on the target, transportation, and metabolism of antiseizure medication (ASM). Typically, following oral administration of ASMs, the drug undergoes absorption in the gut, metabolism in the liver, and passage across the blood-brain barrier to reach its mechanistic target and exert its therapeutic effect. Genetic variants of efflux transporters in the gut, such as glycoprotein-P, can actively pump the drug back into the intestinal lumen, reducing its absorption. Similarly, variants of glycoprotein-P at the blood-brain barrier may facilitate the drug’s return to the bloodstream, decreasing its concentration in the brain. Genetic variants can also induce changes in metabolizing enzymes, such as cytochrome P450, in the liver. This can affect the rate of drug metabolism, leading to variable concentrations in the blood, differences in excretion, and variations in availability at its target site. Consequently, this may alter the effectiveness of ASMs. Additionally, genetic variants within the brain may modify neuronal receptors, compromising ASM’s ability to bind effectively and consequently diminishing its efficacy. *The figures were created with BioRender.com.
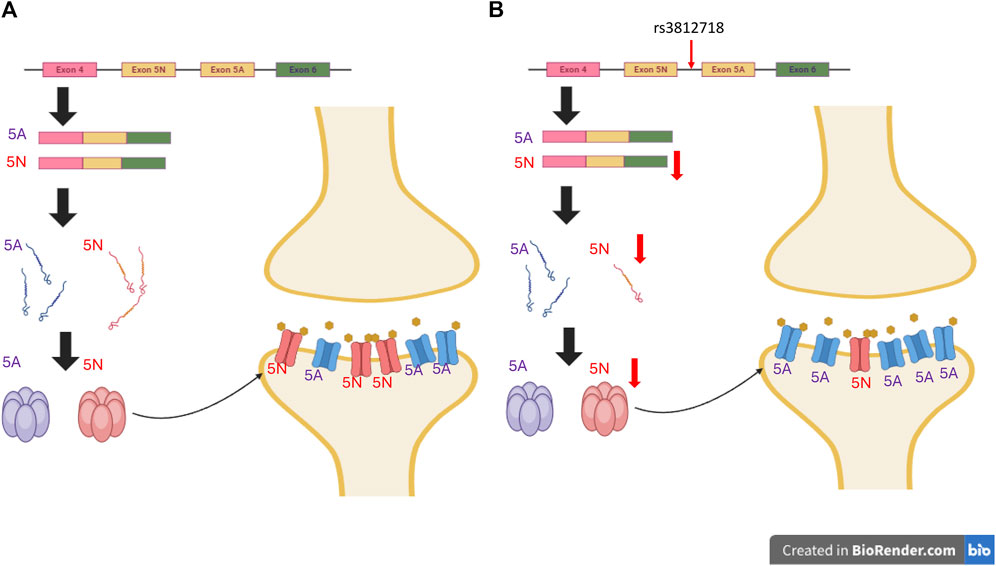
Figure 2. Impact of Non-Coding Genetic Variants on Antiseizure Medication Response. Genetic variants in non-coding regions, particularly introns, have the potential to disrupt typical splicing patterns. This disruption can lead to changes in protein expression levels and alter receptor functionality, consequently influencing responses to ASMs. The example provided in this Figure 2 focuses on SCN1A rs3812718. (A) The typical expression of SCN1A includes exon 5A and 5N. While 5N is more sensitive to sodium channel-blocking ASMs. (B) Conversely, within SCN1A carrying the rs3812718 variant, the expression of exon 5N is diminished, consequently reducing the abundance of receptors with heightened sensitivity to sodium channel-blocking ASMs. This decrease in receptors results in fewer channels being blocked, ultimately diminishing the antiseizure efficacy. (The ratio of 5A to 5N depicted in the figure does not accurately reflect the true conditions.). *The figures were created with BioRender.com.
Mechanistic targets of antiseizure medications
ASMs abolish aberrant neuronal excitability during epileptic discharge by altering the function of voltage-gated and ligand-gated ion channels (Meldrum and Rogawski, 2007). Voltage-gated ion channels are one of the main targets of ASMs including sodium channel (SCN) blockers, calcium current inhibitors, and potassium channel openers (Das et al., 2012). SCNs are responsible for the generation and propagation of action potential in neurons, and thus many ASMs act by reducing the high-frequency firing of voltage-dependent SCNs that occurs during a seizure (Abou-Khalil, 2016). Phenytoin (PHT), carbamazepine (CBZ), lamotrigine (LTG), oxcarbazepine (OXC), and lacosamide (LCM) are all SCN blockers (Brodie, 2017). Calcium channels contribute to the burst firing of neurons (Cain and Snutch, 2010) and control the release of presynaptic transmitters (Imbrici et al., 2004). Of the currently available ASMs, the main action of ethosuximide (Coulter et al., 1989), gabapentin (GBP) (Gee et al., 1996), and pregabalin (PGB) (Belliotti et al., 2005) is considered to be as blockers of calcium channels. The antiseizure effect of other ASMs may also in part be through the blockade of calcium channels, including PHT, phenobarbital, CBZ, OXC, zonisamide (ZNS), LTG, felbamate, topiramate (TPM), and valproate (VPA) (Abou-Khalil, 2019). Potassium channels are responsible for repolarizing the cell membrane and regulating the balance of excitability and resting of the neuron (Benarroch, 2009). Retigabine is currently the only ASM with the main action of enhancing resting membrane potential by opening potassium channels, however, it is not widely available due to discoloration of skin and conjunctiva (Gunthorpe et al., 2012). Other ASMs which may also modify potassium currents include ethosuximide (Leresche et al., 1998), LTG (Huang et al., 2004), and levetiracetam (LEV) (Madeja et al., 2003).
Several ligand-gated ion channels are also targets for ASMs, including gamma-aminobutyric acid (GABA) receptors and ionotropic glutamate receptors. GABA is the main fast inhibitory neurotransmitter, and it acts on GABAA and GABAB receptors in the CNS to terminate the bursting neuronal firing during seizures (Belelli et al., 2009). ASMs in this category, such as barbiturates and benzodiazepines can be used to treat status epilepticus (Czuczwar and Patsalos, 2001). Some ASMs modulate the disposition of GABA by inhibiting its catabolism, such as vigabatrin (Jung et al., 1977), or re-uptake, such as tiagabine (Krogsgaard-Larsen et al., 1987). Ionotropic glutamate receptors consist of three major forms, namely α-amino-3-hydroxy-5-methyl- 4-isoxazole propionic acid (AMPA), N-methyl-D-aspartate (NMDA), and kainate receptors, all of which they mediate the excitatory neurotransmission of glutamate (Reiner and Levitz, 2018). Perampanel (PER), a non-competitive AMPA receptor antagonist (Plosker, 2012), is currently the only ASM in this category.
Carbonic anhydrase (CA) regulates the flux of bicarbonate to maintain ion and pH homeostasis in neurons (Krishnamurthy et al., 2008). Inhibition of CA has been postulated to contribute to some of the antiseizure effects of acetazolamide, TPM, and ZNS (Das et al., 2012). The exact mechanism of how these CA inhibitors halt seizures is still unknown and no pharmacogenetic studies of variants in CA genes have been conducted to date.
LEV and brivaracetam share the same target, synaptic vesicle protein 2A (SV2A) (Rossi et al., 2022), and they exert their antiseizure effects by regulating exocytosis from synaptic vesicles and inhibiting the release of presynaptic neurotransmitters.
There are also broad-spectrum ASMs with multiple targets, such as ZNS (SCN blockade, calcium blocker, and CA inhibitor) (Brodie et al., 2012) and TPM (SCN blockade, GABA enhancer, CA inhibitor, and AMPA blockade) (Gryder and Rogawski, 2003), and ASMs with still unclear mechanisms, such as VPA (Romoli et al., 2019). Even for ASMs with known major targets, additional antiseizure mechanisms could arise from interactions with other minor targets (Sills and Rogawski, 2020). As a result, genetic variations of multiple receptors could result in the varied responses to ASMs, especially in patients taking polytherapy for seizure control.
With advances in the understanding of epileptogenesis, researchers are exploring innovative medications that target the pathogenic pathways implicated in seizure development or progression, beyond ion channels. These promising treatments show potential in effectively controlling seizures, and two are either undergoing human studies or are already in use. Cholesterol is metabolized to 24-hydroxycholesterol in the CNS by the cholesterol 24-hydroxylase (CH24H) enzyme (Popiolek et al., 2020). Lowering the cholesterol level in glial cells can result in an increased extracellular glutamate level (Tian et al., 2010) that may contribute to the genesis of seizures (Dejakaisaya et al., 2021). Soticlestat (TAK-935), a selective inhibitor of CH24H, demonstrated efficacy in decreasing seizure frequency among patients with Dravet syndrome during a phase 2 clinical trial (Hahn et al., 2022). Another example is the inhibition of mammalian target of rapamycin (mTOR). The mTOR pathway participates in cellular signaling and plays a crucial role in neuronal development (Lipton and Sahin, 2014). Tuberous sclerosis complex and focal cortical dysplasia are associated with hyperactivation of the mTOR pathway (Crino, 2015). Everolimus, a TOR inhibitor, can reduce seizure frequency in patients with tuberous sclerosis complex (Samueli et al., 2016). Numerous compounds are currently undergoing rigorous cellular and animal studies. For an in-depth review of the latest advances in epilepsy treatment targeting novel pathways, refer to Belete (2023).
Advances in computational bioinformatics have opened a new avenue for discovering novel targets for ASMs. In a recent study, data from genome-wide association studies for epilepsy were combined with data from proteome-wide association studies, transcriptome-wide association studies, and chemical-related gene set enrichment analysis, with the aim of identifying promising new targets for seizure treatment (Lu et al., 2023). Five genes were found to be significantly associated with epilepsy, and consequently may be new candidate epilepsy genes. This study showcased the potential of using bioinformatics techniques to identify new mechanisms potentially relevant to biological processes.
Association studies of genetic variants among mechanistic targets
We conducted a systematic search of PubMed to find relevant articles regarding the response to ASMs and the mechanistic targets previously discussed. We also included data from our recent studies, which not only enhanced the breadth of the review but also contributed original insights to enhance the overall understanding of the subject. Initially, we searched for keywords including “genetic polymorphisms” and “drug resistance epilepsy” in studies published from 2005 to 2023. Studies from any geographic location and including subjects of any ethnicity, gender, and age were included. Case reports were excluded from this review. Non-English articles or abstracts without full text were also excluded. Titles and abstracts of the articles retrieved from the initial search were screened for relevance, and full texts of potentially relevant articles were reviewed. We also extracted and examined the reference lists of these articles for additional relevant papers based on their titles and abstracts. We then searched for studies involving the mechanistic targets of ASMs, including sodium channels, calcium channels, potassium channels, GABA receptors, and ionotropic glutamate receptors. Subsequently, all available full-text articles were evaluated, and those not relevant to the response to ASMs or the mechanistic targets were excluded. The collected data were synthesized and compared to provide an overview of the progress in ASM pharmacogenetics, including conclusions and discussions regarding the limitations and future prospects.
The search returned 223 results. Thirty-five articles were excluded because they were either not relevant to the response to ASMs, focused on ribonucleic acid rather than genes, or were not human studies. The remaining articles contained 38 reviews or meta-analysis articles and 150 articles related to genetic polymorphisms and drug response. After reviewing these 188 articles and their references, 26 were selected, with 13 related to SCNs, two to calcium channels, four to potassium channels, five to GABA receptors, one to synaptic vesicle proteins, and one to ionotropic glutamate receptors. The remaining articles that mentioned the association between drug response and transporter or metabolic genes, although not included in the scope of our review, are also briefly discussed in this article.
Sodium channel gene variations
Since the first introduction of PHT in 1936 (Merritt and Putnam, 1984), SCN blockers have been one of the most popular treatments for both focal and generalized seizures. As a result, the relationship between SCN gene variants and response to ASMs has received much attention.
Most previous studies have focused on single-nucleotide polymorphisms (SNPs) of SCN genes and the dose of SCN blockers. The intronic SCN1A rs3812718 variant is one of the most extensively studied variants. In 2005, Tate et al. (2005) reported that this variant was associated with the maximum dose of PHT and CBZ required to control seizures in European patients. rs3812718 is located between exon 5 and 6 of the SCN1A gene and affects the splicing of exon 5, which results in decreasing the percentage of transcripts containing exon 5N (neonatal form) of SCN1A (Copley, 2004) (Figure 2). By decreasing the amount of the neonatal form of SCN1A, which is more sensitive to SCN blockers (Thompson et al., 2011), the authors suggested that patients with this SNP may need a higher dose or become more resistant to SCN blockers. This association was replicated in one Taiwanese study, where rs3812718 was also associated with a higher maintenance dose of CBZ monotherapy (Hung et al., 2012).
Later reports focused on the association between SNPs in SCN genes and DRE instead of the dose of ASMs. One report regarding Japanese patients with epilepsy found that SCN1A rs3812718 was associated with resistance to CBZ but not the dose of CBZ (Abe et al., 2008). In Chinese patients receiving CBZ or OXC, SCN1A rs3812718 was associated with DRE (Ma et al., 2014). In addition, a Greek study found that SCN1A rs3812718 was associated with resistance to SCN blocker monotherapy (PHT, CBZ, OXC, or LTG) and increased dosing of SCN blockers in patients responsive to SCN blockers (Angelopoulou et al., 2017), however, they did not find an association between SCN1A rs3812718 and the responsive to ASM polytherapy that contained SCN blockers. Other studies have reported conflicting results about the effect of SCN1A rs3812718 on the responsive to SCN blockers. An Italian study found that SCN1A rs3812718 was not associated with the responsive to CBZ/OXC, the dose of CBZ/OXC, or DRE (Manna et al., 2011). In the study, DRE was defined as having at least one seizure per month within 2 years of follow-up under three or more ASMs. A study from northern India also reported that SCN1A rs3812718 was not associated with the response to CBZ/OXC or DRE (Kumari et al., 2013), which they defined as at least four seizures over 1 year under three or more ASMs. In addition, a Spanish study did not find an association between SCN1A rs3812718 and DRE, defined as having at least four seizures under more than three ASMs 1 year before recruitment (Sanchez et al., 2010). However, this study did not specifically focus on the use of SCN blockers.
Besides SCN1A rs3812718, other SNPs of SCN receptor genes have also been associated with drug responsiveness. SCN2A rs17183814 was associated with DRE in Indian patients, defined as at least four seizures in 1 year under three or more ASMs (Lakhan et al., 2009), and an Australian study found that SCN1A rs10188577 was moderately associated with DRE, albeit without statistical significance (Yip et al., 2014).
These studies generally focused on less than or around a dozen SNPs, however, more genetic variants in SCNs are likely to contribute to the response to SCN-blocking ASMs. A study on Hong Kong Chinese patients investigated 27 SNPs in SCN1A, SCN2A, and SCN3A genes, and found that SCN2A rs2304016 was associated with both DRE, defined as one or more seizures per month for 1 year under ASM treatment, and resistance to SCN blockers, defined as patients with DRE when their most recent ASM regimen consisted of SCN-blocking ASMs (Kwan et al., 2008). Another cohort study evaluated 39 polymorphisms in the SCN1A, SCN2A, and SCN3A genes from patients in Malaysia and Hong Kong, and found no association between gene polymorphisms and responsive to CBZ or VPA monotherapy (Haerian et al., 2013). One study involving Taiwanese patients identified rs55742440 in SCN1B, among 21 coding SNPs in SCN1A, SCN1B, SCN2A, and SCN9A, as being associated with resistance to SCN blockers (Lin et al., 2021). This resistance was defined as the failure of the most recently administered SCN blocker to achieve seizure freedom for at least 1 year.
The methodologies in these studies were heterogeneous (Table 1), including the definition of DRE, the specific ASM under investigation, and the inclusion of different genetic variants. These differences make it challenging to compare the results and provide meaningful clinical predictions. However, most of these studies do imply that genetic variants in the SCN genes have an impact on the responsive to SCN-blocking ASMs.
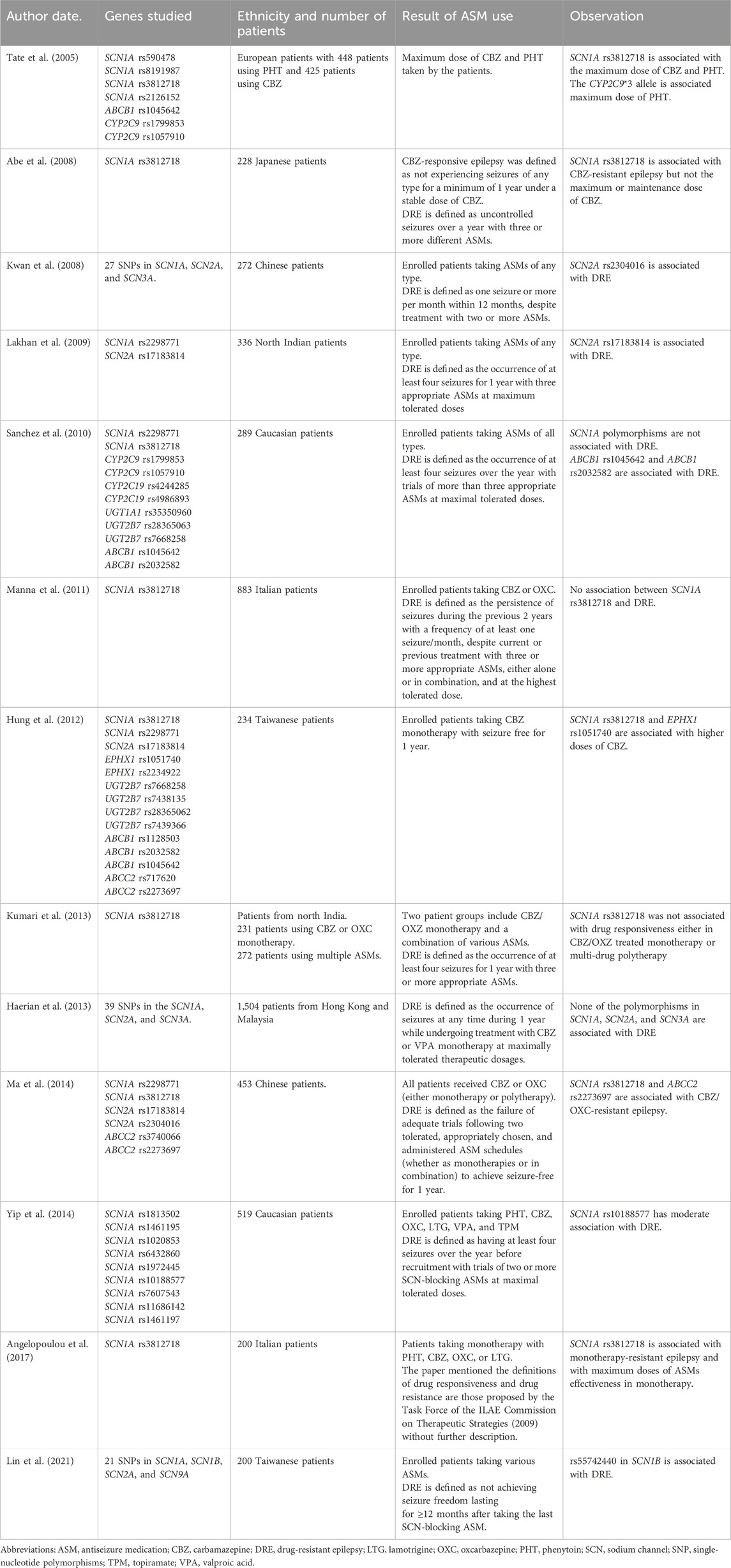
Table 1. Studies focusing on the genetic variants of the sodium channel genes.
Calcium channel gene variations
A Chinese study focusing on the association of DRE and with polymorphisms of calcium channel genes, including 15 SNPs in CACNA1A, CACNA1C, and CACNA1H, found that no SNPs but the TAGAA haplotype of CACNA1A was associated with DRE (Lv et al., 2015). Another study involving Jordanian patients found that CACNG5 rs740805 and GABRA1 rs2279020 were associated with DRE (Al-Eitan et al., 2020). Both of these studies included patients taking various combinations of ASMs, although many ASMs can inhibit calcium channels (Abou-Khalil, 2019). These studies only suggested a relationship between genetic variants of calcium channels and DRE.
Potassium channel gene variations
A Chinese study identified that KCNJ10 rs12402969, among seven other SNPs in KCNJ10, was associated with DRE, defined as experiencing four seizures under at least three ASMs during the previous year (Guo et al., 2015). However, another Chinese study, using the same DRE definition found no association between eight SNPs in KCNA1, KCNA2, and KCNV2 with DRE (Qu et al., 2017). Similarly, a separate Chinese study reported no association between nine SNPs in KCNJ10 and DRE (Zhu et al., 2020). In addition, a Jordanian-Arab study focusing on patients initiating treatment with VPA and CBZ as their first ASM revealed no association between seven SNPs in KCNA1, KCNA2, and KCNV2 and the response to these ASMs (Al-Eitan et al., 2018). Notably, while KCNJ10 rs12402969 was included in the studies by Guo et al. and Zhu et al., it was not considered in the studies by Qu et al. and Al-Eitan et al., leaving the association between this SNP and DRE inconclusive.
With various ASMs included, these studies suggest that genetic variants in potassium channels may have a minimal, if any, effect on DRE. This may not be surprising given that most ASMs do not target potassium channels. Therefore, associations between the response to potassium channel-targeted ASMs and genetic variants in potassium channel genes remain to be fully elucidated. Potassium channel enhancer represents a potential avenue for future ASM development.
Gamma-aminobutyric acid receptor gene variations
The GABRG2 rs211037 variant has been extensively studied across various ethnicities. In an Egyptian study, GABRG2 rs211037 was linked to DRE, defined as the inability to achieve 1 year of seizure freedom while using two ASMs (Abou El Ella et al., 2018). In Chinese patients, resistance to VPA, defined as having seizures with at least 12 months of VPA use, was associated with the heterozygous CT genotype of the GABRG2 rs211037 polymorphism (Lu et al., 2021). In addition, an Indian study found that GABRA1 rs2279020 but not GABRG2 rs211037 was associated with DRE, defined as at least four seizures within 1 year under three ASMs (Kumari et al., 2010). However, a Pakistan study failed to find an association between GABRG2 rs211037 and DRE, defined as no change in seizure frequency under two ASMs (Saleem et al., 2022). Another study from southern India that included four GABAA receptor subunit SNPs also found no association with DRE, defined as having a seizure frequency of 12 per year under at least two ASMs for at least 2 years (Balan et al., 2013). In contrast, a meta-analysis showed that GABRG2 rs211037 was related to DRE in the Asian population (Hu et al., 2023).
These studies did not specifically analyze the types of ASMs taken, except for one study which was limited to VPA, a medication known to enhance the effect of GABA (Loscher, 2002). GABA-enhancing ASMs such as barbiturates and benzodiazepines are predominantly used in patients with status epilepticus (Czuczwar and Patsalos, 2001). Nonetheless, there is currently a lack of studies specifically addressing this aspect.
Synaptic vesicle protein gene variations
A European study evaluated 86 common variants in SV2A, SV2B, and SV2C, but found no associations between these variants and the response to LEV (Lynch et al., 2009). Another study investigated genetic susceptibility to the neuropsychiatric side effects of LEV, and despite conducting genome-wide association analysis and rare-variant analysis, no significant associations were identified (Campbell et al., 2022). However, intriguingly, this study revealed that patients experiencing neuropsychiatric side effects had an elevated polygenic risk score for schizophrenia (Campbell et al., 2022). Taken together, these findings suggest that neither the response to LEV nor the occurrence of psychiatric adverse effects was linked to variability in synaptic vesicle genes, but rather that it was influenced by underlying genetic predispositions.
Ionotropic glutamate receptors
PER, an AMPA receptor antagonist, is currently the only ASM in this category. Lin et al. investigated the association between rare coding genetic variants in glutamate receptor genes and ADR, as well as responsive to PER, in Taiwanese patients undergoing PER treatment (Lin et al., 2023). Resistance to PER was defined as not achieving seizure freedom for 1 year under PER treatment. Although Lin et al. did not discover a direct association between individual genetic variants and drug response, they observed that enrichment of genetic variants within the glutamate receptor gene group was statistically associated with the occurrence of ADRs. Upon further analysis, they found that rare variant enrichment in the glutamate ionotropic receptor delta subunit was nominally associated with ADR. In addition, gene burden analysis revealed a nominal association between GRID1 and ADRs. These findings suggest that studies focusing on genetic variants in mechanistic targets hold the potential not only to predict responsiveness but also to predict the occurrence of ADRs.
In summary, genetic variants in ASM targets beyond SCN (as outlined in Table 2) present conflicting findings. Many of the previous investigations did not specifically target the mechanistic action of ASMs, making it challenging to ascertain whether the response to ASMs was correlated with genetic variants in their mechanistic targets. As relatively little research has investigated the correlation between the response to LEV or PER and their respective targets, further investigations are warranted.
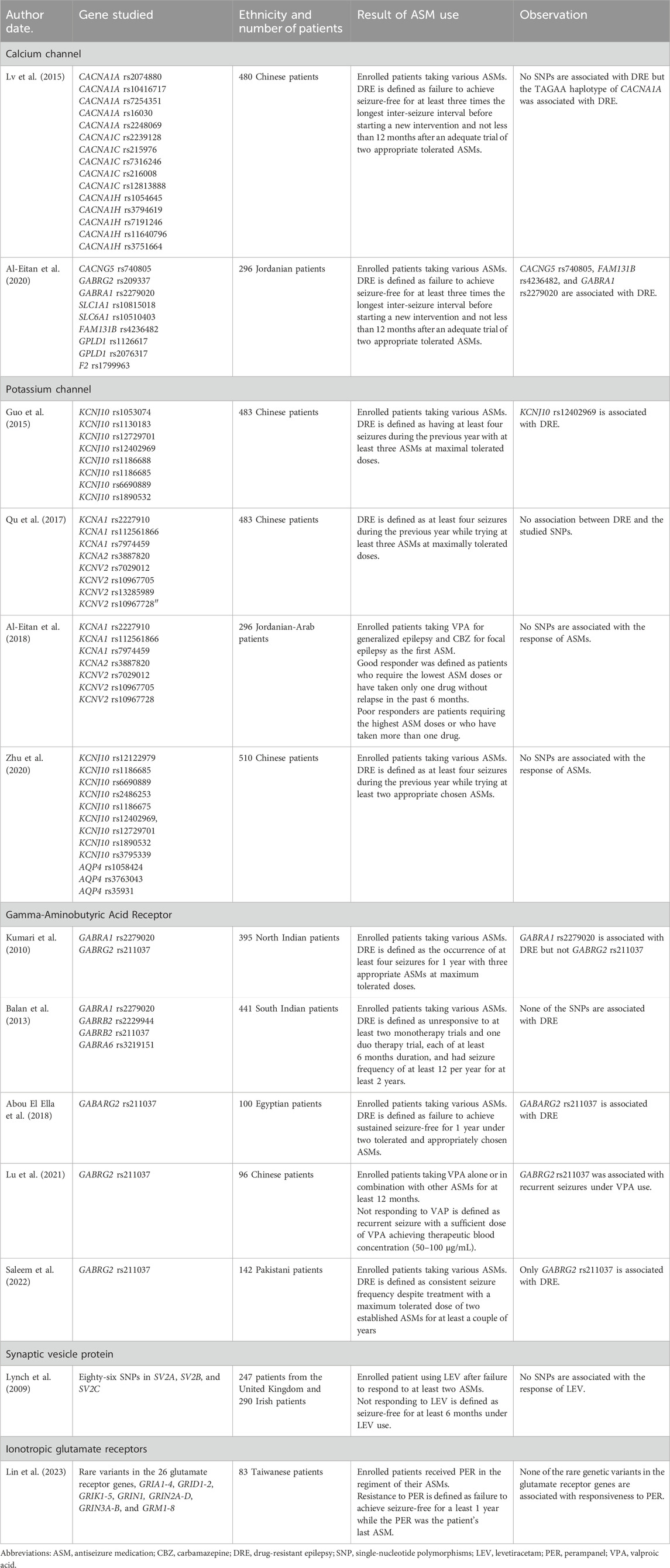
Table 2. Studies focus on genetic variants other than the sodium channel genes.
Other factors related to drug-resistant epilepsy
Besides the mechanistic targets of ASMs, other factors also contribute to DRE, including alterations in the drug delivery system across the brain, and pharmacodynamic modifications that change the absorption or metabolism of ASMs (Loscher et al., 2020). In addition, Mendelian genetic factors may also determine the disease severity and increase refractoriness to ASM treatment, such as developmental epileptic encephalopathy.
Efflux transporters (Figure 1) in the endothelial cells of the blood-brain barrier (BBB) can hamper the ability of ASMs to enter the CNS and decrease their concentration in epileptogenic tissues (Kwan and Brodie, 2005). The ATP binding cassette (ABC) transporter superfamily is the major efflux transporter on the BBB that limits the access of ASMs to the target sites (Qosa et al., 2015). Glycoprotein-P (P-gp), one of these efflux transporters, has been shown to actively remove ASMs from the BBB back into the bloodstream (Miller et al., 2008). Moreover, it has been shown to be over-expressed in epileptogenic foci of the brain (Marchi et al., 2004), further preventing the seizure-suppression effect of ASMs. Studies on the genetic polymorphism of ABC transporter genes and the relationship between the response to ASMs are conflicting (Seo et al., 2006; Ozgon et al., 2008; Meng et al., 2011). For a comprehensive review of the impact of genetic polymorphisms of ABC transporter and the responsive to ASMs, see Leandro et al. (2019).
Drug-metabolizing genes are polymorphic and can influence the biotransformation of many drugs (Meyer and Zanger, 1997). These polymorphisms may result in a higher plasma drug level under standard dosage that leads to the occurrence of ADRs or increases enzyme activity to make the ASMs less effective (Eadie, 1991) (Figure 1). Drug metabolism occurs in two phases. Phase 1 reactions involve the addition of a functional group, mostly hydroxylation, by the cytochrome P450 (CYP) family followed by a phase 2 metabolism of various conjugating reactions that increase hydrophilicity and facilitate renal excretion of the drug. The enzymes included in this process are CYP, UDP-glucuronyl transferase (UGT), epoxide hydroxylase (EPHX), glutathione S-transferase (GST), microsomal GST, N-acetyltransferase (NAT), and sulfatase (SULF) (Ferraro and Buono, 2005). Various studies including polymorphisms in CYP (van der Weide et al., 2001; Lopez-Garcia et al., 2017; Makowska et al., 2021), EPHX (Hung et al., 2012), and UGT (Ma et al., 2015) have been conducted, with conflicting results.
Recently, the EpiPGX Consortium investigated the complex genetic factors associated with responsive to ASMs (Wolking et al., 2020). The study included 1,622 patients and 808,583 genetic variants, which is by far the largest cohort of pharmacogenetic studies regarding ASM. They enrolled patients taking LEV, VPA, and LTG for seizure control and studied rare genetic variants presumed to have functional consequences. A non-presupposed gene-based enrichment analysis did not find associations between any of the coding variants and response to ASM. The gene-set-based enrichment analysis, focusing on ASM target genes and ADME (absorption, distribution, metabolism, and excretion) gene sets, found that patients with resistance to VPA had enrichment of rare genetic variants in the ADME gene set. No associations were found between the response to ASMs for the target genes in all three studied ASMs and ADME gene sets of LEV and LTG. The results suggest that resistance to ASMs may not be the consequence of a single or a few genetic variants, but rather determined by the cumulative effect of various genetic variants that eventually merge to alter the collective effect of drug responsiveness.
Discussion
One of the weaknesses observed in these association studies is the lack of unified definitions of DRE and responsiveness. The ILAE introduced a new definition of DRE in 2010, and studies conducted before this may not have used this definition. It is anticipated that later association studies will adhere to a unified definition until any subsequent changes are made.
In addition to the varied definitions of DRE, the studies were also diverse in their approach. One approach focused on investigating the response to ASMs concerning the genetic variants of their mechanistic targets. Another approach involved examining the association between the response to various combinations of ASMs as a whole and the target of interest. Studies regarding variants in SCNs usually adopted the former approach, concentrating on ASMs that block SCNs, with a minority involving multiple ASMs. These studies could identify potential associations between genetic variants in SCNs and responses to SCN-blocking ASMs. Conversely, studies on calcium channel blockers, potassium channel openers, and GABA receptors typically included ASMs with diverse mechanisms. Consequently, their findings may only have revealed associations between genetic variants of the targets of interest and DRE, which may not necessarily disclose the association between the response to a particular ASM and its target. The identification of pharmacogenetic markers capable of predicting the response to an ASM prior to prescription would be more beneficial than predicting DRE to various ASMs. A more refined study approach that addresses the primary target and its corresponding ASMs is necessary before these findings can inform clinical practice in selecting the appropriate ASM. Given that SCN-blocking ASMs are the most frequently prescribed, studies on targets beyond SCNs may lack sufficient patient numbers to achieve statistical power. This could result in the inclusion of patients taking multiple ASMs for non-SCN blocker investigations. International collaboration in pharmacogenetic association studies could involve more participants and clarify the connection between ASM effectiveness and the genetic variants of their targets.
The aforementioned association studies primarily focused on common variants of the target genes, except for one study which examined rare variants of the glutamate receptor and their impact on response to PER (Lin et al., 2023). Common genetic variants are typically associated with quantitative phenotypes, with each locus contributing partially to various traits (Speliotes et al., 2010). Common variants often do not directly cause functional changes but rather indicate the presence of nearby functional variants due to linkage disequilibrium with observed phenotypes (Bodmer and Bonilla, 2008). In contrast, rare variants are more likely to have direct functional effects (Bodmer and Bonilla, 2008), although they are not as rare as deleterious mutations and have extremely low frequency in the population. These rare variants are evaluated based on computational algorithms suggesting potential functional consequences, particularly in genes relevant to the phenotypic pathophysiology (qualified variants) (Petrovski et al., 2013). Studies investigating the impact of rare variants on response to ASMs remain limited; however, they represent a promising avenue to augment our understanding of the functional consequences at a genetic level. These two types of variants are not mutually exclusive, and they have both been proposed to contribute to specific traits, with common variants modulating the effects of rare variants (Felix et al., 2006) and rare variants directly influencing trait development through functional consequences (Dickson et al., 2010).
Data regarding the functional consequences of these genetic variants are lacking. Therefore, the effect of the genetic variant and response to the drug cannot be determined. Variants found in studies of SNPs may not be the causal variants, since these common variants typically encompass a large number of variants in linkage disequilibrium, only some of which contribute to the phenotype (Weissbrod et al., 2020). For studies on rare variants, although the variants are predicted by computational algorithms, the true effects of these variants on protein structure and function are unknown (Livesey and Marsh, 2020). Understanding the consequences of these genetic variants can enhance our understanding of the reason for different drug responses. As previously discussed, the intronic variant SCN1A rs3812718 is associated with a reduced expression of the neonatal form of SCN1A, which is more sensitive to SCN blockers. This effect may be mitigated by escalating the dosage of SCN blockers (Thompson et al., 2011). The findings from genetic studies along with subsequent functional analyses can offer valuable insights into ASM prescription. This includes not only identifying the most effective medication but also anticipating individual responses to ASMs and adjusting treatment regimens accordingly.
Besides exploring the association of genetic variants and DRE, polygenetic risk score (PRS) offers an additional method for predicting the response to ASs. PRS hypothesizes that the genetic loading of a set of risk variants contributes to the development of a disease or particular trait with the need for little or no knowledge about the underlying mechanism (Lewis and Vassos, 2020). It is calculated using statistical algorithms and quantifies an individual’s genetic predisposition to a specific trait or disease by integrating data from multiple genetic markers across the genome. The PRS has been demonstrated to be a feasible method to predict the psychiatric side effects of LEV treatment for schizophrenia (Campbell et al., 2022). It may also be a reasonable approach to predict responsiveness, since many ASMs may have more than one mechanistic target, or in some cases an unknown mechanism.
By integrating genetic testing into clinical practice, personalized medicine facilitates the more precise selection and dosing of ASMs. This approach not only enhances the efficacy of ASMs but also minimizes the risk of ADRs, thereby improving patient outcomes and overall quality of life. With genetic testing-guided ASM selection, patients may achieve seizure freedom more rapidly. Moreover, this approach enables the identification of patients at risk of developing DRE earlier in their treatment journey, allowing for timely adjustments in treatment strategies or consideration of surgical options.
Conclusion
This review analyzes pharmacogenetic studies, with a specific focus on the mechanistic targets of ASMs. Our findings show that it is currently challenging to definitively conclude that specific genetic variants in drug targets influence the response to ASMs. The existing pharmacogenetic studies often concentrate on a limited number of genetic variations, sometimes without a direct focus on the target of the studied ASMs, or involving a small number of participants. Studies exploring the association between DRE and the pharmacogenetics of ASMs are still in their infancy. Collaborative efforts among multiple centers are imperative to attain sufficient statistical power to draw conclusions regarding the relationship between the efficacy of ASMs and the genetic variants of their targets. In addition, tools such as PRS can be used to predict potential ADRs and integrate them into prediction methodologies. With further research into the pharmacogenetics of ASMs, it may soon be possible to predict the outcomes of specific ASMs by analyzing the genomic data of the patients, thereby eliminating the need for a trial-and-error approach.
Author contributions
C-HL: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing. C-JH: Writing–review and editing, S-YC: Writing–review and editing, Y-TL: Writing–review and editing, M-HT: Conceptualization, Methodology, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was supported by the Chang Gung Medical Foundation (CMRPG8H0751 and CMRPG8K1521).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe, T., Seo, T., Ishitsu, T., Nakagawa, T., Hori, M., and Nakagawa, K. (2008). Association between SCN1A polymorphism and carbamazepine-resistant epilepsy. Br. J. Clin. Pharmacol. 66 (2), 304–307. doi:10.1111/j.1365-2125.2008.03203.x
Abou El Ella, S. S., Tawfik, M. A., Abo El Fotoh, W. M. M., and Soliman, O. A. M. (2018). The genetic variant "C588T" of GABARG2 is linked to childhood idiopathic generalized epilepsy and resistance to antiepileptic drugs.". Seizure 60, 39–43. doi:10.1016/j.seizure.2018.06.004
Abou-Khalil, B. W. (2016). Antiepileptic drugs. Contin. (Minneap Minn) 22, 132–156. doi:10.1212/CON.0000000000000289
Abou-Khalil, B. W. (2019). Update on antiepileptic drugs 2019. Contin. (Minneap Minn) 25 (2), 508–536. doi:10.1212/CON.0000000000000715
Al-Eitan, L. N., Al-Dalala, I. M., Elshammari, A. K., Khreisat, W. H., Nimiri, A. F., Alnaamneh, A. H., et al. (2020). Genetic association of epilepsy and anti-epileptic drugs treatment in Jordanian patients. Pharmgenomics Pers. Med. 13, 503–510. doi:10.2147/PGPM.S273125
Al-Eitan, L. N., Al-Dalalah, I. M., Elshammari, A. K., Khreisat, W. H., and Almasri, A. Y. (2018). The impact of potassium channel gene polymorphisms on antiepileptic drug responsiveness in arab patients with epilepsy. J. Pers. Med. 8 (4), 37. doi:10.3390/jpm8040037
Angelopoulou, C., Veletza, S., Heliopoulos, I., Vadikolias, K., Tripsianis, G., Stathi, C., et al. (2017). Association of SCN1A gene polymorphism with antiepileptic drug responsiveness in the population of Thrace, Greece. Arch. Med. Sci. 13 (1), 138–147. doi:10.5114/aoms.2016.59737
Balan, S., Sathyan, S., Radha, S. K., Joseph, V., Radhakrishnan, K., and Banerjee, M. (2013). GABRG2, rs211037 is associated with epilepsy susceptibility, but not with antiepileptic drug resistance and febrile seizures. Pharmacogenet Genomics 23 (11), 605–610. doi:10.1097/FPC.0000000000000000
Belelli, D., Harrison, N. L., Maguire, J., Macdonald, R. L., Walker, M. C., and Cope, D. W. (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci. 29 (41), 12757–12763. doi:10.1523/JNEUROSCI.3340-09.2009
Belete, T. M. (2023). Recent progress in the development of new antiepileptic drugs with novel targets. Ann. Neurosci. 30 (4), 262–276. doi:10.1177/09727531231185991
Belliotti, T. R., Capiris, T., Ekhato, I. V., Kinsora, J. J., Field, M. J., Heffner, T. G., et al. (2005). Structure-activity relationships of pregabalin and analogues that target the alpha(2)-delta protein. J. Med. Chem. 48 (7), 2294–2307. doi:10.1021/jm049762l
Benarroch, E. E. (2009). Potassium channels: brief overview and implications in epilepsy. Neurology 72 (7), 664–669. doi:10.1212/01.wnl.0000343739.72081.4e
Bodmer, W., and Bonilla, C. (2008). Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40 (6), 695–701. doi:10.1038/ng.f.136
Brodie, M. J. (2017). Sodium Channel blockers in the treatment of epilepsy. CNS Drugs 31 (7), 527–534. doi:10.1007/s40263-017-0441-0
Brodie, M. J., Ben-Menachem, E., Chouette, I., and Giorgi, L. (2012). Zonisamide: its pharmacology, efficacy and safety in clinical trials. Acta Neurol. Scand. Suppl. 126 (194), 19–28. doi:10.1111/ane.12016
Cain, S. M., and Snutch, T. P. (2010). Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 4 (6), 475–482. doi:10.4161/chan.4.6.14106
Campbell, C., McCormack, M., Patel, S., Stapleton, C., Bobbili, D., Krause, R., et al. (2022). A pharmacogenomic assessment of psychiatric adverse drug reactions to levetiracetam. Epilepsia 63 (6), 1563–1570. doi:10.1111/epi.17228
Copley, R. R. (2004). “Evolutionary convergence of alternative splicing in ion channels.” Trends Genet. 20 (4), 171–176. doi:10.1016/j.tig.2004.02.001
Coulter, D. A., Huguenard, J. R., and Prince, D. A. (1989). Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann. Neurol. 25 (6), 582–593. doi:10.1002/ana.410250610
Crino, P. B. (2015). mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 5 (4), a022442. doi:10.1101/cshperspect.a022442
Czuczwar, S. J., and Patsalos, P. N. (2001). The new generation of GABA enhancers. Potential in the treatment of epilepsy. CNS Drugs 15 (5), 339–350. doi:10.2165/00023210-200115050-00001
Das, N., Dhanawat, M., and Shrivastava, S. K. (2012). An overview on antiepileptic drugs. Drug Discov. Ther. 6 (4), 178–193. doi:10.5582/ddt.2012.v6.4.178
Dejakaisaya, H., Kwan, P., and Jones, N. C. (2021). Astrocyte and glutamate involvement in the pathogenesis of epilepsy in Alzheimer's disease. Epilepsia 62 (7), 1485–1493. doi:10.1111/epi.16918
Dickson, S. P., Wang, K., Krantz, I., Hakonarson, H., and Goldstein, D. B. (2010). Rare variants create synthetic genome-wide associations. PLoS Biol. 8 (1), e1000294. doi:10.1371/journal.pbio.1000294
Eadie, M. J. (1991). Formation of active metabolites of anticonvulsant drugs. A review of their pharmacokinetic and therapeutic significance. Clin. Pharmacokinet. 21 (1), 27–41. doi:10.2165/00003088-199121010-00003
Elliott, R. E., Morsi, A., Kalhorn, S. P., Marcus, J., Sellin, J., Kang, M., et al. (2011). Vagus nerve stimulation in 436 consecutive patients with treatment-resistant epilepsy: long-term outcomes and predictors of response. Epilepsy Behav. 20 (1), 57–63. doi:10.1016/j.yebeh.2010.10.017
Felix, R., Bodmer, W., Fearnhead, N. S., van der Merwe, L., Goldberg, P., and Ramesar, R. S. (2006). GSTM1 and GSTT1 polymorphisms as modifiers of age at diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC) in a homogeneous cohort of individuals carrying a single predisposing mutation. Mutat. Res. 602 (1-2), 175–181. doi:10.1016/j.mrfmmm.2006.09.004
Ferraro, T. N., and Buono, R. J. (2005). The relationship between the pharmacology of antiepileptic drugs and human gene variation: an overview. Epilepsy Behav. 7 (1), 18–36. doi:10.1016/j.yebeh.2005.04.010
Franciotta, D., Kwan, P., and Perucca, E. (2009). Genetic basis for idiosyncratic reactions to antiepileptic drugs. Curr. Opin. Neurol. 22 (2), 144–149. doi:10.1097/WCO.0b013e328328f276
Gaston, T. E., and Szaflarski, J. P. (2018). Cannabis for the treatment of epilepsy: an update. Curr. Neurol. Neurosci. Rep. 18 (11), 73. doi:10.1007/s11910-018-0882-y
Gee, N. S., Brown, J. P., Dissanayake, V. U., Offord, J., Thurlow, R., and Woodruff, G. N. (1996). The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 271 (10), 5768–5776. doi:10.1074/jbc.271.10.5768
Gryder, D. S., and Rogawski, M. A. (2003). Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J. Neurosci. 23 (18), 7069–7074. doi:10.1523/JNEUROSCI.23-18-07069.2003
Gunthorpe, M. J., Large, C. H., and Sankar, R. (2012). The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53 (3), 412–424. doi:10.1111/j.1528-1167.2011.03365.x
Guo, Y., Yan, K. P., Qu, Q., Qu, J., Chen, Z. G., Song, T., et al. (2015). Common variants of KCNJ10 are associated with susceptibility and anti-epileptic drug resistance in Chinese genetic generalized epilepsies. PLoS One 10 (4), e0124896. doi:10.1371/journal.pone.0124896
Haerian, B. S., Baum, L., Kwan, P., Tan, H. J., Raymond, A. A., and Mohamed, Z. (2013). SCN1A, SCN2A and SCN3A gene polymorphisms and responsiveness to antiepileptic drugs: a multicenter cohort study and meta-analysis. Pharmacogenomics 14 (10), 1153–1166. doi:10.2217/pgs.13.104
Hahn, C. D., Jiang, Y., Villanueva, V., Zolnowska, M., Arkilo, D., Hsiao, S., et al. (2022). A phase 2, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of soticlestat as adjunctive therapy in pediatric patients with Dravet syndrome or Lennox-Gastaut syndrome (ELEKTRA). Epilepsia 63 (10), 2671–2683. doi:10.1111/epi.17367
Hu, X., Zhao, M., Yang, X., Wang, D., and Wu, Q. (2023). Association between the SLC6A11 rs2304725 and GABRG2 rs211037 polymorphisms and drug-resistant epilepsy: a meta-analysis. Front. Physiol. 14, 1191927. doi:10.3389/fphys.2023.1191927
Huang, C. W., Huang, C. C., Liu, Y. C., and Wu, S. N. (2004). Inhibitory effect of lamotrigine on A-type potassium current in hippocampal neuron-derived H19-7 cells. Epilepsia 45 (7), 729–736. doi:10.1111/j.0013-9580.2004.58403.x
Hung, C. C., Chang, W. L., Ho, J. L., Tai, J. J., Hsieh, T. J., Huang, H. C., et al. (2012). Association of polymorphisms in EPHX1, UGT2B7, ABCB1, ABCC2, SCN1A and SCN2A genes with carbamazepine therapy optimization. Pharmacogenomics 13 (2), 159–169. doi:10.2217/pgs.11.141
Hung, S. I., Chung, W. H., Liu, Z. S., Chen, C. H., Hsih, M. S., Hui, R. C., et al. (2010). Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 11 (3), 349–356. doi:10.2217/pgs.09.162
Imbrici, P., Jaffe, S. L., Eunson, L. H., Davies, N. P., Herd, C., Robertson, R., et al. (2004). Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 127, 2682–2692. doi:10.1093/brain/awh301
Jung, M. J., Lippert, B., Metcalf, B. W., Bohlen, P., and Schechter, P. J. (1977). gamma-Vinyl GABA (4-amino-hex-5-enoic acid), a new selective irreversible inhibitor of GABA-T: effects on brain GABA metabolism in mice. J. Neurochem. 29 (5), 797–802. doi:10.1111/j.1471-4159.1977.tb10721.x
Kimchi-Sarfaty, C., Oh, J. M., Kim, I. W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science 315 (5811), 525–528. doi:10.1126/science.1135308
Krishnamurthy, V. M., Kaufman, G. K., Urbach, A. R., Gitlin, I., Gudiksen, K. L., Weibel, D. B., et al. (2008). Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev. 108 (3), 946–1051. doi:10.1021/cr050262p
Krogsgaard-Larsen, P., Falch, E., Larsson, O. M., and Schousboe, A. (1987). GABA uptake inhibitors: relevance to antiepileptic drug research. Epilepsy Res. 1 (2), 77–93. doi:10.1016/0920-1211(87)90012-x
Kumari, R., Lakhan, R., Kalita, J., Misra, U. K., and Mittal, B. (2010). "Association of alpha subunit of GABAA receptor subtype gene polymorphisms with epilepsy susceptibility and drug resistance in north Indian population.". Seizure 19 (4), 237–241. doi:10.1016/j.seizure.2010.02.009
Kumari, R., Lakhan, R., Kumar, S., Garg, R. K., Misra, U. K., Kalita, J., et al. (2013). SCN1AIVS5-91G-->A polymorphism is associated with susceptibility to epilepsy but not with drug responsiveness. Biochimie 95 (6), 1350–1353. doi:10.1016/j.biochi.2013.02.006
Kwan, P., Arzimanoglou, A., Berg, A. T., Brodie, M. J., Allen Hauser, W., Mathern, G., et al. (2010). Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia 51 (6), 1069–1077. doi:10.1111/j.1528-1167.2009.02397.x
Kwan, P., and Brodie, M. J. (2005). Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia 46 (2), 224–235. doi:10.1111/j.0013-9580.2005.31904.x
Kwan, P., Poon, W. S., Ng, H. K., Kang, D. E., Wong, V., Ng, P. W., et al. (2008). "Multidrug resistance in epilepsy and polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, and SCN3A: correlation among phenotype, genotype, and mRNA expression.". Pharmacogenet Genomics 18 (11), 989–998. doi:10.1097/FPC.0b013e3283117d67
Kwan, P., and Sander, J. W. (2004). The natural history of epilepsy: an epidemiological view. J. Neurol. Neurosurg. Psychiatry 75 (10), 1376–1381. doi:10.1136/jnnp.2004.045690
Lakhan, R., Kumari, R., Misra, U. K., Kalita, J., Pradhan, S., and Mittal, B. (2009). "Differential role of sodium channels SCN1A and SCN2A gene polymorphisms with epilepsy and multiple drug resistance in the north Indian population. Br. J. Clin. Pharmacol. 68 (2), 214–220. doi:10.1111/j.1365-2125.2009.03437.x
Leandro, K., Bicker, J., Alves, G., Falcao, A., and Fortuna, A. (2019). ABC transporters in drug-resistant epilepsy: mechanisms of upregulation and therapeutic approaches. Pharmacol. Res. 144, 357–376. doi:10.1016/j.phrs.2019.04.031
Leresche, N., Parri, H. R., Erdemli, G., Guyon, A., Turner, J. P., Williams, S. R., et al. (1998). On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. J. Neurosci. 18 (13), 4842–4853. doi:10.1523/JNEUROSCI.18-13-04842.1998
Lewis, C. M., and Vassos, E. (2020). Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12 (1), 44. doi:10.1186/s13073-020-00742-5
Lin, C.-H., Ho, C.-J., Chen, S.-Y., Lu, Y.-T., and Tsai, M.-H. (2023). A pharmacogenetic study of perampanel: association between rare variants of glutamate receptor genes and outcomes. Front. Genet. 14, 1215493. doi:10.3389/fgene.2023.1215493
Lin, C. H., Ho, C. J., Lu, Y. T., and Tsai, M. H. (2021). Response to Sodium Channel blocking Antiseizure medications and coding polymorphisms of Sodium Channel genes in Taiwanese epilepsy patients. BMC Neurol. 21 (1), 367. doi:10.1186/s12883-021-02395-2
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84 (2), 275–291. doi:10.1016/j.neuron.2014.09.034
Livesey, B. J., and Marsh, J. A. (2020). Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16 (7), e9380. doi:10.15252/msb.20199380
Lopez-Garcia, M. A., Feria-Romero, I. A., Serrano, H., Rayo-Mares, D., Fagiolino, P., Vazquez, M., et al. (2017). Influence of genetic variants of CYP2D6, CYP2C9, CYP2C19 and CYP3A4 on antiepileptic drug metabolism in pediatric patients with refractory epilepsy. Pharmacol. Rep. 69 (3), 504–511. doi:10.1016/j.pharep.2017.01.007
Loscher, W. (2002). Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 16 (10), 669–694. doi:10.2165/00023210-200216100-00003
Loscher, W., Potschka, H., Sisodiya, S. M., and Vezzani, A. (2020). Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol. Rev. 72 (3), 606–638. doi:10.1124/pr.120.019539
Lu, J., Xia, H., Li, W., Shen, X., Guo, H., Zhang, J., et al. (2021). Genetic polymorphism of GABRG2 rs211037 is associated with drug response and adverse drug reactions to valproic acid in Chinese southern children with epilepsy. Pharmgenomics Pers. Med. 14, 1141–1150. doi:10.2147/PGPM.S329594
Lu, M., Feng, R., Zhang, C., Xiao, Y., and Yin, C. (2023). Identifying novel drug targets for epilepsy through a brain transcriptome-wide association study and protein-wide association study with chemical-gene-interaction analysis. Mol. Neurobiol. 60 (9), 5055–5066. doi:10.1007/s12035-023-03382-z
Lv, N., Qu, J., Long, H., Zhou, L., Cao, Y., Long, L., et al. (2015). Association study between polymorphisms in the CACNA1A, CACNA1C, and CACNA1H genes and drug-resistant epilepsy in the Chinese Han population. Seizure 30, 64–69. doi:10.1016/j.seizure.2015.05.013
Lynch, J. M., Tate, S. K., Kinirons, P., Weale, M. E., Cavalleri, G. L., Depondt, C., et al. (2009). No major role of common SV2A variation for predisposition or levetiracetam response in epilepsy. Epilepsy Res. 83 (1), 44–51. doi:10.1016/j.eplepsyres.2008.09.003
Ma, C. L., Wu, X. Y., Jiao, Z., Hong, Z., Wu, Z. Y., and Zhong, M. K. (2015). SCN1A, ABCC2 and UGT2B7 gene polymorphisms in association with individualized oxcarbazepine therapy. Pharmacogenomics 16 (4), 347–360. doi:10.2217/pgs.14.186
Ma, C. L., Wu, X. Y., Zheng, J., Wu, Z. Y., Hong, Z., and Zhong, M. K. (2014). Association of SCN1A, SCN2A and ABCC2 gene polymorphisms with the response to antiepileptic drugs in Chinese Han patients with epilepsy. Pharmacogenomics 15 (10), 1323–1336. doi:10.2217/pgs.14.89
Madeja, M., Margineanu, D. G., Gorji, A., Siep, E., Boerrigter, P., Klitgaard, H., et al. (2003). Reduction of voltage-operated potassium currents by levetiracetam: a novel antiepileptic mechanism of action? Neuropharmacology 45 (5), 661–671. doi:10.1016/s0028-3908(03)00248-x
Makowska, M., Smolarz, B., Brys, M., Forma, E., and Romanowicz, H. (2021). An association between the rs1799853 and rs1057910 polymorphisms of CYP2C9, the rs4244285 polymorphism of CYP2C19 and the prevalence rates of drug-resistant epilepsy in children. Int. J. Neurosci. 131 (12), 1147–1154. doi:10.1080/00207454.2020.1781110
Manna, I., Gambardella, A., Bianchi, A., Striano, P., Tozzi, R., Aguglia, U., et al. (2011). A functional polymorphism in the SCN1A gene does not influence antiepileptic drug responsiveness in Italian patients with focal epilepsy. Epilepsia 52 (5), e40–e44. doi:10.1111/j.1528-1167.2011.03097.x
Marchi, N., Hallene, K. L., Kight, K. M., Cucullo, L., Moddel, G., Bingaman, W., et al. (2004). Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med. 2, 37. doi:10.1186/1741-7015-2-37
Martin, K., Jackson, C. F., Levy, R. G., and Cooper, P. N. (2016). Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2, CD001903. doi:10.1002/14651858.CD001903.pub3
McCorry, D., Chadwick, D., and Marson, A. (2004). Current drug treatment of epilepsy in adults. Lancet Neurol. 3 (12), 729–735. doi:10.1016/S1474-4422(04)00935-4
Meldrum, B. S., and Rogawski, M. A. (2007). Molecular targets for antiepileptic drug development. Neurotherapeutics 4 (1), 18–61. doi:10.1016/j.nurt.2006.11.010
Meng, H., Guo, G., Ren, J., Zhou, H., Ge, Y., and Guo, Y. (2011). Effects of ABCB1 polymorphisms on plasma carbamazepine concentrations and pharmacoresistance in Chinese patients with epilepsy. Epilepsy Behav. 21 (1), 27–30. doi:10.1016/j.yebeh.2011.02.015
Merritt, H. H., and Putnam, T. J. (1984). Landmark article Sept 17, 1938: sodium diphenyl hydantoinate in the treatment of convulsive disorders. By H. Houston Merritt and Tracy J. Putnam. JAMA 251 (8), 1062–1067. doi:10.1001/jama.251.8.1062
Meyer, U. A., and Zanger, U. M. (1997). Molecular mechanisms of genetic polymorphisms of drug metabolism. Annu. Rev. Pharmacol. Toxicol. 37, 269–296. doi:10.1146/annurev.pharmtox.37.1.269
Miller, D. S., Bauer, B., and Hartz, A. M. (2008). Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 60 (2), 196–209. doi:10.1124/pr.107.07109
Ozgon, G. O., Bebek, N., Gul, G., and Cine, N. (2008). Association of MDR1 (C3435T) polymorphism and resistance to carbamazepine in epileptic patients from Turkey. Eur. Neurol. 59 (1-2), 67–70. doi:10.1159/000109264
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S., and Goldstein, D. B. (2013). Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9 (8), e1003709. doi:10.1371/journal.pgen.1003709
Plosker, G. L. (2012). Perampanel: as adjunctive therapy in patients with partial-onset seizures. CNS Drugs 26 (12), 1085–1096. doi:10.1007/s40263-012-0021-2
Popiolek, M., Izumi, Y., Hopper, A. T., Dai, J., Miller, S., Shu, H. J., et al. (2020). Effects of CYP46A1 inhibition on long-term-depression in hippocampal slices ex vivo and 24S-hydroxycholesterol levels in mice in vivo. Front. Mol. Neurosci. 13, 568641. doi:10.3389/fnmol.2020.568641
Qosa, H., Miller, D. S., Pasinelli, P., and Trotti, D. (2015). Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 1628 (Pt B), 298–316. doi:10.1016/j.brainres.2015.07.005
Qu, J., Lu, S. H., Lu, Z. L., Xu, P., Xiang, D. X., and Qu, Q. (2017). Pharmacogenetic and case-control study on potassium channel related gene variants and genetic generalized epilepsy. Med. Baltim. 96 (26), e7321. doi:10.1097/MD.0000000000007321
Reiner, A., and Levitz, J. (2018). Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron 98 (6), 1080–1098. doi:10.1016/j.neuron.2018.05.018
Romoli, M., Mazzocchetti, P., D'Alonzo, R., Siliquini, S., Rinaldi, V. E., Verrotti, A., et al. (2019). Valproic acid and epilepsy: from molecular mechanisms to clinical evidences. Curr. Neuropharmacol. 17 (10), 926–946. doi:10.2174/1570159X17666181227165722
Rossi, R., Arjmand, S., Baerentzen, S. L., Gjedde, A., and Landau, A. M. (2022). Synaptic vesicle glycoprotein 2A: features and functions. Front. Neurosci. 16, 864514. doi:10.3389/fnins.2022.864514
Rugg-Gunn, F., Miserocchi, A., and McEvoy, A. (2020). Epilepsy surgery. Pract. Neurol. 20 (1), 4–14. doi:10.1136/practneurol-2019-002192
Saleem, T., Maqbool, H., Sheikh, N., Tayyeb, A., Mukhtar, M., and Ashfaq, A. (2022). GABRG2 C588T polymorphism is associated with idiopathic generalized epilepsy but not with antiepileptic drug resistance in Pakistani cohort. Biomed. Res. Int. 2022, 3460792. doi:10.1155/2022/3460792
Samueli, S., Abraham, K., Dressler, A., Groppel, G., Muhlebner-Fahrngruber, A., Scholl, T., et al. (2016). Efficacy and safety of Everolimus in children with TSC - associated epilepsy - pilot data from an open single-center prospective study. Orphanet J. Rare Dis. 11 (1), 145. doi:10.1186/s13023-016-0530-z
Sanchez, M. B., Herranz, J. L., Leno, C., Arteaga, R., Oterino, A., Valdizan, E. M., et al. (2010). Genetic factors associated with drug-resistance of epilepsy: relevance of stratification by patient age and aetiology of epilepsy. Seizure 19 (2), 93–101. doi:10.1016/j.seizure.2009.12.004
Seo, T., Ishitsu, T., Ueda, N., Nakada, N., Yurube, K., Ueda, K., et al. (2006). ABCB1 polymorphisms influence the response to antiepileptic drugs in Japanese epilepsy patients. Pharmacogenomics 7 (4), 551–561. doi:10.2217/14622416.7.4.551
Sills, G. J., and Rogawski, M. A. (2020). Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 168, 107966. doi:10.1016/j.neuropharm.2020.107966
Speliotes, E. K., Willer, C. J., Berndt, S. I., Monda, K. L., Thorleifsson, G., Jackson, A. U., et al. (2010). Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42 (11), 937–948. doi:10.1038/ng.686
Tate, S. K., Depondt, C., Sisodiya, S. M., Cavalleri, G. L., Schorge, S., Soranzo, N., et al. (2005). Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc. Natl. Acad. Sci. U. S. A. 102 (15), 5507–5512. doi:10.1073/pnas.0407346102
Thompson, C. H., Kahlig, K. M., and George, A. L. (2011). SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52 (5), 1000–1009. doi:10.1111/j.1528-1167.2011.03040.x
Tian, G., Kong, Q., Lai, L., Ray-Chaudhury, A., and Lin, C. L. (2010). Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer's disease. J. Neurochem. 113 (4), 978–989. doi:10.1111/j.1471-4159.2010.06661.x
van der Weide, J., Steijns, L. S., van Weelden, M. J., and de Haan, K. (2001). The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics 11 (4), 287–291. doi:10.1097/00008571-200106000-00002
Vetkas, A., Fomenko, A., Germann, J., Sarica, C., Iorio-Morin, C., Samuel, N., et al. (2022). Deep brain stimulation targets in epilepsy: systematic review and meta-analysis of anterior and centromedian thalamic nuclei and hippocampus. Epilepsia 63 (3), 513–524. doi:10.1111/epi.17157
Weissbrod, O., Hormozdiari, F., Benner, C., Cui, R., Ulirsch, J., Gazal, S., et al. (2020). Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 52 (12), 1355–1363. doi:10.1038/s41588-020-00735-5
Wolking, S., Moreau, C., Nies, A. T., Schaeffeler, E., McCormack, M., Auce, P., et al. (2020). Testing association of rare genetic variants with resistance to three common antiseizure medications. Epilepsia 61 (4), 657–666. doi:10.1111/epi.16467
Yip, T. S., O'Doherty, C., Tan, N. C., Dibbens, L. M., and Suppiah, V. (2014). SCN1A variations and response to multiple antiepileptic drugs. Pharmacogenomics J. 14 (4), 385–389. doi:10.1038/tpj.2013.43
Keywords: antiseizure medication, drug-resistant epilepsy, genetic variants, pharmacogenetic studies, mechanistic targets
Citation: Lin C-H, Ho C-J, Chen S-Y, Lu Y-T and Tsai M-H (2024) Review of pharmacogenetics of antiseizure medications: focusing on genetic variants of mechanistic targets. Front. Pharmacol. 15:1411487. doi: 10.3389/fphar.2024.1411487
Received: 03 April 2024; Accepted: 08 August 2024;
Published: 20 August 2024.
Edited by:
Jacob Tyler Brown, University of Minnesota, United StatesCopyright © 2024 Lin, Ho, Chen, Lu and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng-Han Tsai, bWVuZ2hhbkBjZ21oLm9yZy50dw==