Abstract
Introduction:
Dl-PHPB [potassium 2-(1-hydroxypentyl) benzoate] demonstrates robust neuroprotective effects in preclinical models of Alzheimer’s disease (AD), significantly ameliorating cognitive deficits and pathological hallmarks. However, the underlying mechanism remains largely unclear. The current study primarily focused on elucidating dl-PHPB’s neuroprotective mechanisms and identifying potential targets in preclinical AD models.
Methods:
Comparative proteomic analyses were performed on APP/PS1 mice orally administered either dl-PHPB (30 mg/kg) or vehicle daily for 3 months, alongside vehicle-treated wild-type (WT) non-transgenic littermates as controls. Total proteins were separated using two dimensional difference gel electrophoresis, and differentially expressed protein spots were identified via LC‐MS/MS.
Results and discussion:
Our results revealed 11 altered proteins in the cortex and 10 in the hippocampus between the WT and APP/PS1 groups treated with vehicle. Following dl-PHPB treatment, 12 differentially expressed proteins were identified in the cortex and 9 in the hippocampus of APP/PS1 mice. These proteins are primarily involved in energy metabolism, neuronal structure, protein trafficking, inflammatory and oxidative responses, and amyloid β (Aβ) and Tau processes, among which several proteins were validated as potential therapeutic targets. Notably, the expression levels of cofilin-2 and VDAC1 in APP/PS1 mice were restored to near-normal levels by the treatment with dl-PHPB, memantine, or donepezil, and further clinical validation is required to establish their utility as AD biomarkers for therapeutic efficacy.
1 Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder characterized by neurofibrillary tangles (NFTs), senile plaques (SPs), and neuronal loss. Memantine and Donepezil are two medications commonly employed in clinical practice for the treatment of AD. Memantine, an NMDA receptor antagonist, protects neurons by blocking glutamate-induced excitotoxicity and is prescribed for moderate to severe cases, improving cognition and behavior. Donepezil, a cholinesterase inhibitor, increases acetylcholine levels, enhancing neurotransmission, and is used for mild to moderate cases, slowing memory and functional decline. Current treatments for AD primarily alleviate clinical symptoms but do not halt or delay the progressive deterioration (Breijyeh and Karaman, 2020; Vaz and Silvestre, 2020; Zhang et al., 2021). There is an urgent need to develop novel therapeutic agents to address this debilitating condition.
Dl-PHPB [potassium 2-(1-hydroxypentyl) benzoate], a novel compound synthesized by the Chinese Academy of Medical Sciences, has shown promising results in preclinical studies. Previous research indicated that dl-PHPB ameliorated neurobehavioral deficits in a cerebral ischemia animal model by enhancing cerebral blood flow and reducing infarct volume (Abidi et al., 2023). In neuroblastoma SK-N-SH cells, dl-PHPB mitigates hydrogen peroxide-induced apoptosis through modulation of the protein kinase C (PKC) signaling pathway (Hu et al., 2012). Additionally, dl-PHPB has been demonstrated to significantly prevent neuropathological changes, inhibit oxidative stress, and reduce neuroinflammatory responses (Zhao et al., 2017; Zhao et al., 2013). Dl-PHPB ameliorated memory deficits and reduced oxidative injury in an AD mouse model by activating Nrf2 signaling pathway (Shang et al., 2024). Recently, it has been reported that dl-PHPB improves learning and memory impairments by inhibiting aberrant tau hyperphosphorylation, restoring impaired long-term potentiation (LTP), and protecting hippocampal neurons, synapses, and dystrophic axons in APP/PS1 transgenic mice (Huang et al., 2019; Li et al., 2014; Peng et al., 2014). These findings suggest that dl-PHPB exerts its neuroprotective effects through multiple mechanisms and may represent a promising therapeutic candidate for the treatment of AD.
Proteomics facilitates discovery-driven research rather than hypothesis testing, thereby enabling the information garnered through proteomics to inform subsequent verification studies. The therapeutic impacts of drugs are manifested through direct or indirect protein modifications. Consequently, proteomics has been utilized to elucidate the molecular mechanisms of multi-target drugs, evaluate their efficacy, and screen for potential drug targets (El-Khateeb et al., 2019; Yokota, 2019; Masuda et al., 2021). The current study aimed to explore the molecular mechanisms underlying the neuroprotective effects of dl-PHPB on cognitive function under pathological conditions using pharmacoproteomics. Additionally, we sought to identify potential anti-AD drug targets of dl-PHPB or candidate biomarkers for AD. To accomplish this, we applied a two-dimensional difference gel electrophoresis approach coupled with mass spectrometry to comprehensively profile differentially expressed proteins in brain tissues affected by dl-PHPB treatment. This unbiased discovery, corroborated by Western blotting validation, facilitated the identification of dl-PHPB-induced protein changes and the exploration of potential drug targets, offering novel insights into the development of anti-AD therapies.
2 Materials and methods
2.1 Animals and dl-PHPB administration
Dl-PHPB (purity>98%) was synthesized by the Chinese Academy of Medical Sciences (Figure 1). Memantine and donepezil were procured from Sigma-Aldrich and were individually dissolved in distilled water. APP/PS1 double-transgenic mice (strain name: B6C3-Tg (APPswe, PSEN1dE9) 85Dbo/J) were acquired from the Jackson Laboratory. These mice express a chimeric mouse/human amyloid precursor protein (APP) containing the KM670/671NL Swedish mutations and a mutant human presenilin 1 (PS1) carrying the exon 9-deleted variant, both under the control of mouse prion promoter elements, which direct transgene expression primarily to central nervous system neurons (Jankowsky et al., 2001). The two transgenes cosegregate in these mice. The animals were housed in an environmentally controlled room at 23°C ± 1°C with a 12-h light/dark cycle and had free access to water and food. Mice were stratified by weight and randomized into treatment/control groups using a computer-generated sequence. Treatments were administered by blinded personnel via coded syringes. Behavioral tests were scored by an independent observer unaware of group assignments. Sample size was determined using power analysis (α = 0.05, power = 80%, effect size = 1.5 from prior data, and an additional 2–4 rats were included per group to account for attrition. Through the aforementioned design, potential biases are rigorously controlled, thereby strengthening the credibility and reproducibility of the findings while aligning with ARRIVE guidelines for animal research reporting. All experimental procedures were conducted in accordance with the guidelines of the Experimental Animal Center of Ludong University. APP/PS1 transgenic (Tg) mice and age-matched wild-type (WT) mice were randomly assigned to five groups of 12–16 mice each: untreated WT control (WT + H2O; n = 16), untreated APP/PS1 Tg model (Tg + H2O; n = 14), dl-PHPB-treated Tg group (Tg + dl-PHPB; n = 15), memantine-treated Tg group (Tg + memantine; n = 12), and donepezil-treated Tg group (Tg + donepezil; n = 14). The untreated WT control and Tg model groups received distilled water only. Oral gavage administration began when the mice were 9 months old and continued for 12 weeks. Body weight was monitored biweekly.
FIGURE 1

Chemical structure of dl-PHPB.
2.2 Preparation of brain homogenate
The mice were euthanized by CO2 inhalation following the completion of behavioral testing. The skin was incised along the midline of the cranium to expose the skull. Curved scissors were used to cut along the cranial sutures from the foramen magnum to the nasal direction, followed by careful removal of the skull. The frontal cortex and hippocampus were microdissected according to the mouse brain stereotaxic atlas and standardized protocols from Nature Protocols 2016. These cortical and hippocampal tissues were immediately snap-frozen in liquid nitrogen and stored at −80°C until further analysis. The tissues were homogenized and processed for protein extraction. Each sample was suspended in 2DE-specific lysis buffer [7 M urea, 2 M thiourea, 4% CHAPS, 1% dithiothreitol (DTT), 0.5% IPG buffer (pH 3–10 NL, GE Healthcare), 1% Protease Inhibitor Mix (Roche), 30 mM Tris-HCl, pH 8.5] and subjected to sonication for 1 min with cycles of 3 s on and 3 s off using a Fisher 550 Sonic Dismembrator. The samples were then centrifuged at 20,000 g at 4°C for 60 min to remove cellular debris. The supernatants were ultrafiltered at 15,000 g for 30 min to eliminate salts and other impurities and were resuspended in 2DE-specific lysis buffer. The resulting protein solutions were collected and stored at −80°C until use. Protein concentrations were quantified using a 2-D Quant Kit (GE Healthcare, United States) according to the manufacturer’s instructions, and separate proteomic analyses were conducted for the cortex and hippocampus.
2.3 Step-down passive avoidance test
The impact of dl-PHPB on memory impairment in mice was investigated using a step-down passive avoidance test as previously described (Li et al., 2014; Peng et al., 2014). To minimize locomotor-related confounders, standardized speed measurements were performed on all mice before grouping, and animals with notable locomotor variability were excluded. The apparatus comprised a wooden box measuring 15 × 15 × 30 cm. The floor of the box consisted of parallel stainless steel bars, with a wooden insulating platform (4 × 4 × 4 cm) positioned at the center of the grid floor. When a mouse was placed on the grid floor, it received an inescapable intermittent electric shock (1 s, 36 V), while the platform served as a refuge. The experiment spanned 3 days. On the first day, the mouse was acclimated to the box for 5 min without any electric shock. On the second day, acquisition trials were conducted, during which an electric foot shock (36 V) was administered to the animal’s paws through the grid floor. Upon receiving the shock, the mouse sought refuge on the platform. The training session lasted 3 min. Twenty-four hours after the acquisition trial, the retention test was performed. The error times (frequency of the mouse stepping down from the platform and receiving a shock) and the escape latency (time taken by the mouse to step down from the platform onto the grid floor) were recorded as indicators of retention. If the mouse did not step down to the grid floor within 3 min, a ceiling score of 180 s was assigned. DigBehv-SD (Shanghai Jiliang Software Technology) was used to record and analyze them in the avoidance test.
2.4 2DE analysis
Protein samples were loaded onto nonlinear IPG strips (18 cm, pH 3–10 NL, GE Healthcare). Isoelectric focusing was conducted with the following parameters: 50 V for 12 h linearly, 200 V for 1 h linearly, 500 V for 1 h linearly, 1,000 V for 1 h linearly, 8,000 V for 1 h rapidly, and finally reaching a total of 60,000 Vh at 8,000 V. For SDS‒PAGE, the strips were placed on top of a 12.5% SDS‒polyacrylamide gel, and electrophoresis was conducted with the following parameters: 2 W/gel constant power for 1 h, followed by 15 W/gel constant power until the bromophenol blue front reached the bottom of the gel.
2.5 Gel staining and image analysis
After performing 2-DE, the analytical gels were silver-stained using the PlusOne Silver Staining Kit (GE Healthcare). The silver-stained gels were then scanned with an ImageScanner III (GE Healthcare). Image analysis was conducted using ImageMaster 2D Platinum software version 7.0 (GE Healthcare). Protein spots were identified based on the following criteria: a two-fold greater difference in protein expression between the two groups (ratio>2). Statistical significance was assessed using the Student’s t-test with a threshold of FDR-adjusted p < 0.05. The symbols “+” and “−” denote upregulation and downregulation, respectively.
2.6 Protein identification by nano-HPLC-ESI-MS/MS
The differentially expressed protein spots were manually excised, reduced, alkylated, and subsequently digested using sequence-grade modified trypsin (Promega Corporation, United States). LC‒MS/MS analysis was conducted using a Surveyor MS Plus HPLC system coupled to a ThermoFinnigan LTQ linear ion trap mass spectrometer (ThermoFisher Corporation, San Jose, CA). The resulting tryptic peptides were loaded onto a trap column (300SB-C18, 5 × 0.3 mm, 5 μm particle size) (Agilent Technologies, Santa Clara, CA) connected via a zero-dead-volume union to a self-packed analytical column (C18, 100 × 0.1 mm, 3 μm particle size) (SunChrom, Germany). Peptides were eluted with a linear acetonitrile gradient at a flow rate of 500 nL/min. Data-dependent scanning was employed to select the five most abundant ions from a full-scan mass spectrum for fragmentation by collision-induced dissociation. MS data were analyzed using SEQUEST against the NCBI Reference Sequence mouse protein database, and the results were filtered, sorted, and displayed using Bioworks 3.2. Protein lists were filtered based on the following criteria: peptide Xcorr values >1.90 (for +1 charge), >2.75 (for +2 charge), >3.75 (for +3 charge), peptide ΔCN >0.1, and protein probabilities <0.001. Each identified protein required at least two unique peptides.
2.7 Western blotting
Seven proteins—pyruvate dehydrogenase E1 component subunit α (PDHE1α), voltage-dependent anion-selective channel protein 1 (VDAC1), dihydropyrimidinase-related protein 2 (DRP-2), cofilin-2, peroxiredoxin-6, peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin 1), and cathepsin-B—were validated by Western blot analysis. Equal amounts of protein (40 μg) were separated on a 10% polyacrylamide gel, transferred onto a polyvinylidene difluoride (PVDF) membrane, and incubated with primary antibodies overnight. The following antibodies were used: mouse anti-β-actin (1:10,000; Sigma), rabbit anti-PDHE1α (1:200; Santa Cruz Biotechnology), rabbit anti-VDAC1 (1:1,000; Abcam), rabbit anti-DRP-2 (1:1,000; Cell Signaling Technology), mouse anti-cofilin-2 (1:200; Santa Cruz Biotechnology), rabbit anti-peroxiredoxin-6 (1:1,000; Abcam), rabbit anti-Pin 1 (1:200; Santa Cruz Biotechnology), and rabbit anti-cathepsin B (1:200; Santa Cruz Biotechnology). The signals were detected using an enhanced chemiluminescence (ECL) system (LAS-3000 Luminescent Image Analyzer, Fujifilm) and quantified using Quantity One software. The values were normalized to the intensity of β-actin.
2.8 Statistical analysis
The normalized volume of each spot in proteomics was compared between the two groups using Student’s t-test. To control family-wise error rate, Benjamini–Hochberg false discovery rate (FDR) correction was applied to all pairwise comparisons (q < 0.05). Expression changes were considered biologically significant only when meeting both criteria: (a) FDR-adjusted p < 0.05 and (b) fold-change>2. In Western blot analysis and behavioral testing results, one-way analysis of variance (ANOVA) was conducted using SPSS 13.0 software. Data were presented as means±SEMs, and statistical significance was set at p < 0.05.
3 Results
3.1 Dl-PHPB ameliorated memory deficits in APP/PS1 transgenic mice
In this study, we assessed whether dl-PHPB could mitigate behavioral deficits in APP/PS1 mice using the step-down passive avoidance test. Following a 3-month oral administration of dl-PHPB, memantine, or donepezil, behavioral tests were conducted. Escape latency and error times were recorded during the retention test session. A significant difference in escape latency and error times (p < 0.01) was observed between vehicle-treated WT and vehicle-treated Tg groups, indicating that APP/PS1 mice exhibited pronounced memory deficits. Specifically, the escape latency results showed that vehicle-treated Tg mice spent significantly less time on the platform compared to vehicle-treated WT mice (Tg: 16.9 ± 3.8 s; WT: 130.1 ± 15.6 s; Figure 2A). Treatment with dl-PHPB, memantine, and donepezil significantly prolonged escape latency (Tg + dl-PHPB: 139.7 ± 12.3 s; Tg + Memantine: 125.3 ± 21.4 s; Tg + Donepezil: 84.5 ± 18.7 s; Figure 2A) in APP/PS1 mice. Additionally, the vehicle-treated Tg group made more errors (Tg: 4.0 ± 0.5; WT: 1.0 ± 0.3; Figure 2B) compared to the vehicle-treated WT group, and long-term oral treatment with dl-PHPB, memantine, and donepezil reduced the error frequency (Tg + dl-PHPB: 0.8 ± 0.3; Tg + Memantine: 1.3 ± 0.5; Tg + Donepezil: 1.6 ± 0.3; Figure 2B) in APP/PS1 mice.
FIGURE 2
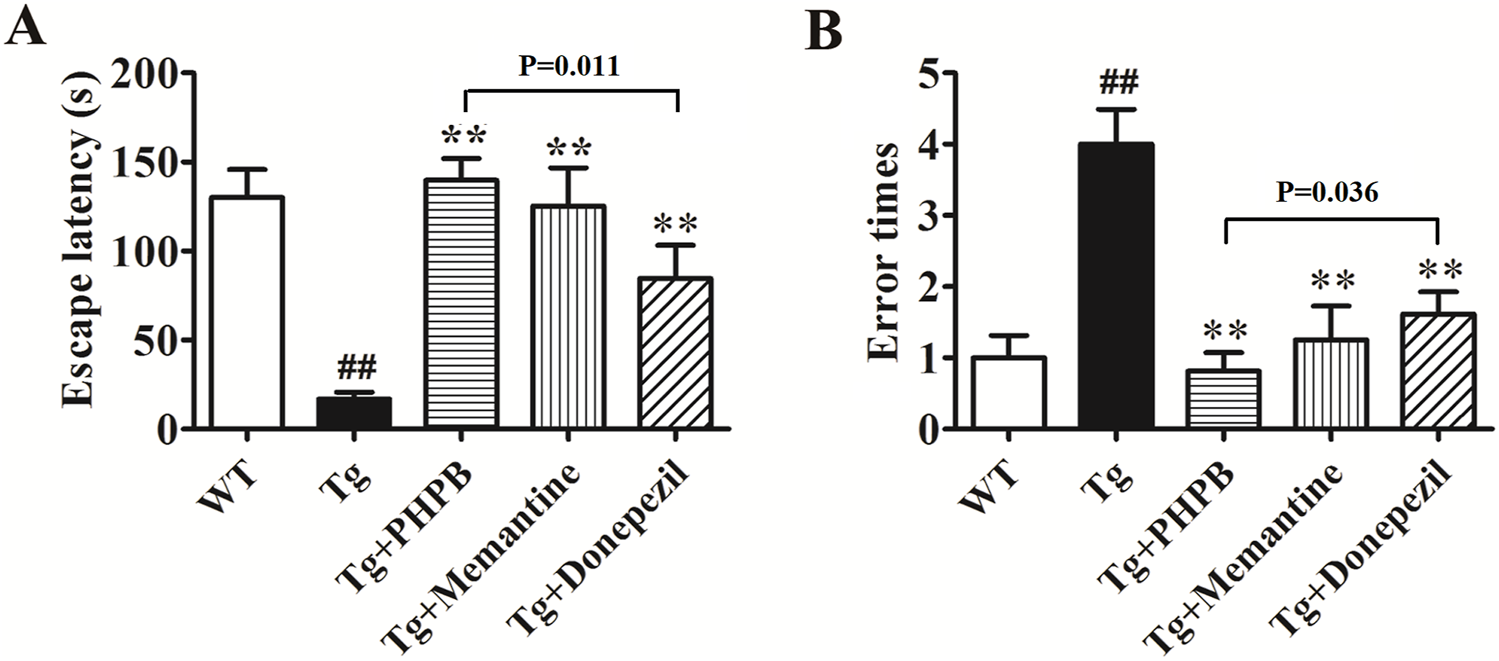
Long-term dl-PHPB treatment ameliorated spatial learning and memory deficits in APP/PS1 mice. The latency (A) and error times (B) were evaluated using the step-down passive avoidance test. Compared with vehicle treatment, dl-PHPB, memantine, and donepezil treatments significantly attenuated learning and memory deficits in APP/PS1 mice. Statistical analysis: Between-group differences were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Data were presented as mean ± SEM (n = 12–16). ##p < 0.01 vs. vehicle-treated WT group; **p < 0.01 vs. vehicle-treated APP/PS1 group.
3.2 Pharmacoproteomic analysis of dl-PHPB in APP/PS1 mice
Proteomic analyses using 2DE, in-gel trypsin digestion, and LC‒MS/MS were conducted on homogenates from the cortex and hippocampus of WT mice provided with drinking water and APP/PS1 mice given either drinking water or water supplemented with dl-PHPB for a period of 3 months. The comparisons included: (i) APP/PS1+H2O vs. WT + H2O and (ii) APP/PS1+dl-PHPB vs. APP/PS1+H2O. Protein spots that exhibited differential expression and were identified through LC‒MS/MS are labeled in Figure 3 and detailed in Tables 1–4. Identified proteins were classified based on their functions, which primarily encompassed the regulation of energy metabolism, neuronal structure and protein trafficking, inflammatory and oxidative responses, and Aβ and Tau processes (Table 5).
FIGURE 3
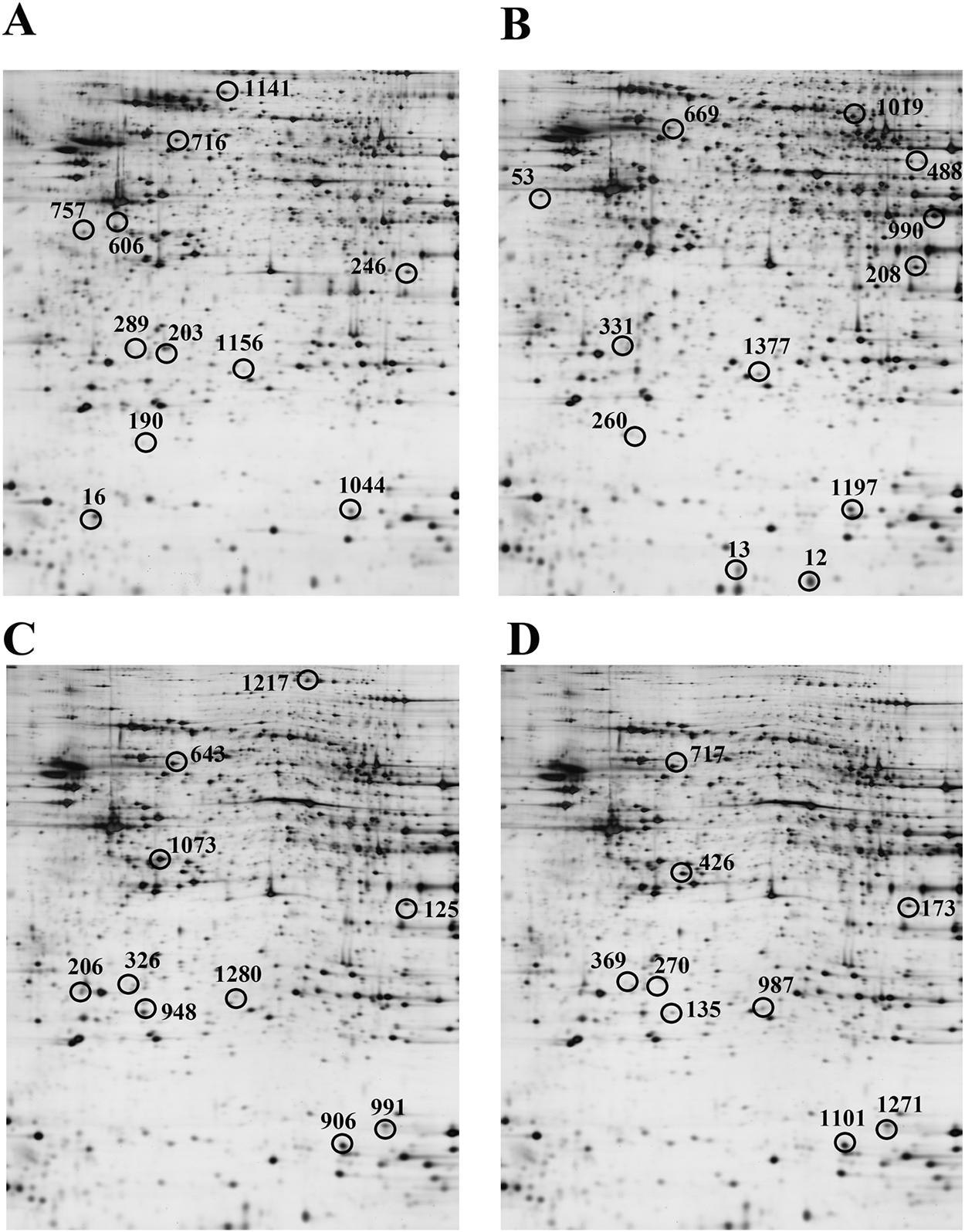
Representative 2D gel images of differentially expressed proteins in the cortex (A,B) and hippocampus (C,D) across groups. A and C, APP/PS1+H2O vs. WT + H2O. B and D, APP/PS1+dl-PHPB vs. APP/PS1+H2O. Approximately 120 µg of protein was loaded per gel to detect protein expression. n = 10. Differentially expressed protein spots (ratio>2, q < 0.05) were labeled in the images. Detailed identification information was shown in Tables 1–4, respectively.
TABLE 1
| Spot No.a | Protein identity | Accession GI no. | Sequence Coverage (%) | Score | pIb | MWc | Variation ratiod |
|---|---|---|---|---|---|---|---|
| 16 | astrocytic phosphoprotein FEA-15 isoform 2 | 21426847 | 36.6 | 40.2 | 4.80 | 15054.0 | −2.01 |
| 190 | peroxiredoxin-6 | 6671549 | 56.1 | 110.3 | 5.96 | 24826.4 | +2.66 |
| 203 | malate dehydrogenase | 254540027 | 30.9 | 130.2 | 6.16 | 36510.9 | −2.43 |
| 246 | pyruvate dehydrogenase E1 component subunit α (PDHE1α) | 6679261 | 25.7 | 80.3 | 8.49 | 43198.6 | −2.01 |
| 289 | cathepsin B | 309152 | 28.8 | 70.2 | 5.51 | 37279.6 | +3.22 |
| 606 | glial fibrillary acidic protein isoform 1 | 196115327 | 47.7 | 288.3 | 5.31 | 49364.2 | +3.34 |
| glial fibrillary acidic protein isoform 2 | 84000448 | 46.5 | 288.3 | 5.14 | 49899.7 | ||
| 716 | dihydropyrimidinase-related protein 2 (DRP-2) | 40254595 | 23.4 | 60.2 | 5.93 | 62278.1 | +2.12 |
| 757 | γ-enolase | 7305027 | 27.6 | 90.3 | 4.84 | 47296.3 | −2.27 |
| 1044 | peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin 1) | 20139259 | 57.9 | 180.2 | 7.97 | 17971.2 | −2.75 |
| 1141 | ezrin | 83921618 | 12.6 | 80.2 | 5.76 | 69406.3 | +2.16 |
| 1156 | voltage-dependent anion-selective channel protein 1 (VDAC1) | 6755963 | 16.8 | 90.3 | 8.86 | 30755.3 | +2.78 |
Identification data of cortical proteins significantly altered in APP/PS1 vs. WT mice (H2O-treated).
Spot numbers correspond to those shown in Figure 3A.
pI, isoelectric point.
MW, molecular mass.
“+” and “–” represent “upregulation” and “downregulation”, respectively. FDR-adjusted p < 0.05.
TABLE 2
| Spot No.a | Protein identity | Accession GI no. | Sequence Coverage (%) | Score | pIb | MWc | Variation ratiod |
|---|---|---|---|---|---|---|---|
| 125 | pyruvate dehydrogenase E1 component subunit α (PDHE1α) | 6,679,261 | 23.9 | 60.2 | 8.49 | 43198.6 | −2.21 |
| 206 | triosephosphate isomerase | 226958349 | 26.7 | 100.3 | 4.74 | 32192.5 | −2.03 |
| 326 | cathepsin B | 309152 | 40.5 | 140.2 | 5.51 | 37279.6 | +3.69 |
| 643 | dihydropyrimidinase-related protein 2 (DRP-2) | 40254595 | 29.1 | 70.3 | 5.93 | 62278.1 | +2.32 |
| 906 | peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin 1) | 20139259 | 46.6 | 120.2 | 7.97 | 17971.2 | −2.45 |
| 948 | pyridoxal kinase | 26006861 | 12.9 | 30.2 | 5.86 | 35015.0 | −2.56 |
| 991 | cofilin-2 | 6671746 | 18.9 | 30.2 | 8.12 | 18709.5 | +2.49 |
| 1073 | D-lactate dehydrogenase, mitochondrial | 23506790 | 13.5 | 40.2 | 6.16 | 51847.6 | −2.06 |
| 1217 | dynamin-1 | 116063570 | 30.3 | 180.3 | 7.61 | 97283.7 | −2.77 |
| 1280 | voltage-dependent anion-selective channel protein 1 (VDAC1) | 6755963 | 61.0 | 328.3 | 8.86 | 30755.3 | +3.82 |
Identification data of hippocampal proteins significantly altered in APP/PS1 vs. WT mice (H2O-treated).
Spot numbers correspond to those shown in Figure 3C.
pI, isoelectric point.
MW, molecular mass.
“+” and “–” represent “upregulation” and “downregulation”, respectively. FDR-adjusted p < 0.05.
TABLE 3
| Spot No.a | Protein identity | Accession GI no. | Sequence Coverage (%) | Score | pIb | MWc | Variation ratiod |
|---|---|---|---|---|---|---|---|
| 12 | histidine triad nucleotide-binding protein 1 | 33468857 | 20.4 | 20.2 | 6.41 | 13776.8 | +2.01 |
| 13 | fatty acid-binding protein | 6754450 | 29.2 | 38.2 | 6.15 | 15137.3 | +2.24 |
| 53 | tubulin | 34740335 | 6.65 | 20.2 | 4.81 | 50151.3 | −2.17 |
| 208 | pyruvate dehydrogenase E1 component subunit α (PDHE1α) | 6679261 | 28.1 | 100.3 | 8.49 | 43198.6 | +2.68 |
| 260 | peroxiredoxin-6 | 6671549 | 40.7 | 90.3 | 5.96 | 24826.4 | +2.33 |
| 331 | cathepsin B | 309152 | 14.3 | 30.2 | 5.51 | 37279.6 | −2.10 |
| 488 | isocitrate dehydrogenase | 1236984 | 15.5 | 40.2 | 8.89 | 58749.1 | +2.02 |
| 669 | dihydropyrimidinase-related protein 2 (DRP-2) | 40254595 | 38.6 | 150.2 | 5.93 | 62278.1 | −2.24 |
| 990 | dihydrolipoamide succinyltransferase, mitochondrial | 21313536 | 26.7 | 80.2 | 9.10 | 48994.5 | +2.66 |
| 1019 | dihydropyrimidinase-related protein 5 | 12746424 | 22.8 | 80.2 | 6.65 | 61516.1 | +2.12 |
| 1197 | peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin 1) | 20139259 | 38.4 | 108.3 | 7.97 | 17971.2 | +3.01 |
| 1377 | voltage-dependent anion-selective channel protein 1 (VDAC1) | 6755963 | 36.5 | 180.3 | 8.86 | 30755.3 | −3.08 |
Identification data of cortical proteins significantly altered by dl-PHPB vs. H2O in APP/PS1 mice.
Spot numbers correspond to those shown in Figure 3B.
pI, isoelectric point.
MW, molecular mass.
“+” and “–” represent “upregulation” and “downregulation”, respectively. FDR-adjusted p < 0.05.
TABLE 4
| Spot No.a | Protein identity | Accession GI no. | Sequence Coverage (%) | Score | pIb | MWc | Variation ratiod |
|---|---|---|---|---|---|---|---|
| 135 | carbonic anhydrase 2 | 157951596 | 34.9 | 50.3 | 6.54 | 29032.3 | −2.25 |
| 173 | pyruvate dehydrogenase E1 component subunit α (PDHE1α) | 6679261 | 20.4 | 50.3 | 8.49 | 43198.6 | +2.41 |
| 270 | malate dehydrogenase | 254540027 | 21.7 | 90.2 | 6.16 | 36510.9 | +2.07 |
| 369 | cathepsin B | 309152 | 34.3 | 160.2 | 5.51 | 37279.6 | −3.13 |
| 426 | α-enolase | 158853992 | 27.6 | 88.3 | 6.70 | 47618.1 | +2.54 |
| 717 | dihydropyrimidinase-related protein 2 (DRP-2) | 40254595 | 33.5 | 110.2 | 5.93 | 62278.1 | −2.21 |
| 987 | voltage-dependent anion-selective channel protein 1 (VDAC1) | 6755963 | 50.2 | 252.4 | 8.86 | 30755.3 | −2.52 |
| 1101 | peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin 1) | 20139259 | 54.3 | 150.2 | 7.97 | 17971.2 | +2.19 |
| 1271 | cofilin-2 | 6671746 | 18.9 | 20.2 | 8.12 | 18709.5 | −2.96 |
Identification data of hippocampal proteins significantly altered by dl-PHPB vs. H2O in APP/PS1 mice.
Spot numbers correspond to those shown in Figure 3D.
pI, isoelectric point.
MW, molecular mass.
“+” and “–” represent “upregulation” and “downregulation”, respectively. FDR-adjusted p < 0.05.
TABLE 5
| Function | Identified proteins |
|---|---|
| Energy metabolism | VDAC1; PDHE1α; D-lactate dehychgenase, mitochondrial; malate dehydrogenase; α-enolase; γ-enolase; triosephosphate isomerase; isocitrate dehydrogenase; dihydrolipoamide succinyltransferase, mitochondrial |
| Neuronal structure and protein trafficking | DRP-2; cofilin-2; tubulin; dihydropyrimidinase-related protein 5; dynamin-1; histidine triad nucleotide-binding protein 1; carbonic anhydrase 2; fatty acid-binding protein |
| Inflammatory and oxidative response | peroxiredoxin-6; stress-70 protein; ezrin, astrocytic phosphoprotein FEA-15 isoform 2; glial fibrillary acidic protein |
| Aβ and Tau process | Pin 1; cathepsin B |
| Others | pyridoxal kinase |
Functional roles attributed to identified proteins.
We identified 11 differentially expressed proteins in the cortex and 10 in the hippocampus between vehicle-treated WT and vehicle-treated APP/PS1 mice (Tables 1, 2). In the cortex, 6 proteins (peroxiredoxin-6, cathepsin B, glial fibrillary acidic protein, DRP-2, ezrin, and VDAC1 showed increased expression, while 5 proteins (astrocytic phosphoprotein FEA-15 isoform 2, malate dehydrogenase, PDHE1α, γ-enolase, and Pin 1) exhibited decreased expression. In the hippocampus of APP/PS1 mice, compared to WT mice, 4 proteins (cathepsin B, DRP-2, cofilin-2, and VDAC1) were upregulated, whereas 6 proteins (PDHE1α, triosephosphate isomerase, Pin 1, pyridoxal kinase, D-lactate dehydrogenase, and mitochondrial dynamin-1) were downregulated.
Proteomic analyses revealed differential expression of 12 cortical and 9 hippocampal proteins between dl-PHPB- and vehicle-treated APP/PS1 mice. In the cortex, the expression levels of histidine triad nucleotide-binding protein 1, fatty acid-binding protein, PDHE1α, peroxiredoxin-6, isocitrate dehydrogenase, dihydrolipoamide succinyltransferase, dihydropyrimidinase-related protein 5, and Pin 1 were significantly elevated, while those of tubulin, cathepsin B, DRP-2, and VDAC1 were significantly reduced in dl-PHPB-treated APP/PS1 Tg mice compared to vehicle-treated APP/PS1 Tg mice. In the hippocampus, four proteins (PDHE1α, malate dehydrogenase, α-enolase, and Pin 1) showed significant increases, whereas five proteins (carbonic anhydrase 2, cathepsin B, DRP-2, VDAC1, and cofilin-2) exhibited decreased levels following dl-PHPB treatment.
3.3 Western blotting analysis
Western blotting analysis was conducted to validate alterations in the protein expression of PDHE1α, VDAC1, DRP-2, cofilin-2, peroxiredoxin-6, Pin 1, and cathepsin B in APP/PS1 mice following long-term dl-PHPB treatment. As illustrated in Figure 4, the levels of VDAC1, DRP-2, and cathepsin B were significantly elevated in the cortex and hippocampus of vehicle-treated APP/PS1 mice compared to WT controls. Conversely, dl-PHPB treatment markedly reduced the expression of these proteins in APP/PS1 mice relative to vehicle-treated APP/PS1 mice (Figures 4B,C,G). Similarly, dl-PHPB treatment significantly decreased hippocampal cofilin-2 levels but had no significant effect on cortical cofilin-2 levels in APP/PS1 mice (Figure 4D). Compared to WT mice, a substantial decrease in PDHE1α and Pin 1 expression was observed in the cortex and hippocampus of APP/PS1 mice, and this reduction was significantly reversed by dl-PHPB treatment (Figures 4A,F). Notably, the expression levels of peroxiredoxin-6 were significantly higher in the cortex and hippocampus of APP/PS1 mice compared to WT controls, and dl-PHPB treatment further increased this expression (Figure 4E). The Western blot results were generally consistent with the proteomic analysis, thereby validating the reliability of the proteomics method.
FIGURE 4
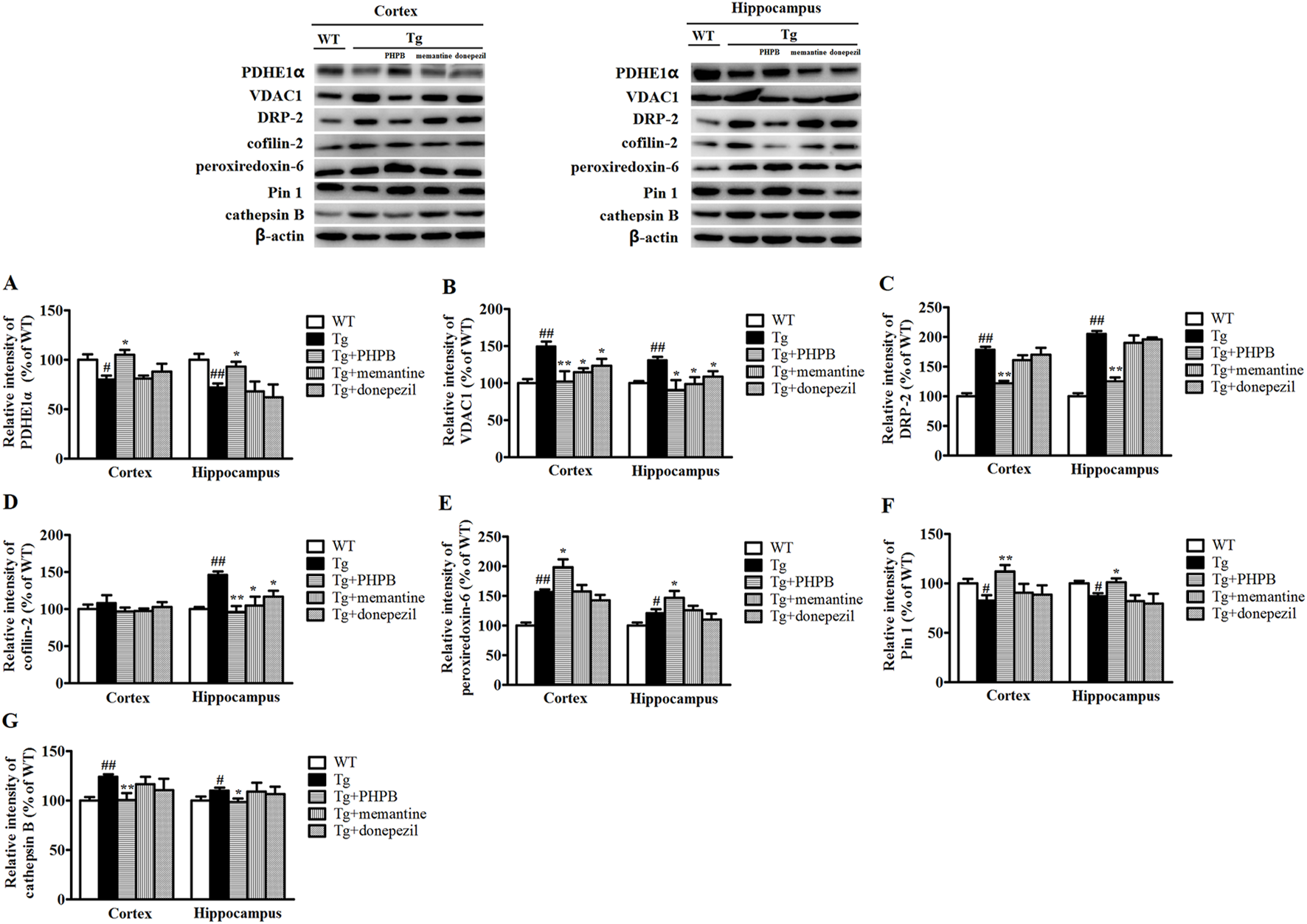
Western blot analysis of several proteins with altered abundance in the cortex or hippocampus of WT + H2O, APP/PS1+H2O, APP/PS1+dl-PHPB, APP/PS1+memantine, and APP/PS1+donepezil mice. The changes in PDHE1α (A), VDAC1 (B), DRP-2 (C), cofilin-2 (D), peroxiredoxin-6 (E), Pin 1 (F), and cathepsin B (G) detected by Western blotting were consistent with the proteomic data. The Western blot images correspond to: lane 1, WT mice treated with normal drinking water; lane 2, APP/PS1 mice treated with normal drinking water; lane 3, APP/PS1 mice treated with dl-PHPB; lane 4, APP/PS1 mice treated with memantine; and lane 5, APP/PS1 mice treated with donepezil. Quantified results were normalized to β-actin expression. Values were expressed as percentages relative to vehicle-treated WT mice (WT + H2O) (set to 100%) and presented as the group mean ± SEM. Statistical analysis: Between-group differences were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. n = 8–10 mice per group. #p < 0.05, ##p < 0.01 versus the vehicle-treated WT group. *p < 0.05, **p < 0.01 versus the vehicle-treated APP/PS1 group.
To examine the impact of positive AD drugs on differentially expressed proteins, mice in the treatment groups were orally administered memantine and donepezil, respectively. Quantitative analysis of Western blot bands revealed a significant increase in VDAC1 levels in both the cortex and hippocampus, as well as a notable rise in cofilin-2 levels specifically in the hippocampus. These increases were markedly attenuated by memantine or donepezil treatments (Figures 4B,D). Furthermore, Western blot results showed no statistically significant alterations in the protein levels of PDHE1α, DRP-2, peroxiredoxin-6, Pin-1, and cathepsin B following either memantine or donepezil treatment, which may be attributed to their single-target mechanisms (Figures 4A,C,E–G).
4 Discussion
The hallmark of AD is cognitive decline associated with Aβ deposition. Extensive research has demonstrated that APP/PS1 transgenic mice exhibit robust amyloid plaque pathology akin to that observed in AD and develop age-related memory deficits (Trinchese et al., 2004). The step-down passive avoidance task, a fear-motivated test, is commonly employed to assess learning and memory in rodent models of central nervous system disorders. Notably, this test revealed that oral administration of dl-PHPB for 3 months significantly ameliorated memory impairment in APP/PS1 mice. These protective effects on learning and memory are consistent with our previous findings (Li et al., 2014; Peng et al., 2014).
In the present study, we conducted a comprehensive proteomic analysis of the cortex and hippocampus in vehicle-treated WT, vehicle-treated, and dl-PHPB-treated APP/PS1 mice. Analysis of over 1,300 spots on 2DE gels identified 11 differentially expressed spots in the cortex and 10 in the hippocampus between the WT + H2O and APP/PS1+H2O groups; 12 spots were different in the cortex and 9 in the hippocampus between the APP/PS1+dl-PHPB and APP/PS1+H2O groups. Western blot analysis confirmed the proteomic findings. The identified proteins play roles in energy metabolism, neuronal structure, protein trafficking, inflammatory and oxidative responses, as well as Aβ and tau processes.
Disturbances in energy metabolism have been well-documented in AD brains (Hoyer, 2004). This is substantiated by a concurrent reduction in glucose utilization and the expression of glycolytic enzymes (Hoyer et al., 1991). In our present investigation, we observed a marked downregulation of key metabolic proteins, including PDHE1α, malate dehydrogenase, D-lactate dehydrogenase, γ-enolase, and triosephosphate isomerase, in APP/PS1 mice relative to WT mice. These reductions imply a potential impairment in energy metabolism, which could compromise the cognitive function of the brain, given its substantial reliance on glucose for ATP production. PDHE1α is an essential component of pyruvate dehydrogenase, governing its activity (Korotchkina et al., 1995). As the rate-limiting enzyme in oxygen-dependent energy production, pyruvate dehydrogenase catalyzes the conversion of pyruvate to acetyl coenzyme A, bridging cytosolic glycolysis to the mitochondrial citric acid cycle (Pithukpakorn, 2005). Consequently, PDHE1α plays a pivotal role in mitochondrial energetics. Research has indicated a decline in PDHE1α protein levels and activity in the early stages of AD model mice and in rats with traumatic brain injury (Sharma et al., 2009; Yao et al., 2009; Xing et al., 2009). In AD neurons, the deficiency of PDHE1α results in diminished glucose aerobic oxidation, leading to reduced oxidative phosphorylation and energy failure (Stacpoole, 2012). Furthermore, the reduction in PDHE1α diminishes the production of acetyl coenzyme A, thereby decreasing the synthesis of the neurotransmitter acetylcholine (Ronowska et al., 2018). Our findings suggest that treatment with dl-PHPB significantly upregulates PDHE1α and other glucose-metabolizing enzymes (α-enolase, malate dehydrogenase, isocitrate dehydrogenase, and dihydrolipoyl succinyltransferase), indicating that dl-PHPB may enhance energy metabolism in APP/PS1 mouse brains, ultimately ameliorating cognitive function.
In addition, our findings indicate that the expression of VDAC1 was markedly elevated in APP/PS1 mice compared to WT mice, consistent with our prior research (Sun et al., 2014). Previous studies have demonstrated that VDAC1 expression is significantly upregulated in the cerebral cortices of 6-, 12-, and 24-month-old APP transgenic mice relative to age-matched WT controls (Manczak and Reddy, 2012). Furthermore, elevated VDAC1 levels have been documented in the affected brain regions of AD postmortem samples (Yoo et al., 2001; Manczak and Reddy, 2012) and in the serum of AD patients (Sun et al., 2014). VDAC1 is recognized as a key regulator of mitochondrial function (Lemasters and Holmuhamedov, 2006; Choudhary et al., 2014; Shoshan-Barmatz et al., 2018), and plays a critical role in various cellular processes, particularly mitochondrial energy metabolism. It is hypothesized that elevated VDAC1 directly interacts with Aβ and phosphorylated tau in neurons, leading to the obstruction of mitochondrial pores and disruption of metabolite transport, which ultimately results in mitochondrial dysfunction and impaired ATP supply to nerve terminals (Manczak and Reddy, 2012). In the present study, we observed that treatment with dl-PHPB, memantine, or donepezil significantly reduced VDAC1 levels in the cortex and hippocampus of APP/PS1 mice compared to vehicle-treated APP/PS1 mice. By decreasing VDAC1 levels, normal mitochondrial pore dynamics can be restored, thereby enhancing energy production.
Neuronal and synaptic degeneration are key pathological features of AD; therefore, proteins involved in neuronal structure and trafficking likely play crucial roles in AD pathogenesis. DRP-2, also known as collapsin response mediator protein 2 (CRMP-2), is implicated in neurite growth, guidance, and neuronal development and polarity (O'Donnell et al., 2009; Ip et al., 2014). Our findings demonstrated that DRP-2 expression was significantly elevated in the cortex and hippocampus of vehicle-treated APP/PS1 mice compared to vehicle-treated WT mice. DRP-2 has been observed to colocalize with neurofibrillary tangles in the cortex of AD patients (Yoshida et al., 1998) and can be phosphorylated by cyclin-dependent kinase 5 (CDK5), glycogen synthase kinase 3β (GSK3β), Rho kinase, and calmodulin-dependent protein kinase II (CaMKII), which inhibit axonal growth and vesicle trafficking (Gu et al., 2000; Yoshimura et al., 2005; Schmidt and Strittmatter, 2007; Hou et al., 2009). Additionally, increased phosphorylation of DRP-2 has been reported to precede AD pathology (Cole et al., 2007). In our study, the overexpression of DRP-2 may be associated with neuritic reorganization and the formation of dystrophic neurites surrounding amyloid plaques. Treatment with dl-PHPB significantly reduced the elevated DRP-2 expression in APP/PS1 mice, potentially contributing to the maintenance of neurite cytoskeletal integrity.
Cofilin, the principal actin-depolymerizing factor, plays a critical role in learning and memory (Kang and Woo, 2019). Prior researches have demonstrated that cofilin and actin can form rod-like structures around amyloid plaques, disrupting distal neurite function and ultimately leading to neuritic atrophy and cell death (Minamide et al., 2000; Bamburg and Bernstein, 2016). Moreover, cofilin regulates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptor-dependent dendritic spine plasticity (Pontrello et al., 2012; Gu et al., 2010), thereby influencing learning and memory. Recent studies have indicated that cofilin activation exacerbates tau pathology by impairing tau-mediated microtubule dynamics, thus affecting learning and memory (Woo et al., 2019). In AD, cofilin has been reported to mediate neuronal apoptosis through p53 translocation and phospholipase D1 (PLD1) regulation (Liu et al., 2017). Our previous findings have shown a substantial increase in cofilin-2 levels in the hippocampus of APP/PS1 mice (Sun et al., 2014). In the present study, long-term treatment with dl-PHPB markedly prevented this increase in hippocampal cofilin-2 levels. Furthermore, both memantine and donepezil significantly reduced hippocampal cofilin-2 levels in APP/PS1 mice. Collectively, these results suggest that cofilin-2 is a promising drug target and may be valuable for identifying potential therapeutic agents. Compounds that inhibit cofilin-2 could potentially alleviate learning and memory deficits in AD.
In recent decades, numerous studies have consistently demonstrated the critical role of oxidative stress and inflammatory responses in the pathogenesis of AD (Kosenko et al., 2014). Peroxiredoxin-6, a major antioxidant enzyme in human neural tissue, has been shown to play a significant role in neuroprotection (Power et al., 2008). Recent evidence indicates that the peroxidase activity of peroxiredoxin-6 protects PC12 cells from Aβ25-35-induced neurotoxicity (Kim et al., 2013), and that the upregulation of peroxiredoxin-6 can modulate astroglial and microglial activation around Aβ plaques, thereby mediating the protective function of Aβ proteostasis (Pankiewicz et al., 2020). In our current study, we observed a significant elevation in the peroxiredoxin-6 protein level in the cortex and hippocampus of APP/PS1 mice compared to WT mice. Consistent with these findings, increased levels of peroxiredoxin-6 have been reported in the brain tissues of amyloid transgenic mouse models (Guerreiro et al., 2008) and AD patients (Sultana et al., 2007; Power et al., 2008). This increased expression of peroxiredoxin-6 may represent a compensatory and protective response to the oxidative stress induced by Aβ accumulation. Our results indicate that peroxiredoxin-6 was upregulated following dl-PHPB treatment, suggesting that dl-PHPB may exert its therapeutic effects in APP/PS1 mice through the antioxidant properties of peroxiredoxin-6.
The major pathologic characteristics of AD encompass the accumulation of intracellular neurofibrillary tangles, the deposition of extracellular senile plaques, and neuronal loss. Pin 1 levels are significantly diminished in the cortex and hippocampus of APP/PS1 mice, a finding consistent with prior studies in AD brains (Sultana et al., 2006; Sultana et al., 2007). Pin 1 prevents tau hyperphosphorylation by inhibiting GSK3β activity (Ma et al., 2012) and promotes tau dephosphorylation through interaction with protein phosphatase 2A (PP2A) (Zhou et al., 2000), thereby restoring its function in microtubule binding. Recent studies have demonstrated that Pin 1 facilitates the non-amyloidogenic processing of APP by accelerating its cis and trans conformational changes, thus protecting against Aβ toxicity (Pastorino et al., 2012). Moreover, Pin 1 interacts with several proteins involved in cell cycle regulation (Zhou et al., 2000; Chen et al., 2012). Consequently, a decline in Pin 1 protein levels may disrupt the balanced phosphorylation state of tau proteins, leading to Aβ deposition and neuronal loss in AD (Wang et al., 2020). In our study, the reduction in Pin 1 levels was reversed by long-term treatment with dl-PHPB, indicating that dl-PHPB may exert therapeutic effects by modulating tau phosphorylation, Aβ production, and neuronal apoptosis.
Lysosomal activation is prominently observed in the pathology of AD (Bendiske and Bahr, 2003). Cathepsin B, a lysosomal cysteine protease, plays a crucial role in degrading proteins that enter the lysosomal system and has been implicated in Aβ processing and AD pathogenesis (Haque et al., 2008; Hook et al., 2020). Specifically, cathepsin B cleaves APP at the β-secretase cleavage site and enhances β-secretase-mediated cleavage of APP, contributing to the production of neurotoxic Aβ peptides (Klein et al., 2009; Schechter and Ziv, 2011). Moreover, genetic knockout (Hook et al., 2009; Kindy et al., 2012) and chemical inhibition (Hook et al., 2008; Cho et al., 2013) studies in cellular and animal models of AD have demonstrated that inhibiting cathepsin B reduces Aβ burden and improves cognitive function. Our current proteomic analysis indicates that dl-PHPB significantly decreases the expression of cathepsin B in the cortex and hippocampus of APP/PS1 mice. Previous research from our group has shown that dl-PHPB treatment markedly reduces Aβ plaque deposition in the brain tissues of APP/PS1 mice (Peng et al., 2014). Therefore, we propose that dl-PHPB may exert neuroprotective effects against Aβ plaques, at least in part, by downregulating cathepsin B levels.
Memantine, an NMDA receptor antagonist, and donepezil, an acetylcholinesterase inhibitor, are currently utilized to treat AD. As a result, they were selected as positive controls to evaluate the therapeutic efficacy of dl-PHPB in the step-down passive avoidance test. In this study, treatment with memantine, donepezil, and dl-PHPB significantly alleviated cognitive dysfunction in APP/PS1 mice. Furthermore, Western blot analysis indicated that most AD-related proteins examined in this study (PDHE1α, DRP-2, peroxiredoxin-6, Pin 1, and cathepsin B) were not modulated by memantine or donepezil, likely due to their nature as single-target drugs. However, the altered expression levels of cofilin-2 and VDAC1 in the brains of APP/PS1 mice were restored to near-normal levels by treatment with dl-PHPB, memantine, and donepezil. This suggests that cofilin-2 and VDAC1 may be involved in common AD-related signaling pathways regulated by these three drugs. Therefore, they are more likely to serve as potential biomarkers for assessing therapeutic efficacy during drug development.
In conclusion, proteomics is recognized as a valuable tool for analyzing protein alterations in complex diseases such as AD. By employing a 2DE-based proteomic approach, we have successfully identified novel candidate proteins involved in AD development and delineated potential intervention targets of dl-PHPB in the cortex and hippocampus of an AD mouse model. Notably, dl-PHPB was found to mitigate the changes in many proteins that were differentially expressed in APP/PS1 mice, suggesting that dl-PHPB may restore these alterations towards normal levels. Due to its capacity to target multiple pathways, dl-PHPB can enhance mitochondrial function, reduce oxidative stress, increase synaptic plasticity, maintain normal neuronal connections, and inhibit tau phosphorylation, Aβ production, and cell apoptosis. Treatment with dl-PHPB, memantine, and donepezil, respectively, could reverse the expression of VDAC1 and cofilin 2 to near-normal levels, indicating that these two proteins may serve as candidate biomarkers for assessing therapeutic efficacy in AD. While this study advances our understanding of VDAC1 and cofilin-2 in AD, several limitations warrant consideration. First, our findings are derived from preclinical models; thus, validation in human-derived samples or longitudinal clinical cohorts is essential to confirm translational relevance. Further studies should: (1) define the precise molecular interplay between VDAC1-mediated mitochondrial permeability and cofilin-2-regulated actin dynamics in AD pathogenesis, (2) validate their diagnostic/therapeutic utility as therapeutic targets or biomarkers in clinical cohorts, and (3) assess the therapeutic potential of pharmacological modulation strategies targeting the VDAC1/cofilin-2 pathway in AD treatment.
Our findings suggest dl-PHPB’s therapeutic potential in enhancing cognitive function in APP/PS1 mice, but several study limitations exist. First, the mechanistic insights derived from proteomic analyses remain correlative, and causality requires further validation using targeted experimental approaches such as pathway-specific inhibitors or genetic models. Second, the current study focused on a single dose; future work should explore dose-dependent effects. Third, although our behavioral tests showed significant improvements, additional neuropathological markers (e.g., synaptic plasticity, Aβ plaque load) could strengthen the conclusions. Finally, the generalizability of our findings may be limited to the specific APP/PS1 mice used here, and validation in aged or other transgenic models with advanced neurodegeneration would be valuable.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the ethics Committee on Animal Experimentation of Ludong University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YS: Writing – original draft, Writing – review and editing. GB: Writing – original draft, Writing – review and editing. KY: Writing – review and editing. YF: Writing – review and editing. HS: Writing – review and editing. LX: Writing – review and editing. HG: Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Our study was supported by the National Science Foundation of China (No. 82104470; No. 81600672), Science and Technology Development Project of the Second Affiliated Hospital of Shandong Medical University (No. 2024FYM162), China Postdoctoral Science Foundation (No. 2022M720337), Natural Science Foundation of Shandong (No. ZR2020QH131), and the High-End Talent Team Construction Foundation (No. 108-10000318).
Conflict of interest
Author YS was employed by Beijing Handian Pharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abidi S. M. S. Sharma C. Randhawa S. Shukla A. K. Acharya A. (2023). A review on nanotechnological perspective of “the amyloid cascade hypothesis” for neurodegenerative diseases. Int. J. Biol. Macromol.253, 126821. 10.1016/j.ijbiomac.2023.126821
2
Bamburg J. R. Bernstein B. W. (2016). Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton73, 477–497. 10.1002/cm.21282
3
Bendiske J. Bahr B. A. (2003). Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease?J. Neuropathol. Exp. Neurol.62, 451–463. 10.1093/jnen/62.5.451
4
Breijyeh Z. Karaman R. (2020). Comprehensive review on Alzheimer's disease: causes and treatment. Molecules25, 5789. 10.3390/molecules25245789
5
Chen H. Z. Li L. Wang W. J. Du X. D. Wen Q. He J. P. et al (2012). Prolyl isomerase Pin1 stabilizes and activates orphan nuclear receptor TR3 to promote mitogenesis. Oncogene31, 2876–2887. 10.1038/onc.2011.463
6
Cho K. Yoon S. Y. Choi J. E. Kang H. J. Jang H. Y. Kim D. H. (2013). CA-074Me, a cathepsin B inhibitor, decreases APP accumulation and protects primary rat cortical neurons treated with okadaic acid. Neurosci. Lett.548, 222–227. 10.1016/j.neulet.2013.05.056
7
Choudhary O. P. Paz A. Adelman J. L. Colletier J. P. Abramson J. Grabe M. (2014). Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol.21, 626–632. 10.1038/nsmb.2841
8
Cole A. R. Noble W. VAN Aalten L. Plattner F. Meimaridou R. Hogan D. et al (2007). Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer's disease progression. J. Neurochem.103, 1132–1144. 10.1111/j.1471-4159.2007.04829.x
9
EL-Khateeb E. Vasilogianni A. M. Alrubia S. AL-Majdoub Z. M. Couto N. Howard M. et al (2019). Quantitative mass spectrometry-based proteomics in the era of model-informed drug development: applications in translational pharmacology and recommendations for best practice. Pharmacol. Ther.203, 107397. 10.1016/j.pharmthera.2019.107397
10
Guerreiro N. Staufenbiel M. Gomez-Mancilla B. (2008). Proteomic 2-D DIGE profiling of APP23 transgenic mice brain from pre-plaque and plaque phenotypes. J. Alzheimers Dis.13, 17–30. 10.3233/jad-2008-13102
11
Gu J. Lee C. W. Fan Y. Komlos D. Tang X. Sun C. et al (2010). ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci.13, 1208–1215. 10.1038/nn.2634
12
Gu Y. Hamajima N. Ihara Y. (2000). Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry39, 4267–4275. 10.1021/bi992323h
13
Haque A. Banik N. L. Ray S. K. (2008). New insights into the roles of endolysosomal cathepsins in the pathogenesis of Alzheimer’s disease: cathepsin inhibitors as potential therapeutics. CNS Neurol. Disord. Drug Targets7, 270–277. 10.2174/187152708784936653
14
Hook V. Y. Kindy M. Hook G. (2008). Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem.283, 7745–7753. 10.1074/jbc.M708362200
15
Hook V. Y. Kindy M. Reinheckel T. Peters C. Hook G. (2009). Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem. Biophys. Res. Commun.386, 284–288. 10.1016/j.bbrc.2009.05.131
16
Hook V. Yoon M. Mosier C. Ito G. Podvin S. Head B. P. et al (2020). Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochimica Biophysica Acta. Proteins Proteomics1868, 140428. 10.1016/j.bbapap.2020.140428
17
Hou S. T. Jiang S. X. Aylsworth A. Ferguson G. Slinn J. Hu H. et al (2009). CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J. Neurochem.111, 870–881. 10.1111/j.1471-4159.2009.06375.x
18
Hoyer S. (2004). Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv. Exp. Med. Biol.541, 135–152. 10.1007/978-1-4419-8969-7_8
19
Hoyer S. Nitsch R. Oesterreich K. (1991). Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J. Neural Transm. Park Dis. Dement. Sect.3, 1–14. 10.1007/BF02251132
20
Huang L. Zhang Y. Peng Y. Zhao Z. Zhou Y. Wang X. et al (2019). Protective effect of potassium 2-(l-hydroxypentyl)-benzoate on hippocampal neurons, synapses and dystrophic axons in APP/PS1 mice. Psychopharmacol. Berl.236, 2761–2771. 10.1007/s00213-019-05251-x
21
Hu Y. Peng Y. Long Y. Xu S. Feng N. Wang L. et al (2012). Potassium 2-(1-hydroxypentyl)-benzoate attenuated hydrogen peroxide-induced apoptosis in neuroblastoma SK-N-SH cells. Eur. J. Pharmacol.680, 49–54. 10.1016/j.ejphar.2012.01.031
22
Ip J. P. Fu A. K. Ip N. Y. (2014). CRMP2: functional roles in neural development and therapeutic potential in neurological diseases. Neuroscientist20, 589–598. 10.1177/1073858413514278
23
Jankowsky J. L. Slunt H. H. Ratovitski T. Jenkins N. A. Copeland N. G. Borchelt D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng.17, 157–165. 10.1016/s1389-0344(01)00067-3
24
Kang D. E. Woo J. A. (2019). Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s disease. J. Alzheimer's Dis. JAD72, S131–S144. 10.3233/JAD-190585
25
Kim I. K. Lee K. J. Rhee S. Seo S. B. Pak J. H. (2013). Protective effects of peroxiredoxin 6 overexpression on amyloid beta-induced apoptosis in PC12 cells. Free Radic. Res.47, 836–846. 10.3109/10715762.2013.833330
26
Kindy M. S. Yu J. Zhu H. EL-Amouri S. S. Hook V. Hook G. R. (2012). Deletion of the cathepsin B gene improves memory deficits in a transgenic ALZHeimer’s disease mouse model expressing AβPP containing the wild-type β-secretase site sequence. J. Alzheimers Dis.29, 827–840. 10.3233/JAD-2012-111604
27
Klein D. M. Felsenstein K. M. Brenneman D. E. (2009). Cathepsins B and L differentially regulate amyloid precursor protein processing. J. Pharmacol. Exp. Ther.328, 813–821. 10.1124/jpet.108.147082
28
Korotchkina L. G. Tucker M. M. Thekkumkara T. J. Madhusudhan K. T. Pons G. Kim H. et al (1995). Overexpression and characterization of human tetrameric pyruvate dehydrogenase and its individual subunits. Protein Expr. Purif.6, 79–90. 10.1006/prep.1995.1011
29
Kosenko E. A. Solomadin I. N. Tikhonova L. A. Reddy V. P. Aliev G. Kaminsky Y. G. (2014). Pathogenesis of Alzheimer disease: role of oxidative stress, amyloid-beta peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol. Disord. Drug Targets13, 112–119. 10.2174/18715273113126660130
30
Lemasters J. J. Holmuhamedov E. (2006). Voltage-dependent anion channel (VDAC) as mitochondrial governator--thinking outside the box. Biochim. Biophys. Acta1762, 181–190. 10.1016/j.bbadis.2005.10.006
31
Li P. P. Wang W. P. Liu Z. H. Xu S. F. Lu W. W. Wang L. et al (2014). Potassium 2-(1-hydroxypentyl)-benzoate promotes long-term potentiation in Aβ1-42-injected rats and APP/PS1 transgenic mice. Acta Pharmacol. Sin.35, 869–878. 10.1038/aps.2014.29
32
Liu T. Wang F. Lepochat P. Woo J. A. Bukhari M. Z. Hong K. W. et al (2017). Cofilin-mediated neuronal apoptosis via p53 translocation and PLD1 regulation. Sci. Rep.7, 11532. 10.1038/s41598-017-09996-3
33
Ma S. L. Pastorino L. Zhou X. Z. Lu K. P. (2012). Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3β (GSK3β) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem.287, 6969–6973. 10.1074/jbc.C111.298596
34
Manczak M. Reddy P. H. (2012). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum. Mol. Genet.21, 5131–5146. 10.1093/hmg/dds360
35
Masuda T. Mori A. Ito S. Ohtsuki S. (2021). Quantitative and targeted proteomics-based identification and validation of drug efficacy biomarkers. Drug Metab. Pharmacokinet.36, 100361. 10.1016/j.dmpk.2020.09.006
36
Minamide L. S. Striegl A. M. Boyle J. A. Meberg P. J. Bamburg J. R. (2000). Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol.2, 628–636. 10.1038/35023579
37
O'Donnell M. Chance R. K. Bashaw G. J. (2009). Axon growth and guidance: receptor regulation and signal transduction. Annu. Rev. Neurosci.32, 383–412. 10.1146/annurev.neuro.051508.135614
38
Pankiewicz J. E. Diaz J. R. Marta-Ariza M. Lizinczyk A. M. Franco L. A. Sadowski M. J. (2020). Peroxiredoxin 6 mediates protective function of astrocytes in Aβ proteostasis. Mol. Neurodegener.15, 50. 10.1186/s13024-020-00401-8
39
Pastorino L. Ma S. L. Balastik M. Huang P. Pandya D. Nicholson L. et al (2012). Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AβPP. J. Alzheimers Dis.30, 277–297. 10.3233/JAD-2012-111259
40
Peng Y. Hu Y. Xu S. Rong X. Li J. Li P. et al (2014). Potassium 2-(1-hydroxypentyl)-benzoate improves memory deficits and attenuates amyloid and tau pathologies in a mouse model of Alzheimer's disease. J. Pharmacol. Exp. Ther.350, 361–374. 10.1124/jpet.114.213140
41
Pithukpakorn M. (2005). Disorders of pyruvate metabolism and the tricarboxylic acid cycle. Mol. Genet. Metab.85, 243–246. 10.1016/j.ymgme.2005.06.006
42
Pontrello C. G. Sun M. Y. Lin A. Fiacco T. A. Defea K. A. Ethell I. M. (2012). Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc. Natl. Acad. Sci. U. S. A.109, E442–E451. 10.1073/pnas.1118803109
43
Power J. H. Asad S. Chataway T. K. Chegini F. Manavis J. Temlett J. A. et al (2008). Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol.115, 611–622. 10.1007/s00401-008-0373-3
44
Ronowska A. Szutowicz A. Bielarczyk H. Gul-Hinc S. Klimaszewska-Lata J. Dys A. et al (2018). The regulatory effects of acetyl-CoA distribution in the healthy and diseased brain. Front. Cell. Neurosci.12, 169. 10.3389/fncel.2018.00169
45
Schechter I. Ziv E. (2011). Cathepsins S, B and L with aminopeptidases display beta-secretase activity associated with the pathogenesis of Alzheimer's disease. Biol. Chem.392, 555–569. 10.1515/BC.2011.054
46
Schmidt E. F. Strittmatter S. M. (2007). The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol.600, 1–11. 10.1007/978-0-387-70956-7_1
47
Shang N. Y. Huang L. J. Lan J. Q. Kang Y. Y. Tang J. S. Wang H. Y. et al (2024). PHPB ameliorates memory deficits and reduces oxidative injury in Alzheimer’s disease mouse model by activating Nrf2 signaling pathway. Acta Pharmacol. Sin.45, 1142–1159. 10.1038/s41401-024-01240-9
48
Sharma P. Benford B. Li Z. Z. Ling G. S. (2009). Role of pyruvate dehydrogenase complex in traumatic brain injury and Measurement of pyruvate dehydrogenase enzyme by dipstick test. J. Emerg. Trauma Shock2, 67–72. 10.4103/0974-2700.50739
49
Shoshan-Barmatz V. Nahon-Crystal E. Shteinfer-Kuzmine A. Gupta R. (2018). VDAC1, mitochondrial dysfunction, and Alzheimer's disease. Pharmacol. Res.131, 87–101. 10.1016/j.phrs.2018.03.010
50
Stacpoole P. W. (2012). The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell11, 371–377. 10.1111/j.1474-9726.2012.00805.x
51
Sultana R. Boyd-Kimball D. Cai J. Pierce W. M. Klein J. B. Merchant M. et al (2007). Proteomics analysis of the Alzheimer's disease hippocampal proteome. J. Alzheimers Dis.11, 153–164. 10.3233/jad-2007-11203
52
Sultana R. Boyd-Kimball D. Poon H. F. Cai J. Pierce W. M. Klein J. B. et al (2006). Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol. Aging27, 918–925. 10.1016/j.neurobiolaging.2005.05.005
53
Sun Y. Rong X. Lu W. Peng Y. Li J. Xu S. et al (2014). Translational study of Alzheimer's disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J. Alzheimers Dis.45, 269–282. 10.3233/JAD-142805
54
Trinchese F. Liu S. Battaglia F. Walter S. Mathews P. M. Arancio O. (2004). Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol.55, 801–814. 10.1002/ana.20101
55
Vaz M. Silvestre S. (2020). Alzheimer's disease: recent treatment strategies. Eur. J. Pharmacol.887, 173554. 10.1016/j.ejphar.2020.173554
56
Wang L. Zhou Y. Chen D. Lee T. H. (2020). Peptidyl-prolyl cis/trans isomerase Pin1 and Alzheimer's disease. Front. cell Dev. Biol.8, 355. 10.3389/fcell.2020.00355
57
Woo J. A. Liu T. Fang C. C. Cazzaro S. Kee T. Lepochat P. et al (2019). Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol.2, 112. 10.1038/s42003-019-0359-9
58
Xing G. Ren M. Watson W. D. O'Neill J. T. Verma A. (2009). Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: a mechanism of dysregulated glucose metabolism. Neurosci. Lett.454, 38–42. 10.1016/j.neulet.2009.01.047
59
Yao J. Irwin R. W. Zhao L. Nilsen J. Hamilton R. T. Brinton R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A.106, 14670–14675. 10.1073/pnas.0903563106
60
Yokota H. (2019). Applications of proteomics in pharmaceutical research and development. Biochim. Biophys. Acta Proteins Proteom1867, 17–21. 10.1016/j.bbapap.2018.05.008
61
Yoo B. C. Fountoulakis M. Cairns N. Lubec G. (2001). Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer’s disease and Down syndrome. Electrophoresis22, 172–179. 10.1002/1522-2683(200101)22:1<172::AID-ELPS172>3.0.CO;2-P
62
Yoshida H. Watanabe A. Ihara Y. (1998). Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. J. Biol. Chem.273, 9761–9768. 10.1074/jbc.273.16.9761
63
Yoshimura T. Kawano Y. Arimura N. Kawabata S. Kikuchi A. Kaibuchi K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell120, 137–149. 10.1016/j.cell.2004.11.012
64
Zhang F. Zhong R. J. Cheng C. Li S. LE W. D. (2021). New therapeutics beyond amyloid-beta and tau for the treatment of Alzheimer’s disease. Acta Pharmacol. Sin.42, 1382–1389. 10.1038/s41401-020-00565-5
65
Zhao C. Hou W. Lei H. Huang L. Wang S. Cui D. et al (2017). Potassium 2-(l-hydroxypentyl)-benzoate attenuates neuroinflammatory responses and upregulates heme oxygenase-1 in systemic lipopolysaccharide-induced inflammation in mice. Acta Pharm. Sin. B7, 470–478. 10.1016/j.apsb.2017.04.007
66
Zhao W. Xu S. Peng Y. Ji X. Cao D. Li J. et al (2013). Potassium 2-(1-hydroxypentyl)-benzoate improves learning and memory deficits in chronic cerebral hypoperfused rats. Neurosci. Lett.541, 155–160. 10.1016/j.neulet.2013.01.053
67
Zhou X. Z. Kops O. Werner A. Lu P. J. Shen M. Stoller G. et al (2000). Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell6, 873–883. 10.1016/s1097-2765(05)00083-3
Summary
Keywords
Alzheimer’s disease, LC-MS/MS, proteomics, dl-PHPB, biomarker
Citation
Sun Y, Bai G, Yang K, Feng Y, Sun H, Xian L and Gao H (2025) Multi-target neuroprotection by dl-PHPB in APP/PS1 mice: a proteomic analysis. Front. Pharmacol. 16:1554168. doi: 10.3389/fphar.2025.1554168
Received
01 January 2025
Accepted
11 April 2025
Published
24 April 2025
Volume
16 - 2025
Edited by
Francisco Lopez-Munoz, Camilo José Cela University, Spain
Reviewed by
Mauro Cunha Xavier Pinto, Universidade Federal de Goiás, Brazil
Tariq Maqbool, University of Kashmir, India
Updates
Copyright
© 2025 Sun, Bai, Yang, Feng, Sun, Xian and Gao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingni Sun, 17805459157@163.com
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.