 Tao Zhou1
Tao Zhou1 Haichuan Wang2*
Haichuan Wang2*- 1Department of Pharmaceutical and Medical equipment, Bayi Orthopedic Hospital, China RongTong Medical Healthcare Group Co. Ltd., Chengdu, China
- 2Department of Paediatrics, Sichuan Academy of Medical Science & Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
The homeostasis of glutamate, the primary excitatory neurotransmitter in the brain and is crucial for normal brain function. The mitochondrial enzyme glutamate dehydrogenase (GDH) connects the multifunctional amino acid glutamate, which is intimately related to glutamate metabolism, to the Krebs cycle. As a result, GDH reglutes the synthesis and uptake of the chemical messenger glutamate in neuroendocrine cells, playing a crucial role in the metabolism of proteins and carbohydrates. Nonetheless, brain ageing and numerous neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease, have been linked to GDH malfunction or dysregulation. In this review, we summarize the dynamics of GDH levels in the ageing brain and provide additional details about the role of GDH in the ageing brain. Understanding the metabolic mechanisms underlying glutamate homeostasis in the aging brain and how GDH regulates glutamate-dependent metabolic processes at synapses may lead to novel therapeutic approaches for neurodegenerative and psychiatric disorders, potentially slowing the aging process and promoting brain regeneration.
Introduction
Glutamate dehydrogenase (GDH) is expressed mostly in the brain, liver, kidney, and pancreas and is located in the mitochondrial matrix of humans. As a hexameric structure, functional GDH consists of two sets of trimers, each containing approximately 500 amino acids. Each monomer has a binding site for guanosine triphosphate (GTP) and another for nicotinamide adenine diphosphate hydride (NADH)/adenosine diphosphate (ADP); this structure allows six GTP molecules, six ADP molecules, and six NADH molecules to bind to the GDH hexamer at its allosteric binding sites. When a substrate interacts with two GDH trimers, a projecting domain that resembles an antenna connects the subunits (Nassar et al., 2019).
Numerous small molecules allosterically regulate GDH in animals. GTP and ATP are the primary allosteric inhibitors. GTP binds to the allosteric domain above the catalytic domain at the bottom of the antenna to block GDH catalytic activity and stop the release of glutamate oxidative products, which is a rate-limiting step. By inhibiting the release of NAD(P)H, an oxidative product of glutamate, NADH can bind to the NADH/ADP binding site and strengthen the inhibitory effect of GTP on GDH activity. Furthermore, diethylstilbestrol (DES), palmitoyl-CoA, and steroid hormones are also inhibitors of mammalian GDH, although the sites where they bind are unclear. Finally, green tea contains a polyphenol called epigallocatechin gallate (EGCG), which likewise functions as an allosteric inhibitor by competing with ADP (Li et al., 2014). In contrast, leucine and ADP are the main allosteric activators of GDH. In contrast to GTP, ADP activates GDH. By binding to the NADH/ADP binding site and preventing GTP binding, ADP can activate GDH. Leucine is both an allosteric activator and a catalytic substrate of GDH; although it has a distinct binding site, its activation mechanism is comparable to that of ADP (Li et al., 2014).
GDH expression and function
GDH activity varies greatly in various mammalian tissues (Botman et al., 2014; Treberg et al., 2014). High enzyme levels are present in the liver, brain, kidney, pancreas, adrenal glands, and placenta (Rothe and Storm-Mathisen, 1995; Spanaki et al., 2014) (Figure 1). The liver, where the enzyme comprises approximately 1% of the total protein, has the highest GDH-specific activity; GDH activity is lower in other mammalian organs (Botman et al., 2014; Treberg et al., 2014). According to estimates, the GDH level in the human kidney and brain is between 20% and 25% of that in the liver (Spanaki et al., 2010). Recently, studies have employed IHC to examine human tissues using antibodies that detect both human GDH isoenzymes. They discovered that all of the hepatocytes in the human liver express GDH quite abundantly. All cells of the pancreatic parenchyma in humans, including acinar cells and the endocrine cells of Langerhans islets, express GDH (Spanaki et al., 2014). In addition, the epithelial cells of the proximal convoluted tubules are where GDH is mostly localized in the human renal cortex (Spanaki et al., 2014; Spanaki et al., 2012). Finally, GDH is present in both Sertoli and Leydig cells in the human testis (Spanaki et al., 2014; Spanaki et al., 2010). However, spermatogonia, spermatocytes, and spermatozoa, i.e., germ cells, are negative for GDH expression (Spanaki et al., 2014; Spanaki et al., 2010).
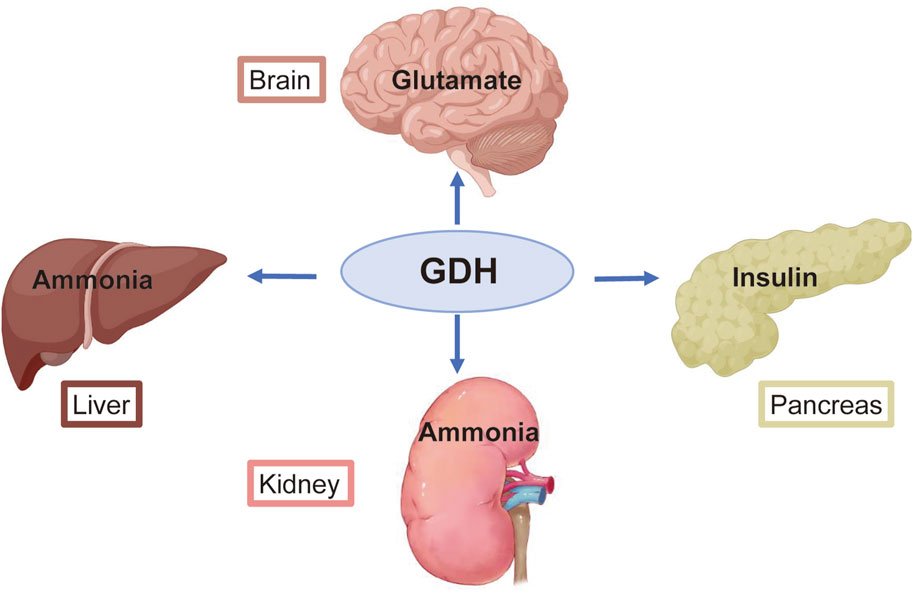
Figure 1. Primary roles of glutamate dehydrogenase (GDH) in several organs. GDH helps the brain recycle glutamate and regulates the production of the primary excitatory neurotransmitter. GDH is essential for the metabolism of ammonia in the liver and kidney. GDH activity affects the rate of insulin release from pancreatic β-cells.
Under normal circumstances, GDH is a major regulator of the metabolism of amino acids and ammonia in the human pancreas, liver, and brain. GDH uses NAD(P)+ as a coenzyme to catalyze the oxidative deamination of glutamate to produce alpha-ketoglutaric acid (α-KG) and ammonia via the following reversible reactions: glutamate + NAD(P)+ ↔ α-KG + NH3 + NAD(P)H. α-KG is used in the tricarboxylic acid (TCA) cycle to produce adenosine triphosphate (ATP) (Figure 2). In islet β cells, the brain, and renal tubular cells, the reaction is directed primarily towards the oxidative deamination of glutamate because these cells and tissues contain high glutamate and low α-KG/NH3 levels (Komlos et al., 2013).
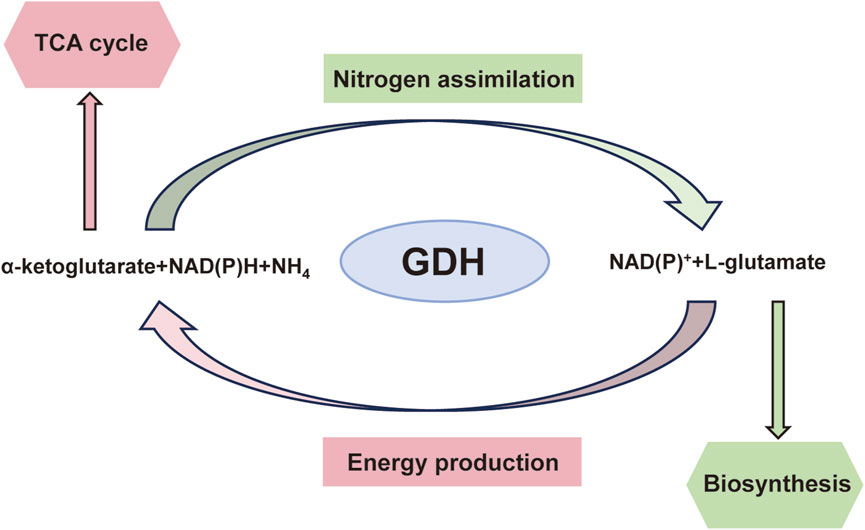
Figure 2. Diagrammatic representation of glutamate dehydrogenase-catalysed processes and their overall metabolic importance.
GDH aids in preserving the NH4+ concentration in the kidneys and several other organs (Cooper and Jeitner, 2016; Treberg et al., 2010). Additionally, the enzyme plays a role in the mechanism by which pancreatic β-cells secrete insulin (Sener et al., 1980). GLUD1 mutations reduce the sensitivity of the enzyme to its inhibitor GTP, which fuels hyperinsulinaemia/hyperammonaemia (HI/HA) syndrome, a condition characterized by elevated blood NH4+ levels and low glucose levels (De Lonlay et al., 2001; MacMullen et al., 2001). Pancreatic β-cells expressing mutant GDH produce 2-oxoglutarate in a dysregulated manner, which is linked to the release of insulin (Fahien and Macdonald, 2011).
Mutations in short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD), which is involved in the oxidation of fatty acids, can also result in hyperinsulaemia and hypoglycaemia (Kapoor et al., 2009; Molven et al., 2004). Recently, GDH from pancreatic islets was shown to be bound by SCHAD, which inhibits its activity (Li et al., 2010). However, mutated SCHAD is incapable of binding to GDH. The subsequent activation of pancreatic GDH causes incorrect insulin release (Li et al., 2010), comparable to that observed with GLUD1 mutations, which impair GDH inhibition by GTP (Nissim, 1999; Treberg et al., 2010).
Research on ADP-ribosylation suggested that this alteration also plays a part in mediating the effect of GDH on insulin synthesis. For example, GDH in pancreatic β-cells is rendered inactive by SIRT4-dependent ADP-ribosylation, which restricts the release of insulin (Haigis et al., 2006). Under low-glucose conditions, the inactivation of GDH may be reversed, leading to an increase in blood insulin levels and pancreatic GDH activity. Under calorie restriction, reduced ADP-ribosylation of GDH was observed, which was linked to increased insulin levels and increased GDH activity (Guan and Xiong, 2011).
Genetics of GDH
The well-preserved 45-kb gene GLUD1, which is divided into 13 exons, encodes GDH (Michaelidis et al., 1993). While GLUD1, which is found only in hominoids, is expressed in multiple tissues (Plaitakis et al., 1980), its isoform, GLUD2, is expressed exclusively in the brain and testicular tissues (Shashidharan et al., 1994). The sole isoform in rodents that encodes GDH is GLUD1 (Burki and Kaessmann, 2004). Recent studies have revealed that astrocytes and testicular supporting cells exhibit significant levels of GLUD2 expression (Spanaki et al., 2010). GLUD2 is an intronless X-linked gene (Shashidharan et al., 1994) that evolved fewer than 23 million years ago via the retrotransposition of a spliced mRNA from the GLUD1 gene, which has an intron, in hominoid progenitors. The alterations in amino acids that confer the distinct brain-specific characteristics of the GLUD2-derived enzyme occurred when the size of the brains of ancestors of humans and great apes increased. By permitting greater neurotransmitter transit and clearance, GLUD2 may have contributed to improved brain function in both humans and apes. Therefore, GLUD2 may have played a role in hominoid evolution and the development of an increased cognitive capacity (Burki and Kaessmann, 2004).
The pH dependence of human GDH isoenzymes (hGDH1 and hGDH2), which are encoded by distinct genes (GLUD1 and GLUD2), varies. In the 2-oxoglutarate amination process, hGDH2 shifts the pH optimum from 8.0 to 7.5, in contrast to hGDH1 (Kanavouras et al., 2007; Plaitakis et al., 2003). This distinction is thought to be significant in astrocytes, whose absorption of synaptic glutamate linked to OH ion counter-transport acidifies their cytoplasm and mitochondrial matrix (Poitry et al., 2000).
ADP activates both isoenzymes; however, ADP activation of hGDH2 is more noticeable. The two enzymes also differ in their nucleotide-dependent regulation (Plaitakis et al., 2000). The amplitude of the effect at saturating ADP concentrations (approximately 1 mM) is ten times greater for hGDH2 than for hGDH1; specifically, the SC50 value for ADP, the ADP concentration eliciting 50% of the maximum activation, is roughly three times higher for hGDH2 (58.7 µM) than for hGDH1 (17.0 µM) (Kanavouras et al., 2007). Similarly, the impact of leucine, another well-known allosteric activator, is approximately ten times greater for hGDH2 than for hGDH1, even though the two hGDH isoenzymes' SC50 values and their decreased activity with the addition of ADP are comparable (Kanavouras et al., 2007).
Cho et al. reported the kinetics of the two different GDH forms from the bovine brain (bGDH1 and bGDH2) (Cho et al., 1995). The authors use the terms isoform and isoprotein as synonyms for bGDH1 and bGDH2. We refer to these GDHs as the enzyme forms since the structural information to categorize them as isoforms or isoenzymes using the previously stated categories is currently insufficient. bGDH1 and bGDH2 were separated from the whole homogenate (Cho et al., 1995). The kinetic characteristics of bGDH1 and bGDH2 were measured at a fixed ADP concentration (1 mM) and are similar to those of the hGDH1 and hGDH2 isoenzymes at intermediate ADP saturation (0.1–0.25 mM). Specifically, both hGDH1 and bGDH1 have a higher Km for glutamate and a lower Km for 2-oxoglutarate than do hGDH2 and bGDH2. The two human and bovine isoenzymes also exhibit the same differences in the Km for ammonium, and under similar conditions, allosteric activation by ADP was more evident for bGDH2 than for hGDH2 (Cho et al., 1995). In addition, the high sensitivity of the hGDH2 isoenzyme to dilution, as previously mentioned, is also reflected in the higher Vmax for bGDH1 than for bGDH2 (202 and 124 μmol/min per mg of protein, respectively, in the presence of 1 mM ADP). Overall, kinetic studies indicate that the two enzyme forms of GDH identified in the bovine brain are functionally similar to the two human GDH isoenzymes.
GDH in brain development
Since glutamate is the primary excitatory neurotransmitter in the central nervous system (CNS), mitochondrial function and glutamate metabolism are closely related in the brain (Baudry and Lynch, 1979). Specifically, mitochondria control the energy state of the brain, the use of metabolic substrates, and the detoxification of excess neurotransmitters.
Target neurons are depolarized by glutamate produced by active synapses via certain receptors (Mayer et al., 1984; Spreafico et al., 1994). Glutamate is a quick-acting neurotransmitter, but it also has long-lasting effects on the structure and function of neurons as a signalling molecule. Glutamate neurotransmission plays a significant role in modulating synaptic activity during nervous system development (Cohen and Greenberg, 2008; Ghiani et al., 2007), as well as in learning and memory construction and the gathering and storing of new knowledge (Cotman et al., 1988; McEntee and Crook, 1993).
Enzyme histochemistry has shown that GDH is present in rat hippocampal dendritic layers and that GDH activity increases in tandem with the postnatal maturation of synaptic structures (Schünzel and Wolf, 1982). Similar increases in neuronal GDH staining were observed in subsequent studies of the rat hippocampal region during postnatal development, namely, in the stratum lacunosum-moleculare and the molecular layer of the dentate gyrus in the hippocampus (Kugler and Schleyer, 2004; Rothe and Schünzel, 1990). Significant increases in GDH levels have also been observed in the rat cerebellum in the late postnatal stage, an increase that is believed to be a reflection of synaptogenesis and neuronal development (Wolf and Schünzel, 1987; Wolf et al., 1986). In particular, GDH activity increases in the cerebellar cortex in the molecular layer, granule cell bodies, internal granule cell layer, cerebellar glomeruli, and Purkinje cell perikarya to a lesser extent. Studies that have measured GDH activity and exogenous glutamate use in primary neuronal cultures from the rat ventral mesencephalon have shown that GDH activity is approximately four times higher in mature (12-day-old) cultures than in immature (4-day-old) cultures and have identified a significant correlation between exogenous glutamate use (added to the medium at 1.2 mM) and GDH activity in cultured cells (Plaitakis and Shashidharan, 2000). Furthermore, compared with mature cultures, immature cultures secrete significantly more alanine and aspartate into the medium, indicating that the transamination route is the primary mechanism by which exogenous glutamate is metabolized in these cells. Together with the previously mentioned results showing significant increases in GDH activity in the growing brain, these observations imply that the growth of nerve terminals and synaptic connections coincides with significant increases in glutamate use (away from transamination).
GDH in the ageing brain
Developmental synaptic pruning, which is essential for honing neurons, progressively results in cumulative, nonpathological synaptic degradation over time, which has an irreversible effect on all maturing organisms. The capacity of an individual to successfully adjust to their surroundings is inevitably hampered by this natural ageing process (Diniz and Crestani, 2023). Ageing is a major risk factor for the development of mental and neurological diseases (Cole et al., 2019; Hou et al., 2019). Changes in neuroplasticity or cellular changes that directly affect plasticity pathways may be partially responsible for the loss of cognitive performance associated with ageing. Numerous studies have shown that as people age, the glutamatergic system changes, which can result in synaptic dysfunction, neuronal damage, or even death (Cox et al., 2022; Segovia et al., 2001). Chronic inflammation plays a major role in neurodegenerative disorders and brain ageing (Godbout and Johnson, 2009; Hirsch and Hunot, 2009). Accordingly, research has shown that as mice age, both wild-type (Wt) and transgenic (Tg) mice with CNS-Glud1 overexpression may exhibit elevated expression of genes linked to neuroinflammation (Wang et al., 2014). Another study revealed that only Tg model mice exhibited a chronic inflammatory response when the hippocampal regions of 9-month-old CNS-Glud1 Tg and WT mice were examined (Wang et al., 2010). These findings could suggest a possible connection between neuroinflammation in the ageing brain and variations in GDH expression.
As mentioned previously, GDH activity fluctuates throughout life and differs across various brain regions. Modest GDH activity is detected in the central and peripheral nervous systems of rats at birth. Nonetheless, GDH activity increases significantly during the postnatal developmental stage, with earlier increases in the medulla and a rough doubling in the cerebellar cortex and fivefold increase in the hippocampus (Rothe et al., 1983). The development of brain regions with high glutamatergic activity results in elevated GDH activity (Rothe et al., 1983). On the other hand, a different study revealed that as male albino rats aged, the tissues throughout the brain presented a decrease in GDH activity (Rajeswari and Radha, 1984). GDH reactivity in the hippocampal regions of juvenile, adult, and elderly rats has also been assessed. In young rats, the stratum oriens of the CA1–CA4 regions in the hippocampus has the highest enzymatic reactivity. The molecular layer of the fascia dentata and the layer of mossy fibres are next in descending order. In practically every stratum analysed, adult rats exhibit stronger enzymatic reactivity than do juvenile rats. Except for the stratum oriens of the CA1–CA4 fields, where enzymatic reactivity is higher in older rats than in adult rats, GDH reactivity is lower in older rats than in adult rats (Bronzetti et al., 1990). Similarly, previous research has shown that in rats, GDH activity is detected earlier in the medulla oblongata and pons than in other brain regions (Leong and Clark, 1984). In adults, these areas present the greatest activity, with the midbrain and hypothalamus being the second and third most affected areas, respectively.
Studies in humans and rodents have consistently shown a loss of synaptic connections and neurons along glutamatergic neurotransmission pathways with aging (Francis, 2003; Morrison and Hof, 2007). These losses mostly impact areas of the brain, such as the cerebral cortex and hippocampus, which are also affected in Tg mice that overexpress Glud1. Because glutamate transport into glial cells or neurons is reduced in the brains of old mice, the loss of synapses and neurons in these areas results in increased amounts of extracellular glutamate (Nickell et al., 2005; Zoia et al., 2004). Tg mice overexpressing Glud1 have been used to investigate the connection between the ageing process and glutamate-induced metabolic responses (Choi et al., 2014). The findings of that study revealed that the hippocampus and striatum of Tg mice differed from those of WT mice at different ages in terms of the amount of neurochemicals, including lactate (Choi et al., 2014).
Neurons appear to be specifically susceptible to glutamate release and overproduction, as well as ageing. Wang et al. reported that Glud1 Tg and WT mice have different gene expression patterns (Wang et al., 2014); additionally, regardless of whether a mouse was Tg or WT, dynamic changes in the expression of certain genes in the hippocampus were observed during ageing through comparisons across several age periods spanning from 10 days to 20 months of life. Overall, the hippocampal transcriptome was more significantly affected by Glud1 overexpression in mature and aged mice than in young mice. Importantly, the Gene Ontology (GO) categories of genes with altered expression in Tg mice overlapped with a significant number of GO categories of genes with altered expression in WT mice during ageing, even though the changes in gene expression in the hippocampus of Tg and WT mice did not always follow the same pattern. The expression of genes linked to growth, neurogenesis, process elongation, and neuronal migration decreased in both Tg and WT animals during the developmental, maturation, and ageing phases; additionally, genes involved in neuroinflammation, voltage-gated channel function, and synaptic transmission modulation were activated. In a different investigation, Michaelis et al. observed that in the CA3 area of the hippocampus, Glud1 overexpression does not result in any age-related changes in dendritic structure or neuronal population numbers (Michaelis et al., 2011). In another study, which contrasted anterograde axoplasmic transport in WT mice with that in Tg mice overexpressing the Glud1 gene in CNS neurons, the authors reported that Tg animals presented increased rates of anterograde axoplasmic transport in olfactory neurons in vivo and in hippocampal neurons in brain slices ex vivo (Lee et al., 2024). Given the links between neurodegeneration and disrupted axonal transport (Berth and Lloyd, 2023) and the ageing process (Milde et al., 2015), additional research is urgently needed.
Notably, GDH plays an epigenetic role in neuronal plasticity. Research has indicated that the α-KG-consuming protein Tet3 (the primary α-KG-dependent dioxygenase in neurons that converts 5-methyl-dC into 5-hydroxymethyl-dC and then into 5-formyl- and 5-carboxy-dC) reroutes mitochondrial GDH, which converts glutamate into α-KG in an NAD + -dependent manner, to the nucleus, indicating the onsite production of α-KG. Additionally, glutamate dehydrogenase has a stimulatory effect on Tet3 demethylation activity in neurons, and neuronal activation increases the level of α-KG. Overall, the GDH–Tet3 interaction may play a role in epigenetic changes during neural plasticity (Traube et al., 2021).
The cellular dysfunction observed during ageing has been partially attributed to impaired mitochondrial activity (Javadpour et al., 2024; Miwa et al., 2022). Studies have shown that GDH activity can differ among various populations of brain mitochondria, in addition to differences in GDH levels and activity across different brain areas. Subcellular fractionation results revealed that the nonsynaptic mitochondria of the medulla oblongata have approximately double the GDH-specific activity of those of the striatum and cortex; in contrast, the GDH activity in synaptic mitochondria is essentially consistent throughout various rat brain areas (Leong and Clark, 1984). Glutamate metabolism has been assessed in a variety of mitochondrial populations in the brain at different ages during the ischaemia and postischemic healing stages (Ferrari et al., 2018); different cerebral mitochondria exhibit varying levels of GDH activity, indicating different responses to ischaemia, postischemic recovery, and ageing. The ability of glutamate to increase synaptic adaptability during the recirculation phase of brain ischaemia may account for the diverse impacts on brain mitochondria. Therefore, depending on where they are located in the presynaptic and postsynaptic compartments, separate nonsynaptic and intrasynaptic mitochondria have variable metabolic properties that result from differing energy needs. However, in another study, GDH activity was shown to decrease with age in both synaptic and nonsynaptic mitochondrial populations in the brains of rats (Deshmukh and Patel, 1980) (Figure 3).
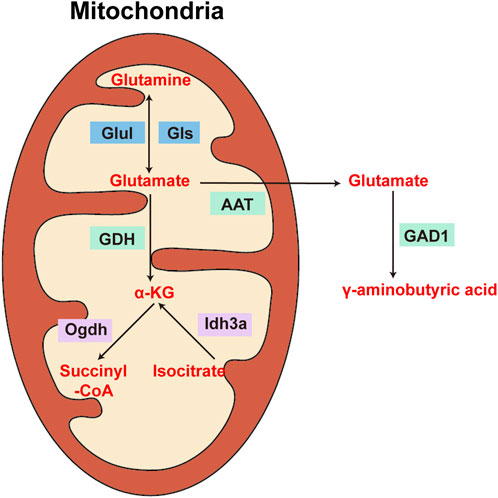
Figure 3. Diagram of the enzymes involved in the metabolism of glutamate, glutamine, α-ketoglutarate, and γ-aminobutyric acid in the brain. Glul: glutaminase; Gls: glutamine synthetase; GDH: glutamate dehydrogenase; Ogdh: α-ketoglutarate dehydrogenase; Idh3a: isocitrate dehydrogenase subunit three alpha; GAD1: glutamate decarboxylase 1.
Changes in GDH levels have been observed in brain cells and other cell types with ageing, findings that may be associated with changes in CNS metabolism. According to previous studies, leukocyte GDH activity is lower in older people than in younger people, suggesting that GDH activity may play a role in ageing (Kravos and Malesic, 2010). However, another study revealed that the overall activity of GDH in lymphocytes increases with age, which may represent an adaptive reaction related to glutamatergic neuron loss (Iwatsuji et al., 1989). The regulatory mechanism of GDH is most likely linked to the regulation of glutamine metabolism (Iwatsuji et al., 1989). However, numerous animal studies have shown that GDH activity in the brain increases with age and decreases with senescence (Mahapatro and Patnaik, 1993). However, GDH activity in the rat liver does not vary with age (Prabhakaram and Singh, 1988). These findings suggest that different cell types exhibit different changes in GDH activity as they age (Figure 4).
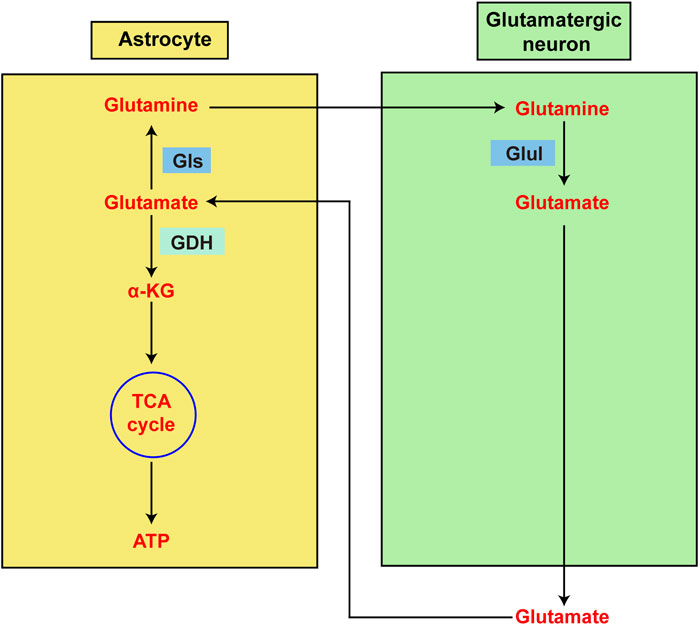
Figure 4. Diagram showing the metabolism of glutamate in astrocytes and glutamatergic neurons. Glutamate is the primary excitatory neurotransmitter in the central nervous system (CNS). However, glutamate is also intimately linked to energy metabolism in the brain. Astrocytes are primarily responsible for intersynaptic glutamate elimination following glutamatergic transmission. Glutamate can be recycled back to neurons after being aminated to glutamine by astrocytes. Alternatively, glutamate dehydrogenase (GDH) may deaminate glutamate to α-ketoglutarate prior to continued oxidation in the TCA cycle, which promotes ATP production.
Mammalian GDH is modulated by high levels of progesterone and oestrogen, which also exert greater inhibitory effects on human GDH2 than on human GDH1. Oestrogens produced by astrocytes may play a role in controlling glutamate metabolism in the brain through their efficient interaction with human GDH2 (Borompokas et al., 2010). One of the variables affecting how GDH levels change as people age is sex. GDH activity was assessed in leukocytes from males and females from various age groups (Kravos and Malesic, 2010). The results revealed that leukocyte GDH activity decreased with age in both sexes, especially after the age of 50 years. Furthermore, after the age of 60 years, both sexes experienced accelerated decreases in GDH activity. GDH activity declined more rapidly in women between the ages of 20 and 50 than during menopause and showed a more uniform decline in men with aging. According to the results from that study, ageing may be exacerbated by a slow decrease in leukocyte GDH activity throughout life, and the brain appears to experience similar phenomena. Additional research on the importance of the effects of hormones on GDH activity and differences in GDH activity patterns between sexes is needed.
Finally, research has shown that an uncommon GLUD2 gene variant, T1492G, which causes a gain-of-function Ser445Ala substitution in hGDH2, is linked to the onset of early-onset Parkinson’s disease in some populations (Plaitakis et al., 2013). This variant has a significantly greater sensitivity to oestrogen-induced inhibition but is even more resistant to GTP inhibition than wild-type hGDH2. In female patients, the consequent regulation of hyperactive GDH by oestrogens may protect against the early onset of PD (Plaitakis et al., 2013). The positive results of oestrogen therapy in animal PD models provide more evidence supporting this hypothesis (Gillies and McArthur, 2010). In addition to a marked loss of neurons and reductions in the numbers of dendritic spines and axon terminals, transgenic mice overexpressing GDH also presented elevated levels and release of glutamate in the brain (Bao et al., 2009). Additionally, increased GDH expression led to the upregulation of numerous genes, including those linked to cellular damage, inflammation, oxidative stress, Parkinson’s disease, and Huntington’s disease (Wang et al., 2010). Early AD was reported to lead to a substantial increase in the quantity of 3-nitrotyrosine residues in proteins, including nitrosylated GDH, whose function is significantly diminished in some patients with early AD (Reed et al., 2009). However, additional research revealed that the GDH protein level in the brain is elevated (Burbaeva et al., 2005) and that the plasma of AD patients exhibits increased levels of GDH activity (Miulli et al., 1993).
Conclusion
GDH is an essential metabolic enzyme that links glutamate to numerous other metabolic pathways. The roles of GDH dysregulation and dysfunction in a variety of illnesses, particularly neurodegenerative diseases, have received increasing attention in recent years. More investigations are needed to fully understand the effects of this enzyme, especially with respect to the neuroplasticity of the ageing human brain. We have examined the functions of GDH in the ageing brain in this review. We believe that GDH plays a key role in neuronal degenerative pathways. Future research efforts should consider human studies, even if the significance of GDH in ageing has been described. Promoting neuronal regeneration may help prevent neurodegenerative diseases. Future studies may uncover novel approaches for using GDH in their detection and treatment. Thus, developing new techniques to cure mental and neurological illnesses, slow the ageing process, and promote neuroplasticity may be possible.
Author contributions
TZ: Data curation, Writing – original draft. HW: Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors acknowledge the Department of Pharmaceutical and Medical Equipment, Bayi Orthopaedic Hospital, and the Department of Paediatrics, Sichuan Academy of Medical Science and Sichuan Provincial People’s Hospital, for supporting our work.
Conflict of interest
Author TZ was employed by China RongTong Medical Healthcare Group Co. Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bao, X., Pal, R., Hascup, K., Wang, Y., Wang, W., Xu, W., et al. (2009). Transgenic expression of Glud1 (glutamate dehydrogenase 1) in neurons: in vivo model of enhanced glutamate release, altered synaptic plasticity, and selective neuronal vulnerability. J. Neurosci. 29, 13929–13944. doi:10.1523/JNEUROSCI.4413-09.2009
Baudry, M., and Lynch, G. (1979). Regulation of glutamate receptors by cations. Nature 282, 748–750. doi:10.1038/282748a0
Berth, S. H., and Lloyd, T. E. (2023). Disruption of axonal transport in neurodegeneration. J. Clin. Invest 133, e168554. doi:10.1172/JCI168554
Borompokas, N., Papachatzaki, K., Kanavouras, V., Mastorodemos, I., Zaganas, C., Spanaki, A., et al. (2010). Estrogen modification of human glutamate dehydrogenases is linked to enzyme activation state. J. Biol. Chem. 285, 31380–31387. doi:10.1074/jbc.M110.146084
Botman, D., Tigchelaar, W., and Van Noorden, C. J. (2014). Determination of glutamate dehydrogenase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem Cytochem 62, 802–812. doi:10.1369/0022155414549071
Bronzetti, E., Felici L Fau - Amenta, F., and Amenta, F. (1990). Age-related changes of glutamate dehydrogenase in the rat hippocampus: an enzyme histochemical study. Arch. Gerontol. Geriatr. 10, 221–229. doi:10.1016/0167-4943(90)90023-y
Burbaeva, G., Boksha, I., Tereshkina, E. B., Savushkina, O. K., Starodubtseva, L. I., and Turishcheva, M. S. (2005). Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer's disease patients. Neurochem. Res. 30, 1443–1451. doi:10.1007/s11064-005-8654-x
Burki, F., and Kaessmann, H. (2004). Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet. 36, 1061–1063. doi:10.1038/ng1431
Cho, S. W., Lee, J., and Choi, S. Y. (1995). Two soluble forms of glutamate dehydrogenase isoproteins from bovine brain. Eur. J. Biochem. 233, 340–346. doi:10.1111/j.1432-1033.1995.340_1.x
Choi, Y., Lee, T., Wang, D., Hui, X., Wang, M., Brooks, K., et al. (2014). Metabolism changes during aging in the hippocampus and striatum of glud1 (glutamate dehydrogenase 1) transgenic mice. Neurochem. Res. 39, 446–455. doi:10.1007/s11064-014-1239-9
Cohen, S., and Greenberg, M. E. (2008). Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 24, 183–209. doi:10.1146/annurev.cellbio.24.110707.175235
Cole, J. H., Marioni, R. E., Harris, S. E., and Deary, I. J. (2019). Brain age and other bodily 'ages': implications for neuropsychiatry. Mol. Psychiatry 24, 266–281. doi:10.1038/s41380-018-0098-1
Cooper, A. J., and Jeitner, T. M. (2016). Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 6, 16. doi:10.3390/biom6020016
Cotman, C. W., Monaghan, A. H., and Ganong, A. H. (1988). Excitatory amino acid neurotransmission: NMDA receptors and Hebb-type synaptic plasticity. Annu. Rev. Neurosci. 11, 61–80. doi:10.1146/annurev.ne.11.030188.000425
Cox, M. F., Hascup, E. R., Bartke, A., and Hascup, K. N. (2022). Friend or foe? Defining the role of glutamate in aging and Alzheimer's disease. Front. Aging 3, 929474. doi:10.3389/fragi.2022.929474
De Lonlay, P., Benelli, C., Fouque, F., Aral, B., Dionisi-Vici, C., Touati, G., et al. (2001). Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr. Res. 50, 353–357. doi:10.1203/00006450-200109000-00010
Deshmukh, D. R., and Patel, M. S. (1980). Age-dependent changes in glutamate oxidation by non-synaptic and synaptic mitochondria from rat brain. Mech. Ageing Dev. 13, 75–81. doi:10.1016/0047-6374(80)90131-1
Diniz, O., and Crestani, O. (2023). The times they are a-changin': a proposal on how brain flexibility goes beyond the obvious to include the concepts of “upward” and “downward” to neuroplasticity. Mol. Psychiatry 28, 977–992. doi:10.1038/s41380-022-01931-x
Fahien, L. A., and Macdonald, M. J. (2011). The complex mechanism of glutamate dehydrogenase in insulin secretion. Diabetes 60, 2450–2454. doi:10.2337/db10-1150
Ferrari, O., Gorini, A., Hoyer, S., and Villa, O. (2018). Glutamate metabolism in cerebral mitochondria after ischemia and post-ischemic recovery during aging: relationships with brain energy metabolism. J. Neurochem. 146, 416–428. doi:10.1111/jnc.14464
Francis, P. T. (2003). Glutamatergic systems in Alzheimer's disease. Int. J. Geriatr. Psychiatry 18, S15–S21. doi:10.1002/gps.934
Ghiani, C. A., Sforza, D. M., Malvar, J. S., Seksenyan, A., Cole, R., Smith, D. J., et al. (2007). The title is “Genetic program of neuronal differentiation and growth induced by specific activation of NMDA receptors. Neurochem. Res. 32, 363–376. doi:10.1007/s11064-006-9213-9
Gillies, G. E., and McArthur, S. (2010). Independent influences of sex steroids of systemic and central origin in a rat model of Parkinson's disease: a contribution to sex-specific neuroprotection by estrogens. Horm. Behav. 57, 23–34. doi:10.1016/j.yhbeh.2009.06.002
Godbout, J. P., and Johnson, R. W. (2009). Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol. Allergy Clin. North Am. 29, 321–337. doi:10.1016/j.iac.2009.02.007
Guan, K. L., and Xiong, Y. (2011). Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116. doi:10.1016/j.tibs.2010.09.003
Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., et al. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954. doi:10.1016/j.cell.2006.06.057
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi:10.1016/S1474-4422(09)70062-6
Hou, O., Dan, X., Babbar, M., Wei, Y., Hasselbalch, O., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi:10.1038/s41582-019-0244-7
Iwatsuji, K., Nakamura, S., Fau - Kameyama, M., and Kameyama, M. (1989). Lymphocyte glutamate dehydrogenase activity in normal aging and neurological diseases. Gerontology 35, 218–224. doi:10.1159/000213026
Javadpour, P., Abbaszadeh, F., Ahmadiani, A., Rezaei, O., and Ghasemi, O. (2024). Mitochondrial transportation, transplantation, and subsequent immune response in Alzheimer's disease: an update. Mol. Neurobiol. 61, 7151–7167. doi:10.1007/s12035-024-04009-7
Kanavouras, K., Mastorodemos, V., Borompokas, N., Spanaki, C., and Plaitakis, A. (2007). Properties and molecular evolution of human GLUD2 (neural and testicular tissue-specific) glutamate dehydrogenase. J. Neurosci. Res. 85, 3398–3406. doi:10.1002/jnr.21576
Kapoor, R. R., James, C., Flanagan, S. E., Ellard, S., Eaton, S., and Hussain, K. (2009). 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J. Clin. Endocrinol. Metab. 94, 2221–2225. doi:10.1210/jc.2009-0423
Komlos, D., Zhuo, Y., Ricupero, C. L., Hart, R. P., Liu, A. Y. C., Firestein, B. L., et al. (2013). Glutamate dehydrogenase 1 and SIRT4 regulate glial development. Glia 61, 394–408. doi:10.1002/glia.22442
Kravos, M., and Malesic, I. (2010). Glutamate dehydrogenase activity in leukocytes and ageing. Neurodegener. Dis. 7, 239–242. doi:10.1159/000273582
Kugler, P., and Schleyer, V. (2004). Developmental expression of glutamate transporters and glutamate dehydrogenase in astrocytes of the postnatal rat hippocampus. Hippocampus 14, 975–985. doi:10.1002/hipo.20015
Lee, O., Kim, J., Choi, O., Pal, R., Hui, D., Marcario, J. K., et al. (2024). Increases in anterograde axoplasmic transport in neurons of the hyper-glutamatergic, glutamate dehydrogenase 1 (Glud1) transgenic mouse: effects of glutamate receptors on transport. J. Neurochem. 168, 719–727. doi:10.1111/jnc.16035
Leong, S. F., and Clark, J. B. (1984). Regional development of glutamate dehydrogenase in the rat brain. J. Neurochem. 43, 106–111. doi:10.1111/j.1471-4159.1984.tb06684.x
Li, C., Chen, P., Palladino, A., Narayan, S., Russell, L. K., Sayed, S., et al. (2010). Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J. Biol. Chem. 285, 31806–31818. doi:10.1074/jbc.M110.123638
Li, M., Allen, A., Stanley, C. A., and Smith, T. J. (2014). Glutamate dehydrogenase: structure, allosteric regulation, and role in insulin homeostasis. Neurochem. Res. 39, 433–445. doi:10.1007/s11064-013-1173-2
MacMullen, C., Fang, J., Hsu, B. Y., Kelly, A., de Lonlay-Debeney, P., Saudubray, J. M., et al. (2001). Hyperinsulinism/hyperammonemia syndrome in children with regulatory mutations in the inhibitory guanosine triphosphate-binding domain of glutamate dehydrogenase. J. Clin. Endocrinol. Metab. 86, 1782–1787. doi:10.1210/jcem.86.4.7414
Mahapatro, N., and Patnaik, B. K. (1993). Age-dependent changes in some aspects of glutamate metabolism in the brain of the teleost, Channa punctatus. I. Ammonia and glutamine contents and glutamate dehydrogenase activity. Mech. Ageing Dev. 68, 47–57. doi:10.1016/0047-6374(93)90139-i
Mayer, M. L., Westbrook, G. L., and Guthrie, P. B. (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263. doi:10.1038/309261a0
McEntee, W. J., and Crook, T. H. (1993). Glutamate: its role in learning, memory, and the aging brain. Psychopharmacol. Berl. 111, 391–401. doi:10.1007/BF02253527
Michaelidis, T. M., Moschonas, N. K., and Papamatheakis, J. (1993). The human glutamate dehydrogenase gene family: gene organization and structural characterization. Genomics 16, 150–160. doi:10.1006/geno.1993.1152
Michaelis, E. K., Pal, R., Bao, X., Hascup, K. N., Wang, Y., Wang, W. T., et al. (2011). Neuronal Glud1 (glutamate dehydrogenase 1) over-expressing mice: increased glutamate formation and synaptic release, loss of synaptic activity, and adaptive changes in genomic expression. Neurochem. Int. 59, 473–481. doi:10.1016/j.neuint.2011.03.003
Milde, S., Adalbert, R., Elaman, M. H., and Coleman, M. P. (2015). Axonal transport declines with age in two distinct phases separated by a period of relative stability. Neurobiol. Aging 36, 971–981. doi:10.1016/j.neurobiolaging.2014.09.018
Miulli, D. E., Norwell, D. Y., and Schwartz, F. N. (1993). Plasma concentrations of glutamate and its metabolites in patients with Alzheimer's disease. J. Am. Osteopath Assoc. 93, 670–676. doi:10.7556/jaoa.1993.93.6.670
Miwa, S., Kashyap, S., Chini, E., and von Zglinicki, T. (2022). Mitochondrial dysfunction in cell senescence and aging. J. Clin. Invest 132, e158447. doi:10.1172/JCI158447
Molven, A., Matre, G., Duran, M., Wanders, R. J., Rishaug, U., Njølstad, P. R., et al. (2004). Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 53, 221–227. doi:10.2337/diabetes.53.1.221
Morrison, J. H., and Hof, P. R. (2007). Life and death of neurons in the aging cerebral cortex. Int. Rev. Neurobiol. 81, 41–57. doi:10.1016/S0074-7742(06)81004-4
Nassar, O. M., Wong, K. Y., Lynch, G. C., Smith, T. J., and Pettitt, B.A.-O. (2019). Allosteric discrimination at the NADH/ADP regulatory site of glutamate dehydrogenase. Protein Sci. 28, 2080–2088. doi:10.1002/pro.3748
Nickell, J., Allen, J., and Gerhardt, G. A. (2005). Age-related changes in the dynamics of potassium-evoked L-glutamate release in the striatum of Fischer 344 rats. J. Neural Transm. (Vienna) 112, 87–96. doi:10.1007/s00702-004-0151-x
Nissim, I. (1999). Newer aspects of glutamine/glutamate metabolism: the role of acute pH changes. Am. J. Physiol. 277, 493–497. doi:10.1152/ajprenal.1999.277.4.F493
Plaitakis, A., Metaxari, M., and Shashidharan, P. (2000). Nerve tissue-specific (GLUD2) and housekeeping (GLUD1) human glutamate dehydrogenases are regulated by distinct allosteric mechanisms: implications for biologic function. J. Neurochem. 75, 1862–1869. doi:10.1046/j.1471-4159.2000.0751862.x
Plaitakis, A., Nicklas, W. J., and Desnick, R. J. (1980). Glutamate dehydrogenase deficiency in three patients with spinocerebellar syndrome. Ann. Neurol. 7, 297–303. doi:10.1002/ana.410070403
Plaitakis, A., and Shashidharan, P. (2000). Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: implications for the pathogenesis of Parkinson’s disease. J. Neurol. 247, 25–35. doi:10.1007/pl00007757
Plaitakis, A., Spanaki, C., Mastorodemos, V., and Zaganas, I. (2003). Study of structure-function relationships in human glutamate dehydrogenases reveals novel molecular mechanisms for the regulation of the nerve tissue-specific (GLUD2) isoenzyme. Neurochem. Int. 43, 401–410. doi:10.1016/s0197-0186(03)00028-7
Plaitakis, A., Zaganas, I., and Spanaki, C. (2013). Deregulation of glutamate dehydrogenase in human neurologic disorders. J. Neurosci. Res. 91, 1007–1017. doi:10.1002/jnr.23176
Poitry, S., Poitry-Yamate, C., Ueberfeld, J., MacLeish, P. R., and Tsacopoulos, M. (2000). Mechanisms of glutamate metabolic signaling in retinal glial (Müller) cells. J. Neurosci. 20, 1809–1821. doi:10.1523/JNEUROSCI.20-05-01809.2000
Prabhakaram, M., and Singh, S. N. (1988). Properties of purified liver glutamate dehydrogenase of aging rats. Arch. Gerontol. Geriatr. 7, 297–309. doi:10.1016/0167-4943(88)90013-1
Rajeswari, T. S., and Radha, E. (1984). Metabolism of the glutamate group of amino acids in rat brain as a function of age. Mech. Ageing Dev. 24, 139–149. doi:10.1016/0047-6374(84)90066-6
Reed, T. T., Pierce, W. M., Turner, D. M., Markesbery, W. R., and Allan Butterfield, D. (2009). Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J. Cell Mol. Med. 13, 2019–2029. doi:10.1111/j.1582-4934.2008.00478.x
Rothe, F., Schmidt, W., and Wolf, G. (1983). Postnatal changes in the activity of glutamate dehydrogenase and aspartate aminotransferase in the rat nervous system with special reference to the glutamate transmitter metabolism. Brain Res. 313, 67–74. doi:10.1016/0165-3806(83)90202-x
Rothe, F., and Schünzel, G. (1990). Immunohistochemical demonstration of glutamate dehydrogenase in the postnatally developing rat hippocampal formation and cerebellar cortex: comparison to activity staining. Neuroscience 39, 419–429. doi:10.1016/0306-4522(90)90278-c
Rothe, F., and Storm-Mathisen, J. (1995). Quantitative ultrastructural localization of glutamate dehydrogenase in the rat cerebellar cortex. Neuroscience 64, iii–xvi. doi:10.1016/0306-4522(94)e0200-n
Schünzel, G., and Wolf, G. (1982). Topographic and quantitative characteristics of glutamate dehydrogenase of the hippocampus formation during the postnatal development of the rat brain. Comparative studies on succinate and alpha-glycerophosphate dehydrogenase with special reference to putatively glutamatergic structures. Acta histochem. 71, 145–151. doi:10.1016/s0065-1281(82)80028-7
Segovia, G., Porras A Fau - Del Arco, A., Del Arco A Fau - Mora, F., and Mora, F. (2001). Glutamatergic neurotransmission in aging: a critical perspective. Mech. Ageing Dev. 122, 1–29. doi:10.1016/s0047-6374(00)00225-6
Sener, A., and Malaisse, W. J. (1980). Stimulation of insulin release by L-glutamine. Mol. Cell Biochem. 33, 157–159. doi:10.1007/BF00225288
Shashidharan, P., Kresovali, A., Papamatheakis, J., and Plaitakis, A. (1994). Novel human glutamate dehydrogenase expressed in neural and testicular tissues and encoded by an X-linked intronless gene. J. Biol. Chem. 269, 16971–16976.
Spanaki, C., Kleopa, K. A., and Plaitakis, A. (2010). Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J. Biol. Chem. 285, 16748–16756. doi:10.1074/jbc.M109.092999
Spanaki, C., Kounoupa, Z., and Plaitakis, A. (2012). The complex regulation of human glud1 and glud2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem. Int. 61, 470–481. doi:10.1016/j.neuint.2012.05.020
Spanaki, C., Petraki, Z., Drakos, E., and Plaitakis, A. (2014). Heterogeneous cellular distribution of glutamate dehydrogenase in brain and in non-neural tissues. Neurochem. Res. 39, 500–515. doi:10.1007/s11064-013-1235-5
Spreafico, R., Frassoni, C., Arcelli, P., Battaglia, G., Wenthold, R. J., and De Biasi, S. (1994). Distribution of AMPA selective glutamate receptors in the thalamus of adult rats and during postnatal development. A light and ultrastructural immunocytochemical study. Brain Res. Dev. Brain Res. 82, 231–244. doi:10.1016/0165-3806(94)90166-x
Traube, F., Özdemir, D., Sahin, H., Scheel, C., Glück, A. F., Geserich, A. S., et al. (2021). Redirected nuclear glutamate dehydrogenase supplies Tet3 with α-ketoglutarate in neurons. Nat. Commun. 12, 4100. doi:10.1038/s41467-021-24353-9
Treberg, J. R., Clow, K. A., Greene, K. A., Brosnan, M. E., and Brosnan, J. T. (2010). Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am. J. Physiol. Endocrinol. Metab. 298, E1219–E1225. doi:10.1152/ajpendo.00028.2010
Treberg, J. R., Pandey, U., and Weihrauch, D. (2014). Intertissue differences for the role of glutamate dehydrogenase in metabolism. Neurochem. Res. 39, 516–526. doi:10.1007/s11064-013-0998-z
Wang, X., Hui, D., Pal, R., Hafez, M. M., Ahmed, M. M., Yahya, A. A., et al. (2014). Gene expression patterns in the hippocampus during the development and aging of Glud1 (Glutamate Dehydrogenase 1) transgenic and wild type mice. BMC Neurosci. 15, 37. doi:10.1186/1471-2202-15-37
Wang, X., Pal, R., Agbas, A., and Michaelis, E. K. (2010). Transcriptomic responses in mouse brain exposed to chronic excess of the. BMC Genomics 11, 360. doi:10.1186/1471-2164-11-360
Wolf, G., and Schünzel, G. (1987). Glutamate dehydrogenase in aminoacidergic structures of the postnatally developing rat cerebellum. Neurosci. Lett. 78, 7–11. doi:10.1016/0304-3940(87)90552-0
Wolf, G., Schünzel, G., and Rothe, F. (1986). Histochemical demonstration of sodium-dependent glutamate uptake in brain tissues by glutamate dehydrogenase reaction. Exp. Brain Res. 62, 659–662. doi:10.1007/BF00236047
Keywords: glutamate dehydrogenase, ageing brain, Parkinson’s disease, Alzheimer’s disease, glutamate metabolism
Citation: Zhou T and Wang H (2025) The role of glutamate dehydrogenase in the ageing brain. Front. Pharmacol. 16:1586655. doi: 10.3389/fphar.2025.1586655
Received: 03 March 2025; Accepted: 14 April 2025;
Published: 28 April 2025.
Edited by:
Jiu Chen, Nanjing University, ChinaReviewed by:
Maria Bayliak, Vasyl Stefanyk Precarpathian National University, UkraineImran Ahmd Khan, Jamia Hamdard University, India
Copyright © 2025 Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haichuan Wang, d2hjMTg1ODI1MjQxODJAMTYzLmNvbQ==