Abstract
The application of deep learning algorithms in protein structure prediction has greatly influenced drug discovery and development. Accurate protein structures are crucial for understanding biological processes and designing effective therapeutics. Traditionally, experimental methods like X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy have been the gold standard for determining protein structures. However, these approaches are often costly, inefficient, and time-consuming. At the same time, the number of known protein sequences far exceeds the number of experimentally determined structures, creating a gap that necessitates the use of computational approaches. Deep learning has emerged as a promising solution to address this challenge over the past decade. This review provides a comprehensive guide to applying deep learning methodologies and tools in protein structure prediction. We initially outline the databases related to the protein structure prediction, then delve into the recently developed large language models as well as state-of-the-art deep learning-based methods. The review concludes with a perspective on the future of predicting protein structure, highlighting potential challenges and opportunities.
1 Introduction
Proteins are one of the most important chemicals in animals and the material basis of living organisms. Proteins undertake various vital activities of living organisms, such as material transport, energy conversion, and catalytic reactions. A protein molecule is composed of several different amino acids, and there are 20 different types of amino acids that undergo dehydration condensation chemical reactions to form peptide bonds, which in turn form a sequence of amino acids linked from the beginning to the end. Then, transformations, such as helices, folding, and chemical reactions, result in the formation of proteins that are complex in both physical space and structure (Wang and Dunbrack Jr 2003). Protein structure can be divided into four levels, as shown in Figure 1. The primary structure of a protein is the linear sequence of amino acids, which is determined by the nucleotide sequence of the corresponding gene. The peptide chain results from dehydration condensation between amino acids to form peptide bonds. The number of polypeptide chains, the order of amino acid arrangement, and the number and positions of the bonds of peptide chains determine the primary structure of a protein. Hydrogen bonds are formed by the atoms between the residues of the peptide chain, which results in changes in the local structure of the peptide chain. Protein structure of proteins refers to the regular coiling or folding patterns formed by the polypeptide backbone in localized regions, which is stabilized by hydrogen bonds between backbone groups. Common secondary structural motifs include the alpha-helices and beta-sheet (Onofrio et al., 2014). The secondary structure is only related to the spatial position of the backbone atoms of the main chain and not to the position of the side chains (R groups). Protein tertiary structure is formed by the interaction of distant side chains in the protein secondary structure, and the three-dimensional (3D) spatial arrangement of the main chain and side chains after folding and coiling constitutes the protein’s tertiary structure. It gives rise to two major molecular shapes called fibrous and globular. Globular protein structures can be divided into four structural classes (i.e., main alpha-structure, main beta-structures, alpha/beta-structures, alpha + beta-structures). The function of a protein is largely determined by its tertiary structure, which fully describes its 3D shape. The function of a protein is largely determined by its tertiary structure, which fully describes its 3D shape. While the native state of globular proteins corresponds to a thermodynamically stable energy minimum under physiological conditions, pathological aggregates such as amyloids can occupy deeper energy minima stabilized by cross- sheet interactions. This complexity in the energy landscape makes protein structure prediction particularly challenging (Majid and Khan, 2023). The quaternary structure of proteins refers to the architecture of a complex formed by two or more protein molecules, known as protein subunits, interacting through non-covalent bonds. The problem of protein structure prediction focuses on the transformation from amino acid sequence to protein 3D structure (Anfinsen, 1973). The problem of protein structure prediction focuses on the transformation from amino acid sequence to protein 3D structure. While Anfinsen’s dogma established that the native structure of a protein is determined by its amino acid sequence, the Levinthal paradox highlights a fundamental challenge in this process (Levinthal, 1968). Cyrus Levinthal pointed out that if a protein were to sample all possible conformations randomly to find its native structure, it would take an astronomically long time given the enormous number of possible conformations. However, proteins in nature fold reliably in microseconds to seconds. This paradox demonstrates the inherent complexity of the protein folding process, while simultaneously suggesting that protein folding must proceed along specific pathways rather than through random conformational searches. This theoretical framework has motivated scientists to develop a wide range of approaches for protein structure prediction. A comprehensive and in-depth analysis of the multitude of protein sequences, along with the mining of concealed information, holds profound significance in the fields of modern biology, medicine, and pharmaceuticals (Kuhlman and Bradley, 2019; Li et al., 2021). Due to the extremely rapid growth of protein data and the large scale of data, traditional experimental methods such as NMR and X-ray diffraction to obtain protein structures have the limitations of long cycle time, high cost, and high intermediate product requirements, and the rate of access to resolved protein structures using experimental methods is much slower than the explosive growth of protein sequences (Ladd et al., 1977; Sorgen, 2005). As of 2022, according to the TrEMBL database (Consortium, 2020), there are over 200 million sequence entries, with only 200,000 known protein structures according to the Protein Data Bank (PDB) database (Compton, 2003). It is not feasible to extract protein structure information from experimental methods alone, and a method that enables rapid and accurate prediction of protein structure based on amino acid sequence information needs to be explored. Protein structure prediction approaches can be classified into three categories: template-based modeling (TBM), template-free modeling (TFM), and ab initio. First, TBM approaches rely on identifying and using known protein structures as templates, typically through sequence or structural homology. Second, TFM approaches encompass both traditional (e.g., TrRosetta) and modern AI-based approaches (e.g., AlphaFold3). While commonly referred to as “template-free”, the modern AI-based approaches still rely heavily on comparative analysis and training data from the Protein Data Bank (PDB). It is important to emphasize that current AI-based approaches do not explicitly use templates, but their models are indirectly dependent on known structural information, as they are trained on large-scale PDB data. Despite their remarkable success, these AI-based tools show significant limitations when predicting structures of proteins that lack homologous counterparts in the PDB. Finally, the third category, ab initio, represents the true “free modeling” approach. Unlike TBM and TFM, ab initio approaches are based purely on physicochemical principles and do not rely on existing structural information. The specific steps involved in the three protein structure prediction approaches are illustrated in Figure 2. TBM tools is well represented by MODELLER Webb and Sali (2016) and SwissPDBViewer Guex and Peitsch (1997), where MODELLER implements multi-template modeling to integrate local structural features from multiple homologous templates, while SwissPDBViewer provides comprehensive tools for protein structure visualization and analysis. TBM involves comparing the target sequence with a suitable template structure and then selecting the model with the best match while considering mutations, deletions, and insertions that may be present in the target template structure (Kong et al., 2021; Wu and Xu, 2021; Kong et al., 2022; Weißenow et al., 2022; Sun et al., 2013). The specific steps are as follows. Step 1 involves identifying a homologous protein structure that serves as a template for the target protein. It is crucial that the target sequence and the template sequence share a sequence identity of at least 30%. Step 2 entails creating a sequence alignment between the target sequence and the template sequence. This alignment lays the foundation for accurately mapping the amino acids from the target sequence to their corresponding positions in the template structure. In step 3, through the sequence alignment, amino acids from the target sequence are replaced into the spatial positions of corresponding amino acids in the template structure. This replacement and modeling process is facilitated by homology modeling software, which utilizes the alignment to predict the three-dimensional structure of the target protein. Step 4, the generated structural model undergoes a quality assessment to evaluate its accuracy and reliability. Based on the assessment results, the sequence alignment may be adjusted or corrected, followed by a reiteration of the model building process. This cycle of model building and quality evaluation continues until the model meets the required quality standards. Finally, in step 5, the 3D structure is then refined at the atomic level to obtain the final predicted model. TBM is based on the distance between the target protein structure and the template protein structure, which can be subdivided into comparative modeling and threading. Comparative modelling, also known as Homology modeling, is designed for target proteins with near-homologous templates, and templates can be usually identified by sequence-based comparisons. Threading, also known as fold recognition, operates under the premise that dissimilar amino acid sequences can map onto similar protein structures. Protein threading involves comparing a target sequence andor a hidden Markov model Karplus (2009) against one or more protein structures to identify the best matching sequence-structure template pair. Consequently, threading can effectively identify similar folds or structural motifs in a target sequence, even when sequence similarity is minimal. As different threading programs are trained with different scoring functions and matching algorithms, the template recognition and matching results are often different for the same query sequence. However, establishing the best sequence–template pairing is very challenging, especially when only remotely related templates to the target protein are available.
FIGURE 1
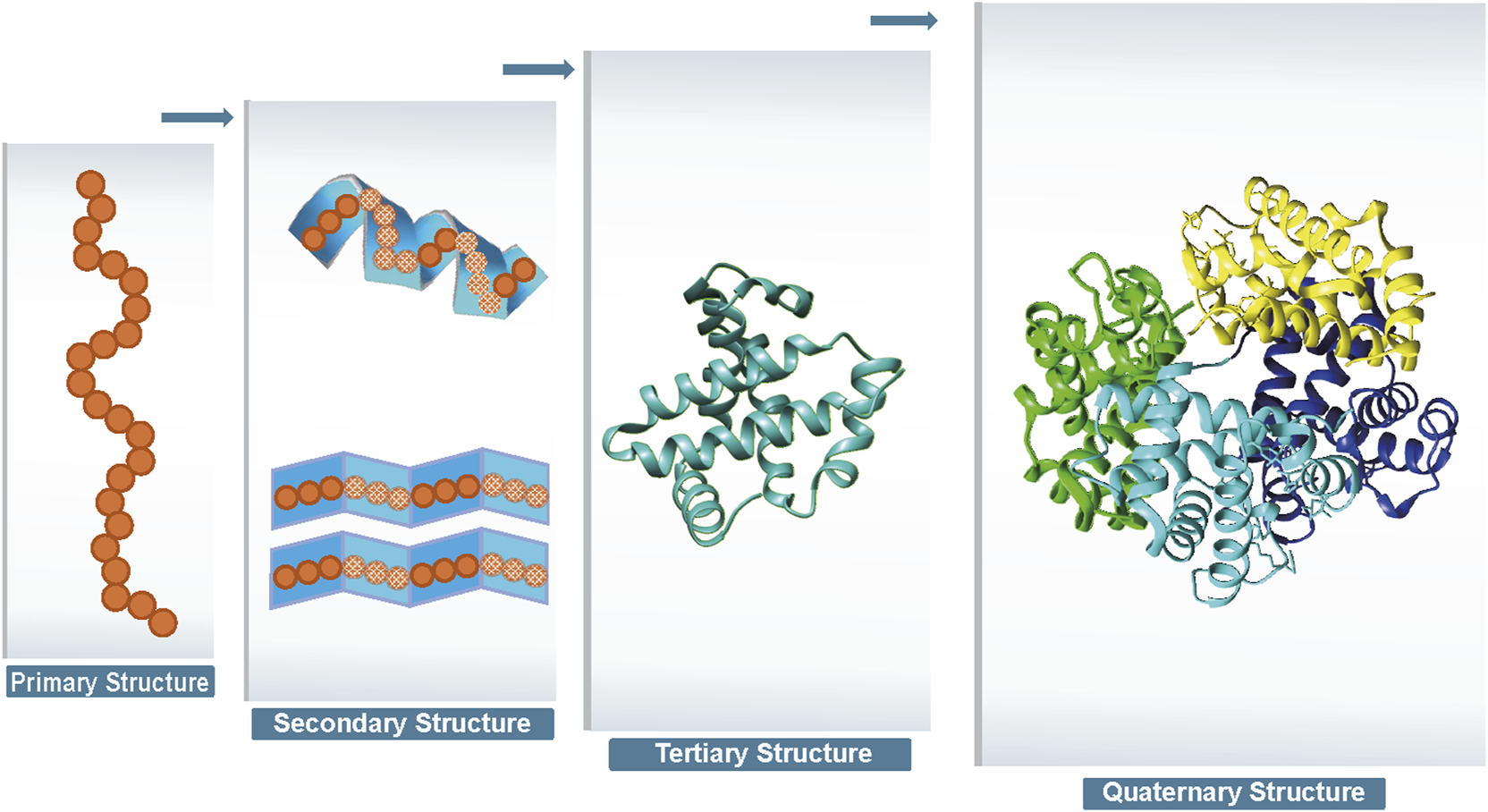
Four levels of protein structure.
FIGURE 2
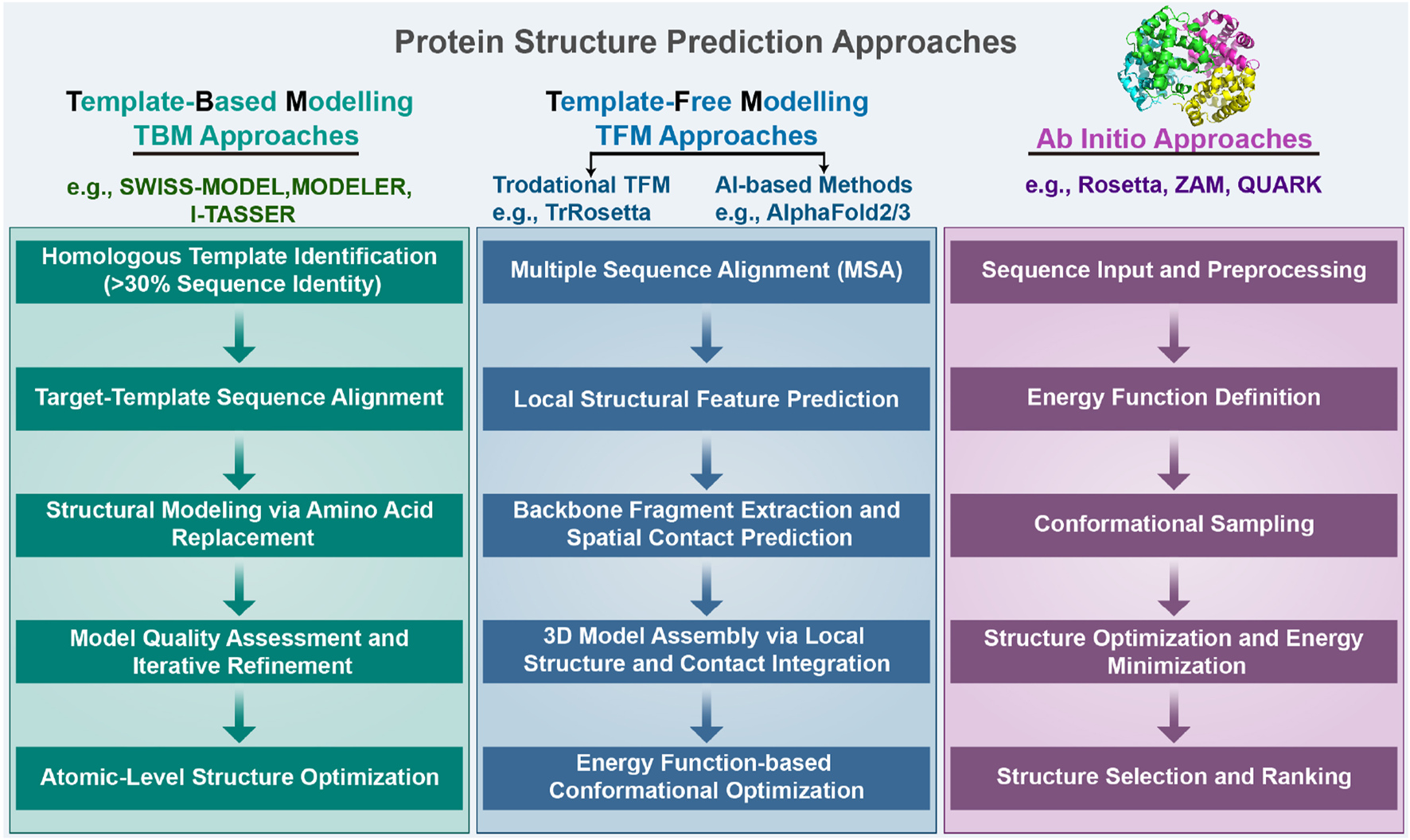
We categorize protein structure prediction approaches into three types: template-based modeling (TBM), template-free modeling (TFM), and ab initio. Detailed steps of these three approaches are provided in the Introduction section.
TFM predicts the structure of a protein directly from the sequence without using global template information by using only amino acid sequence information and without reference to any protein template (Xu and Wang, 2019; Senior et al., 2019; de Oliveira et al., 2021; Hou et al., 2019; Chen et al., 2020). The steps for this process are delineated as follows. Step 1 involves performing multiple sequence alignments (MSAs) between target proteins and their homologous sequences. This process gathers information about amino acid alterations between the homologous sequences and discerns correlation patterns of sequence changes occurring at varied positions. Step 2: Target protein sequences and multiple sequence comparisons are used to construct the basis for predicting local structural frameworks, including torsion angles and secondary structures. Step 3: Backbone fragments are extracted from proteins predicted to have similar local structures and are used for model building, and based on the mutations in multiple sequence comparisons, residue pairs that may be in spatial contact can also be predicted. Step 4: 3D models of protein structures are built by prediction of local structure and disability contacts, which includes gradient-based optimization, distance geometry, and fragment assembly. Step 5: Based on the large search space, the model is improved using the energy function to identify low-energy conformational groups by comparing them with each other. Given the water-soluble nature of amino acids in proteins, physically standard molecular dynamics potential energy functions are used to model protein folding; the protein structure is most stable when the energy is at its lowest. Fragment assembly, a highly effective approach in free modeling, starts by identifying short structural fragments from unrelated proteins. Fragment lengths can be discrete or continuous, and the fragments are mainly based on the comparison of local structural features extracted from the template, such as secondary structure, solvent accessibility, twist angle, and other similarities. Fragment assembly simulation is performed by replacing the main chain structure of a specific region of the simulated structure with the structure of the selected fragment, which can be of the desired bond length, angle, or other component. The replaced fragment can be extracted directly from the fragment itself. Constructing models through fragment assembly reduces the entropy of the conformational search space while ensuring that the local structure of the model is well-formed. TFM is more time-consuming than TBM, as it requires the creation of a model from a random conformation. Although some optimization algorithms such as gradient descent have made progress, there is still a disparity between TFM and TBM in terms of accuracy.
Ab initio approaches rely entirely on physicochemical principles (such as molecular mechanics force fields and energy minimization) and conformational search algorithms, without depending on any known structural data (including training data) for prediction (Pierri et al., 2008). This approach is based on Anfinsen’s thermodynamic hypothesis, which states that a native structure corresponds to the global free energy minimum under a given set of conditions. Among the notable ab initio tools, Rosetta (Rohl et al., 2004) employs a technique called Monte Carlo with Minimization to explore the conformational space of a protein and predict its three-dimensional structure from the amino acid sequence. This method iteratively optimizes to find the lowest energy conformation. QUARK (Xu and Zhang, 2012) stands as another representative tool, specifically designed for ab initio protein structure prediction and peptide folding simulation. QUARK models are built from small fragments (1–20 residues long) by replica-exchange Monte Carlo simulation under the guidance of an atomic-level knowledge-based force field. Despite its capabilities, the tool is constrained by computational limitations, being applicable only to proteins shorter than 200 amino acids and requiring over 48 h for structure prediction. While these approaches are computationally expensive and have certain limitations, they offer unique value in understanding the fundamental physical principles of protein folding, particularly for novel proteins lacking homologous templates in structural databases (Dill et al., 2008).
The structure of this review is as follows. Section 1 describes the generation of protein structures and the two types of modeling approaches currently available for protein structure prediction. Section 2 articulates the necessity for protein structure prediction and explores the potential influence of deep learning in this domain. Section 3 lists publicly available databases in the field. Section 4 discusses in detail the contribution of deep learning in this area of study. Section 5 summarizes our work, offering a perspective on the potential future directions of the field.
2 Current status of protein structure prediction research
The determination of protein structures has led to a greater understanding of the foundations of biology. Physical experiments such as X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy have helped obtain protein structures (Ladd et al., 1977; Sorgen, 2005; Cheng et al., 2021), but there is still a large and growing gap between the number of proteins and the number of known protein structures. The study of protein structure is necessary, and protein structure prediction is an important area of research in biology. For example,: protein structure prediction is a crucial part of protein design. It has been widely used for the evaluation of designed candidate proteins with topology or symmetry constraints (Huang and Li, 2023). The pathological features of some diseases are also related to proteins, the two primary pathological features of Alzheimer’s disease are the accumulation of Amyloid-beta plaques and hyperphosphorylated tau (p-tau) protein, which form neurofibrillary tangles (Lin R.-R. et al., 2023). When an organism is infected by microorganisms such as parasites, bacteria, or viruses, certain proteins play a key role in the immune response by acting as antibodies. These proteins are involved in detecting and neutralizing the pathogens, helping the organism defend against the disease. When an organism is treated with a drug, specific proteins can serve as target receptors. The drug molecule interacts with these receptors, allowing the drug to bind to the correct protein in the body, thereby triggering the appropriate chemical reaction and producing the desired therapeutic effect to treat the disease. In this regard, the prediction of protein structure is a prerequisite for research on drug reuse, disease treatment, and protein function (Pan et al., 2022; Gligorijević et al., 2021; Xu et al., 2022; Tang et al., 2021). As a basis for the prediction of drug–target, drug–disease (Yang et al., 2020b; Liu et al., 2020; Meng et al., 2022), and target–disease associations in the study of protein function and drug repositioning and based on the widening gap between the number of known proteins and the number of actual proteins, protein structure prediction requires more powerful deep learning for research. Artificial intelligence (AI) is broadly affecting many aspects of various fields and addressing diverse tasks and problems in place of humans (Huang et al., 2023; Fu et al., 2024). Deep learning is gradually becoming a key technique in research areas such as computer vision, speech recognition, and natural language processing (Esteva et al., 2021, Chai et al., 2021; Cochero et al., 2022; Santhanavijayan et al., 2021; Shahamiri, 2021; Alsayadi et al., 2021; Pandey et al., 2022; Lauriola et al., 2022; Wahab et al., 2021; Yang et al., 2022; Ye et al., 2022; Li et al., 2022). With advances in deep learning algorithms and increased computing power, great progress has been made in biomedical fields such as predicting protein structures, single-cell technologies (Wen et al., 2022), and cancer research (Gu et al., 2020; Ji et al., 2023; Shi et al., 2022). CASP is an international competition to assess the state of the art in modeling protein structures from amino acid sequences, with the aim of advancing the problem of computing 3D structures of proteins from amino acid sequence information (Qian et al., 2018). With the development of deep learning techniques, more than half of the teams involved in CASP 14 used deep learning algorithms in the protein structure prediction task, we employ data analysis of submissions to the CASP14 and CASP15 competitions as evidence. Our findings indicate that within CASP14, a total of 88 papers explicitly reported the use of deep learning methodologies, compared to 40 papers that did not incorporate such technologies. Similarly, in CASP15, 68 submissions were identified as utilizing deep learning approaches, whereas 16 submissions were found to abstain from applying deep learning techniques. The DL-based AlphaFold2 in CASP14 can accurately predict the 3D structures of 98.5% of human proteins. It is even considered to be the second-largest breakthrough in life sciences after the human genome project (Xu Y. et al., 2021). This demonstrates the excellent learning capability of deep learning and accelerates the development of the field of bioinformatics.
AlphaFold3, further extends these capabilities by modeling interactions between proteins and diverse biomolecules (e.g., DNA, RNA, ligands) with atomic precision (Abramson et al., 2024), demonstrating remarkable success in predicting protein complexes and multi-domain assemblies. However, the performance of AI-based tools like AlphaFold is inherently constrained by the limitations of the PDB. Recent studies highlight that the PDB’s restricted size and structural bias may lead to overfitting and memorization effects in deep learning models. Chakravarty et al. (2024) While AlphaFold3 improves upon its predecessor in capturing biomolecular interactions, it still struggles with dynamic systems such as fold-switching proteins. For example, AlphaFold3 failed to predict the experimentally observed dimeric conformation of human XCL1, instead generating a domain-swapped structure inconsistent with evolutionary restraints. This underscores a critical issue: the PDB predominantly contains static, thermodynamically stable conformations, with limited representation of dynamic or multi-state proteins. Consequently, even advanced models exhibit modest success rates for known fold-switching cases within their training sets and perform poorly on novel conformations These limitations emphasize that current AI tools remain dependent on the structural diversity present in training data, calling for expanded databases with transient states and hybrid approaches integrating physical modeling.
3 Summary of databases
The exponential growth of protein-related data, driven by advancements in genome sequencing and proteomic techniques, presents significant opportunities for computational protein structure prediction methods to reveal novel protein structures. The Protein Data Bank wwPDB consortium (2018) (PDB) serves as a repository for experimentally determined 3D structures, primarily focusing on proteins, nucleic acids, and biological macromolecules. As of 2023, the PDB contains an impressive collection of 214,108 structures. The Universal Protein Resource (UniProt) Consortium U. (2019) is a comprehensive database that offers detailed information on protein sequences and functional annotations. It consists of three databases: UniProtKB Boutet et al. (2007), UniParc, and UniRef. Among these, UniProtKB is the largest component, providing known protein sequences and related annotation information. UniParc serves as an archive library, housing copies of all known protein sequences, while UniRef is a protein clustering database that groups similar proteins, offering representative sequences and related annotations. Table 1 summarizes the databases involved in protein structure prediction.
TABLE 1
| Type | Name | Description | URL | API |
|---|---|---|---|---|
| In vitro determined structure | PDB Berman et al. (2000) | PDB is an archive of experimentally-determined structures of proteins, nucleic acids, and complex assemblies | www.rcsb.org | Yes |
| MMDB Madej et al. (2014) | MMDB contains experimentally resolved structures of proteins, RNA, and DNA, derived from the PDB, with value-added features | www.ncbi.nlm.nih.gov/Structure/MMDB/mmdb.shtml | Yes | |
| SCOP Murzin et al. (1995) | A database of protein structural domains based on their evolutionary and structural relationships | https://scop.mrc-lmb.cam.ac.uk/ | No | |
| BMRB Ulrich et al. (2007) | A repository for experimental and derived data of biomolecular NMR studies | https://bmrb.io/ | Yes | |
| In silico predicted structure | AlphaFold Protein Structure Database Jumper et al. (2021) | This platform provides open access to over 200 million protein structure predictions, aiming to accelerate scientific research | https://alphafold.ebi.ac.uk/ | No |
| Sequence | UniProt Consortium (2019b) | A comprehensive protein sequence and functional annotation database, consisting of UniProtKB, UniParc, and UniRef. | www.uniprot.org/ | Yes |
| ModBase Pieper et al. (2014) | A database of comparative protein structure models based on the sequences of proteins with known structures | https://modbase.compbio.ucsf.edu/ | No | |
| ProteinNet AlQuraishi (2019) | A standardized data set for machine learning of protein structure | https://github.com/aqlaboratory/proteinnet | No | |
| ENA Amid et al. (2020) | ENA provides a comprehensive record of the world’s nucleotide sequencing information, covering raw sequencing data, sequence assembly information and functional annotation | www.ebi.ac.uk/ena | Yes | |
| GenBank Benson et al. (2012) | A database of nucleotide sequences that is maintained by the National Center for Biotechnology Information (NCBI) | www.ncbi.nlm.nih.gov/genbank/ | Yes | |
| HSSP Dodge et al. (1998) | A database of alignments of protein sequences and their secondary structures, which can be used for the prediction of protein structures | https://swift.cmbi.umcn.nL/gv/hssp/ | No | |
| Family | Pfam Mistry et al. (2021) | It is a comprehensive collection of protein domains and families, represented as multiple sequence alignments and as profile hidden Markov models | https://pfam.xfam.org/ | Yes |
| HOMSTRAD Mizuguchi et al. (1998) | A curated database of protein structure alignments for homologous families | www-cryst.bioc.cam.ac.uk/homstrad/ | No | |
| SUPERFAMILY Gough and Chothia (2002) | A database of protein domains and their relationships, based on hidden Markov models (HMMs) and structural alignments | https://supfam.org/ | No | |
| PROSITE Sigrist et al. (2010) | A database of protein families, domains, and functional sites, which are annotated with information about their structure, function, and evolutionary history | https://prosite.expasy.org/ | NO | |
| Family | InterPro Mitchell et al. (2015) | A database of protein families, domains, and functional sites, which integrates information from several different databases and predictive algorithms | www.ebi.ac.uk/interpro/ | Yes |
| SMART Letunic et al. (2015) | A database of protein domains and families, providing information about the sequence, structure, and function of these domains and families | https://smart.embl-heidelberg.de/ | Yes | |
| Interaction | DIP Salwinski et al. (2004) | Catalogs experimentally determined interactions between proteins | https://dip.doe-mbi.ucla.edu/ | No |
| STRING Szklarczyk et al. (2019) | A database of protein-protein interactions and functional associations, which integrates experimental and computational data from multiple sources | https://string-db.org/ | Yes | |
| BioGRID Oughtred et al. (2019) | A database of protein-protein and genetic interactions, integrating experimental data from high-throughput screens and literature curation | https://thebiogrid.org/ | Yes | |
| Function | CATH Sillitoe et al. (2021) | CATH is a hierarchical classification of protein domains based on their structures and functions | www.cathdb.info/ | No |
| PIR Wu et al. (2002) | An integrated public resource of functional annotation of protein data to aid in the exploration of the protein universe | https://pir.georgetown.edu/ | Yes | |
| Hybrid | NKAB Berman et al. (2022) | This tool offers searching, reporting, statistical analysis, mapping, and visualization for all experimentally determined 3D structures involving nucleic acids, maintained by both NDB and PDB. | https://nakb.org/ | No |
| SWISS-MODEL Waterhouse et al. (2018) | A web-based protein structure homology-modeling server that uses a template-based approach to generate three-dimensional models of proteins | swissmodel.expasy.org/ | Yes | |
| PSPC Moult et al. (2018) | It provides resources for the community-wide Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiments | https://predictioncenter.org/ | No | |
| EVcouplings Hopf et al. (2019) | A platform for predicting protein structure, function, and mutations using evolutionary sequence covariation | https://evcouplings.org/ | No | |
| PMP Haas et al. (2013) | PMP provides a single, consistent interface to various sources of computational models of protein structure | https://proteinmodelportal.org/ | Yes | |
| PRISM server Baspinar et al. (2014) | A server for predicting protein-protein interactions and modeling their 3D complexes | https://prism.ccbb.ku.edu.tr/ | No | |
| SGC Lee et al. (2009) | A not-for-profit organization that aims to determine the three-dimensional structures of proteins of medical relevance | www.thesgc.org/ | No | |
| Protein Atlas Thul and Lindskog (2018) | It aims to map all the human proteins in cells, tissues and organs using integration of various omics technologies | www.proteinatlas.org/ | Yes | |
| COFACTOR Zhang et al. (2017) | A protein function prediction tool that uses multiple sources of data to generate predictions, including protein-protein interaction data, gene ontology terms, and sequence information | https://zhanglab.ccmb.med.umich.edu/COFACTOR/ | No | |
| HMMER Finn et al. (2011) | It is a tool for searching protein sequences against a database of HMMs of protein families and domains | https://hmmer.org/ | Yes | |
| Hybrid | Robetta Kim et al. (2004) | A protein structure prediction server that uses comparative modeling, de novo modeling, and structure-based protein function prediction | https://robetta.bakerlab.org/ | No |
| MODELLER Webb and Sali (2016) | A software package for protein structure prediction, which uses comparative modeling to generate 3D models of protein structures based on homology to known structures | https://salilab.org/modeller/ | Yes | |
| Phyre2 Kelley et al. (2015) | A web-based service for protein modeling, prediction, and analysis | www.sbg.bio.ic.ac.uk/phyre2 | No | |
| PconsFold Yang et al. (2020a) | A tool for protein structure prediction, which uses a probabilistic approach to generate models that are more accurate and reliable than those generated by traditional methods | https://toolkit.tuebingen.mpg.de/tools/pcons-fold | Yes | |
| ModFOLD McGuffin (2008) | A tool for protein structure prediction, which uses an ensemble approach to generate models that are more accurate and reliable than those generated by individual methods | www.reading.ac.uk/bioinf/ModFOLD/ | Yes | |
| PIP Ofran and Rost (2003) | A tool for protein-protein interaction prediction, which uses a machine learning approach to predict the interaction partners of a query protein based on its sequence and structural features | www.pip-tools.org/ | Yes | |
| MolIDE Canutescu and Dunbrack Jr (2005) | A tool for interactive molecular visualization and analysis, providing a user-friendly interface for exploring protein structures and their interactions | https://dunbrack.fccc.edu/molide/molide.php | No | |
| PRODIGY Xue et al. (2016) | A tool for predicting protein-ligand binding affinity, using a physics-based approach to model the thermodynamics and kinetics of the binding process | https://milou.science.uu.nL/services/PRODIGY/ | Yes | |
| Reactome Fabregat et al. (2018) | A bioinformatics tool for visualizing, interpreting, and analyzing pathway knowledge | https://reactome.org/ | Yes | |
| HHblits Remmert et al. (2012) | A tool for protein sequence alignment, using a profile hidden Markov model (HMM) to align query sequences against a database of HMMs for protein families and domains | https://toolkit.tuebingen.mpg.de/tools/hhblits | Yes | |
| PISA server Krissinel and Henrick (2007) | A server for analyzing protein-protein and protein-ligand interactions, providing information about the geometry, energetics, and surface area of these interactions | www.ebi.ac.uk/pdbe/pisa/ | Yes | |
| CRISPR Grissa et al. (2007) | A database of CRISPR/Cas systems, providing information about the classification, function, and diversity of these systems in bacteria and archaea | https://crispr.i2bc.paris-saclay.fr/ | No |
The databases involved in protein structure prediction.
Accessing publicly available datasets is essential for leveraging data in deep learning models. Therefore, ensuring easy downloads or APIs for dataset availability is crucial. Researchers have the flexibility to select inputs from diverse data sources and conduct cross-database comparative analyses. Protein structure prediction, a complex and challenging task, requires a range of databases. These databases can be broadly categorized into six types: protein sequence, structure, family, interaction, function, and hybrid methods databases. The PDB exemplifies a protein structure database, offering experimentally determined 3D structures of proteins (Burley et al., 2017). The quality and quantity of structural databases directly determine the level of development and optimization of structural prediction methods. More structural data allows for more accurate algorithm training and testing, thereby improving the accuracy of predictive models. However, it’s also important to consider complementary datasets providing sequence information, protein associations, family details, and functional annotations. Sequence databases like UniProt Consortium U. (2019) and RefSeq O’Leary et al. (2016) database at NCBI contain amino acid sequences of numerous proteins, serving as the foundation for protein structure prediction. The breadth and depth of sequence databases directly affect the accuracy and feasibility of protein structure prediction. More abundant sequence data means a higher chance of finding sequences highly similar to the target protein, thereby increasing the success rate of structure prediction. Family databases classify proteins based on sequence and structural similarities. By providing structural information of similar family members, the family database supports accurate model prediction on unknown proteins. In addition, the functional information of the family database provides strong support for annotation and prediction of protein functions. The widely used Pfam database focuses on protein families, while InterPro(Paysan-Lafosse et al., 2023) integrates multiple databases, including Pfam, ProSite Sigrist et al. (2012), and PRINTS Attwood et al. (2000). Additionally, protein family databases like SMART (Letunic et al., 2012), CDD (Lu et al., 2020), and PROSITE Sigrist et al. (2012) greatly contribute to understanding protein structure-function relationships. Interaction databases such as STRING Szklarczyk et al. (2021), I2D Brown and Jurisica (2005) and BioGrID Oughtred et al. (2021) provide valuable information on protein-protein interactions, including functional and regulatory associations. Interaction information enriches the context of structure prediction, allowing researchers to not only predict the structure of individual proteins but also to predict the interactions and arrangements of proteins within complexes. Function databases like CATH Sillitoe et al. (2021), PIR Wu et al. (2002) and Gene Ontology Consortium G. O. (2019), aid in comprehending the connection between protein structure and function. Functional information provides important clues for predicting structures, especially in predicting protein functional domains and active sites, helping to improve the relevance and accuracy of structure predictions. Hybrid methods databases, including ModBase (Pieper et al., 2014), Robetta Kim et al. (2004), and SWISS-MODEL Waterhouse et al. (2018), offer integrated tools and resources that combine multiple approaches. The comprehensive information from hybrid databases makes structure prediction more holistic and accurate, especially for complex prediction tasks that require an integrated consideration of sequence features, structural patterns, and functional information.
4 Advances in deep learning for protein structure prediction
Machine learning techniques have contributed substantially to the generation of innovative concepts in the field of protein structure prediction, resulting in notable advancements. Most machine learning methods for protein structure prediction have focused on methods based on co-evolution (Bonetta and Valentino, 2020; S Bernardes, 2013; Zhang and Zhang, 2019; Yang et al., 2014). The accuracy of these methods depends on the number of homologous protein sequences available in the database. Protein structure prediction is challenging when there are no target proteins with homologous protein sequences in the database. Machine learning models with simpler structures are unable to predict them accurately, whereas deep learning can learn deeper and more complex structural features; thus, deep learning models are considered for protein structure prediction. Deep learning methods can be utilized to integrate and extract features from these various data sources, allowing for accurate and efficient prediction of protein structures. Table 2 summarizes the deep learning models used in the protein structure prediction. Table 3 lists the available online web servers for protein structure prediction. Integrating data from multiple sources can lead to more accurate predictions of protein structure and function. One example is the AlphaFold algorithm, which combines PDB data, protein sequence data from UniProt, and multiple sequence alignment data from publicly available databases. The utilization of these data sources in combination with deep learning approaches has led to significant advancements in the field of protein structure prediction, offering new avenues for drug discovery and protein engineering. We next analyze and summarize deep learning models such as deep neural networks, convolutional neural networks, recurrent neural networks, graph neural networks, and deep residual neural networks for protein structure prediction.
TABLE 2
Models used for protein structure prediction.
TABLE 3
| Web server | URL |
|---|---|
| AlphaFold3 | https://alphafoldserver.com/ |
| D-I-TASSER | https://zhanggroup.org/D-I-TASSER/ |
| Robetta | https://robetta.bakerlab.org |
| I-TASSER | https://zhanggroup.org/I-TASSER/ |
| Phyre2 | http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index |
| Modeller | https://salilab.org/modeller/ |
| SWISS-MODEL | https://swissmodel.expasy.org |
| ModeBase | https://modbase.compbio.ucsf.edu/modweb/ |
| DMFold | https://zhanggroup.org/DMFold/ |
Web servers available for protein structure prediction.
4.1 Deep neural networks
Deep neural networks (DNNs) are also called multilayer perceptrons. The layers within a DNN can be divided into three categories: the input layer, the hidden layer, and the output layer, with nodes fully connected between the layers. The framework of the model is shown in Figure 3a. The amino acid sequence is input to the DNN after one-hot encoding or embedding representation, and after the processing through the hidden layers, a number of protein structures are finally output, with the structure having the highest score as the final prediction.DNNs have been used by many researchers to model protein structure prediction. Aaron Hein et al. used artificial neural networks (ANNs) to optimize the encoding of protein primary sequence structure, which helps in the prediction of protein secondary structure and protein tertiary structure, thus improving the quality of protein structure prediction Hein et al. (2021). John Jumper et al. designed important DNN-based protein 3D structure model, called MULTICOM. MULTICOM is an automated protein structure prediction system that involves three major components: contact distance prediction based on deep convolutional neural networks, distance-driven template-free modeling, and protein model ranking that’s empowered by deep learning and contact prediction (Hou et al., 2019; Hou et al., 2020; Wang et al., 2010). In general, deep neural networks can assist in predicting protein primary, secondary, and tertiary structures. These networks have shown promise in optimizing predictions for primary and secondary structures (Senior et al., 2019; Du et al., 2021; Timmons and Hewage, 2021; Senior et al., 2020; Mulnaes et al., 2020; Ju et al., 2021). While deep neural networks (DNNs) have demonstrated remarkable success in predicting secondary and tertiary structures of globular proteins, These methods excel when evolutionary or structural homologs exist in training datasets (e.g., the PDB), leveraging coevolutionary patterns to infer folds. However, their efficacy diminishes for non-globular proteins, such as intrinsically disordered proteins (IDPs) or fold-switching systems, where training data are sparse or conformational diversity is critical. For example, AlphaFold often mispredicts alternative folds or dynamic conformations due to overreliance on static training-set structures (Chakravarty et al., 2025). Similarly, DNNs struggle with membrane proteins and IDPs, where sequence-structure relationships diverge from globular paradigms (Agarwal and McShan, 2024).
FIGURE 3
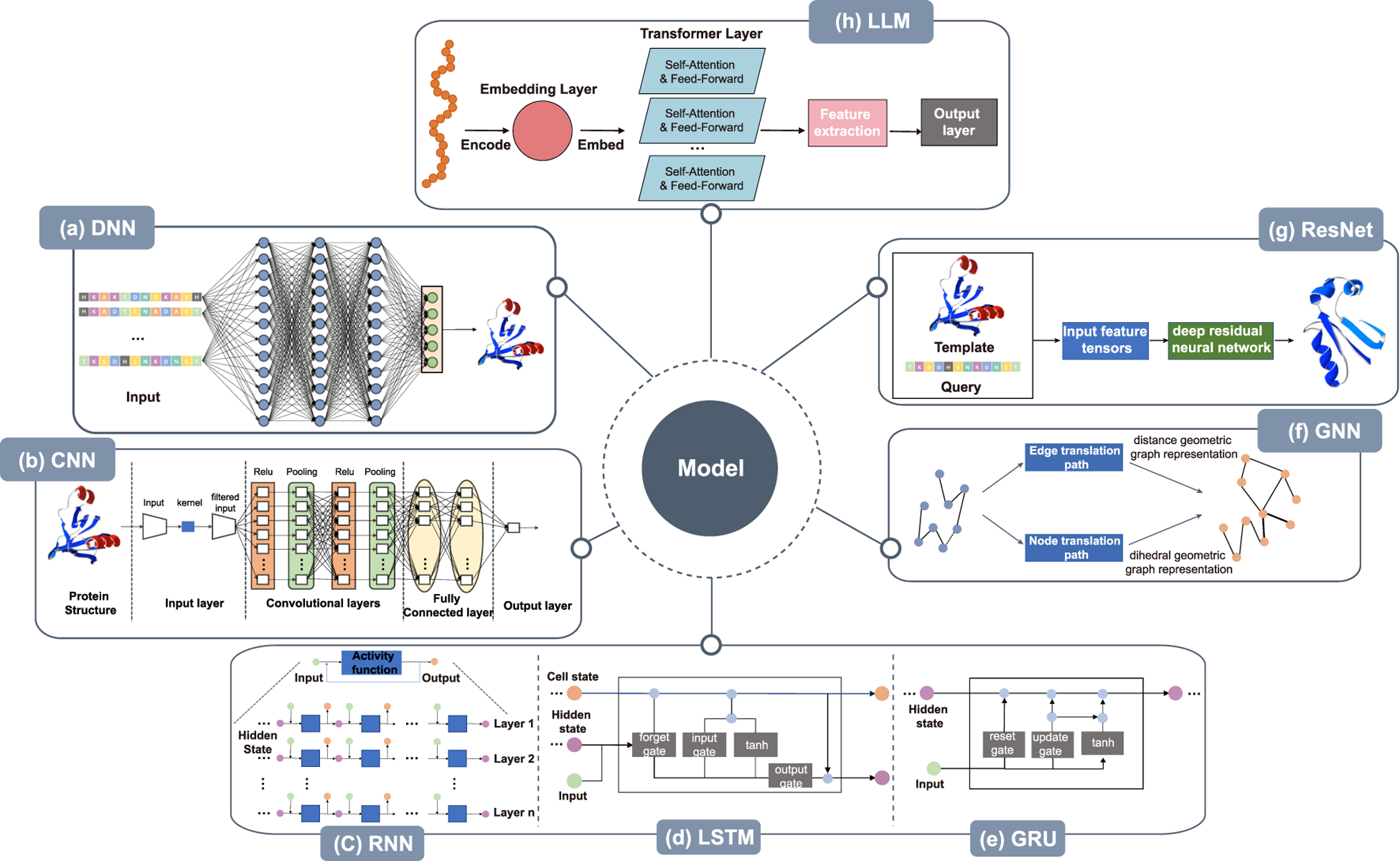
Architecture of deep learning models. (a) DNN takes the protein sequence as input and outputs the protein structure after processing through several hidden layers. (b) The CNN takes the protein structure as input for pre-processing, then, within the convolution layer, features are extracted by convolutional operations to reduce noise and pool data features remain unchanged while the data is compressed to reduce overfitting. After several rounds of convolution and pooling operations, the data is compressed. At the same time, the data is abstracted into features with higher information content, and finally, through the fully connected layer, the results are obtained. (c) RNN takes protein sequence data as input and increases the number of layers of the network for vertical expansion, using chaining and recursion to finally obtain prediction results. (d) LSTM can solve the long-term dependency problem found in general RNN, as well as issues such as long-term memory and gradients in back propagation. (e) GRU, a variation of LSTM, runs more efficiently than LSTM networks. GRU can achieve comparable results and can improve training efficiency to a great extent. (f) The amino acid sequence of the protein is used as input in GNN to abstract the protein structure as a graph structure. The features of nodes and edges are extracted by edge embedding and node embedding to obtain edge translation path and node translation path. In node translation path, each amino acid is considered as a node within a graph, with the node’s feature vector typically encompassing the physicochemical properties of the amino acid. The translation of edges focuses on the interactions between amino acids in the protein sequence. In GNNs, edges represent the relationships between nodes (amino acids), and by updating the weights of these edges, it’s possible to capture these interactions, thereby reflecting the three-dimensional structural characteristics of proteins in the graph. The geometry of the 3D protein backbone structure is then predicted after the distance geometric graph representation and the dihedral geometric graph representation, respectively. (g) Deep residual neural network takes protein templates and query sequences as input and predicts the protein 3D structure by the input feature tensor. (h) Large language models train a processed data, often using techniques like transfer learning from pre-trained models.
4.2 Convolutional neural networks
A convolutional neural network (CNN) is a type of neural network. It is a feed-forward neural network with a deep structure and convolutional calculation. The structure of the CNN is shown in Figure 3b. The amino acid sequences are converted to a two-dimensional matrix as input after being represented by solo thermal encoding or embedding and pre-processed using strategies such as normalization, and principal components analysis. It then enters the convolution layer, where the convolutional operations extract features, enhance signal characteristics, and reduce noise. Following this, after pooling, the data features remain unchanged while the data is compressed, thereby reducing overfitting. After several rounds of convolution and pooling operations, the input data is abstracted into features with higher information content. These then enter the fully connected layer to generate prediction results based on the final extracted data features.
Gabriel Cretin et al., leveraging the capabilities of deep neural networks, proposed the PYTHIA method. This approach incorporates a deep residual incidence neural network with a convolutional block attention module to predict the local conformation of a protein directly from the amino acid sequence Cretin et al. (2021). TBM, which aims to construct structural models by replicating and refining the structural framework of other known proteins, is an accurate method for protein structure prediction. However, it is challenging to identify distant homologous templates, and as a result, the accuracy of TBM rapidly decreases when the evolutionary relationship between the target and the template diminishes. Lupeng Kong et al. proposed a novel deep learning method, named ProALIGN, that predicts accurate sequence–template comparisons (Kong et al., 2022). Protein alignment are represented as a binary matrix, after which a deep convolutional neural network is employed to predict the optimal permutation from the query protein and its template. This method can enhance the accuracy of matching target proteins from the TBM method, with the template proteins in the protein database. This improves subsequent protein structure prediction and enhances the overall accuracy of protein structure prediction. Protein secondary structure prediction is crucial for studying protein structure and function. Both traditional machine learning methods and deep learning neural networks have been utilized, and have made great progress in approaching the theoretical limits. Shiyang Long et al. constructed a contextual convolutional neural network (Contextnet) with high accuracy on the JPred and CASP13 datasets (Long and Tian, 2019). The CNN also successfully integrated 1D structural features, 2D contact information, and 3D structural quality scores to improve protein model quality assessment, where contact prediction using convolutional neural networks was first shown to consistently improve protein model rankings. Convolutional neural networks can predict tertiary structures directly from protein as well as structural sequences, and Timmons et al. proposed the use of neural networks and simulated annealing algorithms to predict tertiary sequences from peptide primary sequences to help accelerate the peptide drug design process (Timmons and Hewage, 2021). A large convolutional residual neural network proposed by Jinbo Xu et al. can predict the correctly folded structures of 26 of the 32 free model targets of CASP13 and L/5 long-range contacts with an accuracy of over 80% Xu J. et al. (2021a).
4.3 Recurrent neural networks
Recurrent Neural Networks (RNNs) are a class of neural networks designed to handle sequential data, performing recursive operations along the sequence’s evolution direction. All nodes (recurrent units) are connected in a chain-like manner. The RNN can also be expanded vertically by increasing the number of layers of the network as in other neural networks, as shown in Figure 3c. Long short-term memory (LSTM) is a temporal recurrent neural network designed to solve the long-term dependency problem of general RNNs, all of which have a chained form of repeating neural network modules. A recurrent unit is a type of RNN. Like LSTM, it was proposed to solve problems such as long-term memory and gradients in back propagation. The LSTM model is shown in Figure 3d. A GRU is a simple variant of LSTM. It is simpler in structure, is no less effective, and is more efficient in operation than LSTM networks, making it a popular network structure at present. The GRU can achieve comparable results, which can improve the training efficiency to a great extent. Its structure is shown in Figure 3e. Proteins exhibit strong sequential characteristics at the primary structure level, and models such as RNN, LSTM, and GRU can predict their 3D tertiary structures based on this sequence information.
Protein secondary structure provides crucial structural insights, and its accurate prediction from primary sequences is pivotal in protein research. The local interactions and neighboring residues in the primary sequence determine the secondary structure formation. RNNs, LSTM networks, and GRUs have demonstrated remarkable performance in predicting protein secondary structures from amino acid residue information and capturing long-range interactions. Yanbu Guo et al. proposed 2D convolutional bidirectional recurrent neural networks (2C-BRNNs) (Guo et al., 2018) to improve the accuracy of secondary structure prediction by extracting discriminative local interactions between amino acid residues and then further capturing the interactions between amino acid residues using bidirectional gated recurrent units or bidirectional LSTM. AK Sharma et al. proposed the use of deep RNNs to predict the secondary structure of proteins from primary sequences. Bidirectional LSTM models (Sharma and Srivastava, 2021) have been used to extract past and unknown residue information from primary sequences, on which the description and understanding of protein structure rely heavily. In protein NMR studies, it is more convenient to predict the secondary structure from chemical shifts than from inter-nuclear distances. Zhiwei Miao et al. proposed a deep neural network based on bi-directional LSTM (Miao et al., 2021) to predict the 3-state secondary structure of proteins using the NMR chemical shifts of the backbone. Wei Zhong et al. proposed clustered RNNs as a method for protein tertiary structure prediction (Zhong and Gu, 2020) using RNNs from multiple sample clusters organized in a hierarchical tree structure to learn local sequence–structure relationships at different granularity levels. Their model can learn the non-linear sequence–structure relationships of proteins more effectively than a single machine learning model. Understanding protein sequence–structure relationships is key to using sequence information to predict the 3D structure of proteins. J Antony et al. combined LSTM and bidirectional LSTM neural network architectures for predicting the tertiary structure of proteins from primary sequences (Antony et al., 2021), and their results showed that bidirectional LSTM networks with primary sequence and site-specific scoring matrix data as input had high accuracy. Lina Yang et al. were able to better handle long sequences by building a GRU neural network that can handle long sequences for learning long-term dependencies well (Yang L. et al., 2020). They combined batch normalization with GRU to construct a new network, and a position-specific scoring matrix was used to correlate with other features to build a completely new feature set, thus effectively improving prediction accuracy.
4.4 Graph neural networks
Graph neural networks (GNNs) have become a research hotspot in areas such as natural language processing, computer vision, and traffic prediction. Graph convolutional networks have shown practical utility in the field of bioinformatics. The protein backbone holds proteins together and produces the tertiary structure of a protein. The amino acid sequence of a protein is used as input to predict the geometry of the 3D protein backbone structure. This is a sequence-to-structure prediction task that abstracts the protein structure to that of a graph, extracting the features of the nodes and edges, in the process shown in Figure 3f.
Determining the three dimensions of a protein from its sequence is one of the most challenging problems in biology. Geometric deep learning has been highly successful in the fields of social networking, chemistry, and computer graphics. Although it is natural to render protein structures as 3D shapes, few existing studies have examined protein structures directly as graphs. Tian Xia et al. explored the geometric deep learning and proposed a graphical neural network architecture to address these challenges (Xia and Ku, 2021). The proposed protein geometric GNN models distance geometric representations and dihedral geometric representations by geometric graphical convolution. This study shed new light on the study of protein 3D structures. The authors validated the effectiveness of GNNs on multiple datasets. AlphaFold2 and related systems use deep learning to predict protein structures from co-evolutionary relationships encoded in MSAs. Despite recent dramatic improvements in accuracy, the following challenges remain: (i) predicting proteins that cannot generate MSAs templates and that evolve rapidly; (ii) rapidly exploring designed structures; and (iii) understanding the rules of spontaneous polypeptide folding in solution. Ratul Chowdhury et al. reported the development of an end-to-end distinguishable recursive geometric network (RGN) that can predict protein structures without using MSAs from an individual protein sequence to predict protein structure (Chowdhury et al., 2022). Compared to AlphaFold2, the RGN is superior in predicting distal protein structures. The prediction of protein secondary structure based on amino acids is important for gathering information about protein features and their mechanisms, such as the catalytic function of enzymes, biochemical reactions, and DNA replication. Tamzid Hasan Nahid et al. proposed a new technique for predicting protein secondary structure using GNNs Nahid et al. (2021). First, a graph is drawn from a dataset using primary sequences (amino acids). The entire graph is then iterated sequentially using a GNN to summarize the information of neighboring nodes. The method has high accuracy in the prediction of eight states of protein secondary structure.
4.5 Deep residual neural networks
Deep learning networks can improve the learning efficiency by increasing the number of layers, but the classification and recognition prediction of deeper networks are not improved by increasing the number of layers. Rather, the gradient disappears due to the stacking of layers. Deep residual neural networks can deepen the network and solve the gradient disappearance problem at the same time. Figure 3g shows the protein 3D structure predicted by ResNet after inputting the template structure and query sequence to the feature tensors.
Protein structure prediction (PSP) is considered to be a complex problem in computational biology. Although co-evolution-based approaches have made significant progress in PSP, it is still a challenging and unsolved problem. Predicting contacts and distances between residues from co-evolutionary data using deep learning has greatly advanced protein structure prediction (Wu and Xu, 2021). F Wu et al. proposed a new method, New Deep Learning Threader (ND Threader), to refine sequence–template alignments from predicted protein distances. It is a good premise for protein structure prediction. The method is based on TBM and uses an integration of deep ResNet (residual neural network) and conditional random field to align query proteins to templates without using any distance information. The sequence–template alignment and input to deep ResNet were then used to predict the interatomic distance distribution, and a 3D model was constructed using PyRosetta. A deep residual network was developed by Jianyi Yang et al. to predict the direction of residuals in addition to distance (Yang et al., 2020a). This model assigned higher probabilities to newly designed proteins and helped identify the key residues that determine folding. The method is expected to be used for a wide range of protein structure prediction and design problems. S. Geethu et al. proposed a new method for predicting inter-residue distances and dihedral angles using a deep ResNet architecture designed to generate an average of 125 homologous sequences from a set of custom sequence databases (Geethu and Vimina, 2021). These sequences were used to generate input features. As a result of the neural network, a structure library was generated, from which the lowest potential structure was selected as the final predicted 3D protein structure. H Zhang et al. showed that a new TBM approach, called ThreaderAI (Zhang and Shen, 2020), improved protein tertiary structure prediction. ThreaderAI formulated the task of querying sequence to template alignment as a computer vision and a classical pixel classification problem and applied deep residual neural networks for the prediction. ThreaderAI first uses deep learning to predict the probability matrix of residue–residue alignment by integrating sequence profiles, predicted sequence structural features, and predicted residue–residue contacts and then builds a template–query alignment by applying a dynamic programming algorithm to the probability matrix using the aligned template for structure prediction with high accuracy.
4.6 Transformer
The transformer is a deep learning architecture that has gained widespread popularity in natural language processing tasks, particularly in the context of machine translation. The key innovation of the transformer is its ability to capture long-range dependencies between input sequences, which is particularly relevant in the case of protein sequences, where long-range interactions between amino acids are critical to determining the final structure (Jiang et al., 2023). The multi-head attention mechanism, on the other hand, enables the model to attend to different parts of the sequence simultaneously, allowing it to capture both local and global features of the protein. For example, Alphafold2, the researchers utilized a combination of the transformer architecture and multi-head attention mechanism, along with other innovations such as the use of distance constraints and evolutionary information, to predict protein structures with unprecedented accuracy. Similarly, other models such as RosettaFold and ESMfold have also incorporated the transformer architecture and multi-head attention mechanism, with impressive results. The quality of the input MSAs is therefore a key factor in determining whether a high-accuracy model can be produced. DMFold algorithm (Zheng et al., 2024), which excelled in the protein complex structure prediction category of the recent CASP15 competition by integrating DeepMSA2 with the state-of-the-art AlphaFold2 modeling approach. Compared with existing MSA construction methods, one of the major advantages of DeepMSA2 lies in the iterative search and model-based preselection strategy, which can result in MSAs with more balanced alignment coverage and homologous diversity. The 2024 release of AlphaFold3 by DeepMind Abramson et al., 2024) represents a revolutionary breakthrough in biomolecular structure prediction. Unlike its predecessor AlphaFold2 which focused solely on protein structures, AlphaFold3 achieves end-to-end joint prediction of proteins, nucleic acids (DNA/RNA), small molecule ligands, and their complexes. The architecture replaces AlphaFold2’s Evoformer with a Pairformer module, reducing reliance on multiple sequence alignments (MSAs) while improving data utilization efficiency. The framework introduces a geometric diffusion model that enables probabilistic sampling of complex conformations, significantly enhancing the modeling capability for flexible interfaces and allosteric effects, thereby extending its applicability to a broader range of biomolecules.
4.7 Diffusion-based model
Although AlphaFold2 and alternative models such as RoseTTAFold Baek et al. (2021), ESMFold Lin et al. (2022), and OmegaFold (Wu et al., 2022) are widely considered to have successfully addressed the challenge of predicting protein structures from sequences, their effectiveness is primarily limited to globular proteins with a clear counterpart or homologous crystallized protein in the training dataset (the PDB). These models are developed and trained as deterministic mappings from input (sequence or MSA) to output (structure), which limits their ability to model structural ensembles. As generative models, diffusion-based models learn an iterative, stochastic generative process that model multimodal data distributions and generate samples efficiently (Watson et al., 2022), applied in many domains, including molecules generation (Hoogeboom et al., 2022), protein-ligand complex structure generation (Nakata et al., 2023) and protein structure generation (Anand and Achim, 2022; Trippe et al., 2022; Wu et al., 2024; Fu et al., 2023). There have been a limited number of diffusion models designed for forward problems involving protein structures (Nakata et al., 2023; Qiao et al., 2022). However, a recent study firstly designed a diffusion generative modeling framework (called EignFold) for protein structure prediction from a fixed protein sequence. EignFold (Jing et al., 2023) is a novel harmonic diffusion process that models the molecule as a system of harmonic oscillators and explored the application of diffusion modeling to protein structural ensembles, aiming to develop a tool for modern structure prediction frameworks.
4.8 Large language model
Large language models (LLMs) are built on a transformer with many parameters which enable the model to better understand the relationships between different elements of the input Høie et al. (2022). They have recently applied to machine translation, question answering, language-image pre-training with emerging functionalities, such as performing higher-level reasoning and generate lifelike images and text. Recent advances have proved the power of large language models in processing the protein sequence databases (Lin Z. et al., 2023; Wu et al., 2022; Fang et al., 2022). The primary, secondary, tertiary, and quaternary of protein structures bear an analogy to the letters, words, sentences, and texts of human language Hu et al. (2022). These characteristics of reused and rearranged of modular elements significantly benefit the development of protein large-scale language models. For example, Meta AI, FAIR Team developed a high accuracy end-to-end atomic level protein structure prediction method using the individual sequence of a protein, called ESMFold. ESMFold has up to 15 billion parameters and is the largest protein language model to date (Lin et al., 2022). Different from the AlphaFold2, RoseTTAFold and other related models that use deep learning and MSAs (Jumper et al., 2021; Yang et al., 2020a; Baek et al., 2021), Chowdhury et al., proposed an end-to-end protein language model (named AminoBERT) (Chowdhury et al., 2022) using single protein sequences. These protein large language models open up new possibilities for protein structure prediction, especially proteins that have not been structurally characterized before.
4.9 Applications in disease-related protein structure prediction
Deep learning methods for protein structure prediction have demonstrated significant practical value in addressing urgent public health challenges. A notable example is the application of AlphaFold during the COVID-19 pandemic. In early 2020, when SARS-CoV-2 was first emerging, DeepMind rapidly deployed AlphaFold to predict structures of several understudied viral proteins, including the membrane protein, protein 3a, Nsp2, Nsp4, Nsp6, and Papain-like proteinaseTeam (2020). These predictions were released to the scientific community before experimental structures were available, providing valuable insights for understanding viral mechanisms and accelerating therapeutic development. The accuracy of these predictions was later validated when the experimental structure of ORF3a protein was determined, confirming AlphaFold’s ability to predict novel protein folds accurately. As the pandemic evolved, deep learning methods continued to provide crucial insights into new variants. Yang et al. utilized AlphaFold2 to predict the structures of S, M, and N proteins in the Omicron variant, with particular emphasis on analyzing the structural alterations in the RBD and NTD regions of the S protein and their potential implications for viral transmission and immune evasion. This study provided crucial structural insights for the development of vaccines and therapeutic strategies targeting the Omicron variant (Yang et al., 2021). More recent applications have extended to other emerging diseases. For instance, Sahu et al. employed RoseTTAFold to predict protein structures of Monkeypox virus targets. By combining these structural predictions with computational drug screening, they identified potential FDA-approved drugs that could be repurposed to target these viral proteins (Sahu et al., 2023). This approach demonstrates how AI-powered structure prediction can accelerate the drug discovery process by enabling rapid identification of therapeutic candidates for emerging diseases. These applications highlight how deep learning methods have transformed from purely academic tools into practical solutions for urgent public health challenges, enabling rapid response to emerging diseases through structure-based drug discovery and therapeutic development.
5 Model validation
Deep learning models are often evaluated by cross-validation (Berrar, 2019; Liu et al., 2021), in which the original observation dataset is divided into a training set for model training and a separate set for evaluating model performance (Zhong and Gu, 2020; Cretin et al., 2021; Akbar et al., 2021; Xu et al., 2020). The most commonly used cross-validation methods include hold-out cross-validation (Ziggah et al., 2019), k-fold cross-validation (Wong and Yeh, 2019), and leave-one-out cross-validation (LOOCV) Magnusson et al. (2020). Hold-out cross-validation splits the dataset into two mutually exclusive sets; that is, the training and test sets have no cross-over samples. This requires that the number of samples in the training set is at least 50% of the total number of samples. However, there are limitations to hold-out cross-validation, as this validation method only performs one division, and when the division of the dataset is not performed randomly, the evaluation results are subject to chance. This can lead to underfitting or overfitting when the training and test sets are not evenly distributed. K-fold cross-validation is a widely used cross-validation technique. It divides the dataset into k equally-sized, mutually exclusive sets at random. Then, the k sets are used as the test set and the rest as the training set, and the final validation result is averaged after k validations. As each data appear once in the validation set and k-1 times in the training set, this will significantly reduce underfitting. The majority of the data in the dataset is used for training, and the possibility of overfitting is also reduced. LOOCV is a special type of k-fold cross-validation. In LOOCV, the value of k is the number of samples in the dataset. One sample at a time is used as the test set and the rest as the training set, which provides the closest expectation to training on the entire test set. LOOCV, being the most objective method, is therefore used by many researchers to test the ability of various prediction methods. However, when the number of proteins in a given set is not large enough, the sequential exclusion of each protein from the set may result in a severe loss of information. In such cases, the leave-one-out test cannot be utilized.
Q3 accuracy and Q8 accuracy are among the most frequently used evaluation metrics by researchers in protein secondary structure prediction Drori et al. (2018), protein structures are diverse, but the torsion angles and hydrogen bonds in protein structures are repetitive, allowing the classification of protein residues into relatively few structural categories. In the 1980s, the Dictionary of Secondary Structure Patterns (DSSP) proposed eight residue categories, which were later combined into three categories in order to ease the difficulty of protein structure prediction.where m = 3 and m = 8 is referred as and accuracy, respectively. is the total number of residues, and is correctly predicted number of residues in state i Equation 1. Thus, Q8 and Q3 provide the overall percentage of trimers and octamers that have their residues correctly predicted. The root mean square deviation (RMSD) Maiorov and Crippen (1994) is a traditional and commonly used metric for assessing the quality of predicted structures.where n is the number of atoms in protein, and are the corresponding distances between the ith and jth atoms Equation 2. The RMSD calculates the average distance between equivalent atom pairs in two best stacked protein structures. Typically, only backbone atoms are involved in the RMSD calculation.
TM-score (Zhang and Skolnick, 2005) is a metric for assessing the topological similarity of protein structures. TM-score weights smaller distance errors more heavily than larger distance errors, making the score values more sensitive to global folding similarity compared to local structural variation.Here, is the number of residues in the aligned regions, is the length of the target protein, is the distance between corresponding residues in the target and predicted structures, and is a normalization factor adjusted based on the target protein’s length, facilitating comparisons across proteins of different sizes Equation 3. The TM-score introduces a length-dependent scale to normalize the distance error, making the size of the TM-score independent of the length of the random structure pair, thus allowing the TM-score to refine traditional metrics such as RMSD. The Global Distance Test (GDT-score) is calculated based on the largest set of residue-residue pairs that fall within a defined distance from the demarcation line given the superimposed structure (Chen and Cheng, 2022). The Global Distance Test Total Score (GDT_TS) (Li et al., 2016) is a threshold-based measure that determines the topology.Each score is calculated as the percentage of atoms in the predicted structure that are within the corresponding distance threshold from the native structure, multiplied by 100. , , and are the scores for the 1.00Å, 2.00Å, 3.00Å, and 4.00Å thresholds, respectively Equation 4. GDT_TS is the most widely used scoring method to assess the overall quality of a model after CASP4. GDT scores typically range from 0 to 100, with higher scores indicating a more perfectly constructed target backbone conformation. The metric also shows a strong dependence on protein length. GDT_TS allows comparison of results within and between experiments, and this focus on similarity allows the measure to distinguish models that are poor, but contain locally correct fragments, from those that are globally wrong in a way that other related measures cannot.
In recent years, researchers have developed numerous quality assessment (QA) methods to evaluate the accuracy of predictions for protein quaternary structures. One widely used metric is LDDT/pLDDT (Local Distance Difference Test/Probability Local Distance Difference Test), which measures the local structural similarity between predicted and native structures. This metric calculates the difference between predicted and native structures at each residue and averages these differences over the entire structure. LDDT/pLDDT has proven effective in evaluating the accuracy of predicted protein structures, especially when experimental data is unavailable. Another metric, DockQ Score, is specifically designed to evaluate the accuracy of predicted protein-protein docking structures. This composite score considers various aspects of the predicted structure, including shape complementarity, electrostatics, and desolvation energy. DockQ Score has also demonstrated effectiveness in evaluating the accuracy of predicted protein-protein docking structures when compared to other available metrics. For example, studies such as Chen et al. (2022) and Chen et al. (2023) have utilized these metrics to assess protein structure predictions.
6 Conclusion
Protein structure prediction has a crucial role in bioinformatics because protein structure determines protein function. The study of protein structure is fundamental to research areas such as drug repositioning, disease treatment, and protein function. The study of protein structure prediction has received increasing attention from researchers with the continuous development of deep learning techniques and especially the use of deep learning models to accomplish protein structure prediction tasks. This paper provides a summary of protein structure prediction based on deep learning, and it is easy to see that the addition of deep learning methods has made a significant contribution to protein structure prediction. As shown in Figure 4, in the coming years, we are likely to see more advances in protein structure prediction.
FIGURE 4
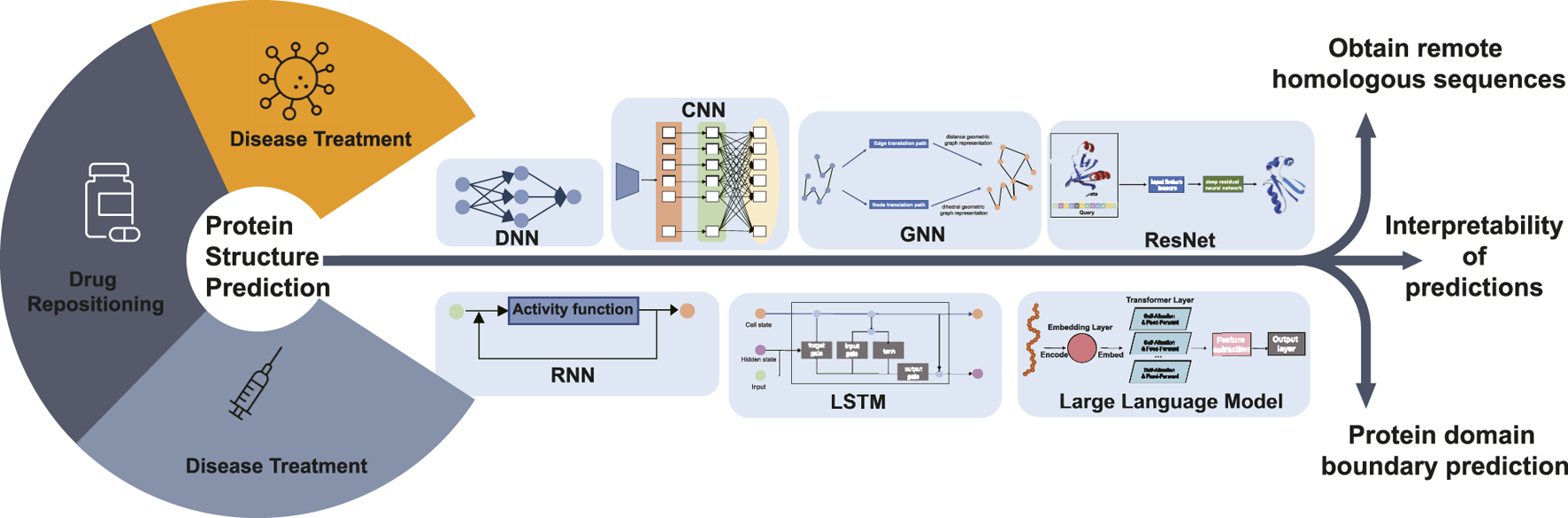
Diagram illustrating the future research hotspots and application scope of deep learning-based protein structure prediction. Protein structure prediction is the basis for disease diagnosis, drug repositioning, and vaccine development research. Future research can predict the 3D structure of proteins, including obtaining remote homologous sequences, interpretability of protein structure predictions, and protein domain boundary prediction, by DNN, CNN, GNN, RNN, LSTM, ResNet, and LLM deep learning algorithms.
For template modeling, all protein structure prediction methods take multiple sequence alignment as input. Therefore, access to homologous sequences becomes the main reason for improved prediction results. Searching for remote homologous sequences in databases can be a difficult research problem. Finding sequence information in major sequence databases to generate multiple sequence comparisons and using more powerful search algorithms to generate multiple sequence comparisons quickly offer possibilities in this field.
Deep learning models are often considered black-box systems because the internal decision-making processes are not easily interpretable to humans. While the models consistently make predictions, the intricate workings of how input data is transformed into predictions can be complex and not straightforward to understand. This is also true in bioinformatics, so research into the interpretability of deep learning models could enhance the interpretability of the sequence-to-structure process in protein structure prediction. In addition, the sequence–structure–function relationship of proteins can be complemented and refined.
The task of single-domain protein structure prediction has been accomplished to some extent, and the correct assignment of domain boundaries from sequences is a key step toward accurate multi-domain protein structure prediction. Future work could be carried out in the area of deep learning-based prediction of multi-domain protein structures. Zheng et al. (2021) developed a contact-based domain boundary prediction algorithm, FUpred, for detecting protein domain boundaries, which could be a new trend in protein structure research. The majority of protein structure prediction methods, including AlphaFold2, focus primarily on predicting static protein structures, which is a significant limitation, but proteins undergo conformational changes and flexible motions under physiological conditions, which are important for their functions. As a unified structure prediction tool, AlphaFold3 has significantly expanded its prediction scope, capable of not only predicting protein structures but also the structures and interactions of various biomolecules including DNA, RNA, antibodies, small molecules. While achieving such comprehensive coverage, AlphaFold3 has also improved prediction accuracy, particularly demonstrating significant advances in predicting protein-DNA complexes and protein-antibody complexes. However, AlphaFold3 still faces certain limitations. Approximately 4.4% of predictions exhibit chirality mismatches or steric clashes, particularly prominent in large complexes. Furthermore, similar to its predecessors, AlphaFold3 primarily focuses on predicting static conformations and may not fully capture protein dynamic transitions. These challenges indicate that despite major breakthroughs in deep learning-based structure prediction, there remains substantial room for improvement in enhancing prediction accuracy and molecular dynamics simulation. Structure prediction is only the first step. Challenges remain in using these structures for better functional annotation and designing new proteins. In summary, despite the breakthrough, there is still ample room for improvement in protein structure prediction, such as handling complex cases, capturing dynamic features, improving time efficiency, and reducing reliance on experimental data. Future efforts are needed to address these challenges and push the boundaries of this field.
With the advancing development of protein structure prediction techniques, they will play an even more important role in biology and medicine. For example, in the development of vaccines, proteins act as scaffolds for immunogens. In disease treatment, proteins act as receptors that bind to drugs for pharmacological responses and act as drug carriers that integrate multiple targeting cues. Proteins are designed to make drugs active in specific environments to reduce side effects. The importance of protein structure prediction is reflected in the need for protein structures to support research in all of these application areas. As protein structure data grow exponentially and provide a larger platform for protein structure prediction, the possibility of using these data to create new methodological techniques is opened.
7 Key points
A comprehensive review and summary of datasets involved in protein structure prediction, providing an up-to-date overview of available resources in this field.
Protein structure prediction based on deep learning has been receiving increasing attention. These approaches can capture underlying features and grasp the complex structures of amino acid sequence information.
Evaluation metrics are clearly listed for the evaluation of computational protein structure prediction model.
There are several challenges to future trends for protein structure prediction, including homologous sequences generation, interpretable deep learning approaches and automation pipeline.
Statements
Author contributions
YM: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing. ZZ: Data curation, Investigation, Writing–original draft, Writing–review and editing. CZ: Data curation, Investigation, Resources, Writing–review and editing. XT: Supervision, Writing–review and editing. XH: Supervision, Writing–review and editing. GT: Supervision, Writing–review and editing. Jialiang Yang: Writing–review and editing YY: Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work is supported by the National Natural Science Foundation of China (Grant Nos. 62302156, and 62402349), the Natural Science Foundation of Hunan Province (Grant No. 2023JJ40180), the Natural Science Foundation of Hubei Province (Grant No. 2024AFB127), Wuhan Textile University Foundation (Grant Nos. 20230612 and 2024309), the Funding National Natural Science Foundation of China (grant no. 62162025) and Hainan Provincial Natural Science Foundation of China (grant no. 621QN0887, 122RC653).
Conflict of interest
JY is employed by the company Geneis Beijing Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abramson J. Adler J. Dunger J. Evans R. Green T. Pritzel A. et al (2024). Accurate structure prediction of biomolecular interactions with alphafold 3. Nature630, 493–500. 10.1038/s41586-024-07487-w
2
Agarwal V. McShan A. C. (2024). The power and pitfalls of alphafold2 for structure prediction beyond rigid globular proteins. Nat. Chem. Biol.20, 950–959. 10.1038/s41589-024-01638-w
3
Akbar S. Pardasani K. R. Panda N. R. (2021). Pso based neuro-fuzzy model for secondary structure prediction of protein. Neural Process. Lett.53, 4593–4612. 10.1007/s11063-021-10615-6
4
AlQuraishi M. (2019). Proteinnet: a standardized data set for machine learning of protein structure. BMC Bioinforma.20, 1–10. 10.1186/s12859-019-2932-0
5
Alsayadi H. A. Abdelhamid A. A. Hegazy I. Fayed Z. T. (2021). Arabic speech recognition using end-to-end deep learning. IET Signal Process.15, 521–534. 10.1049/sil2.12057
6
Amid C. Alako B. T. Balavenkataraman Kadhirvelu V. Burdett T. Burgin J. Fan J. et al (2020). The european nucleotide archive in 2019. Nucleic acids Res.48, D70–D76. 10.1093/nar/gkz1063
7
Anand N. Achim T. (2022). Protein structure and sequence generation with equivariant denoising diffusion probabilistic models. arXiv preprint arXiv:2205.15019.
8
Anfinsen C. B. (1973). Principles that govern the folding of protein chains. Science181, 223–230. 10.1126/science.181.4096.223
9
Antony J. Penikalapati A. Reddy J. V. K. Pournami P. Jayaraj P. (2021). Towards protein tertiary structure prediction using LSTM/BLSTM. Springer, 65–77.
10
Attwood T. K. Croning M. D. Flower D. R. Lewis A. Mabey J. Scordis P. et al (2000). Prints-s: the database formerly known as prints. Nucleic Acids Res.28, 225–227. 10.1093/nar/28.1.225
11
Baek M. DiMaio F. Anishchenko I. Dauparas J. Ovchinnikov S. Lee G. R. et al (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science373, 871–876. 10.1126/science.abj8754
12
Baspinar A. Cukuroglu E. Nussinov R. Keskin O. Gursoy A. (2014). Prism: a web server and repository for prediction of protein–protein interactions and modeling their 3d complexes. Nucleic acids Res.42, W285–W289. 10.1093/nar/gku397
13
Benson D. A. Cavanaugh M. Clark K. Karsch-Mizrachi I. Lipman D. J. Ostell J. et al (2012). Genbank. Nucleic acids Res.41, D36–D42. 10.1093/nar/gks1195
14
Berman H. M. Lawson C. L. Schneider B. (2022). Developing community resources for nucleic acid structures. Life12, 540. 10.3390/life12040540
15
Berman H. M. Westbrook J. Feng Z. Gilliland G. Bhat T. N. Weissig H. et al (2000). The protein data bank. Nucleic acids Res.28, 235–242. 10.1093/nar/28.1.235
16
Berrar D. (2019). Cross-validation, 507, 522. 10.1201/9780429341830-29
17
Bonetta R. Valentino G. (2020). Machine learning techniques for protein function prediction. Proteins Struct. Funct. Bioinforma.88, 397–413. 10.1002/prot.25832
18
Boutet E. Lieberherr D. Tognolli M. Schneider M. Bairoch A. (2007). UniProtKB/Swiss-Prot: the manually annotated section of the UniProt KnowledgeBase. Springer, 89–112.
19
Brown K. R. Jurisica I. (2005). Online predicted human interaction database. Bioinformatics21, 2076–2082. 10.1093/bioinformatics/bti273
20
Burley S. K. Berman H. M. Kleywegt G. J. Markley J. L. Nakamura H. Velankar S. (2017). Protein data bank (pdb): the single global macromolecular structure archive. Protein Crystallogr. methods Protoc.1607, 627–641. 10.1007/978-1-4939-7000-1_26
21
Canutescu A. A. Dunbrack Jr R. L. (2005). Mollde: a homology modeling framework you can click with. Bioinformatics21, 2914–2916. 10.1093/bioinformatics/bti438
22
Chai J. Zeng H. Li A. Ngai E. W. (2021). Deep learning in computer vision: a critical review of emerging techniques and application scenarios. Mach. Learn. Appl.6, 100134. 10.1016/j.mlwa.2021.100134
23
Chakravarty D. Lee M. Porter L. L. (2025). Proteins with alternative folds reveal blind spots in alphafold-based protein structure prediction. Curr. Opin. Struct. Biol.90, 102973. 10.1016/j.sbi.2024.102973
24
Chakravarty D. Schafer J. W. Chen E. A. Thole J. F. Ronish L. A. Lee M. et al (2024). Alphafold predictions of fold-switched conformations are driven by structure memorization. Nat. Commun.15, 7296. 10.1038/s41467-024-51801-z
25
Chen C. Chen X. Morehead A. Wu T. Cheng J. (2023). 3d-equivariant graph neural networks for protein model quality assessment. Bioinformatics39, btad030. 10.1093/bioinformatics/btad030
26
Chen X. Akhter N. Guo Z. Wu T. Hou J. Shehu A. et al (2020). “Deep ranking in template-free protein structure prediction,” in Proceedings of the 11th ACM international conference on bioinformatics, computational biology and health informatics (New York, NY, USA: Association for Computing Machinery). 10.1145/3388440.3412469
27
Chen X. Cheng J. (2022). Distema: distance map-based estimation of single protein model accuracy with attentive 2d convolutional neural network. BMC Bioinforma.23, 141. 10.1186/s12859-022-04683-1
28
Chen X. Morehead A. Liu J. Cheng J. (2022). Dproq: a gated-graph transformer for protein complex structure assessment. arXiv Prepr. arXiv:2205.10627.
29
Cheng J. Li B. Si L. Zhang X. (2021). Determining structures in a native environment using single-particle cryoelectron microscopy images. Innovation2, 100166. 10.1016/j.xinn.2021.100166
30
Chowdhury R. Bouatta N. Biswas S. Floristean C. Kharkar A. Roy K. et al (2022). Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol.40, 1617–1623. 10.1038/s41587-022-01432-w
31
Cochero J. Pattori L. Balsalobre A. Ceccarelli S. Marti G. (2022). A convolutional neural network to recognize chagas disease vectors using mobile phone images. Ecol. Inf.68, 101587. 10.1016/j.ecoinf.2022.101587
32
Compton C. C. (2003). Colorectal carcinoma: diagnostic, prognostic, and molecular features. Mod. Pathol.16, 376–388. 10.1097/01.MP.0000062859.46942.93
33
Consortium G. O. (2019a). The gene ontology resource: 20 years and still going strong. Nucleic acids Res.47, D330–D338. 10.1093/nar/gky1055
34
Consortium T. U. (2020). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res.49, D480–D489. 10.1093/nar/gkaa1100
35
Consortium U. (2019b). Uniprot: a worldwide hub of protein knowledge. Nucleic acids Res.47, D506–D515. 10.1093/nar/gky1049
36
Cretin G. Galochkina T. de Brevern A. G. Gelly J.-C. (2021). Pythia: deep learning approach for local protein conformation prediction. Int. J. Mol. Sci.22, 8831. 10.3390/ijms22168831
37
de Oliveira G. B. Pedrini H. Dias Z. (2021). Ensemble of template-free and template-based classifiers for protein secondary structure prediction. Int. J. Mol. Sci.22, 11449. 10.3390/ijms222111449
38
Dill K. A. Ozkan S. B. Shell M. S. Weikl T. R. (2008). The protein folding problem. Annu. Rev. Biophys.37, 289–316. 10.1146/annurev.biophys.37.092707.153558
39
Dodge C. Schneider R. Sander C. (1998). The hssp database of protein structure—sequence alignments and family profiles. Nucleic Acids Res.26, 313–315. 10.1093/nar/26.1.313
40
Drori I. Dwivedi I. Shrestha P. Wan J. Wang Y. He Y. et al (2018). High quality prediction of protein q8 secondary structure by diverse neural network architectures. arXiv Prepr. arXiv:1811.07143.
41
Du Z. Su H. Wang W. Ye L. Wei H. Peng Z. et al (2021). The trrosetta server for fast and accurate protein structure prediction. Nat. Protoc.16, 5634–5651. 10.1038/s41596-021-00628-9
42
Esteva A. Chou K. Yeung S. Naik N. Madani A. Mottaghi A. et al (2021). Deep learning-enabled medical computer vision. NPJ Digit. Med.4 (5), 5. 10.1038/s41746-020-00376-2
43
Fabregat A. Jupe S. Matthews L. Sidiropoulos K. Gillespie M. Garapati P. et al (2018). The reactome pathway knowledgebase. Nucleic acids Res.46, D649–D655. 10.1093/nar/gkx1132
44
Fang X. Wang F. Liu L. He J. Lin D. Xiang Y. et al (2022). Helixfold-single: msa-free protein structure prediction by using protein language model as an alternative. arXiv preprint arXiv:2207.13921.
45
Finn R. D. Clements J. Eddy S. R. (2011). Hmmer web server: interactive sequence similarity searching. Nucleic acids Res.39, W29–W37. 10.1093/nar/gkr367
46
Fu C. Yan K. Wang L. Au W. Y. McThrow M. Komikado T. et al (2023). A latent diffusion model for protein structure generation. arXiv preprint arXiv:2305.04120.
47
Fu Y. Ma L. Wan S. Ge S. Yang Z. (2024). A novel clinical artificial intelligence model for disease detection via retinal imaging. Innovation5, 100575. 10.1016/j.xinn.2024.100575
48
Geethu S. Vimina E. (2021). Improved 3-d protein structure predictions using deep resnet model. protein J.40, 669–681. 10.1007/s10930-021-10016-7
49
Gligorijević V. Renfrew P. D. Kosciolek T. Leman J. K. Berenberg D. Vatanen T. et al (2021). Structure-based protein function prediction using graph convolutional networks. Nat. Commun.12, 3168. 10.1038/s41467-021-23303-9
50
Gough J. Chothia C. (2002). Superfamily: hmms representing all proteins of known structure. scop sequence searches, alignments and genome assignments. Nucleic acids Res.30, 268–272. 10.1093/nar/30.1.268
51
Grissa I. Vergnaud G. Pourcel C. (2007). The crisprdb database and tools to display crisprs and to generate dictionaries of spacers and repeats. BMC Bioinforma.8, 1–10. 10.1186/1471-2105-8-172
52
Gu C. Shi X. Dai C. Shen F. Rocco G. Chen J. et al (2020). Rna m6a modification in cancers: molecular mechanisms and potential clinical applications. innovation1, 100066. 10.1016/j.xinn.2020.100066
53
Guex N. Peitsch M. C. (1997). Swiss-model and the swiss-pdb viewer: an environment for comparative protein modeling. electrophoresis18, 2714–2723. 10.1002/elps.1150181505
54
Guo Y. Wang B. Li W. Yang B. (2018). Protein secondary structure prediction improved by recurrent neural networks integrated with two-dimensional convolutional neural networks. J. Bioinforma. Comput. Biol.16, 1850021. 10.1142/S021972001850021X
55
Haas J. Roth S. Arnold K. Kiefer F. Schmidt T. Bordoli L. et al (2013). The protein model portal—a comprehensive resource for protein structure and model information. Database2013, bat031. 10.1093/database/bat031
56
Hein A. Cole C. Valafar H. (2021). An investigation in optimal encoding of protein primary sequence for structure prediction by artificial neural networks. Springer, 685–699.
57
Høie M. H. Kiehl E. N. Petersen B. Nielsen M. Winther O. Nielsen H. et al (2022). Netsurfp-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic acids Res.50, W510–W515. 10.1093/nar/gkac439
58
Hoogeboom E. Satorras V. G. Vignac C. Welling M. (2022). Equivariant diffusion for molecule generation in 3d
59
Hopf T. A. Green A. G. Schubert B. Mersmann S. Schärfe C. P. Ingraham J. B. et al (2019). The evcouplings python framework for coevolutionary sequence analysis. Bioinformatics35, 1582–1584. 10.1093/bioinformatics/bty862
60
Hou J. Wu T. Cao R. Cheng J. (2019). Protein tertiary structure modeling driven by deep learning and contact distance prediction in casp13. Proteins Struct. Funct. Bioinforma.87, 1165–1178. 10.1002/prot.25697
61
Hou J. Wu T. Guo Z. Quadir F. Cheng J. (2020). The multicom protein structure prediction server empowered by deep learning and contact distance prediction. Protein Struct. Predict.2165, 13–26. 10.1007/978-1-0716-0708-4_2
62
Hu B. Xia J. Zheng J. Tan C. Huang Y. Xu Y. et al (2022). Protein language models and structure prediction: connection and progression. arXiv Prepr. arXiv:2211.16742.
63
Huang T. Li Y. (2023). Current progress, challenges, and future perspectives of language models for protein representation and protein design. Innovation4, 100446. 10.1016/j.xinn.2023.100446
64
Huang T. Xu H. Wang H. Huang H. Xu Y. Li B. et al (2023). Artificial intelligence for medicine: progress, challenges, and perspectives. Innovation Med.1, 100030. 10.59717/j.xinn-med.2023.100030
65
Ji L. Zhang H. Tian G. Xi S. Chu Y. Zhang Y. et al (2023). Tumor microenvironment interplay amid microbial community, host gene expression and pathological features elucidates cancer heterogeneity and prognosis risk. Innovation Life1, 100028–1–100028–10. 10.59717/j.xinn-life.2023.100028
66
Jiang T. T. Fang L. Wang K. (2023). Deciphering “the language of nature”: a transformer-based language model for deleterious mutations in proteins. Innovation4, 100487. 10.1016/j.xinn.2023.100487
67
Jing B. Erives E. Pao-Huang P. Corso G. Berger B. Jaakkola T. (2023). Eigenfold: generative protein structure prediction with diffusion models. arXiv preprint arXiv:2304.02198.
68
Ju F. Zhu J. Shao B. Kong L. Liu T.-Y. Zheng W.-M. et al (2021). Copulanet: learning residue co-evolution directly from multiple sequence alignment for protein structure prediction. Nat. Commun.12, 2535. 10.1038/s41467-021-22869-8
69
Jumper J. Evans R. Pritzel A. Green T. Figurnov M. Ronneberger O. et al (2021). Highly accurate protein structure prediction with alphafold. Nature596, 583–589. 10.1038/s41586-021-03819-2
70
Kandathil S. M. Greener J. G. Jones D. T. (2019). Prediction of interresidue contacts with deepmetapsicov in casp13. Proteins Struct. Funct. Bioinforma.87, 1092–1099. 10.1002/prot.25779
71
Karplus K. (2009). Sam-t08, hmm-based protein structure prediction. Nucleic acids Res.37, W492–W497. 10.1093/nar/gkp403
72
Kelley L. A. Mezulis S. Yates C. M. Wass M. N. Sternberg M. J. (2015). The phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc.10, 845–858. 10.1038/nprot.2015.053
73
Kim D. E. Chivian D. Baker D. (2004). Protein structure prediction and analysis using the robetta server. Nucleic acids Res.32, W526–W531. 10.1093/nar/gkh468
74
Kong L. Ju F. Zhang H. Sun S. Bu D. (2021). Falcon2: a web server for high-quality prediction of protein tertiary structures. BMC Bioinforma.22, 1–14. 10.1186/s12859-021-04353-8
75
Kong L. Ju F. Zheng W.-M. Zhu J. Sun S. Xu J. et al (2022). Proalign: directly learning alignments for protein structure prediction via exploiting context-specific alignment motifs. J. Comput. Biol.29, 92–105. 10.1089/cmb.2021.0430
76
Krissinel E. Henrick K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol.372, 774–797. 10.1016/j.jmb.2007.05.022
77
Kuhlman B. Bradley P. (2019). Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol.20, 681–697. 10.1038/s41580-019-0163-x
78
Ladd M. F. C. Palmer R. A. Palmer R. A. (1977). Structure determination by X-ray crystallography, 233. Springer.
79
Lauriola I. Lavelli A. Aiolli F. (2022). An introduction to deep learning in natural language processing: models, techniques, and tools. Neurocomputing470, 443–456. 10.1016/j.neucom.2021.05.103
80
Lee W. H. Atienza-Herrero J. Abagyan R. Marsden B. D. (2009). Sc-structural biology and human health: a new approach to publishing structural biology results. PLoS One4, e7675. 10.1371/journal.pone.0007675
81
Letunic I. Doerks T. Bork P. (2012). Smart 7: recent updates to the protein domain annotation resource. Nucleic acids Res.40, D302–D305. 10.1093/nar/gkr931
82
Letunic I. Doerks T. Bork P. (2015). Smart: recent updates, new developments and status in 2015. Nucleic acids Res.43, D257–D260. 10.1093/nar/gku949
83
Levinthal C. (1968). Are there pathways for protein folding?J. de chimie physique65, 44–45. 10.1051/jcp/1968650044
84
Li H. Yu Z. Zhao Q. Zhong T. Lai P. (2022). Accelerating deep learning with high energy efficiency: from microchip to physical systems. Innovation3, 100252. 10.1016/j.xinn.2022.100252
85
Li T. Huang T. Guo C. Wang A. Shi X. Mo X. et al (2021). Genomic variation, origin tracing, and vaccine development of sars-cov-2: a systematic review. Innovation2, 100116. 10.1016/j.xinn.2021.100116
86
Li W. Schaeffer R. D. Otwinowski Z. Grishin N. V. (2016). Estimation of uncertainties in the global distance test (gdt_ts) for casp models. PloS one11, e0154786. 10.1371/journal.pone.0154786
87
Lin R.-R. Huang H.-F. Tao Q.-Q. (2023a). Advancing the battle against alzheimer’s disease: a focus ontargeting tau pathology by antisense oligonucleotide. Innovation Med.1, 100020. 10.59717/j.xinn-med.2023.100020
88
Lin Z. Akin H. Rao R. Hie B. Zhu Z. Lu W. et al (2022). Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv2022, 500902.
89
Lin Z. Akin H. Rao R. Hie B. Zhu Z. Lu W. et al (2023b). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science379, 1123–1130. 10.1126/science.ade2574
90
Liu C. Wei D. Xiang J. Ren F. Huang L. Lang J. et al (2020). An improved anticancer drug-response prediction based on an ensemble method integrating matrix completion and ridge regression. Mol. Therapy-Nucleic Acids21, 676–686. 10.1016/j.omtn.2020.07.003
91
Liu H. Qiu C. Wang B. Bing P. Tian G. Zhang X. et al (2021). Evaluating dna methylation, gene expression, somatic mutation, and their combinations in inferring tumor tissue-of-origin. Front. cell Dev. Biol.9, 619330. 10.3389/fcell.2021.619330
92
Long S. Tian P. (2019). Protein secondary structure prediction with context convolutional neural network. RSC Adv.9, 38391–38396. 10.1039/c9ra05218f
93
Lu S. Wang J. Chitsaz F. Derbyshire M. K. Geer R. C. Gonzales N. R. et al (2020). Cdd/sparcle: the conserved domain database in 2020. Nucleic acids Res.48, D265–D268. 10.1093/nar/gkz991
94
Madej T. Lanczycki C. J. Zhang D. Thiessen P. A. Geer R. C. Marchler-Bauer A. et al (2014). Mmdb and vast+: tracking structural similarities between macromolecular complexes. Nucleic acids Res.42, D297–D303. 10.1093/nar/gkt1208
95
Magnusson M. Vehtari A. Jonasson J. Andersen M. (2020). “Leave-one-out cross-validation for bayesian model comparison in large data,” in International conference on artificial intelligence and statistics. (PMLR), 341–351.
96
Maiorov V. N. Crippen G. M. (1994). Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol.235, 625–634. 10.1006/jmbi.1994.1017
97
Majid N. Khan R. H. (2023). Protein aggregation: consequences, mechanism, characterization and inhibitory strategies. Int. J. Biol. Macromol.242, 125123. 10.1016/j.ijbiomac.2023.125123
98
McGuffin L. J. (2008). The modfold server for the quality assessment of protein structural models. Bioinformatics24, 586–587. 10.1093/bioinformatics/btn014
99
Meng Y. Lu C. Jin M. Xu J. Zeng X. Yang J. (2022). A weighted bilinear neural collaborative filtering approach for drug repositioning. Briefings Bioinforma.23, bbab581. 10.1093/bib/bbab581
100
Miao Z. Wang Q. Xiao X. Kamal G. M. Song L. Zhang X. et al (2021). Csi-lstm: a web server to predict protein secondary structure using bidirectional long short term memory and nmr chemical shifts. J. Biomol. NMR75, 393–400. 10.1007/s10858-021-00383-9
101
Mistry J. Chuguransky S. Williams L. Qureshi M. Salazar G. A. Sonnhammer E. L. et al (2021). Pfam: the protein families database in 2021. Nucleic acids Res.49, D412–D419. 10.1093/nar/gkaa913
102
Mitchell A. Chang H.-Y. Daugherty L. Fraser M. Hunter S. Lopez R. et al (2015). The interpro protein families database: the classification resource after 15 years. Nucleic acids Res.43, D213–D221. 10.1093/nar/gku1243
103
Mizuguchi K. Deane C. M. Blundell T. L. Overington J. P. (1998). Homstrad: a database of protein structure alignments for homologous families. Protein Sci.7, 2469–2471. 10.1002/pro.5560071126
104
Moult J. Fidelis K. Kryshtafovych A. Schwede T. Tramontano A. (2018). Critical assessment of methods of protein structure prediction (casp)—round xii. Proteins Struct. Funct. Bioinforma.86, 7–15. 10.1002/prot.25415
105
Mulnaes D. Porta N. Clemens R. Apanasenko I. Reiners J. Gremer L. et al (2020). Topmodel: template-based protein structure prediction at low sequence identity using top-down consensus and deep neural networks. J. Chem. theory Comput.16, 1953–1967. 10.1021/acs.jctc.9b00825
106
Murzin A. G. Brenner S. E. Hubbard T. Chothia C. (1995). Scop: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol.247, 536–540. 10.1006/jmbi.1995.0159
107
Nahid T. H. Jui F. A. Shill P. C. (2021). “Protein secondary structure prediction using graph neural network,” in 2021 5th international conference on electrical information and communication technology (EICT), 1–6. 10.1109/EICT54103.2021.9733590
108
Nakata S. Mori Y. Tanaka S. (2023). End-to-end protein–ligand complex structure generation with diffusion-based generative models. BMC Bioinforma.24, 233. 10.1186/s12859-023-05354-5
109
O’Connell J. Li Z. Hanson J. Heffernan R. Lyons J. Paliwal K. et al (2018). Spin2: predicting sequence profiles from protein structures using deep neural networks. Proteins Struct. Funct. Bioinforma.86, 629–633. 10.1002/prot.25489
110
Ofran Y. Rost B. (2003). Predicted protein–protein interaction sites from local sequence information. FEBS Lett.544, 236–239. 10.1016/s0014-5793(03)00456-3
111
O’Leary N. A. Wright M. W. Brister J. R. Ciufo S. Haddad D. McVeigh R. et al (2016). Reference sequence (refseq) database at ncbi: current status, taxonomic expansion, and functional annotation. Nucleic acids Res.44, D733–D745. 10.1093/nar/gkv1189
112
Onofrio A. Parisi G. Punzi G. Todisco S. Di Noia M. A. Bossis F. et al (2014). Distance-dependent hydrophobic–hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys.16, 18907–18917. 10.1039/c4cp01131g
113
Oughtred R. Rust J. Chang C. Breitkreutz B. Stark C. Willems A. et al (2021). The biogrid database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci.30, 187–200. 10.1002/pro.3978
114
Oughtred R. Stark C. Breitkreutz B.-J. Rust J. Boucher L. Chang C. et al (2019). The biogrid interaction database: 2019 update. Nucleic acids Res.47, D529–D541. 10.1093/nar/gky1079
115
Pan X. Lin X. Cao D. Zeng X. Yu P. S. He L. et al (2022). Deep learning for drug repurposing: methods, databases, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci.12, e1597. 10.1002/wcms.1597
116
Pandey B. Pandey D. K. Mishra B. P. Rhmann W. (2022). A comprehensive survey of deep learning in the field of medical imaging and medical natural language processing: challenges and research directions. J. King Saud University-Computer Inf. Sci.34, 5083–5099. 10.1016/j.jksuci.2021.01.007
117
Paysan-Lafosse T. Blum M. Chuguransky S. Grego T. Pinto B. L. Salazar G. A. et al (2023). Interpro in 2022. Nucleic acids Res.51, D418–D427. 10.1093/nar/gkac993
118
Pieper U. Webb B. M. Dong G. Q. Schneidman-Duhovny D. Fan H. Kim S. J. et al (2014). Modbase, a database of annotated comparative protein structure models and associated resources. Nucleic acids Res.42, D336–D346. 10.1093/nar/gkt1144
119
Pierri C. L. De Grassi A. Turi A. (2008). Lattices for ab initio protein structure prediction. Proteins Struct. Funct. Bioinforma.73, 351–361. 10.1002/prot.22070
120
Qian J. Fang D. Lu H. Cao Y. Zhang J. Ding R. et al (2018). Tanshinone iia promotes il2-mediated sw480 colorectal cancer cell apoptosis by triggering inf2-related mitochondrial fission and activating the mst1-hippo pathway. Biomed. and Pharmacother.108, 1658–1669. 10.1016/j.biopha.2018.09.170
121
Qiao Z. Nie W. Vahdat A. Miller III T. F. Anandkumar A. (2022). Dynamic-backbone protein-ligand structure prediction with multiscale generative diffusion models. arXiv Prepr. arXiv:2209.15171.
122
Remmert M. Biegert A. Hauser A. Söding J. (2012). Hhblits: lightning-fast iterative protein sequence searching by hmm-hmm alignment. Nat. methods9, 173–175. 10.1038/nmeth.1818
123
Rohl C. A. Strauss C. E. Misura K. M. Baker D. (2004). Protein structure prediction using rosetta. Methods Enzym. (Elsevier)383, 66–93. 10.1016/S0076-6879(04)83004-0
124
Sahu A. Gaur M. Mahanandia N. C. Subudhi E. Swain R. P. Subudhi B. B. (2023). Identification of core therapeutic targets for monkeypox virus and repurposing potential of drugs against them: an in silico approach. Comput. Biol. Med.161, 106971. 10.1016/j.compbiomed.2023.106971
125
Salwinski L. Miller C. S. Smith A. J. Pettit F. K. Bowie J. U. Eisenberg D. (2004). The database of interacting proteins: 2004 update. Nucleic acids Res.32, D449–D451. 10.1093/nar/gkh086
126
Santhanavijayan A. Naresh Kumar D. Deepak G. (2021). “A semantic-aware strategy for automatic speech recognition incorporating deep learning models,” in Intelligent system design: proceedings of intelligent system design: India 2019 (Springer), 247–254.
127
S Bernardes J. Pedreira C. E. (2013). A review of protein function prediction under machine learning perspective. Recent Pat. Biotechnol.7, 122–141. 10.2174/18722083113079990006
128
Senior A. W. Evans R. Jumper J. Kirkpatrick J. Sifre L. Green T. et al (2019). Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (casp13). Proteins Struct. Funct. Bioinforma.87, 1141–1148. 10.1002/prot.25834
129
Senior A. W. Evans R. Jumper J. Kirkpatrick J. Sifre L. Green T. et al (2020). Improved protein structure prediction using potentials from deep learning. Nature577, 706–710. 10.1038/s41586-019-1923-7
130
Shahamiri S. R. (2021). Speech vision: an end-to-end deep learning-based dysarthric automatic speech recognition system. IEEE Trans. Neural Syst. Rehabilitation Eng.29, 852–861. 10.1109/TNSRE.2021.3076778
131
Sharma A. K. Srivastava R. (2021). Protein secondary structure prediction using character bi-gram embedding and bi-lstm. Curr. Bioinforma.16, 333–338. 10.2174/1574893615999200601122840
132
Shi X. Young S. Cai K. Yang J. Morahan G. (2022). Cancer susceptibility genes: update and systematic perspectives. Innovation3, 100277. 10.1016/j.xinn.2022.100277
133
Sigrist C. J. Cerutti L. De Castro E. Langendijk-Genevaux P. S. Bulliard V. Bairoch A. et al (2010). Prosite, a protein domain database for functional characterization and annotation. Nucleic acids Res.38, D161–D166. 10.1093/nar/gkp885
134
Sigrist C. J. De Castro E. Cerutti L. Cuche B. A. Hulo N. Bridge A. et al (2012). New and continuing developments at prosite. Nucleic acids Res.41, D344–D347. 10.1093/nar/gks1067
135
Sillitoe I. Bordin N. Dawson N. Waman V. P. Ashford P. Scholes H. M. et al (2021). Cath: increased structural coverage of functional space. Nucleic acids Res.49, D266–D273. 10.1093/nar/gkaa1079
136
Sorgen P. L. (2005). How to solve a protein structure by nuclear magnetic resonance—the Connexin43 carboxyl terminal domain. Springer, 948–958.
137
Stahl K. Graziadei A. Dau T. Brock O. Rappsilber J. (2023). Protein structure prediction with in-cell photo-crosslinking mass spectrometry and deep learning. Nat. Biotechnol.41, 1810–1819. 10.1038/s41587-023-01704-z
138
Stern J. Hedelius B. Fisher O. Billings W. M. Della Corte D. (2021). Evaluation of deep neural network prospr for accurate protein distance predictions on casp14 targets. Int. J. Mol. Sci.22, 12835. 10.3390/ijms222312835
139
Sun H. Yang J. Zhang T. Long L.-P. Jia K. Yang G. et al (2013). Using sequence data to infer the antigenicity of influenza virus. MBio4, e00230. 10.1128/mbio.00230–13
140
Szelogowski D. (2023). Deep learning for protein structure prediction: advancements in structural bioinformatics. bioRxiv2023 (04. 26.538026).
141
Szklarczyk D. Gable A. L. Lyon D. Junge A. Wyder S. Huerta-Cepas J. et al (2019). String v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids Res.47, D607–D613. 10.1093/nar/gky1131
142
Szklarczyk D. Gable A. L. Nastou K. C. Lyon D. Kirsch R. Pyysalo S. et al (2021). The string database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic acids Res.49, D605–D612. 10.1093/nar/gkaa1074
143
Tang X. Cai L. Meng Y. Xu J. Lu C. Yang J. (2021). Indicator regularized non-negative matrix factorization method-based drug repurposing for covid-19. Front. Immunol.11, 603615. 10.3389/fimmu.2020.603615
144
Team A. (2020). Computational predictions of protein structures associated with covid-19. Deep. Website K.417, Y453.
145
Thul P. J. Lindskog C. (2018). The human protein atlas: a spatial map of the human proteome. Protein Sci.27, 233–244. 10.1002/pro.3307
146
Timmons P. B. Hewage C. M. (2021). Apptest is a novel protocol for the automatic prediction of peptide tertiary structures. Briefings Bioinforma.22, bbab308. 10.1093/bib/bbab308
147
Trippe B. L. Yim J. Tischer D. Baker D. Broderick T. Barzilay R. et al (2022). Diffusion probabilistic modeling of protein backbones in 3d for the motif-scaffolding problem. arXiv Prepr. arXiv:2206.04119.
148
Ulrich E. L. Akutsu H. Doreleijers J. F. Harano Y. Ioannidis Y. E. Lin J. et al (2007). Biomagresbank. Nucleic acids Res.36, D402–D408. 10.1093/nar/gkm957
149
Wahab A. Tayara H. Xuan Z. Chong K. T. (2021). Dna sequences performs as natural language processing by exploiting deep learning algorithm for the identification of n4-methylcytosine. Sci. Rep.11, 212. 10.1038/s41598-020-80430-x
150
Wang G. Dunbrack Jr R. L. (2003). Pisces: a protein sequence culling server. Bioinformatics19, 1589–1591. 10.1093/bioinformatics/btg224
151
Wang Z. Eickholt J. Cheng J. (2010). Multicom: a multi-level combination approach to protein structure prediction and its assessments in casp8. Bioinformatics26, 882–888. 10.1093/bioinformatics/btq058
152
Waterhouse A. Bertoni M. Bienert S. Studer G. Tauriello G. Gumienny R. et al (2018). Swiss-model: homology modelling of protein structures and complexes. Nucleic acids Res.46, W296–W303. 10.1093/nar/gky427
153
Watson J. L. Juergens D. Bennett N. R. Trippe B. L. Yim J. Eisenach H. E. et al (2022). Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models. BioRxiv12.
154
Watson J. L. Juergens D. Bennett N. R. Trippe B. L. Yim J. Eisenach H. E. et al (2023). De novo design of protein structure and function with rfdiffusion. Nature620, 1089–1100. 10.1038/s41586-023-06415-8
155
Webb B. Sali A. (2016). Comparative protein structure modeling using modeller. Curr. Protoc. Bioinforma.54, 5.6.1–5.6.37. 10.1002/cpbi.3
156
Weißenow K. Heinzinger M. Rost B. (2022). Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction. Structure30, 1169–1177. e4. 10.1016/j.str.2022.05.001
157
Wen L. Li G. Huang T. Geng W. Pei H. Yang J. et al (2022). Single-cell technologies: from research to application. Innovation3, 100342. 10.1016/j.xinn.2022.100342
158
Wong T.-T. Yeh P.-Y. (2019). Reliable accuracy estimates from k-fold cross validation. IEEE Trans. Knowl. Data Eng.32, 1586–1594. 10.1109/tkde.2019.2912815
159
Wu C. H. Huang H. Arminski L. Castro-Alvear J. Chen Y. Hu Z.-Z. et al (2002). The protein information resource: an integrated public resource of functional annotation of proteins. Nucleic acids Res.30, 35–37. 10.1093/nar/30.1.35
160
Wu F. Xu J. (2021). Deep template-based protein structure prediction. PLoS Comput. Biol.17, e1008954. 10.1371/journal.pcbi.1008954
161
Wu K. E. Yang K. K. van den Berg R. Alamdari S. Zou J. Y. Lu A. X. et al (2024). Protein structure generation via folding diffusion. Nat. Commun.15, 1059. 10.1038/s41467-024-45051-2
162
Wu R. Ding F. Wang R. Shen R. Zhang X. Luo S. et al (2022). High-resolution de novo structure prediction from primary sequence. BioRxiv2022 (07. 21.500999).
163
wwPDB consortium (2018). Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res.47, D520–D528. 10.1093/nar/gky949
164
Xia T. Ku W.-S. (2021). “Geometric graph representation learning on protein structure prediction,” in Proceedings of the 27th ACM SIGKDD conference on knowledge discovery and data mining (New York, NY, USA: Association for Computing Machinery), 1873–1883. 10.1145/3447548.3467323
165
Xu D. Zhang Y. (2012). Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinforma.80, 1715–1735. 10.1002/prot.24065
166
Xu J. Cai L. Liao B. Zhu W. Yang J. (2020). Cmf-impute: an accurate imputation tool for single-cell rna-seq data. Bioinformatics36, 5563–5564. 10.1093/bioinformatics/btaa664
167
Xu J. Mcpartlon M. Li J. (2021a). Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell.3, 601–609. 10.1038/s42256-021-00348-5
168
Xu J. Meng Y. Peng L. Cai L. Tang X. Liang Y. et al (2022). Computational drug repositioning using similarity constrained weight regularization matrix factorization: a case of covid-19. J. Cell. Mol. Med.26, 3772–3782. 10.1111/jcmm.17412
169
Xu J. Wang S. (2019). Analysis of distance-based protein structure prediction by deep learning in casp13. Proteins Struct. Funct. Bioinforma.87, 1069–1081. 10.1002/prot.25810
170
Xu Y. Liu X. Cao X. Huang C. Liu E. Qian S. et al (2021b). Artificial intelligence: a powerful paradigm for scientific research. Innovation2, 100179. 10.1016/j.xinn.2021.100179
171
Xue L. C. Rodrigues J. P. Kastritis P. L. Bonvin A. M. Vangone A. (2016). Prodigy: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics32, 3676–3678. 10.1093/bioinformatics/btw514
172
Yang J. Anishchenko I. Park H. Peng Z. Ovchinnikov S. Baker D. (2020a). Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci.117, 1496–1503. 10.1073/pnas.1914677117
173
Yang J. Ju J. Guo L. Ji B. Shi S. Yang Z. et al (2022). Prediction of her2-positive breast cancer recurrence and metastasis risk from histopathological images and clinical information via multimodal deep learning. Comput. Struct. Biotechnol. J.20, 333–342. 10.1016/j.csbj.2021.12.028
174
Yang J. Peng S. Zhang B. Houten S. Schadt E. Zhu J. et al (2020b). Human geroprotector discovery by targeting the converging subnetworks of aging and age-related diseases. Geroscience42, 353–372. 10.1007/s11357-019-00106-x
175
Yang J. Zhang T. Wan X.-F. (2014). Sequence-based antigenic change prediction by a sparse learning method incorporating co-evolutionary information. PloS one9, e106660. 10.1371/journal.pone.0106660
176
Yang L. Wei P. Zhong C. Li X. Tang Y. Y. (2020c). Protein structure prediction based on bn-gru method. Int. J. Wavelets, Multiresolution Inf. Process.18, 2050045. 10.1142/s0219691320500459
177
Yang Q. Syed A. A. S. Fahira A. Shi Y. (2021). Structural analysis of the sars-cov-2 omicron variant proteins. Research.
178
Ye Z. Zhang Y. Liang Y. Lang J. Zhang X. Zang G. et al (2022). Cervical cancer metastasis and recurrence risk prediction based on deep convolutional neural network. Curr. Bioinforma.17, 164–173. 10.2174/1574893616666210708143556
179
Zhang C. Freddolino P. L. Zhang Y. (2017). Cofactor: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic acids Res.45, W291–W299. 10.1093/nar/gkx366
180
Zhang H. Shen Y. (2020). Template-based prediction of protein structure with deep learning. BMC genomics21, 878–879. 10.1186/s12864-020-07249-8
181
Zhang T.-H. Zhang S.-W. (2019). Advances in the prediction of protein subcellular locations with machine learning. Curr. Bioinforma.14, 406–421. 10.2174/1574893614666181217145156
182
Zhang Y. Skolnick J. (2005). Tm-align: a protein structure alignment algorithm based on the tm-score. Nucleic acids Res.33, 2302–2309. 10.1093/nar/gki524
183
Zheng W. Li Y. Zhang C. Zhou X. Pearce R. Bell E. W. et al (2021). Protein structure prediction using deep learning distance and hydrogen-bonding restraints in casp14. Proteins Struct. Funct. Bioinforma.89, 1734–1751. 10.1002/prot.26193
184
Zheng W. Wuyun Q. Li Y. Zhang C. Freddolino P. L. Zhang Y. (2024). Improving deep learning protein monomer and complex structure prediction using deepmsa2 with huge metagenomics data. Nat. Methods21, 279–289. 10.1038/s41592-023-02130-4
185
Zhong W. Gu F. (2020). Predicting local protein 3d structures using clustering deep recurrent neural network. IEEE/ACM Trans. Comput. Biol. Bioinforma.19, 593–604. 10.1109/TCBB.2020.3005972
186
Ziggah Y. Y. Youjian H. Tierra A. R. Laari P. B. (2019). Coordinate transformation between global and local datums based on artificial neural network with K-fold cross-validation: a case study, Ghana. Earth Sci. Res. J.23, 67–77. 10.15446/esrj.v23n1.63860
Summary
Keywords
protein structure prediction, deep learning, large language model, protein structure databases, evaluation index
Citation
Meng Y, Zhang Z, Zhou C, Tang X, Hu X, Tian G, Yang J and Yao Y (2025) Protein structure prediction via deep learning: an in-depth review. Front. Pharmacol. 16:1498662. doi: 10.3389/fphar.2025.1498662
Received
30 September 2024
Accepted
28 February 2025
Published
03 April 2025
Volume
16 - 2025
Edited by
Sajjad Gharaghani, University of Tehran, Iran
Reviewed by
Ciro Leonardo Pierri, University of Bari Aldo Moro, Italy
Minh Nguyen, Bioinformatics Institute (A∗STAR), Singapore
Updates
Copyright
© 2025 Meng, Zhang, Zhou, Tang, Hu, Tian, Yang and Yao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinrong Hu, hxr@wtu.edu.cn; Jialiang Yang, yangjl@geneis.cn; Yuhua Yao, yaoyuhua@hainnu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.