 Jie Gao
Jie Gao Yuhao Wang
Yuhao Wang Ruoqi Wang2
Ruoqi Wang2- 1China Institute of Atomic Energy, Beijing, China
- 2China Institute for Radiation Protection/National Atomic Energy Agency Nuclear Technology (Nonclinical Evaluation of Radiopharmaceuticals) Research and Development Center/CNNC Key Laboratory on Radiotoxicology and Radiopharmaceutical Preclinical Evaluation, Taiyuan, Shanxi, China
- 3First Hospital of Shanxi Medical University, Taiyuan, Shanxi, China
- 4China Isotope and Radiation Corporation, Beijing, China
Objective: This study aims to improve the biodistribution of probes and enhance tumor targeting through 68Ga/177Lu-labeled optimized probes, thereby providing better tumor detection and assessment in PET imaging while also exploring their therapeutic effects on tumors.
Methods: The physicochemical properties of PARPi probes were optimized through polyethylene glycol (PEG) modification. The tumor inhibition effect of the novel probes was validated through the assessment of in vitro affinity, uptake, in vivo distribution, and tumor targeting of the PARPi probes. Based on the distribution results, OLINDA/EXM radiation dose estimation was then performed to optimize the clinically administered dose.
Results: In the study, a novel PARP-targeted imaging agent, DOTA-PEG-PARPi, was designed and optimized, demonstrating sufficient in vivo stability. The results of in vitro trials showed strong affinity and uptake of PEG-PARPi in pancreatic cancer tumor cells. SPECT/CT imaging revealed significant radioactive accumulation, notable uptake, and prolonged retention time in PSN-1 tumors. Tissue distribution results showed that tumor uptake peaked 3 h after administration. According to dose estimation, the highest absorbed dose was observed in the pancreas of female adults.
Conclusion: The PEG-modified PARPi probe not only retained high affinity and targeting capability but also significantly improved retention time during in vivo trials.
Introduction
Genomic instability in tumors is significantly increased due to endogenous and exogenous factors, such as breast cancer 1/breast cancer 2 (BRCA1/BRCA2) gene mutations and oxidative stress (Chen et al., 2022). Among various DNA repair factors, the overexpression of poly ADP-ribose polymerase-1 (PARP-1) family members in tumor cells has drawn particular attention (Ke et al., 2022). PARP-1 plays a crucial role in identifying DNA damage and maintaining genomic integrity (Safdar et al., 2024; Steffen et al., 2014). The inhibition of PARP-1 expression can disrupt DNA repair and replication processes in cancer cells, making them more susceptible to death (Bhamidipati et al., 2023; Soung and Chung, 2023). The United States Food and Drug Administration (FDA) has approved multiple PARP inhibitors for the treatment of BRCA mutation-positive breast cancer (Mccann and Hurvitz, 2018) and advanced ovarian cancer (Colombo et al., 2018). Nevertheless, current studies on the application of PARP-1-targeting probes in tumor therapy remain limited, necessitating further research, exploration, and validation of their potential clinical value (Li et al., 2023).
Due to the extensive application of PARP inhibitors in the treatment of ovarian and breast cancer, PARP imaging technology holds promise for providing more optimized treatment options for patients with these cancers (Slade, 2020; Cortesi et al., 2021). Recently, several research teams have demonstrated the potential of 18F-labeled PARP inhibitors in PARP imaging through preclinical studies (Reiner et al., 2011; Zhou et al., 2018; Mcdonald et al., 2021). Most of these compounds share structural similarities with olaparib and have shown positive efficacy in the treatment of breast and pancreatic cancers. However, the current 18F-labeled PARP inhibitors are primarily metabolized by the liver and excreted via bile, which potentially cause radioactive accumulation in the liver and other abdominal organs, thereby affecting image quality and hindering lesion identification, particularly at metastatic sites commonly observed in pancreatic and ovarian cancers (Young et al., 2024). The inhibitors encounter resistance challenges similar to those faced by other targeted anticancer drugs. Some patients even exhibit primary resistance at the early stages of treatment, significantly limiting the clinical application of PARPis (Young et al., 2024; Wang et al., 2023). To overcome these limitations, this study explored new labeling strategies, improved chemical properties of compounds, and developed more effective PARP inhibitors to provide better tumor detection and assessment in PET imaging. The utilization of PARP-1-targeting probes to detect PARP-1 expression in tumor cells has potential value for identifying patient populations suitable for PARPi-targeted therapy and improving treatment efficacy (Dibitetto et al., 2024; Bhin et al., 2023; Wang et al., 2019). Furthermore, such probes play a vital role in predicting therapeutic responses and quantifying tumor treatment outcomes. Therefore, it is essential to develop methods for evaluating PARP expression and activity to help identify patients who may benefit from these treatments.
Materials and methods
Synthesis and radiolabeling
68Ga/177Lu-DOTA-PEG-PARPi was synthesized, as described in Figure 1. Compound 1 (209.14 mg, 570.82 umol, 1.1 eq), EDCI (129.32 mg, 674.61 umol, 1.3 eq), and DIPEA (257.29 uL, 1.56 mmol, 3 eq) were added to a solution of compound 2 (200 mg, 518.93 umol, 1.0 eq) in DCM (20 mL). The reaction mixture was diluted with DCM and washed with 0.5 M HCl aqueous solution. The aqueous layer was further extracted once with DCM and stirred overnight at room temperature. The combined organic layers were washed with saturated sodium bicarbonate and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to produce crude compound 3 (568 mg, 99.44%) as a white-foamed solid, which can be used without further purification.

Figure 1. Synthesis of the designed DOTA-PEG-PARPi.
Piperidine (PIP, 2 mL) was added to a solution of compound 3 (568 mg, 774.07 µmol, 1.0 eq) in DMF (8 mL). The reaction mixture was stirred at room temperature for 1.5 h. TLC showed that the reaction was completed. The reaction mixture was washed several times with Et2O and PE to yield crude compound 4 (374 mg, 94.45%) as a light yellow oil.
Compound 4 (96.46 mg, 188.57 µmol, 1.2 eq), HATU (89.62 mg, 235.71 µmol, 1.5 eq), and DIPEA (77.91 µL, 471.42 µmol, 3 eq) were added to a solution of compound 5 (90 mg, 157.14 µmol, 1.0 eq) in DCM (4 mL). The reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with DCM and washed with H2O, and the aqueous layer was further extracted once with DCM. The combined organic layers were washed with saturated sodium bicarbonate and brine, dried over Na2SO4, and filtered. The crude product obtained after vacuum evaporation was purified by column chromatography (DCM: MeOH = 100:1 to 10:1) and Prep-TLC (twice) to yield compound 6 (115 mg, 68.63%) as a white solid.
A measure of 5 M HCl (in 1,4-dioxane) (2 mL) was added to a solution of compound 6 (110 mg, 103.16 umol, 1.0 eq) in DCM (2 mL). The reaction mixture was stirred overnight at room temperature. TLC and HPLC showed that the reaction was completed. The reaction mixture was precipitated with Et2O2, and the solid was dried to yield DOTA-PEG-PARPi (82.3 mg, 88.84%) as a white solid.
68GaCl3 dissolved in an acetate buffer system (0.5 M, pH = 4.7) was mixed with DOTA-PEG-PARPi at 95°C for 15 min and then passed through an activated C18 column to purify the 68Ga-DOTA-PEG-PARPi solution. The solution was diluted with physiological saline, and its radiochemical purity was determined using radio-TLC [NH4Ac/MeOH = 1:1 DTPA solution or 0.5 M citric acid buffer (pH5.0), where chromatographic paper is iTLC-SG] and radio-HPLC. The process of 177LuCl3 radiolabeling is similar to those described above.
Cell culture and tumor models
A549, AsPC-1, PSN-1, ISN-1, and U87MG cells were purchased from ACTT and cultured as a monolayer using DMEM (Gibco, Carlsbad, United States) and RPMI 1640 medium (Gibco, Carlsbad, United States) containing 10% FBS (Gibco, Carlsbad, United States) and 1% penicillin–streptomycin (Invitrogen, Carlsbad, United States) at 37°C in a humidified atmosphere with 5% CO2 and 95% air.
Balb/c nude mice aged 6–8 weeks were purchased from Charles River (Beijing, China) and kept in specific pathogen-free (SPF) conditions during the experimental period. All mice had free access to food and water. All animal experimental procedures were performed in accordance with the guidelines and approved by the Animal Care and Use Committee of the China Institute for Radiation Protection. Following a one-week acclimation period, PSN-1 cells were inoculated subcutaneously into the right flank of the mice (5 × 106 cells/mice) to establish the model. The mice with tumor diameters >1 cm could be used in this experiment.
68Ga-DOTA-PEG-PARPi in vitro binding assays
A549, AsPC-1, PSN-1, ISN-1, and U87MG cells were cultured in 96-well plates for approximately 24 h. To evaluate the binding selectivity of 68Ga-DOTA-olaparib and 68Ga-DOTA-PEG-PARPi, an unlabeled PARP inhibitor was added at 37°C for 30 min prior to the addition of a 68GA-labeled radiotracer. After the removal of the cell culture medium, cells were washed with PBS, and the 68Ga content in the cell lysate was determined using a γ-counter.
177Lu-DOTA-PEG-PARPi in vitro cell uptake
A549, AsPC-1, PSN-1, ISN-1, and U87MG cells were seeded in 6-well plates (400 × 103/well) for 24 h. Then, the cells were incubated with 177Lu-DOTA-olaparib and 177Lu-DOTA-PEG-PARPi (740 kBq/mL in each well), respectively, at 37°C for 2 h, followed by rinsing three times with PBS to eliminate unbound 177Lu-labeled radiotracers. Cell suspensions were produced after adding pancreatin (0.5 mL). Finally, the γ-counter was used to measure radioactivity.
Radiochemical stability
The in vitro stability of 177Lu-DOTA-PEG-PARPi was evaluated in saline, mouse plasma, and human plasma. In brief, 0.2 mL of 177Lu-DOTA-PEG-PARPi (0.74–2.96 MBq) was incubated with 0.8 mL of saline at room temperature for 1 and 24 h and then evaluated by radio-HPLC to assess the stability of 177Lu-DOTA-PEG-PARPi. Additionally, 177Lu-DOTA-PEG-PARPi (37–74 MBq, 0.2 mL) was added to 0.8 mL of mouse plasma or human plasma, and the solution was incubated at 37°C for 1 and 24 h, respectively. After incubation, 0.2 mL of the above solution was added to 0.4 mL of acetonitrile to precipitate plasma proteins and then swirled. The plasma proteins were subsequently separated by centrifugation at 14,800 rpm for 10 min at 4°C. The supernatant was then analyzed by HPLC. The above experiments were repeated three times.
Micro PET/SPECT/CT imaging
68Ga-DOTA-PEG-PARPi and 177Lu-DOTA-PEG-PARPi imaging were obtained using a microPET/SPECT/CT all-in-one machine (Novel Medical, China).
The tumor-bearing mice (n = 1) were intravenously injected with 68Ga-DOTA-PEG-PARPi via the lateral tail vein at a dose of 3.7 MBq under isoflurane maintenance anesthesia. A 15-min acquisition was performed at 0.5 and 2 h post-injection. Image reconstruction was performed using the 3D ordered subset expectation maximization (OSEM) algorithm based on the Monte Carlo system model. A CT scan with a tube voltage of 80 kV and a tube current of 0.5 mA was performed on mice after PET acquisition. The image reconstruction was performed based on the filtered back projection (FBP) algorithm. Images were analyzed using NMSoft-AIWS software.
The tumor-bearing mice (n = 3) were intravenously injected with 177Lu-DOTA-PEG-PARPi via the lateral tail vein at a dose of 30 MBq under isoflurane maintenance anesthesia. A 30-min acquisition was performed at 1, 3, 6, 24, and 48 h post-injection. Image reconstruction was performed using the 3D OSEM algorithm based on the Monte Carlo system model. A CT scan with a tube voltage of 80 kV and a tube current of 0.5 mA was performed on mice after SPECT acquisition. Image reconstruction was performed based on the FBP algorithm. Images were analyzed using NMSoft-AIWS software. ID%/cc values were obtained by drawing the ROIs of the lesions in the SPECT image and using the automatic thresholding tool.
Ex vivo biodistribution and radiation dosimetry
Biodistribution in the model
177Lu-DOTA-PEG-PARPi was administered via the tail vein. All mice were euthanized via isoflurane inhalation at 3, 24, 48, 96, and 168 h post-injection, respectively. Blood and organs were collected, dissected, and measured, and the radioactivity was quantified using a γ-counter (2480 WIZARD2, PerkinElmer, United States). Organ samples included the pancreas, kidney, heart, liver, spleen, lung, stomach, intestine, bone (femur), muscle, and tumor. Data are expressed as the percentage of the injected dose per gram of tissue (%ID/g).
Radiation dosimetry
The mean absorbed radiation doses were evaluated using the mathematic formalism established by MIRD. The estimated dose for an adult human was computed using OLINDA/EXM 2.2.3 (Hermes Medical Solutions, Sweden) with the mice 177Lu-DOTA-PEG-PARPi pharmacokinetic data as input. It was assumed that the biodistribution of the tracer in mice was the same as in adult humans. The mice biodistribution data (%ID/g) were converted to human whole-organ biodistribution data (%ID/organ) using formula (a):
The calculated total number of decays per gram of tissue was converted to the residence time per gram (MBq*h/MBq/g). Values were then multiplied with the organ mass in the respective male or female MIRD89 phantom to derive the human residence times. The values for human residence times were used as input in OLINDA/EXM.
Treatment of the pancreatic cancer model
A total of 20 tumor-bearing mice were included and randomly assigned to four groups. The tumor-bearing mice (n = 5) that did not receive treatment were set as the vehicle group (control group). The other three groups of tumor-bearing mice were treated with different doses of 177Lu-DOTA-PEG-PARPi (L, low dose; M, medium dose; and H, high dose), that is, a 10 MBq group (n = 5, 10 ± 1.28 MBq), a 20 MBq group (n = 5, 21 ± 1.64 MBq), and a 30 MBq group (n = 5, 29 ± 1.06 MBq). The body weight of these mice was measured twice every week in grams (g) after treatment. The relative body weight (RBW) at day n was calculated according to the formula: RBW = body weight on day n/body weight on day 1. The length (L) and width (W) of the tumor using the same caliper were also recorded in millimeters (mm), and the volume of the tumor was then calculated (V = 0.5 × L × W2; mm3). The relative TV (RTV) was calculated according to the formula: RTV = TV on day n/TV on day 1. At the end of the study, all mice were euthanized, and the organs and tumors were preserved and tested.
Statistical analysis
The radioactivity data, as measured using the γ-counter, are presented as CPM with time–decay correction. All experimental results are presented as the mean ± SEM (standard error of the mean). All statistical analyses were performed with GraphPad 9.0 (Prism, United States). ANOVA was employed to examine statistical differences among groups. Survival analysis was conducted using the Kaplan–Meier method to compare the free-of-event rate among the groups. P-value <0.05 was considered statistically significant.
Results
Radiochemistry
The chemical structures of 68Ga-DOTA-PEG-PARPi and 177Lu-DOTA-PEG-PARPi are shown in Figure 2. Based on radio-HPLC analysis, the radiochemical purity is determined to be more than 95%.
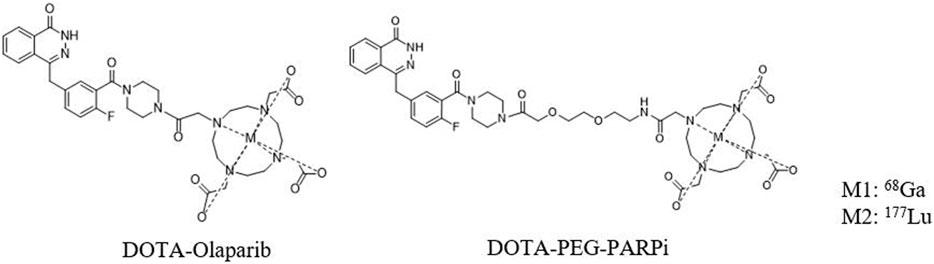
Figure 2. Structure of 68Ga/177Lu- DOTA-olaparib and DOTA-PEG-PARPi.
Binding affinity of 68Ga-DOTA-PEG-PARPi
The binding affinity of 68Ga-DOTA-olaparib and 68Ga-DOTA-PEG-PARPi for PARP-1 was determined in A549, AsPC-1, PSN-1, ISN-1, and U87MG cells, the results of which are shown in Figure 3. The IC50 values for compounds 68Ga-DOTA-olaparib and 68Ga-DOTA-PEG-PARPi in PSN-1 were 1.289E-08 and 9.810E-09, respectively. These results indicate that radiotracers have a high affinity in pancreatic cancer toward PARP-1.
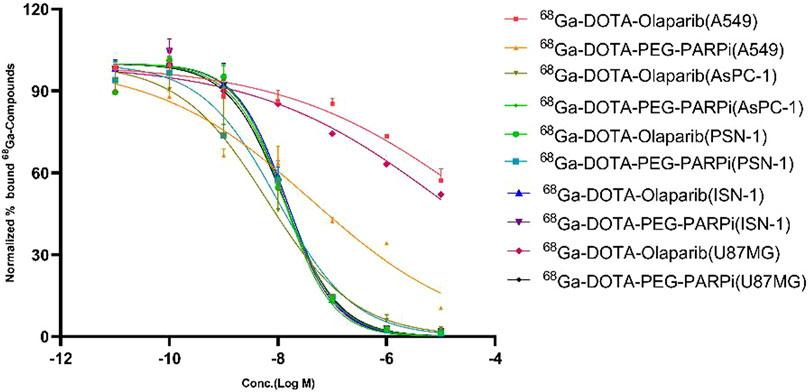
Figure 3. Results of binding affinity and uptake in vitro on DOTA-PEG-PARPi.
Cell uptake of 177Lu-DOTA-PEG-PARPi
As shown in Figure 4, cellular uptake also showed an increasing trend with the increase in time, reaching the maximum at 48 h. The uptake of 177Lu-DOTA-PEG-PARPi by A549, AsPC-1, PSN-1, ISN-1, and U87MG cells at 48 h was 9.45%, 13.42%, and 14.34%, and the uptake of 177Lu-DOTA-olaparib was 8.34%, 9.36%, 8.21%, 8.54%, and 8.46%, respectively. These results indicate that the radiotracer has high clinical application potential for PARP-1.
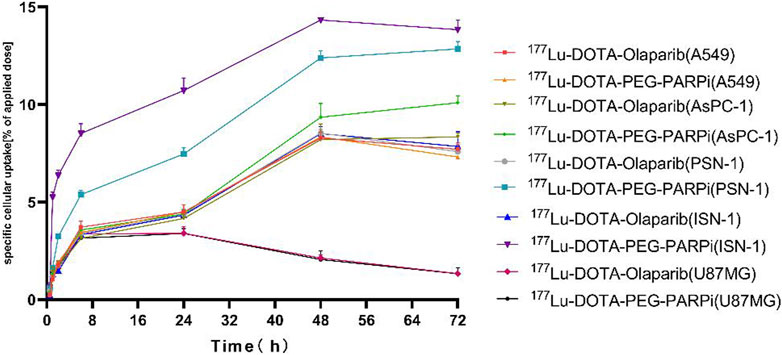
Figure 4. Results of cell uptake in vitro on 177Lu-DOTA-olaparib and 177Lu-DOTA-PEG-PARPi.
Stability of 177Lu-DOTA-PEG-PARPi
The results exhibited satisfactory in vitro stability for 177Lu-DOTA-PEG-PARPi at 1 and 24 h, as evaluated by HPLC analysis. As shown in Figure 5, greater than 95% of the prototype radiopharmaceutical had no degradation in saline (after 0, 4, 24, 48, and 72 h) and mouse plasma and human plasma (after 0 and 24 h) incubation. From the in vitro stability results, it can be concluded that 177Lu-DOTA-PEG-PARPi is stable enough to be used.
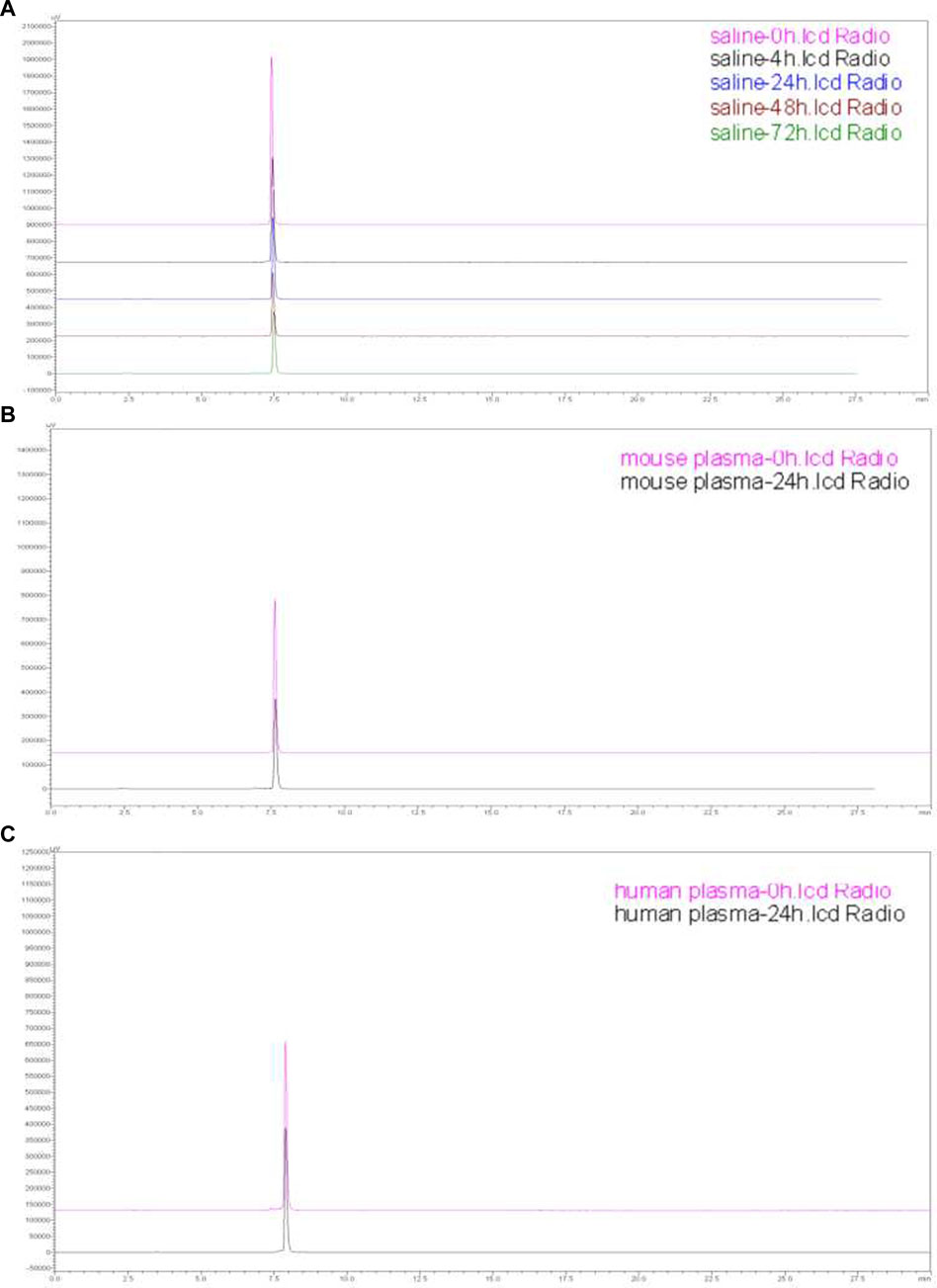
Figure 5. Stability of 177Lu-DOTA-PEG-PARPi. Stability of 177Lu-DOTA-PEG-PARPi in saline (0, 4, 24, 48, and 72 h) (A), mouse plasma (0 and 24 h) (B), and human plasma (0 and 24 h) (C).
Micro SPECT/CT imaging
68Ga-DOTA-PEG-PARPi PET/CT imaging demonstrated significant accumulation in the PSN-1 tumors (Figure 6). SPECT/CT imaging was performed at 48 h after injection of 177Lu-DOTA-PEG-PARPi (Figure 7A). 177Lu-DOTA-PEG-PARPi SPECT/CT imaging showed a significant concentration of radioactivity in the PSN-1 tumors (Figure 7B), suggesting high uptake and prolonged retention time in the tumor. The uptake of radioactive 177Lu-DOTA-PEG-PARPi in the PSN-1 models was approximately 8.88 ± 0.61%ID/cc (1 h), 9.73 ± 0.68%ID/cc (3 h), 11.48 ± 0.56%ID/cc (6 h), 6.29 ± 1.35%ID/cc (24 h), and 1.63 ± 0.38%ID/cc (48 h), as determined by quantitative calculations from the SPECT images. The T/M ratios of 177Lu-DOTA-PEG-PARPi were approximately 9.26 ± 1.37 (1 h), 10.37 ± 1.30 (3 h), 17.97 ± 1.67 (6 h), 41.80 ± 10.07 (24 h), and 21.88 ± 11.58 (48 h).
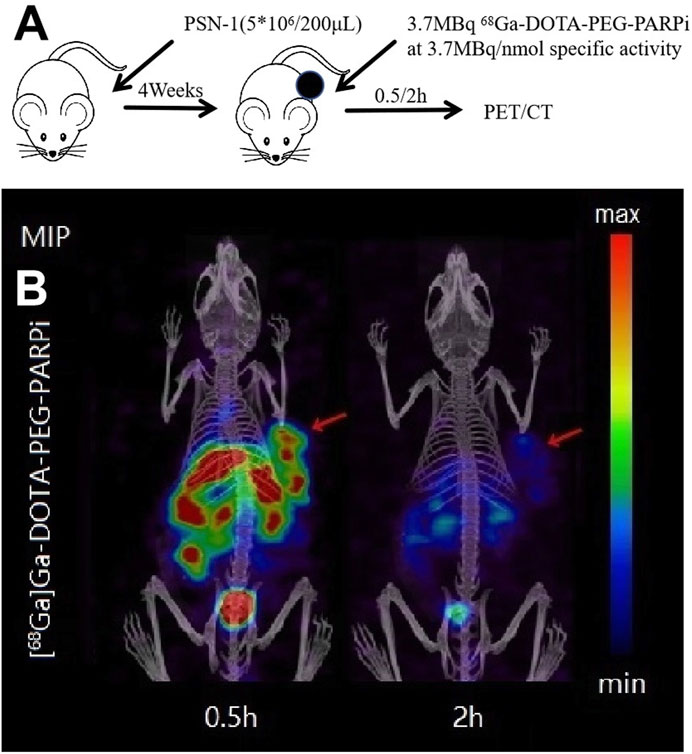
Figure 6. PET/CT result of 68Ga-DOTA-PEG-PARPi in PSN-1 models. Molecule imaging of 68Ga-DOTA-PEG-PARPi (A). Micro PET/CT images of 68Ga-DOTA-PEG-PARPi in PSN-1 models at 0.5, 1, and 2 h after administration (B).
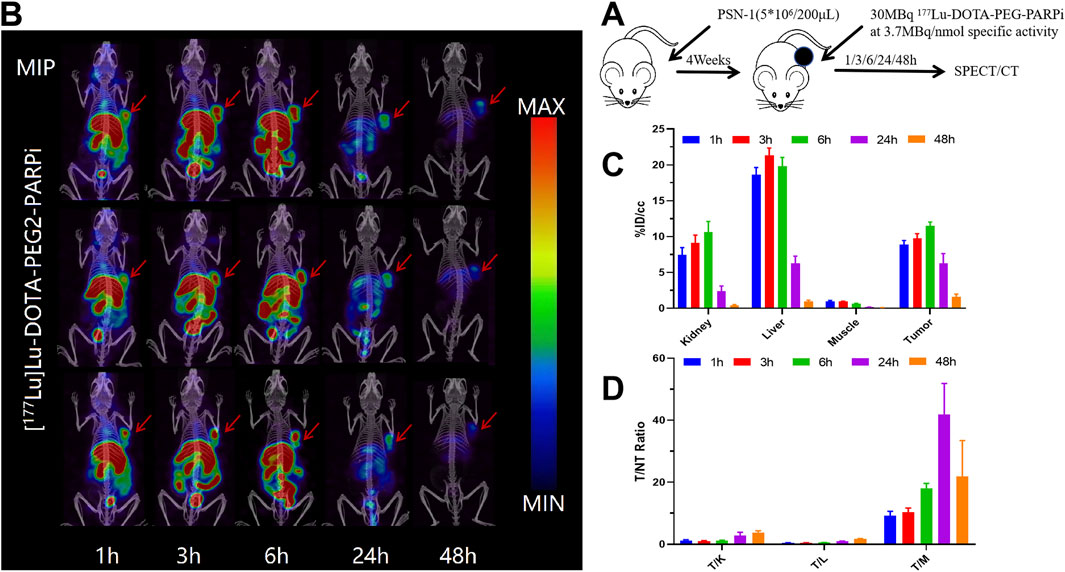
Figure 7. SPECT/CT result of 177Lu-DOTA-PEG-PARPi in PSN-1 models. Molecule imaging of 177Lu-DOTA-PEG-PARPi (A). Micro SPECT/CT images of 177Lu-DOTA-PEG-PARPi in PSN-1 models at 1, 3, 6, 24, and 48 h after administration (B). Ratios of tumor-to-non-target tissue (n = 3). Quantification of the regions of interest (ROIs) in PSN-1 models expressed as %ID/cc (C). T/M (tumor-to-muscle) ratios (D).
Ex vivo biodistribution of PSN-1 models and dosimetry estimates on adult humans
Female tumor xenograft Balb/c nude mice were randomized into BioD timepoint groups at study day 0 based on body weight. On study day 0, mice were intravenously administered 177Lu-DOTA-PEG-PARPi (30 μCi/animal) and euthanized at 3, 24, 48, 96, and 168 h after administration. Ex vivo distribution was evaluated by counting 1 min on a γ-counter, and dosimetry was assessed using OLINDA/EXM software.
The biodistribution of 177Lu-DOTA-PEG-PARPi in PSN-1 tumors and the major organs is shown in Figure 8A. Tumor uptake peaked at 3 h, with an absorption of 102.65% ID/g, and decreased to 40.25% ID/g at 168 h. The liver was the normal organ with the highest uptake, as shown in Figure 8B. Tumor-to-organ uptake ratios of 177Lu-DOTA-PEG-PARPi in PSN-1 models are shown in Figure 8C.
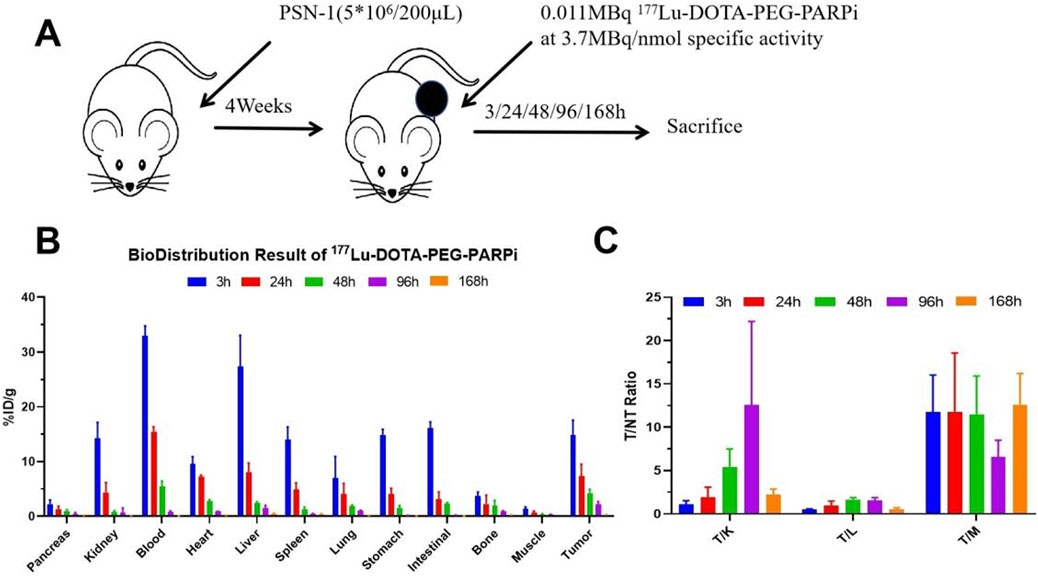
Figure 8. Biodistribution result of 177Lu-DOTA-PEG-PARPi in PSN-1 models. . Biodistribution of 177Lu-DOTAPEG-PARPi in PSN-1 models (*p = 0.046 < 0.05, n = 3) (A). Ratios of tumor-to-non-target tissue (n = 3). Quantification of the regions of interest (ROIs) in PSN-1 models expressed as %ID/g (B). T/M (tumor-to-muscle) ratios (C).
As a result, for an adult female (ICRP 103 adult female), the Spleen exhibited the highest absorbed dose (5.52E-03mGy/MBq), followed by the Red Marrow, lung, and Kidneys (4.36E-03, 2.33E-03, and 2.00E-03 mGy/MBq, respectively). The effective dose was 2.00E-02 mSv/MBq (Table 1).
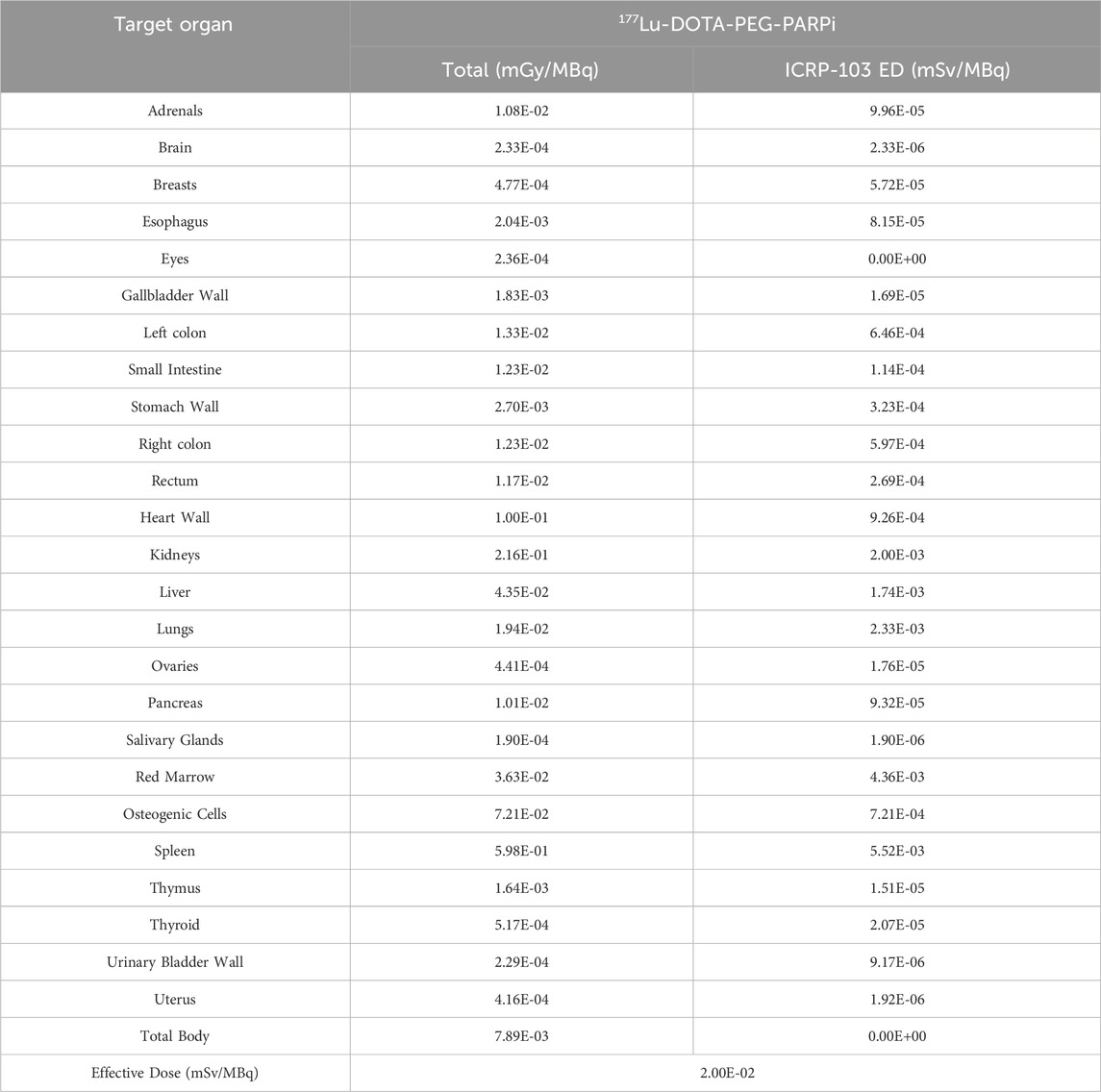
Table 1. The extrapolated dose estimation data and tumor dose estimation data can be included in the supplementary materials.
Effects of 177Lu-DOTA-PEG-PARPi treatment on PSN-1 tumor-bearing mice
The in vivo activity of 177Lu-DOTA-PEG-PARPi was evaluated in a PSN-1 pancreatic cancer xenograft model. Mice were administered 177Lu-DOTA-PEG-PARPi via tail vein injection of either vehicle or 10 MBq, 20 MBq, or 30 MBq total activity with a specific activity of 3.7 mBq/nmol (Figure 9A). The mean tumor volume of the vehicle control reached 2,041 mm3 after 26 days of treatment (day 28 after tumor inoculation). 177Lu-DOTA-PEG-PARPi at 30 mBq/mice as a single dose exhibited moderate antitumor activity, with a final TV of 919 mm3 and a TGI value of 51.56%, which were significantly different from those of the vehicle control group (p < 0.05). 177Lu-DOTA-PEG-PARPi at 20 mBq/mice and 10 mBq/mice as single doses demonstrated lower antitumor activity, with final TVs of 1,365 mm3 and 1,521 mm3 and TGI values of 28.07% and 19.76%, respectively, which were significantly different from those of the vehicle control group (p < 0.05) (Figure 9B). No weight loss was observed in the surviving animals (Figure 9C).
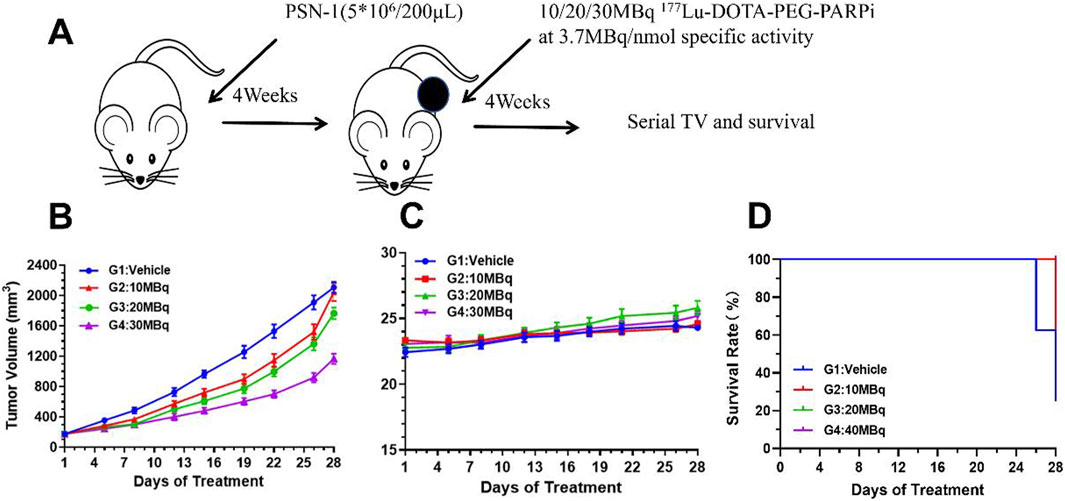
Figure 9. Efficacy result of 177Lu-DOTA-PEG-PARPi in PSN-1 models. The effects of 177Lu-DOTA-PEG-PARPi treatment on PSN-1 tumor-bearing mice. (A) Biodistribution of 177Lu-DOTA-PEG-PARPi in PSN-1 tumor-bearing mice (*p < 0.05, n = 3). (B) Changes in the relative body weight. (C) Changes in the relative lesion size (*p < 0.05, n = 3). (D) Kaplan–Meier curves for four groups (*p < 0.05, n = 3).
Discussion
As a key tool for real-time monitoring of the status of tumor DNA damage repair, PARP-1 PET imaging agents have made remarkable progress in recent years in aspects such as the development of novel imaging agents (e.g., [18F]-PARPZ), personalized treatment strategies (screening patients sensitive to PARP inhibitors), and the combination of multimodal imaging techniques. However, their clinical application still faces challenges such as insufficient specificity (non-specific uptake in normal tissues), complexity of clinical translation (interference from tumor heterogeneity), the absence of standardized quantitative approaches, high costs, and limitations in accessibility. In the future, it is essential to optimize the structure of the imaging agents, combine dynamic imaging with AI analysis, and promote multicenter clinical trials to achieve their widespread application in precision cancer treatment (Boinapally et al., 2023; Cao et al., 2023; Kong et al., 2022; Kostos et al., 2022; Zhang et al., 2024; Zheng et al., 2023).
In the study, we used the PARP-1 inhibitor olaparib as the parent structure to successfully synthesize a novel probe capable of chelating medical radiometals (68Ga and 177Lu). Its potential as a targeted PARP-1 probe in diagnosis and treatment was evaluated through in vitro and in vivo druggability studies. The objective of our study is to develop a PARP-1 theranostic probe with clinical application potential, providing tools for patient screening, therapeutic response prediction, quantitative evaluation of therapeutic effect, and new therapeutic approaches directly targeting PARP-1. To address the instability issues associated with 18F-labeled PARPi probes in vivo, this study utilized the radiometals 68Ga and 177Lu to label PARPi. These two radionuclides exhibit excellent in vivo stability and suitable physical properties, with 68Ga emitting positrons suitable for PET imaging and 177Lu-emitting β-rays suitable for radiotherapy. This labeling strategy aims to develop a PARPi probe with high stability and strong affinity, exploring its clinical application potential.
Additionally, polyethylene glycol (PEG) modification was employed during the labeling process to optimize the physicochemical properties of the probe. PEG modification enhances the water solubility of the probe and reduces immunogenicity and non-specific protein binding, thereby decreasing the liver and spleen uptake and improving the probe biodistribution and tumor-targeting specificity. The experimental results demonstrated that the PEG-modified PARPi probe not only retained very high affinity (IC50 = 6.491E-09) but also significantly reduced liver accumulation in vivo, which is critical for minimizing potential side effects and improving therapeutic safety. To evaluate the efficacy and safety of the novel PARPi probes in vivo, tumor models were established by transplanting these cell lines into immunodeficient mice. These mice were then administered 177Lu- and 68Ga-labeled PARPi probes, and changes in tumor size and body weight were monitored to assess the probes’ tumor-inhibitory effects in vivo. The results showed that both labeled PARPi probes exhibited significant tumor growth inhibition, with the 177Lu-labeled probe demonstrating greater therapeutic potential. In summary, this study developed a novel, stable, and effective PARP-1-targeted probe and validated its potential therapeutic effects in animal models. Future research will focus on further optimizing the probe design to enhance safety and efficacy, ultimately advancing toward clinical application.
Despite the significant progress achieved in this study, several limitations remain. First, the in vitro experiments were conducted with a limited sample size, and key characterizations, such as pharmacokinetics, were not thoroughly analyzed in relation to the drug structure and lipid–water partition coefficients. Second, comprehensive toxicity studies were not conducted, and the maximum tolerated dose (MTD) in animals was not determined, leaving the safety assessment incomplete. Finally, since animal models cannot fully replicate the conditions of pancreatic cancer patients, particularly when pancreatic lesions are located near the liver, β-emitting radionuclides may cause crossfire effects on liver tissue. Consequently, the actual liver dose might have been underestimated in animal experiments, and the available dose estimates may not provide comprehensive information for clinical translation. These challenges need to be addressed in subsequent studies to facilitate the clinical application of PARP-1-targeted probes. With continuous optimization and refinement, it is anticipated that PARP-1-targeted probes can be developed into a precise and effective tool to benefit cancer patients.
Conclusion
In the study, we introduced a chelating group into the lead compound 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one, thus obtaining olaparib derivatives capable of chelating diagnostic and therapeutic radionuclides (gallium-68 and lutetium-177). After radionuclide labeling, a diagnostic and therapeutic probe targeting PARP-1 was obtained. The results showed that lutetium-177-DOTA-polyethylene glycol-PARPi (177Lu-DOTA-PEG-PARPi), gallium-68-DOTA-polyethylene glycol-PARPi (68Ga-DOTA-PEG-PARPi), and polyethylene glycol-PARPi (PEG-PARPi) had high chemical yields, radiochemical yields, and purities. The imaging results demonstrated that 68Ga-DOTA-PEG-PARPi showed significant accumulation in PSN-1 tumors, while 177Lu-DOTA-PEG-PARPi had significant uptake in tumors and a prolonged retention time. This study has demonstrated that PEG-PARPi has the potential to be a promising radiotracer for further clinical applications and, ultimately, for the personalized treatment of pancreatic cancer patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the China Institute for Radiation Protection. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JG: writing–original draft. YW: writing–original draft. RW: writing–original draft. ML: writing–original draft. HW: writing–review and editing. JL: writing–review and editing. JD: writing–review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Shanxi Province (202303021212385, 202303021212386).
Conflict of interest
Author JD was employed by China Isotope and Radiation Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bhamidipati, D., Haro-Silerio, J. I., Yap, T. A., and Ngoi, N. (2023). PARP inhibitors: enhancing efficacy through rational combinations. Br. J. Cancer 129 (6), 904–916. doi:10.1038/s41416-023-02326-7
Bhin, J., Paes Dias, M., Gogola, E., Rolfs, F., Piersma, S. R., de Bruijn, R., et al. (2023). Multi-omics analysis reveals distinct non-reversion mechanisms of PARPi resistance in BRCA1-versus BRCA2-deficient mammary tumors. Cell Rep. 42 (5), 112538. doi:10.1016/j.celrep.2023.112538
Boinapally, S., Alati, S., Jiang, Z., Yan, Y., Lisok, A., Singh, R., et al. (2023). Preclinical evaluation of a new series of albumin-binding (177)Lu-labeled PSMA-based low-molecular-weight radiotherapeutics. Molecules 28 (16), 6158. doi:10.3390/molecules28166158
Cao, Q., Li, L., Zhao, Y., Wang, C., Shi, Y., Tao, X., et al. (2023). PARPi decreased primary ovarian cancer organoid growth through early apoptosis and base excision repair pathway. Cell Transpl. 32, 9636897231187996. doi:10.1177/09636897231187996
Chen, M., Linstra, R., and Van Vugt, M. (2022). Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 1877 (1), 188661. doi:10.1016/j.bbcan.2021.188661
Colombo, I., Lheureux, S., and Oza, A. M. (2018). Rucaparib: a novel PARP inhibitor for BRCA advanced ovarian cancer. Drug Des. Devel Ther. 12, 605–617. doi:10.2147/DDDT.S130809
Cortesi, L., Rugo, H. S., and Jackisch, C. (2021). An overview of PARP inhibitors for the treatment of breast cancer. Target Oncol. 16 (3), 255–282. doi:10.1007/s11523-021-00796-4
Dibitetto, D., Widmer, C. A., and Rottenberg, S. (2024). PARPi, BRCA, and gaps: controversies and future research. Trends Cancer 10 (9), 857–869. doi:10.1016/j.trecan.2024.06.008
Ke, B., Li, A., Fu, H., Kong, C., Liu, T., Zhu, Q., et al. (2022). PARP-1 inhibitors enhance the chemosensitivity of leukemia cells by attenuating NF-кB pathway activity and DNA damage response induced by Idarubicin. Acta Biochim. Biophys. Sin. (Shanghai) 54 (1), 91–98. doi:10.3724/abbs.2021011
Kong, L., Xu, J., Yu, L., Liu, S., Liu, Z., and Xiang, J. (2022). Construction of PARPi resistance-related competing endogenous RNA network. Curr. Genomics 23 (4), 262–274. doi:10.2174/1389202923666220527114108
Kostos, L., Buteau, J. P., Yeung, T., Iulio, J. D., Xie, J., Cardin, A., et al. (2022). AlphaBet: combination of Radium-223 and [(17) (7)Lu]Lu-PSMA-I&T in men with metastatic castration-resistant prostate cancer (clinical trial protocol). Front. Med. (Lausanne) 9, 1059122. doi:10.3389/fmed.2022.1059122
Li, W. H., Wang, F., Song, G. Y., Yu, Q. H., Du, R. P., and Xu, P. (2023). PARP-1: a critical regulator in radioprotection and radiotherapy-mechanisms, challenges, and therapeutic opportunities. Front. Pharmacol. 14, 1198948. doi:10.3389/fphar.2023.1198948
Mccann, K. E., and Hurvitz, S. A. (2018). Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 7, 212540. doi:10.7573/dic.212540
Mcdonald, E. S., Pantel, A. R., Shah, P. D., Farwell, M. D., Clark, A. S., Doot, R. K., et al. (2021). In vivo visualization of PARP inhibitor pharmacodynamics. JCI Insight 6 (8), e146592. doi:10.1172/jci.insight.146592
Reiner, T., Keliher, E. J., Earley, S., Marinelli, B., and Weissleder, R. (2011). Synthesis and in vivo imaging of a 18F-labeled PARP1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew. Chem. Int. Ed. Engl. 50 (8), 1922–1925. doi:10.1002/anie.201006579
Safdar, R., Mishra, A., Shah, G. M., and Ashraf, M. Z. (2024). Poly (ADP-ribose) Polymerase-1 modulations in the genesis of thrombosis. J. Thromb. Thrombolysis 57 (5), 743–753. doi:10.1007/s11239-024-02974-3
Slade, D. (2020). PARP and PARG inhibitors in cancer treatment. Genes Dev. 34 (5-6), 360–394. doi:10.1101/gad.334516.119
Soung, Y. H., and Chung, J. (2023). Combination treatment strategies to overcome PARP inhibitor resistance. Biomolecules 13 (10), 1480. doi:10.3390/biom13101480
Steffen, J. D., Tholey, R. M., Langelier, M. F., Planck, J. L., Schiewer, M. J., Lal, S., et al. (2014). Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res. 74 (1), 31–37. doi:10.1158/0008-5472.CAN-13-1701
Wang, X., Liu, W., Li, K., Chen, K., He, S., Zhang, J., et al. (2023). PET imaging of PARP expression using (68)Ga-labelled inhibitors. Eur. J. Nucl. Med. Mol. Imaging 50 (9), 2606–2620. doi:10.1007/s00259-023-06249-6
Wang, Y., Luo, W., and Wang, Y. (2019). PARP-1 and its associated nucleases in DNA damage response. DNA Repair (Amst) 81, 102651. doi:10.1016/j.dnarep.2019.102651
Young, A. J., Pantel, A. R., Kiani, M., Doot, R. K., Bagheri, S., Pryma, D. A., et al. (2024). Kinetic analysis and metabolism of poly(adenosine diphosphate-ribose) polymerase-1-targeted (18)F-fluorthanatrace PET in breast cancer. J. Nucl. Med. 65, 1862–1868. doi:10.2967/jnumed.124.268254
Zhang, J., Lin, Y., Gao, J., Pan, Y., Hou, G., Guo, C., et al. (2024). Development and biological evaluation of (68)Ga-labeled peptides for potential application in HER2-positive colorectal cancer. Bioorg Chem. 151, 107645. doi:10.1016/j.bioorg.2024.107645
Zheng, Y., Long, T., Peng, N., Zhen, M., Ye, Q., Zhang, Z., et al. (2023). The value of targeting CXCR4 with 68Ga-pentixafor PET/CT for subtyping primary aldosteronism. J. Clin. Endocrinol. Metab. 109 (1), 171–182. doi:10.1210/clinem/dgad421
Keywords: poly ADP-ribose polymerase inhibitor, radioactive probes, pancreatic cancer, preclinical studies, radiopharmaceutical
Citation: Gao J, Wang Y, Wang R, Liu M, Wang H, Li J and Du J (2025) Preclinical studies of a PARP-targeted theranostic radiopharmaceutical for pancreatic cancer. Front. Pharmacol. 16:1576587. doi: 10.3389/fphar.2025.1576587
Received: 14 February 2025; Accepted: 17 March 2025;
Published: 15 May 2025.
Edited by:
Dong Dai, Tianjin Medical University Cancer Institute and Hospital, ChinaReviewed by:
Yiliang Li, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaKuan Hu, China Academy of Chinese Medical Sciences, China
Copyright © 2025 Gao, Wang, Wang, Liu, Wang, Li and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Du, ZHVqaW5AY2lyYy5jb20uY24=