 Julia Macente1†
Julia Macente1† Rodolfo Hernandes Bonan2†
Rodolfo Hernandes Bonan2† Edilainy Rizzieri Caleffi-Marchesini3†
Edilainy Rizzieri Caleffi-Marchesini3† Leonardo Régis Leira Pereira4†Priscila De Freitas Lima5
Leonardo Régis Leira Pereira4†Priscila De Freitas Lima5 Pieter Annaert1,2†
Pieter Annaert1,2† Karel Allegaert6,7,8†
Karel Allegaert6,7,8† Andrea Diniz3*†
Andrea Diniz3*†- 1Drug Delivery and Disposition, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Leuven, Belgium
- 2BioNotus CommV, Niel, Belgium
- 3Pharmacokinetics and Biopharmaceutical Laboratory (PKBio), Department of Pharmacy, State University of Maringa, Maringa, Brazil, Department of Pharmacy, State University of Maringá, Maringá, Paraná, Brazil
- 4Faculty of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto, São Paulo, Brazil
- 5Barão de Mauá University Center, Ribeirão Preto, São Paulo, Brazil
- 6Clinical Pharmacology and Pharmacotherapy, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Leuven, Belgium
- 7Department of Development and Regeneration, KU Leuven, Leuven, Belgium
- 8Department of Hospital Pharmacy, Erasmus University Medical Center, Rotterdam, Netherlands
Introduction: Optimizing levetiracetam (LEV) dosing in children is challenging due to high pharmacokinetic variability, which often necessitates empirical dose titration. This study aimed to develop and verify a physiologically-based pharmacokinetic (PBPK) modeling and simulation to guide and optimize initial LEV dose selection in pediatric patients.
Methods: A whole-body PBPK model for LEV was developed and verified in adults, then scaled and verified in a pediatric population (0.5–12 years). This model was used to simulate various dosing regimens. Subsequently, a multivariate linear regression (MLR) analysis correlated key covariates (dose, regimen, body weight, and glomerular filtration rate) with simulated steady-state peak (Cmax) and trough (Ctr) concentrations to create a practical dosing tool.
Results: The MLR model successfully explained over 90% of the variance (R2 > 0.9) between covariates and simulated plasma concentrations. For a twice-daily regimen, daily doses of 40–60 mg/kg were required to achieve concentrations within a target therapeutic window (e.g., Cmax of 20–46 mg/L). A three-times-daily regimen allowed for a broader effective dose range of 50–80 mg/kg/day, enabling higher total daily doses while maintaining Cmax within a safe range.
Conclusion: The combined PBPK-MLR approach provides a robust, data-driven framework to support rational first-dose prescriptions of LEV in children. This tool has the potential to accelerate therapeutic effects while enhancing treatment individualization. Prospective clinical validation is required to confirm the model predictive performance for drug exposure and, consequently, its impact on therapeutic efficacy.
1 Introduction
Levetiracetam is a widely used second-generation anticonvulsant approved for both adult and pediatric populations as a first-line or adjunctive treatment for various seizure types (Celdran de Castro et al., 2023, UCB Biosciences, 1999). It has a favorable pharmacokinetic profile, including near-complete oral absorption, low plasma protein binding (<10%), and linear kinetics, which generally make its pharmacologic response predictable (Patsalos, 2000). LEV is primarily eliminated renally, with approximately 66% of the dose excreted unchanged in the urine. Its minimal reliance on hepatic metabolism results in a low potential for drug-drug interactions, an advantage in polytherapy regimens commonly used in epilepsy management.
Despite these favorable properties, optimizing LEV dosing in the pediatric population remains challenging. Pediatric patients exhibit higher renal clearance per kilogram compared to adults, necessitating weight-based dosing strategies to achieve similar exposures (Glauser et al., 2002; Pellock JM et al., 2001). This, combined with the dynamic physiological changes occurring from infancy through adolescence, complicates dose selection and has led to widespread off-label use and dosing variability in clinical practice (Franco et al., 2016).
A further complication is the lack of a universally accepted therapeutic window for LEV. Although suggested therapeutic ranges for plasma concentrations have been proposed, the relationship between plasma levels and therapeutic response varies widely between individuals, making routine therapeutic drug monitoring (TDM) uncommon (Sinha et al., 2022). As a result, clinicians often rely on empirical dose titration, a time-consuming approach that may delay effective seizure control. Additionally, LEV has been associated with behavioral adverse events in children, which appear to be more related to the rate of dose escalation than the final maintenance dose. This suggests that reaching therapeutic levels quickly, without aggressive titration, could improve both efficacy and tolerability.
The application of physiologically-based pharmacokinetic (PBPK) modeling to levetiracetam (LEV) has been instrumental in addressing dosing challenges in pediatric populations. For instance, Shao et al. (2023) developed a PBPK model to optimize LEV dosing for Chinese children, finding that standard recommendations may lead to underexposure in this group. Similarly, a model by Maglalang et al. (2024) was extended to account for LEV disposition in infants and children with obesity, offering critical insights for these understudied populations.
While these models have advanced our understanding, there remains a gap in providing clinicians with a practical, quantitative framework to guide initial LEV across a broad pediatric age range. There is a clear need to move beyond empirical titration toward a more rational strategy that safely accelerates the time to therapeutic drug concentrations. Key unanswered questions, such as the optimal frequency of administration (e.g., twice-daily vs. three times a day) and the justification for more assertive starting doses are critical to improve clinical outcomes.
All therapeutic window recommendations are related to the minimal concentration in the body (Ctr), mainly because to get blood samples to quantify, experimentally the maximum concentration (Cmax) is an improbable success. However, for many drugs, Cmax is directly linked to concentration-dependent side effects and toxicity. Relying solely on trough levels could therefore mask potential harm to the patient, making the estimation or avoidance of high peak concentrations a critical aspect of safe and effective therapy, especially for drugs with a narrow therapeutic index.
Therefore, the aim of this study was to develop and verify a whole-body PBPK model for LEV in a pediatric population aged 6 months to 12 years. The model was designed to evaluate different dosing strategies and provide a scientific rationale to support initial dose selection, with the goal of safely and rapidly achieving therapeutic concentrations.
2 Methodology
2.1 Adult PBPK model development
A whole-body PBPK model for LEV was first developed in healthy adult volunteers using GastroPlus software version 9.8 (Simulations Plus, Lancaster, CA). Drug-specific physicochemical and pharmacokinetic parameters (e.g., logP, pKa, molecular weight, solubility, fraction unbound in plasma, intestinal permeability, and blood-to-plasma ratio) were obtained from the literature and used as model inputs (Table 1). Intestinal permeability (25.5 × 10−6 cm/s) was used to define the absorption process. The volume of distribution was predicted using the Poulin and Theil model (Poulin & Theil, 2009). The total clearance (CLtotal) is known to occur via two main routes: approximately 66% through glomerular filtration and 34% through esterase-mediated metabolism (Patsalos, 2000). However, the specific metabolic enzymes responsible for this non-renal clearance remain unidentified. Some studies suggest the involvement of serum esterase (Di, 2019) or hepatic esterase activity (Boberg et al., 2017; Wang et al., 2016), but no consensus has been reached regarding the exact enzymatic pathway. Given this uncertainty, it was assumed in this study that 34% of total clearance is attributed to non-renal pathways (CLNR), without specifying the enzyme system, while the remaining 66% was attributed to net renal clearance, accounting for both glomerular filtration and tubular reabsorption.

Table 1. Drug-dependent parameters for Levetiracetam PBPK model development.
Pharmacokinetic data for LEV, encompassing intravenous and oral administration as well as single and multiple-dose regimens, were collected from the literature for model development and qualification. Only studies that provided clear information on age, sex, height, weight, renal function, dosing, and plasma concentration profiles were included (Supplementary Table S1). Plasma concentration-time profiles were digitally extracted using WebPlotDigitizer 4.2 (Rohatgi, 2021) to enable overlay with model-predicted profiles. For publications lacking reported summary exposure parameters, the area under the curve (AUC) and the maximum concentration (Cmax) were estimated from the average profiles using non-compartmental analysis.
A complete clinical dataset from a previous published study including Brazilian population (Freitas-Lima et al., 2011) was used to support model validation. All subjects gave their written consent, and the protocol was approved by the Ethics Committee of Hospital das Clínicas, Ribeirão Preto School of Medicine, Brazil (Freitas-Lima et al., 2011). This dataset included 15 adult patients with epilepsy (8 males and 7 females; age range: 19–51 years, mean age: 39) who were admitted to the Clinical Research Unit of the Clinical Hospital of Ribeirão Preto Medical School. Pregnant women or those at risk for pregnancy were excluded. Each patient received a single 1000 mg oral dose of levetiracetam (UCB, Italy) in a fasting state with 50 mL of water. Blood samples were collected at pre-dose and at 1, 3, 6-, 9-, 12-, and 24-h post-dose. Plasma LEV concentrations were measured as described in the original publication (Freitas-Lima et al., 2011).
2.2 Model performance verification
The exposure metrics predicted by the PBPK model were compared with in vivo concentration-time profiles after oral or intravenous administration collected from the literature. Virtual populations consisting of 100 subjects per simulation were generated to closely match the characteristics of participants in the corresponding clinical study. Each simulation used the same study design parameters, such as dose, route of administration, age, sex distribution, ethnicity (e.g., White, Japanese or Chinese) and body weight, as reported in the respective studies (Supplementary Table S1).
The suitability of the PBPK model was assessed by comparing the predicted plasma concentration-time profile with the observed clinical data from each study. Model verification was deemed successful when key PK parameters, Cmax and area AUC, were predicted within two-fold range of the corresponding observed values (Jones et al., 2009; Medicines Agency, 2018; Obach et al., 1997). To quantitatively assess model accuracy, the geometric mean fold-error (GMFE) was calculated for both AUC and Cmax using Equation 1.
2.3 Pediatric PBPK model and dose scaling
The verified adult PBPK model was scaled to the pediatric population (infant and children) using the GastroPlus Population Estimates for Age-Related Physiology™ module. Extrapolation from adults to pediatrics was primarily based on differences in glomerular filtration rate (GFR) and unbound plasma protein fraction (fup). Plasma concentration profiles over time for pediatric patients aged 6 months to 12 years receiving doses ranging from 20 to 60 mg/kg were collected from the literature (Fountain et al., 2007; Glauser et al., 2002; Pellock JM et al., 2001) (Supplementary Material, Supplementary Table S1). The pediatric model was considered acceptable when the primary pharmacokinetic parameters (Cmax and AUC) were predicted within a two-fold range of the corresponding observed values.
The qualified pediatric PBPK model was then used to simulate various doses, ranging from 10 to 120 mg/kg, administered either twice a day (BID) or three times a day (TID), across four predefined age groups: a) 0.5–3 years; b) 4–6 years; c) 7–9 years; and d) 10–12 years. These age categories were defined based on the availability of supporting clinical data to optimize age-appropriate dose recommendations. Simulations were performed at steady-state conditions (SS).
Dose suitability was evaluated using two PK targets: through concentration (Ctr) and Cmax at the SS. The plasma therapeutic window for adults assumed was between 5 and 46 mg/L which was established based on previous studies (Patsalos et al., 2008; Reimers et al., 2018; Stepanova and Beran, 2014). The efficacy target was defined as achieving a steady-state Ctr above the lower bound of 5 mg/L and the safety target was defined as maintaining a steady-state Cmax below the upper bound of 46 mg/L. Population simulations were performed as 10 repeated trials, each with 15 virtual pediatric subjects per age group. All graphs were generated using R version 4.4.1, with the ggplot2 package (v. 3.5.1).
2.4 Probability of target attainment (PTA)
The dataset containing the anthropometric characteristics (age, weight, height and sex), dose, Cmax and Ctr at the SS for all simulated pediatric individual, according to Section 2.3, were assumed to evaluate the PTA for pediatric population.
The efficacy target was a Ctr higher than 5 mg/L, and for the safety target was a Cmax lower than 46 mg/L. The PTA was calculated using R version 4.4.1.
2.5 Multivariate linear regression analysis to guide the first dose prescription
From the simulated pediatric trial dataset, variables for each dose, dosing regimen and age group were used as data to construct the multivariate linear regression model. For each virtual patient subject, covariates such as age, weight, height, body surface area (BSA), GFR and daily dose (DD) were included, along with the pharmacokinetic outcomes, Cmax and Ctr at SS. Cmax or Ctr were set as dependent variables, and all other covariates were assessed as potential predictors. A forward stepwise inclusion was employed, with each included factor evaluated for statistical significance (p < 0.05) and multicollinearity. The regression analysis was conducted using the tidyverse package in Rstudio.
The multivariate linear equation has the general model as presented in Equation 2.
where: β represents each coefficient related to each factor x and ɛ is the standard error of regression.
3 Results
3.1 Adult PBPK model
A PBPK model for levetiracetam was developed and verified in healthy adult volunteers after oral or intravenous administration of single/multiple doses at various dose levels. The model was able to predict AUC and Cmax within a two-fold range of observed clinical data used for model development and verification (Figure 1). Predicted-to-observed ratios for both intravenous and oral dosing scenarios are provided in the Supplementary Table S2. The model was considered verified and used as the basis for extrapolation to the pediatrics population.

Figure 1. Predictive performance plots for the levetiracetam PBPK model comparing predicted and observed AUC and Cmax. Symbols represent the performance for adults (circle) and pediatrics from 6 months to 12 years old (triangles). Solid bold line represents the line of unity, dashed lines and solid lines represent the 2.0-fold and 1.5-fold difference, respectively.
3.2 Dose scaling by PBPK model and probability of target attainment
The adult PBPK model was successfully extrapolated to the pediatric population (0.5–12 years old). Model performance was verified against published clinical data, with predicted AUC and Cmax values falling within a 2-fold margin of observed data (Figures 1, 2; Supplementary Table S2; Table S3).

Figure 2. Predicted and observed plasma concentration profiles of LEV. The solid lines represent mean simulated, grey areas between dashed lines represent 5-95th percentiles of predictions and the black circles are observed data from each experimental condition. The observed data were from (A) adult healthy volunteers after a single 15-min IV infusion of 1500 mg (Ramael et al., 2006), (B) adult healthy volunteers after multiple IV administration of 500 mg (Spencer et al., 2011), (C) adult healthy volunteers after a single oral administration of 1000 mg (Freitas-Lima et al., 2011), (D) pediatric patients (6–12 years old) after a single oral administration of 20 mg kg-1 (Pellock et al., 2001).
To guide dose optimization, pharmacokinetic targets were defined based on established clinical evidence. Efficacy is primarily associated with maintaining a sufficient trough concentration (Ctr), while safety is related to avoiding excessively high peak concentrations (Cmax).
Simulations revealed a challenge in achieving the efficacy target with standard dosing. To achieve a Ctr > 5 mg/L, our model indicates that high daily doses are required often exceeding 90 mg/kg for children up to 9 years old and 80 mg/kg for those 10–12 years old. The choice of dosing regimen twice-daily (BID) versus three-times-daily (TID) was critical in balancing efficacy and safety. For BID regimen while lower doses (e.g., 40–70 mg/kg/day) can keep the Cmax within the safety limit, the corresponding Ctr frequently falls below the 5 mg/L efficacy threshold. On the other hand, a TID regimen allows for higher total daily doses required for efficacy while mitigating the risk of high peak concentrations. For example, a DD of 90 mg/kg administered TID can achieve the therapeutic trough while keeping the Cmax manageable.
The Probability of Target Attainment (PTA) analysis confirms this finding (Figures 3, 4). For the BID regimen, the probability of achieving a Ctr > 5 mg/L was low across the tested dose ranges. The TID regimen, however, showed a markedly improved PTA, making it the superior strategy for reliably achieving therapeutic goals in the pediatric population.

Figure 3. Probability of target attainment (PTA) for trough concentration (Ctr), considering the plasma concentration range between 5 and 46 mg/mL. Daily Dose tested were 10, 20, 30, 35, 40, 50, 60, 70 and 80 mg/day.

Figure 4. Probability of target attainment (PTA) for Cmax, considering the plasma concentration above 100 mg/mL. Daily Dose tested were 10, 20, 30, 35, 40, 50, 60, 70 and 80 mg/day.
Finally, these weight-based recommendations are most relevant for children weighing less than 50 kg. For heavier children and adolescents, total daily doses should align with adult recommendations (1000–3000 mg/day) to achieve the desired therapeutic target.
3.3 Multivariate linear regression analysis to guide the first dose prescription
The multivariate linear regression results are presented in Table 2. Only statistically significant variables were kept in the final model, after assessing inter-variable correlations. The significant independent variables were dose, weight, dosing regimen and GFR. When Ctr was the dependent variable, the adjusted R2 indicated that the model explained 72% of the variability in the data. In contrast, when Cmax was the dependent variable, the adjusted R2 demonstrated that the model explained about 94% of the variability. Considering the better predictive performance for Cmax and greater clinical relevance supported by previous studies, only the Cmax-based model was considered.

Table 2. Multivariate linear regression results for Cmax and Ctrough as dependent variable.
The multivariate linear equation has the general model as presented in Equation 2 and the results show the significant regression values for both regimen and the final model for pediatric dose suggestion for the first prescription is presented as Equations 3, 4.
where: CmaxSS is given as mg. L-1; dose is given as mg.kg-1.day-1, WT is weight in kg, and Reg is the Regimen as 1 for BID and 2 for TID and GFR is the glomerular filtration rate in mL.sec-1.
When adjustment errors are removed from Equation 3, it could be simplified as Equation 4.
However, for the clinical routine, only the Ctr is possible to access. Then, the prescriber can define the desirable Cmax by Equation 4 and estimate the Ctr at SS by Equation 5.
where:
4 Discussion
LEV is considered one of the safest antiepileptic drugs, with its primary elimination via glomerular filtration being a key determinant of its favorable safety profile (Patsalos, 2000). In pediatric patients, LEV is often preferred due to its established efficacy, safety, linear pharmacokinetics, and minimal drug-drug interactions (Glauser et al., 2002). Despite its widespread use, optimizing initial dose selection in children remains a challenge. Therefore, this study aimed to develop and qualify a PBPK model to provide a more rational basis for initial dose prescription of levetiracetam in the pediatric population.
The development process began by building a whole-body PBPK model for healthy adults, which was successfully qualified against clinical data from Caucasian, Japanese, and Chinese populations. The model predicted the pharmacokinetic profile of LEV in adults to within a 2-fold error margin of observed in vivo data, establishing a reliable base model for pediatric extrapolation.
Subsequently, the adult model was scaled to a pediatric population (6 months–12 years old). This extrapolation was mechanistically anchored to age-dependent changes in glomerular filtration rate and the unbound plasma protein fraction, a methodology consistent with established pediatric modeling practices (Zhang et al., 2019). Our model incorporated a physiologically scaled GFR, which is dependent on organ size and blood flow. A key refinement was the reduction of the standard GFR value (approx. 120 mL/min/1.73 m2) to account for the known partial tubular reabsorption of LEV (Patsalos, 2000). This adjustment, applied proportionally across all pediatric age groups, was crucial for accurately capturing the drug elimination process. This approach is supported by recent PBPK work from Maglalang et al. (2024), who also noted that LEV renal clearance is substantially lower than the GFR, confirming significant tubular reabsorption (Germovsek et al., 2019; Zhang et al., 2019; Maglalang et al., 2024). As demonstrated in the results, the final model predictive accuracy was critically dependent on this GFR adjustment, thereby verifying our approach (see Supplementary Table S2; Figure 1).
The pediatric PBPK model was able to predict the pharmacokinetic profile of levetiracetam across all ages tested in this study, with a GMFE range of 0.77–1.32. Analysis of the virtual pediatric population showed that children weighing over 50 kg achieved Cmax comparable to adults when receiving the same dose, validating the current weight-based cutoff for adult dosing. The model was then used to explore the safety and efficacy of alternative dosing strategies.
Critically, the model identified the TID regimen as a safer strategy for dose escalation in younger children (0.5–9 years). As shown in Figure 5, this regimen allows for the daily dose to be increased up to 90 mg/kg while maintaining Cmax below the 46 mg/L safety boundary. This finding provides a mechanistic basis for the successful use of high doses (90–120 mg/kg/day) reported in clinical studies by Depositario-Cabacar et al. (2010) and Mandelbaum et al. (2005), where seizure frequency was reduced without significant safety concerns. It also aligns with a case report of a 7-year-old who tolerated 200 mg/kg/day without adverse effects (Kartal, 2017). The crucial clinical message from our findings is that for children aged 0.5–9 years who are not responding sufficiently to standard doses, clinicians could more confidently and safely escalate the dose up to 90 mg/kg/day by switching to a TID regimen. This approach mitigates plasma fluctuations and reduces the risk of peak-concentration-related side effects, allowing for a wider therapeutic dose range than is currently recommended (UCB, Biosciences, 1999) (Depositario-Cabacar et al., 2010).
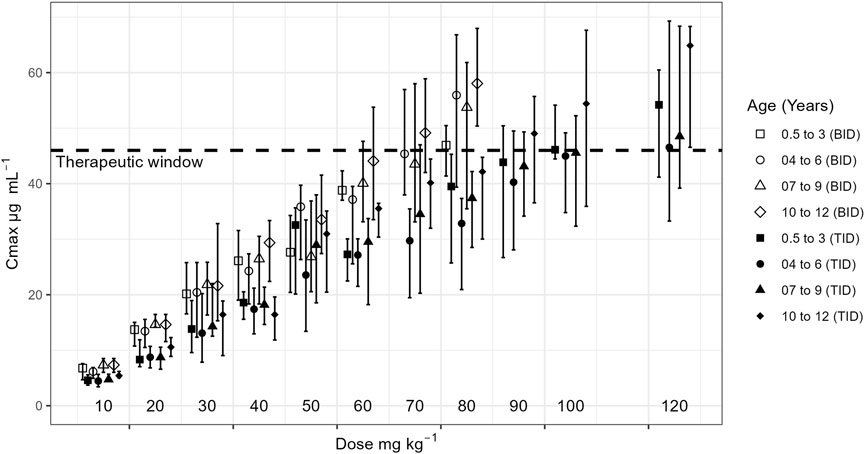
Figure 5. Maximum plasma concentration (Cmax) for each dose by age groups for a BID and TID regimen. Dashed lines represent the therapeutic window.
To further enhance clinical utility, the mechanistic simulation results from the PBPK model were leveraged to develop a simple statistical tool. While traditional population pharmacokinetic (PopPK) modeling is a standard method for creating covariate-based dosing equations from clinical data, our PBPK-generated dataset was better suited for a different purpose. The PBPK model served as the mechanistic engine to generate a rich dataset, and a multivariate linear regression (MLR) model was then applied as a pragmatic ‘translator’ to distill these complex outputs into a simple algebraic formula for clinical use. This statistical approach successfully correlated Cmax with key predictors identified by the PBPK model (body weight, dose, dosing regimen, and GFR), which together explained 94% of the variance in the simulated data. The resulting mathematical correlation (Equation 4) offers a practical tool for estimating an appropriate starting dose for pediatric patients (>0.5 years, <50 kg) on LEV monotherapy. This data-driven approach supports a more aggressive dose initiation strategy; if a concentration within the therapeutic range in desired, the initial titration steps could potentially be bypassed in favor of a starting dose predicted to directly achieve the therapeutic target, accelerating the time to clinical response.
This work compares favorably with other recent PBPK models for pediatric LEV. While our study focused on dose regimen optimization across a broad pediatric age range (0.5–12 years), the model by Maglalang et al. (2024) focused specifically on the effects of obesity and maturation in children from 1 month to 19 years. Their finding that obesity can alter weight-normalized clearance complements our work, suggesting that patient-specific factors like body composition are important considerations. Both studies underscore the central role of renal function in LEV disposition and validate the utility of PBPK modeling in refining pediatric dosage.
The limitations of this study must be acknowledged. The PBPK model was developed for LEV monotherapy in patients without significant comorbidities, whereas clinical practice often involves polytherapy. Potential interactions with other antiepileptic drugs (May et al., 2003; Florek-Luszczki et al., 2014) were not considered. Similarly, as our primary focus was on the maturational effects on clearance, the model was validated in populations with normal renal function, and its performance in patients with renal impairment has not been evaluated. Validating the model against data from subjects with varying degrees of renal impairment would be a valuable future step to further confirm the model mechanistic handling of renal clearance and expand its domain of applicability. Furthermore, our model simplifies renal elimination by empirically scaling the physiological glomerular filtration rate (GFR) downwards to match observed renal clearance, thereby implicitly accounting for tubular reabsorption. While the specific transporters involved in LEV reabsorption are not yet fully elucidated, future PBPK models could incorporate mechanistic transport kinetics (e.g., for Organic Anion Transporters (OAT) or other transporters) as this information becomes available, which would further refine predictive accuracy.
Although the PBPK-based MLR equation provides a simple and powerful tool for clinicians, its own limitations must be understood. The MLR model is a statistical abstraction of the PBPK model’s output, not a mechanistic model itself. Its predictive accuracy is therefore entirely dependent on the validity of the underlying PBPK simulations. Furthermore, its use is restricted to the range of covariates (e.g., age, weight, GFR) evaluated in this study and cannot be extrapolated. As such, it requires validation with new, independent pediatric clinical data before it can be recommended for routine clinical use.
5 Conclusion
In conclusion, the PBPK model developed for levetiracetam in adults and pediatric patients (6 months–12 years old) was successfully verified and accurately predicted the drug pharmacokinetic profile. The pediatric model was subsequently used to explore the safe administration of doses exceeding currently recommended limits. The study demonstrated that therapeutic goals could be achieved with doses between 30 and 60 mg/kg/day using a BID regimen and showed that doses could be safely escalated up to 90 mg/kg/day in younger children by employing a TID regimen. To facilitate clinical translation, a multivariate linear regression model was developed that effectively predicted Cmax based on body weight, dose, regimen, and glomerular filtration rate. The robustness of this framework is founded upon the core PBPK model key methodological correction for renal tubular reabsorption. While these findings provide a more rational approach to LEV dosing, prospective clinical validation is essential to confirm the model predictions and ensure its applicability across diverse clinical scenarios. It is also important to note that our model was developed using data from immediate-release (IR) oral formulations; therefore, this framework is specific to IR levetiracetam and would require re-parameterization to be applied to other formulations, such as extended-release.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
JM: Data curation, Investigation, Methodology, Software, Writing – original draft. RH: Data curation, Investigation, Methodology, Software, Writing – review and editing. EC-M: Investigation, Methodology, Software, Writing – review and editing. LP: Data curation, Investigation, Writing – review and editing. PD: Data curation, Investigation, Writing – review and editing. PA: Investigation, Writing – review and editing. KA: Data curation, Investigation, Methodology, Resources, Supervision, Writing – review and editing. AD: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Writing – review and editing.
Funding
The authors declare that financial support was received for the research and/or publication of this article. The research was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes) [Process n 88887.464109/2019-00] and Araucária Foundation (Paraná State of Brazil) [Process 061/202 – Prot SUS 2020131000109].
Acknowledgements
This research was made possible by the free license of GastroPlus® provided by SimulationPlus-Lancaster-CA and the support of Senior Scientist Maxime Le Merdy. We thank to Frederico Severino Martins for the support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1678960/full#supplementary-material
SUPPLEMENTARY TABLE S3 | Simulated mean maximum concentration (Cmax), steady-state plasma concentration range, and their corresponding coefficient of variation (CV%) in the pediatric population
References
Boberg, M., Vrana, M., Mehrotra, A., Pearce, R. E., Gaedigk, A., Bhatt, D. K., et al. (2017). Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab. Dispos. 45 (2), 216–223. doi:10.1124/dmd.116.072652
Celdran de Castro, A., Nascimento, F. A., Beltran-Corbellini, Á., Toledano, R., Garcia-Morales, I., Gil-Nagel, A., et al. (2023). Levetiracetam, from broad-spectrum use to precision prescription: a narrative review and expert opinion. Seizure Eur. J. Epilepsy 107, 121–131. doi:10.1016/j.seizure.2023.03.017
Depositario-Cabacar, D. T., Peters, J. M., Pong, A. W., Roth, J., Rotenberg, A., Riviello, J. J., et al. (2010). High-dose intravenous levetiracetam for acute seizure exacerbation in children with intractable epilepsy. Epilepsia 51 (7), 1319–1322. doi:10.1111/j.1528-1167.2010.02519.x
Di, L. (2019). The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr. Drug Metab. 20 (2), 91–102. doi:10.2174/1389200219666180821094502
Drugbank. Levetiracetam (2021). Available online at: https://go.drugbank.com/drugs/db01202.
Florek-Luszczki, M., Wlaz, A., and Luszczki, J. J. (2014). Interactions of levetiracetam with carbamazepine, phenytoin, topiramate and vigabatrin in the mouse 6Hz psychomotor seizure model - a type II isobolographic analysis. Eur. J. Pharmacol. 723, 410–418. doi:10.1016/j.ejphar.2013.10.063
Franco, V., Canevini, M. P., Capovilla, G., De Sarro, G., Galimberti, C. A., Gatti, G., et al. (2014). Off-label prescribing of antiepileptic drugs in pharmacoresistant epilepsy: a cross-sectional drug utilization study of tertiary care centers in Italy. CNS Drugs 28 (10), 939–949. doi:10.1007/s40263-014-0189-8
Fountain, N. B., Conry, J. A., Rodríguez-Leyva, I., Gutierrez-Moctezuma, J., Salas, E., Coupez, R., et al. (2007). Prospective assessment of levetiracetam pharmacokinetics during dose escalation in 4- to 12-year-old children with partial-onset seizures on concomitant carbamazepine or valproate. Epilepsy Res. 74 (1), 60–69. (Sarah). doi:10.1016/j.eplepsyres.2006.12.005
Freitas-Lima, P., Alexandre, V., Pereira, L. R. L., Feletti, F., Perucca, E., and Sakamoto, A. C. (2011). Influence of enzyme inducing antiepileptic drugs on the pharmacokinetics of levetiracetam in patients with epilepsy. Epilepsy Res. 94 (1–2), 117–120. doi:10.1016/j.eplepsyres.2011.01.007
Germovsek, E., Barker, C. I. S., Sharland, M., and Standing, J. F. (2019). Pharmacokinetic-pharmacodynamic modeling in pediatric drug development, and the importance of standardized scaling of clearance. Clin. Pharmacokinet. 58(1):39–39152. doi:10.1007/s40262-018-0659-0
Glauser, T. A., Pellock, J. M., Bebin, E. M., Fountain, N. B., Ritter, F. J., Jensen, C. M., et al. (2002). Efficacy and safety of levetiracetam in children with partial seizures: an open-label trial. Epilepsia 43 (5), 518–524. doi:10.1046/j.1528-1157.2002.13101.x
Glauser, T. A., Mitchell, W. G., Weinstock, A., Bebin, M., Chen, D., Coupez, R., et al. (2007). Pharmacokinetics of levetiracetam in infants and young children with epilepsy. Epilepsia 48 (6), 1117–1122. doi:10.1111/j.1528-1167.2007.01090.x
Jones, H. M., Gardner, I. B., and Watson, K. J. (2009). Modelling and PBPK simulation in drug discovery. AAPS J. 11 (1), 155–166. doi:10.1208/s12248-009-9088-1
Maglalang, P. D., Sinha, J., Zimmerman, K., McCann, S., Edginton, A., Hornik, C. P., et al. (2024). Application of physiologically based pharmacokinetic modeling to characterize the effects of age and obesity on the disposition of levetiracetam in the pediatric population. Clin. Pharmacokinet. 63 (6), 885–899. doi:10.1007/s40262-024-01367-2
Mangelings, D., Saevels, J., and Vander Heyden, Y. (2006). Enantiomeric impurity determination of levetiracetam using capillary electrochromatography. J. Sep. Sci. 29 (18), 2827–2836. doi:10.1002/jssc.200600190
Masungi, C., Mensch, J., Van Dijck, A., Borremans, C., Willems, B., Mackie, C., et al. (2008). Parallel artificial membrane permeability assay (PAMPA) combined with a 10-day multiscreen Caco-2 cell culture as a tool for assessing new drug candidates. Pharmazie 63 (3), 194–199. doi:10.1691/ph.2008.7327
May, T. W., Rambeck, B., and Jürgens, U. (2003). Serum concentrations of levetiracetam in epileptic patients: the influence of dose and co-medication. Ther. Drug Monit. 25 (6), 690–699. doi:10.1097/00007691-200312000-00007
Medicines Agency, E. (2018). Committee for medicinal products for human use (CHMP) guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. Available online at: www.ema.europa.eu/contact.
Obach, R. S., Baxter, J. G., Liston, T. E., Silber, B. M., Jones, B. C., MacIntyre, F., et al. (1997). The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 283 (1), 46–58. doi:10.1016/s0022-3565(24)36999-x
Patsalos, P. N. (2000). Pharmacokinetic profile of levetiracetamtoward ideal characteristics. Pharmacol. Ther. 85 (2), 77–85. doi:10.1016/S0163-7258(99)00052-2
Patsalos, P. N., Berry, D. J., Bourgeois, B. F. D., Cloyd, J. C., Glauser, T. A., Johannessen, S. I., et al. (2008). Antiepileptic drugs—best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE commission on therapeutic strategies. Epilepsia 49 (7), 1239–1276. doi:10.1111/j.1528-1167.2008.01561.x
Pellock, J. M., Glauser, T. A., Bebin, E. M., Fountain, N. B., Ritter, F. J., Coupez, R. M., et al. (2001). Pharmacokinetic study of levetiracetam in children. Epilepsia 42 (12), 1574–1579. doi:10.1046/j.1528-1157.2001.41300.x
Petruševska, M., Berglez, S., Krisch, I., Legen, I., Megušar, K., Peternel, L., et al. (2015). Biowaiver monographs for immediate release solid oral dosage forms: Levetiracetam. J. Pharm. Sci. 104 (9), 2676–2687. doi:10.1002/jps.24350
Poulin, P., and Theil, F. P. (2002). Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J. Pharm. Sci. 91 (5), 1358–1370. doi:10.1002/jps.10128
Ramael, S., de Smedt, F., Toublanc, N., Otoul, C., Boulanger, P., Riethuisen, J.-M., et al. (2006). Single-dose bioavailability of levetiracetam intravenous infusion relative to oral tablets and multiple-dose pharmacokinetics and tolerability of levetiracetam intravenous infusion compared with placebo in healthy subjects. Clin. Ther. 28 (5), 734–744. doi:10.1016/j.clinthera.2006.05.004
Reimers, A., Berg, J. A., Burns, M. L., Brodtkorb, E., Johannessen, S. I., and Johannessen Landmark, C. (2018). Reference ranges for antiepileptic drugs revisited: a practical approach to establish national guidelines. Drug Des. Devel Ther. 12, 271–280. doi:10.2147/DDDT.S154388
Rohatgi, A. (2021). WebPlotDigitiser. [Computer Software version 4.5] https://automeris.io/WebPlotDigitizer/.
Shao, W., Shen, C., Wang, W., Sun, H., Wang, X., Geng, K., et al. (2023). Development and validation of physiologically based pharmacokinetic model of levetiracetam to predict exposure and dose optimization in pediatrics. J. Pharm. Sci. 112 (10), 2667–2675. doi:10.1016/j.xphs.2023.03.025
Sinha, J., Karatza, E., and Gonzalez, D. (2022). Physiologically-based pharmacokinetic modeling of oxcarbazepine and levetiracetam during adjunctive antiepileptic therapy in children and adolescents. CPT Pharmacometrics Syst. Pharmacol. 11 (2), 225–239. doi:10.1002/psp4.12750
Stepanova, D., and Beran, R. G. (2014). Measurement of levetiracetam drug levels to assist with seizure control and monitoring of drug interactions with other anti-epileptic medications (AEMs). Seizure 23 (5), 371–376. doi:10.1016/j.seizure.2014.02.003
Strolin Benedetti, M., Whomsley, R., Nicolas, J. M., Young, C., and Baltes, E. (2003). Pharmacokinetics and metabolism of 14C-levetiracetam, a new antiepileptic agent, in healthy volunteers. Eur. J. Clin. Pharmacol. 59 (8–9), 621–630. doi:10.1007/s00228-003-0655-6
Wang, D.-D., Jin, Q., Hou, J., Feng, L., Li, N., Li, S.-Y., et al. (2016). Highly sensitive and selective detection of human carboxylesterase 1 activity by liquid chromatography with fluorescence detection. J. Chromatogr. B 1008, 212–218. doi:10.1016/j.jchromb.2015.11.046
Keywords: levetiracetam, PBPK modeling, pediatric population, dose optimization, epilepsy
Citation: Macente J, Hernandes Bonan R, Caleffi-Marchesini ER, Leira Pereira LR, De Freitas Lima P, Annaert P, Allegaert K and Diniz A (2025) Physiologically-based pharmacokinetic modeling and simulation for initial dose optimization of levetiracetam in pediatrics. Front. Pharmacol. 16:1678960. doi: 10.3389/fphar.2025.1678960
Received: 03 August 2025; Accepted: 10 November 2025;
Published: 04 December 2025.
Edited by:
Jeannine S. McCune, CSL, United StatesCopyright © 2025 Macente, Hernandes Bonan, Caleffi-Marchesini, Leira Pereira, De Freitas Lima, Annaert, Allegaert and Diniz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Diniz, YWRpbml6QHVlbS5icg==
†Orcid: Julia Macente, orcid.org/0000-0002-4452-5867; Rodolfo Hernandes Bonan, orcid.org/0000-0001-5944-2602; Edilainy Rizzieri Caleffi-Marchesini, orcid.org/0000-0002-8806-3158; Leonardo Régis Leira Pereira, orcid.org/0000-0002-8609-1390; Pieter Annaert, orcid.org/0000-0003-3525-7351; Karel Allegaert, orcid.org/0000-0001-9921-5105; Andrea Diniz, orcid.org/0000-0002-9638-9246