

- Department of Neurosciences, Medical University of South Carolina, Charleston, SC, USA
Theories of drug addiction that incorporate various concepts from the fields of learning and memory have led to the idea that classical and operant conditioning principles underlie the compulsiveness of addictive behaviors. Relapse often results from exposure to drug-associated cues, and the ability to extinguish these conditioned behaviors through inhibitory learning could serve as a potential therapeutic approach for those who suffer from addiction. This review will examine the evidence that extinction learning alters neuronal plasticity in specific brain regions and pathways. In particular, subregions of the prefrontal cortex (PFC) and their projections to other brain regions have been shown to differentially modulate drug-seeking and extinction behavior. Additionally, there is a growing body of research demonstrating that manipulation of neuronal plasticity can alter extinction learning. Therefore, the ability to alter plasticity within areas of the PFC through pharmacological manipulation could facilitate the acquisition of extinction and provide a novel intervention to aid in the extinction of drug-related memories.
Once believed to result from an immoral personality or lack of will power, it is now clear that drug addiction is a disease of the nervous system that involves uncontrollable drug intake and compulsive drug-seeking behavior. As such, addiction is characterized by periods of repeated drug use followed by unsuccessful attempts to maintain abstinence. As a chronic relapsing disorder, addiction is associated with numerous brain changes that include signaling pathways, neurotransmitters, and cell mechanisms that overlap with those that mediate normal learning and memory processes. Thus, there have been numerous theories that incorporate mechanisms of learning and memory as a basis for drug addiction (O’Brien et al., 1992; Di Chiara, 1999; Volkow et al., 2002; Kelley, 2004; Wise, 2004; Hyman, 2005; Weiss, 2005). These theories suggest that through basic conditioning principles, certain behaviors and drug-environment associations become “overlearned” and thus contribute to the compulsive behavior of addicts.
In classical Pavlovian conditioning, also referred to as stimulus-outcome conditioning, the presentation of a conditioned stimulus (CS) paired with presentation of an unconditioned stimulus (US) after repeated pairings comes to elicit a conditional response (CR). In a drug context, the repeated pairing of the CS (e.g., environmental cues) with the reinforcing properties of a drug (US) results in the ability of the CS alone to elicit drug-seeking behaviors. Conversely, instrumental conditioning, also referred to as response-outcome conditioning, involves learning through consequences (either positive or negative) that are contingent upon a particular behavior. In a drug context, behaviors that lead to the reinforcing effects of a drug are more likely to be repeated in the future. It is believed that drug-taking behaviors become compulsive and automatic (instrumental conditioning) with repeated drug exposure, and the associations between drugs and specific environmental cues and context become overly salient (classical conditioning). Conditioning processes also play a role in the influence of environments that predict drug availability to induce craving and promote relapse (Childress et al., 1988, 1999; Kalivas and Volkow, 2005).
The ability to suppress drug-seeking behaviors that are heavily influenced by drug memories is a logical therapeutic approach in the prevention of relapse. Extinction is the gradual reduction of a CR when the CS is no longer paired with the US. Functionally, it is observed as a decrease in responding from higher levels observed prior to extinction to lower levels following extinction training. Theoretically, this type of inhibitory training could reduce the occurrence of behaviors that are trademarks of addiction including drug-seeking and relapse. However, current implementations of extinction-based techniques, such as exposure therapy, have a poor record of efficacy (Childress et al., 1993; Conklin and Tiffany, 2002a,b). Therefore, there is a need to better understand the neural mechanisms that underlie extinction learning and develop therapeutic interventions that increase the success rates of cue exposure therapies. This could lead to treatments involving a combination of behavioral training and pharmacological interventions that create a more robust and persistent decrease in cue-induced affective responses to drug memories (Davis et al., 2006). A substantial amount of research has focused on the neurobiological processes that underlie the extinction of conditioned fear and non-drug reinforcers (e.g., food). While the majority of previous work has focused on understanding the mechanisms involved in fear/non-drug extinction, there is an increasing interest in understanding how these principles apply to addiction related behaviors. Results from the fear and non-drug extinction field have greatly informed and helped guide studies in addiction. Therefore, while the focus of this review is on the extinction of drug-seeking behavior, observations from the fear/non-drug extinction field will be incorporated where appropriate.
What is Extinction Learning?
At first glance, the phenomenon of extinction may appear to simply represent a process that involves the unlearning, forgetting, and/or erasure of a previously formed memory (Rescorla and Wagner, 1972). However, a large body of evidence gained over the past several decades provides strong support for the idea originally suggested by Pavlov (1927) that extinction is “new” and “active” learning and is not simply the “unlearning” or erasure of previously formed associations. Many of these studies have been carried out in rodents and involve the extinction of responding for a natural reinforcer such as food. In contrast, studies of extinction learning in addiction typically involve extinction of self-administration of a drug of abuse such as cocaine. These experimental procedures incorporate aspects of both instrumental and classical conditioning to train animals to perform a behavior (e.g., lever pressing) to receive access to a drug and associate discrete cues (e.g., auditory and/or visual) with the drug’s reinforcing effects. Regardless of the type of reinforcer used (e.g., food or drug), extinction is defined in this review as the omission of a previously delivered unconditioned stimuli/reinforcers or the absence of a contingency between a response and reinforcer (Lattal and Lattal, 2012). In addition, while extinction behavior can be observed in both classical and instrumental conditioning paradigms, this review will not attempt to define the neural mechanisms associated with each form of learning.
The idea that extinction involves new learning has great implications for not only understanding how drug memories can have a lasting influence on relapse but also for the development of pharmacological treatments for addiction. The following lines of evidence from studies examining the extinction of drug-related behaviors support the idea that extinction is indeed new learning:
(1) After extinction training, drug-seeking behavior can be reactivated with a single stimulus without the need for additional behavioral training (Sinha et al., 2000; Stewart, 2000, 2003; Sinha, 2001; Shalev et al., 2002; See, 2005; Epstein et al., 2006; Kalivas et al., 2006; Olmstead, 2006).
(2) Drug-seeking can resume after lengthy periods of abstinence or extinction training indicating that the original drug-memory remains and has not simply been deleted (Hammersley, 1992; Tobena et al., 1993; Corty and Coon, 1995; Di Ciano and Everitt, 2004).
(3) Extinction is context-specific (Bouton, 2000, 2002, 2004; Chaudhri et al., 2008; Wells et al., 2011), which suggests that original memory of drug reinforcement is still present even after extinction training.
(4) The retraining of self-administration after extinction is considerably less compared to original training (Carroll, 1998; Grasing et al., 2005).
(5) Extinction learning has been shown to involve classic cellular hallmarks of learning and memory (Crombag and Shaham, 2002; Sutton et al., 2003; Self and Choi, 2004; Self et al., 2004; Knackstedt et al., 2010).
Thus, findings from the literature on addiction support the idea that extinction training is not the removal of a previously formed association but instead involves the generation of a new memory that competes with the initial memory for control of behavior. As such, the original associative and instrumental conditioning that occurs during the early stages of addiction remains intact. Based on similar findings from the fear extinction literature, Quirk et al. (2006) presented a schematic model to illustrate the idea that even though fear behavior decreases, the original fear memory remains. As depicted in Figure 1 the same concept can be mapped onto the processes of addiction such that drug-seeking behavior declines during extinction training, but the drug-memory remains and competes with the newly formed extinction memory for the control of behavior. The formation of new memories during extinction training likely utilizes neural circuitry involved in basic learning and memory process. In the following sections we review studies that have highlighted specific brain regions and mechanisms involved in extinction learning.
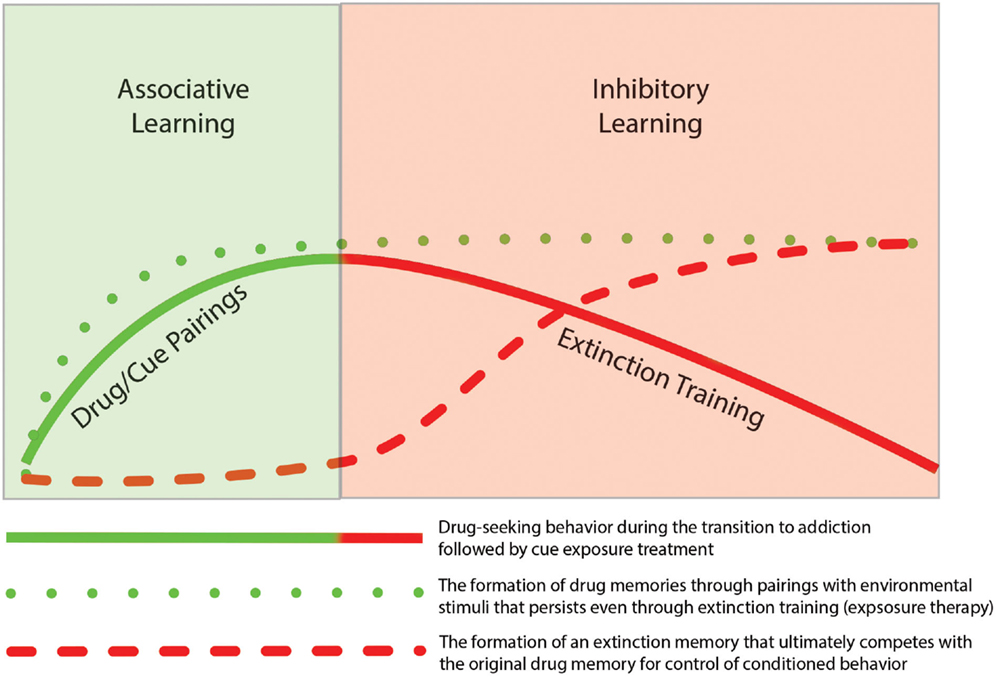
Figure 1. Depiction of the temporal relationship of associative learning of drug-seeking behavior with inhibitory learning during subsequent extinction of the drug-seeking behavior. The initial phase of addiction involves associative learning processes in which drug-taking becomes linked through classic Pavlovian conditioning with drug-related cues (e.g., drug paraphernalia or drug-taking environment). With repeated pairing, this association results in formation of a persistent “drug memory.” This memory trace remains long after discontinuation of drug-taking. The extinction of drug-seeking by pairing unreinforced exposure of drug-related cues, does not result in the deletion of the original drug memory, but instead involves the formation of a new inhibitory “extinction memory.” While this new memory provides inhibitory drive over drug-seeking behavior in the short term, the original drug-memory remains, which may explain the high rate of relapse following behavioral extinction therapies.
Neurocircuitry of the Extinction Learning
While the neurocircuitry of extinction is likely diffuse and involves a distributed network, there is evidence for the involvement of several key brain regions in drug-seeking, fear expression, and extinction behavior that could constitute differential circuits associated with each of these behaviors.
The Prefrontal Cortex
Increasing evidence has implicated the prefrontal cortex (PFC) in the extinction of both fear and drug-seeking behaviors. Anatomically, the rodent PFC is located in the anterior pole of the frontal cortex and is loosely defined as the anterior cingulate (ACC), medial PFC (mPFC), and orbital frontal cortex (OFC). As illustrated in Figure 2, the rodent mPFC can be further subdivided into a dorsal region called the prelimbic (PrL) cortex and a ventral region called the infralimbic (IfL) cortex. These subregions do not have well demarcated structural boundaries that can often make it difficult to clearly delineate these subregions, especially given the small size of the rodent brain. For this reason, investigators often simply divide this area into a dorsomedial PFC that includes the dorsal region of the PrL cortex and much of the overlying ACC, and a ventromedial PFC that includes the IfL cortex and the ventral portions of the PrL cortex (Figure 2). Defining analogous subregions of the PFC of rodents and human brain is also difficult due to the evolutionary expansion of the PFC. Therefore definitions are based not only upon common anatomical circuitry but also upon function. Based upon similarities in thalamic inputs, the rodent PrL region is considered to be equivalent to Brodmann area 32 (pregenual anterior cortex) and the IfL cortex is equivalent to Brodmann area 25 (subgenual anterior cortex) in the human (Figure 2). It should also be noted that the dorsolateral PFC of humans (conservatively defined as areas 9 and 46) is also considered to be equivalent to the rodent mPFC using a functional definition as both regions are involved in working-memory processes.
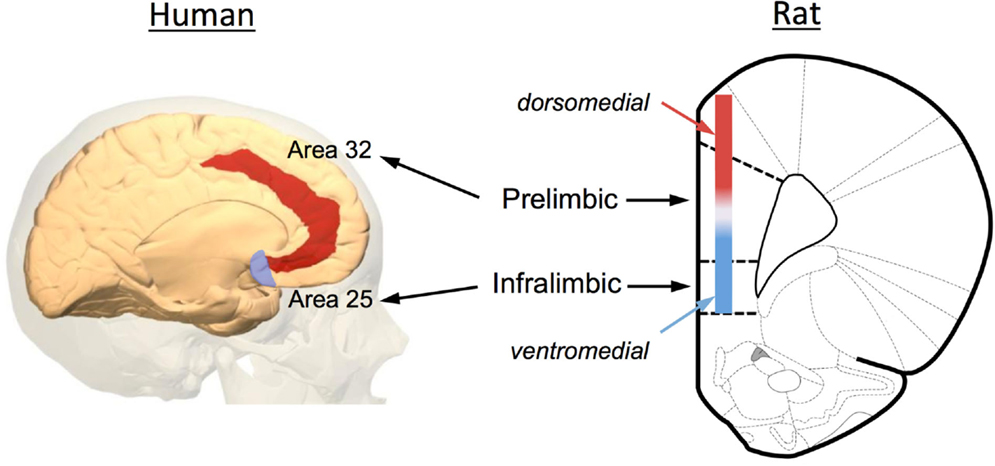
Figure 2. Anatomical depiction showing the location of the prelimbic (PrL) and infralimbic (IfL) subregions of the medial PFC of the rat and their equivalent regions of the human brain. Based upon commonality of thalamic inputs, the rodent PrL region is roughly analogous to Brodmann area 32 while the IfL is roughly analogous to Brodmann area 25. Because of the small size of the rodent brain and the lack of defined borders for the PrL and IfL regions, some investigators simply divide the rodent medial PFC into a dorsomedial and ventromedial region as illustrated in the diagram. The original image of the human brain shown on the left was modified from an image downloaded from Wikipedia (http://en.wikipedia.org/wiki/File:Brodmann_area_32_medial.jpg). The original rat brain image shown on the right was modified from Paxinos and Watson (6th Edition).
{kind=link}
While complex behaviors such as working memory, impulsivity, motivation, and decision-making have often been linked to the cognitive function of the PFC, a number of recent studies have implicated PFC subregions in extinction behavior. In particular, lesion studies have shown that the PrL cortex is necessary for the expression of conditioned fear while the IfL cortex is critical for the expression of extinction behavior (for reviews, see Quirk et al., 2010; Sierra-Mercado et al., 2011; Milad and Quirk, 2012). Drug-seeking behavior has been studied extensively in humans where it has been shown that presentation of drug stimuli significantly increase activation in specific regions of the PFC (for a review, see Goldstein and Volkow, 2011). Several inactivation studies have also implicated the PrL cortex of the rat as a critical component in the circuitry for drug-seeking behavior including cocaine (McFarland and Kalivas, 2001; Capriles et al., 2003; McLaughlin and See, 2003; McFarland et al., 2004; See, 2005; Di Pietro et al., 2006) and heroin (LaLumiere and Kalivas, 2008; Rogers et al., 2008). Additionally, the IfL cortex, which has been studied extensively in fear extinction, has also been implicated in the extinction of drug-seeking behavior (Ovari and Leri, 2008; Peters et al., 2008a,b). As depicted in Figure 3, converging lines of evidence from both the fear- and drug-conditioning fields suggest that the PrL cortex serves as an “on-switch” for conditioned fear expression and drug-seeking, while the IfL cortex functions as an “off-switch” for the expression of extinction behavior (LaLumiere and Kalivas, 2008; Peters et al., 2008a; Quirk and Mueller, 2008; LaLumiere et al., 2010). These subregions of the PFC could thus serve as candidate regions for plasticity-related changes associated with extinction behavior.
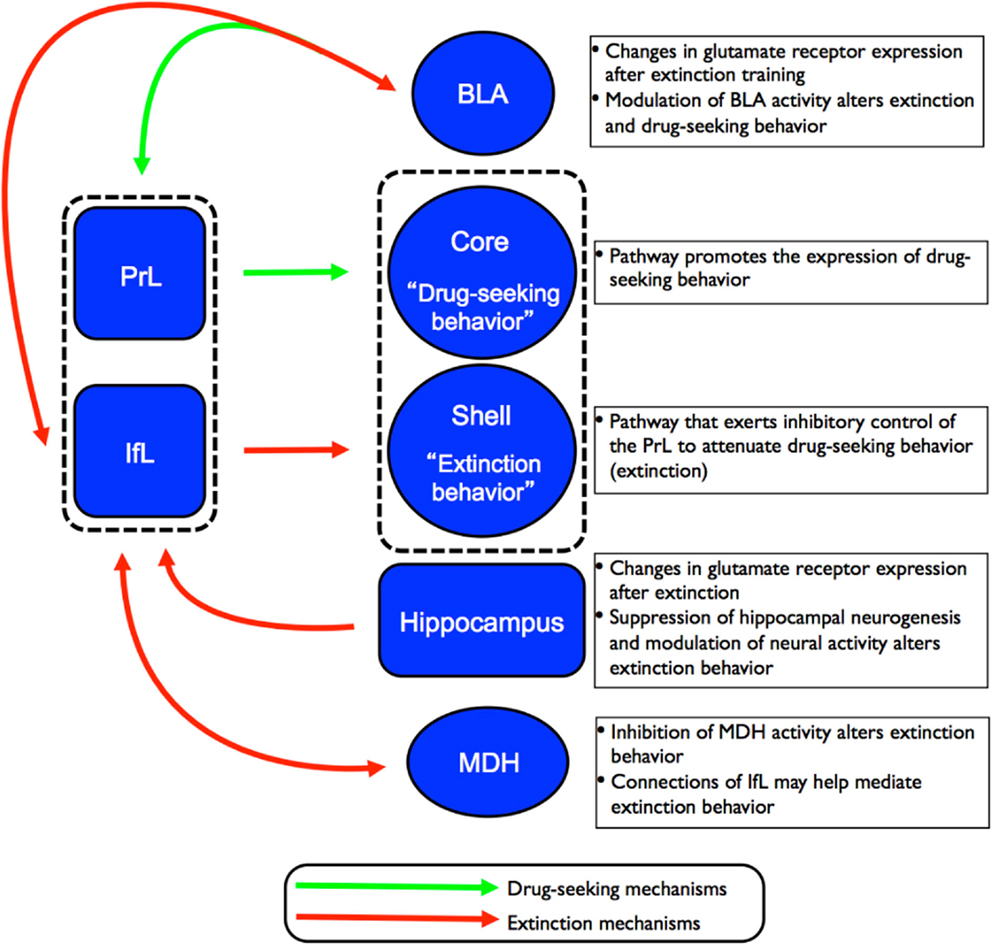
Figure 3. Schematic of the proposed circuitry involved in the drug-seeking and extinction behavior. Projections from the PrL cortex to the NAc core regulates the expression of cocaine-seeking behavior (indicated by green arrows) while projections from the IfL cortex to the NAc shell regulates the expression of extinction behavior (indicated by red arrows). Recent studies have also implicated the involvement of other brain regions such as the hippocampus, MDH, and BLA in the neurocircuitry of extinction of drug-seeking behavior.
Dorsal and Ventral Striatum
Different subregions of the striatum are important for mediating components of reward. The rodent striatum is divided into the dorsal and ventral striatum, and each of these regions can be further subdivided. Due to its involvement in habit learning, the dorsal striatum has been implicated in various aspects of the transition from voluntary behavior to uncontrolled habitual behavior that characterizes drug abuse (Robbins and Everitt, 2002; Weiss, 2005; Izquierdo et al., 2006). In particular, the dorsomedial subregion has been shown to modulate goal-direction actions that transitions to the dorsolateral striatum as these actions become habitual. The ventral striatum or nucleus accumbens (NAc) can be further divided into a lateral “core” and medial “shell” subregion. Through its connections with the PFC, amygdala, hippocampus, and motor regions, the NAc plays a role in guiding emotionally relevant behavioral responses related to the reinforcing properties of drugs and drug-related stimuli (Bonci et al., 2003; Di Chiara and Bassareo, 2007).
Recent studies have also implicated the NAc in extinction of drug-seeking behavior. Cocaine self-administration causes a decrease in tyrosine hydroxylase in the NAc shell which is reversed with extinction training during withdrawal from cocaine (Schmidt et al., 2001). Extinction training also induces an upregulation in the expression of AMPA receptor subunits within the NAc shell (Sutton et al., 2003; Self et al., 2004). More recently, it was shown that inactivation for the NAc shell resulted in the expression of cocaine-seeking behavior, possibly through an interaction with the IfL cortex (Peters et al., 2008a). Similarly, activation of IfL glutamatergic projections with an AMPA receptor positive allosteric modulator reduced cocaine-seeking behavior, and blockade of AMPA activity in the NAc shell attenuated this effect (LaLumiere et al., 2012). Similar findings have been observed with the extinction of ethanol-seeking behavior. For instance, the NAc shell, possibly through interactions with the hypothalamus or the amygdala, helps mediate the expression of extinction behavior (Millan et al., 2010; Millan and McNally, 2011). With regards to the NAc core, extinction training also normalizes cocaine-induced deficits in levels of the GluN1 subunit of the NMDA receptor (Self et al., 2004). Consistent with its role in goal-directed and habitual actions, the dorsal striatum has also been implicated in the extinction of habitual cocaine-seeking behavior (Fuchs et al., 2006). These lines of evidence suggest that there is a significant amount of plasticity that occurs within the dorsal striatum and NAc during extinction learning, and that these regions are central in the neurocircuitry of extinction of drug-seeking behavior.
Amygdala
As is the case with the PFC and the striatum, the amygdala is made up of a complex of different substructures that differentially contribute to extinction of fear- and drug-seeking behavior. The amydaloid complex includes the basal and lateral subregions (collectively known as the basolateral amygdala, BLA), medial amygdala (MeA), central amygdala (CeA), and cortical amygdala (CoA). The amygdala is involved with various learning and memory processes including formation and consolidation of emotional memories (Cahill et al., 2001; LaBar, 2003). The BLA also has an established role in synaptic plasticity associated with emotion-related behaviors, the processing of emotionally relevant stimuli (Cahill et al., 1995; McGaugh, 2004; Phelps et al., 2004; Maren, 2005; LaBar and Cabeza, 2006), and in stimulus-reward associations (Hatfield et al., 1996; Blundell et al., 2001; Baxter and Murray, 2002; Everitt et al., 2003; See, 2005; Balleine and Killcross, 2006). The BLA also plays an integral role in the formation of associations between drugs and environmental cues (Hiroi and White, 1991; Brown and Fibiger, 1993; Whitelaw et al., 1996; Rizos et al., 2005). While there has been a substantial amount of research implicating the BLA in the extinction of fear conditioning (Myers and Davis, 2002, 2007; Quirk et al., 2010; Sierra-Mercado et al., 2011), studies have also implicated this region in the extinction of drug-seeking behavior. For example, enhancement of glutamatergic transmission within the BLA facilities the extinction of a drug-paired conditioned place preference (CPP) (Shidara and Richmond, 2002; Schroeder and Packard, 2004), and given the essential role of the BLA in drug-seeking (See et al., 2003), it is logical to assume that plasticity within this structure may also influence extinction learning.
Hippocampus
The hippocampus is known to play an important role in various forms of learning and spatial/contextual memory and in memory consolidation/retrieval (Neves et al., 2008). The hippocampus is also involved in extinction behavior as evidenced by impairments in context-dependent extinction of fear conditioning that results from inactivation of this brain region (Corcoran and Maren, 2001; Corcoran et al., 2005; Ji and Maren, 2005) and cellular substrate inhibition (Szapiro et al., 2003; Vianna et al., 2003; Power et al., 2006). Similarly, studies have also implicated the hippocampus in the extinction of drug-related behaviors. Electrical stimulation of the ventral subiculum of the hippocampus reinstates cocaine-seeking (Vorel et al., 2001), and inactivation of this region abolishes cocaine drug-seeking (Sun et al., 2005). Neuronal activity within the CA1 and dentate gyrus (DG) has also been shown to change with extinction training of cocaine-associated cues providing further evidence that plasticity within this structure is associated with extinction behavior (Neisewander et al., 2000).
Hypothalamus
A less investigated structure that has recently been implicated in extinction behavior is the hypothalamus. This structure has traditionally been shown to be involved in reward and feeding but its influence on drug-seeking behavior is becoming better understood (for reviews, see Millan et al., 2011; Marchant et al., 2012). The medial dorsal hypothalamus (MDH) is associated with the termination of motivated behaviors and, therefore, is a logical candidate for involvement in extinction learning. In rats trained to self-administer alcohol and then exposed to extinction training, infusion of the inhibitory neuropeptide known as cocaine and amphetamine-regulated transcript (CART) into the MDH prevented the expression of extinction (Marchant et al., 2010). It is important to note that a similar effect was found with the extinction of sucrose-seeking behavior suggesting the mechanisms within the LDH that help regulate extinction may not be unique to drug reinforcers (Millan et al., 2011). To add further support for the role of the MDH in extinction behavior, this region receives extensive projections from the IfL cortex (Thompson and Swanson, 1998; Heidbreder and Groenewegen, 2003). In rats exposed to extinction training after a history of alcohol administration, the expression of extinction is associated with induction of c-Fos expression in retrograde labeled IfL cortical neurons projecting to the MDH (Marchant et al., 2010; Millan et al., 2011). Together, these findings suggest plasticity-related changes in the MDH, and through its connections with the IfL cortex, can mediate the extinction of reward-seeking behavior. These results also identify a brain region to investigate as a novel candidate for the facilitation of extinction behavior.
Based on findings detailed in the preceding sections, there are several key brain regions involved in extinction behavior. The exact details of how these structures interact to form a neurocircuitry that mediates extinction behavior have yet to be fully established. However, converging lines of evidence indicate that subregions of the PFC (and their corresponding projections to subcortical structures) play a major role in the extinction of drug and fear behaviors. Peters et al. (2009) proposed that extinction of drug memories comprises overlapping neural circuitry with that of fear memories. According to the model of the neurocircuitry of fear conditioning, the PrL cortex sends excitatory projections to the BLA that, in turn, promote the expression of conditioned fear via excitation of the CeA. In contrast, the IfL cortex sends excitatory projections to GABAergic inhibitory neurons in the intercalated (ITC) cell masses in the amygdala. This leads to inhibition of the CeA and attenuation of the expression of conditioned fear, and promotes the expression of extinction behavior. In the neurocircuitry of the extinction of drug memories, the PrL cortex also sends excitatory projections to the core region of the NAc where it has been shown to regulate the expression of cocaine-seeking behavior. In contrast, excitatory projections from the IfL cortex to the shell region of the NAc promote the extinction of cocaine-seeking behavior. This proposed circuitry for the extinction of drug behaviors is depicted in Figure 3. What is currently unknown is how structures such as the BLA, hippocampus, and MDH contribute to the established role of the PFC subregions in extinction behavior.
Glutamatergic Mechanisms in Extinction
In recent years, a number of studies have provided a more detailed analysis of the plasticity-related mechanisms that may mediate extinction behavior. Pathways connecting the various brain regions involved in extinction may differentially modulate the expression of drug-seeking and extinction of drug-seeking behavior. For instance, it was observed that there is increased activity of ventromedial PFC neurons in response to presentation of cocaine-related cues during extinction training. Interestingly, when activity in this region was inhibited, there was a corresponding decrease in extinction responding (Koya et al., 2009). Additionally, it has been shown that prefrontal regions have the ability to influence activity in other extinction-related brain structures. For instance, stimulation of IfL cortical output results in an inhibition of pyramidal neurons in the PrL cortex through a feed-forward mechanism (Ferrante et al., 2009). Similar results were found in a study that utilized optogenetic procedures to activate or inhibit specific cell types in isolated brain regions in combination with single-unit recordings of neuronal activity. It was revealed that optogenetic stimulation of viral vector encoding channel rhodopsin 2 (ChR2) excitatory neurons in the IfL cortex produced excitation of IfL cortical pyramidal neurons and also increased their responsiveness to excitatory input from multisensory brain regions (Ji and Neugebauer, 2012). It was further observed that activation of the IfL cortex inhibits PrL output, supporting the suggestion that IfL cortex mediated extinction mechanisms may involve inhibition of PrL cortex output that would ultimately mediate fear expression and possibly drug-seeking. Previous research has also shown that stimulation of the PrL region results in excitation of BLA neurons (Likhtik et al., 2005) and stimulation of the IfL region reduced the responsiveness of CeA neurons to inputs from the insula and BLA (Quirk et al., 2003). While these studies did not directly address extinction of fear expression or drug-seeking behavior, they provide support for how the IfL region of the PFC-through its direct projections to subcortical regions (e.g., amygdala, NAc, and hippocampus)-can mediate extinction behavior. Additionally, the ability of IfL cortical activation to exert inhibitory control over output from pyramidal neurons in the PrL cortex may also impact the expression of fear and drug-seeking behaviors.
The highly persistent nature of drug- and fear-related cues to induce relapse and the ineffectiveness of behavioral therapies to reduce the impact of these cues has led to a focus on understanding the neural mechanisms involved in relapse with the goal that they may be targeted as a means to enhance extinction learning. Studies have pharmacologically manipulated cellular process and substrates in specific brain regions in an attempt to “strengthen” inhibitory learning formed during extinction training. Using various behavioral paradigms such as fear-conditioning procedures and drug-self administration, investigators have begun to uncover plasticity-related mechanisms that facilitate extinction learning. Given the importance of glutamatergic transmission in learning and memory processes, a strong focus has been placed on targeting glutamate-related processes in extinction learning. Manipulation of both ionotropic and metabotropic receptors facilitates the extinction of fear-conditioning and drug-seeking behavior (for reviews, see Cleva et al., 2010; Myers et al., 2011). While blockade of NMDA receptors impairs extinction learning, enhancement of these receptors with the NMDA partial agonist D-cycloserine (DCS) facilitates the acquisition of extinction of conditioned fear and drug-seeking behavior (Myers and Carlezon, 2012). Similarly, modulation of AMPA receptor activity, which like NMDA receptors is also critically involved in synaptic plasticity, can also facilitate extinction learning (Kaplan and Moore, 2011; Myers et al., 2011).
In addition to targeting ionotropic glutamate receptors, activation of mGluR5 have been shown to facilitate extinction learning through a process that may involve enhanced NMDA receptor function. Systemic administration of the mGluR5 positive allosteric modulator CDPPB facilitates extinction of cocaine-seeking behavior in CPP (Gass and Olive, 2009) and self-administration (Cleva et al., 2011) paradigms, but does not alter the extinction of methamphetamine self-administration (Widholm et al., 2011). Further implicating mGluR5 in extinction, studies in mGluR5 knockout mice revealed marked deficits in both contextual and auditory fear extinction (Xu et al., 2009). Additionally, inhibition of mGluR5 prior to extinction learning prevented the recall of extinction learning while localized infusion of a mGluR5 antagonist in the IfL cortex produced a similar effect (Fontanez-Nuin et al., 2011). A recent study also highlighted the importance of group 1 mGluRs in the ventromedial PFC in the extinction of cocaine-seeking behavior. In rats trained to self-administer cocaine, infusion of a mGluR1/5 antagonist into the dorsomedial PFC failed to alter the rate of extinction. In contrast, infusion of a mGluR1/5 agonist had a facilitating effect on extinction of cocaine-seeking behavior (Ben-Shahar et al., 2013). This study also revealed that animals displaying deficits in extinction learning also had a significant reduction in group 1 mGluR function in the ventromedial PFC. Together these intriguing findings provide further support for glutamate-related plasticity in the IfL cortex in extinction learning.
Studies of conditioned fear have shown that inactivation of the rostral BLA (rBLA) slows cocaine cue extinction learning, and it has been suggested that simultaneous activity in the rBLA and hippocampus might be required for the acquisition of cocaine cue extinction learning (Szalay et al., 2011). Another study has shown that inactivation of the BLA not only resulted in a delay in extinction recall of an opiate reward memory, but also caused an increase in the spontaneous firing of neurons in the PrL cortex (Sun and Laviolette, 2012). This suggests that a functional link between the PrL cortex and BLA might modulate the processing of an opiate-related memory. An influence of AMPA receptor activity in the BLA during the extinction of cocaine-seeking behavior has also been reported. It was observed that expression of AMPA receptor subunit GluA1 decreased in the BLA but increased in the ventromedial PFC in response to extinction training (Nic Dhonnchadha et al., 2013), adding further support for a functional connection between the mPFC and amygdala in the extinction of drug-seeking behavior. In the hippocampus, extinction of a morphine-conditioned context was associated with changes in the phosphorylation of AMPA receptors at hippocampal synapses while no changes were observed in animals that were not exposed to extinction training (Billa et al., 2009). Furthermore, suppression of neurogenesis in the adult hippocampus after the acquisition of cocaine self-administration significantly enhanced resistance to extinction (Noonan et al., 2010). Similar to the effects observed in the rBLA, inactivation of the dorsal hippocampus slowed the rate of extinction of a cocaine memory (Szalay et al., 2011). Furthermore, cocaine self-administration training reduces neurogenesis in the DG, an effect that was normalized by extinction training (Deschaux et al., 2012). It was also observed that low frequency stimulation of the hippocampus prevented this extinction-induced normalization of DG neurogenesis. Together, these studies indicate a critical role of plasticity-related changes within the amygdala and hippocampus in the extinction of drug-seeking behavior. Although it has yet to be explored, it is possible that pharmacological manipulation of plasticity within these brain regions could serve to facilitate extinction of conditioned drug-seeking behavior.
Noradrenergic Mechanisms in Extinction
While glutamate-related neurochemical processes have received the most attention in extinction behavior, an emerging area of interest is the role that noradrenergic mechanisms play in extinction learning (for an extensive review, see Mueller and Cahill, 2010). Norepinephrine has been shown to be involved in various aspects of memory, most notably the strengthening of memory formation (McGaugh, 2004). While there has been a renewed interest in the ability of noradrenergic mechanisms to mediate fear extinction, the results have been inconsistent. For example, it has been shown that systemic administration of the beta-adrenergic antagonist propranolol prior to extinction training impaired subsequent retrieval of contextual fear extinction (Ouyang and Thomas, 2005). However, direct infusions of norepinephrine into the amygdala after extinction training facilitated the extinction of contextual fear (Berlau and McGaugh, 2006), suggesting that noradrenergic mechanisms may help mediate the consolidation of extinction learning. It has also been shown that arousal-related norepinephrine release in the IfL cortex is important for the formation of fear extinction memory (Mueller et al., 2008).
There have been several interesting observations regarding the influence of noradrenergic mechanisms on the extinction of drug-seeking behavior. Yohimbine, an alpha2-receptor antagonist that promotes the release of norepinephrine, impairs the extinction of cocaine CPP (Davis et al., 2008) and slows the rate of extinction of cocaine self-administration (Kupferschmidt et al., 2009). Furthermore, infusion of the beta-receptor agonist clenbuterol into the IfL cortex facilitates extinction of cocaine-seeking behavior (LaLumiere et al., 2010). These studies add support to the growing body of evidence that areas of the PFC are heavily involved in extinction behavior, and one possible mechanism could be noradrenergic-related changes in this region. Norepinephrine release alters the cellular properties of target neurons that may enhance excitability and synaptic plasticity and thus promote the formation of an extinction memory (Mueller and Cahill, 2010). Support for this comes from studies showing that norepinephrine enhances intrinsic excitability in the IfL cortex (Barth et al., 2007; Mueller et al., 2008), amygdala (Tully et al., 2007), and hippocampus (Pedreira and Maldonado, 2003).
Epigenetics and Extinction
Epigenetic mechanisms associated with extinction learning have received substantial attention over the pass several years and are providing unique insight into plasticity-related mechanisms of extinction. Epigenetic modification refers to the structural adaptation of chromosomes that results in altered activity states (Bird, 2007; Graff and Tsai, 2013). Epigenetic mechanisms exert lasting control over gene expression without altering the genetic code and may mediate stable changes in brain function (Tsankova et al., 2007). Investigation into the epigenetic regulation of neurobiological adaptations that are associated with psychiatric disorders, including addiction and PTSD, could provide novel approaches to the mechanisms underlying extinction learning.
The formation of long-term memories is thought to correlate with changes in gene expression. Research suggests that epigenetic-related mechanisms, such as histone acetylation/deacytylation and DNA methylation/demethylation, may mediate some of these processes (for a review, see Tsankova et al., 2007). For example, memory deficits in rodents can be recovered with administration of a histone deacetylase (HDAC) inhibitor, while conditioning in rodents is associated with histone protein H3 phosphoacetylation and chromatin remodeling (Levenson and Sweatt, 2005). Furthermore, synaptic plasticity is associated with epigenetic changes and can be promoted with HDAC inhibitors (Levenson et al., 2004). While these data indicate that epigenetic mechanisms are involved during the acquisition of conditioning, evidence also indicates that these same mechanisms may play a role in extinction learning.
In fear conditioning, it has been shown that acetylation and deacetylation of histones can enhance memories formed during conditioning and extinction behavior (Levenson et al., 2004; Bredy et al., 2007; Lattal et al., 2007). The non-selective HDAC inhibitor valproic acid can facilitate not only the acquisition and extinction of conditioned fear, but also the reconsolidation of this memory (Bredy and Barad, 2008). Similar results have been obtained with the HDAC inhibitor vorinostat (Fujita et al., 2012). It has also been shown that deficits in the extinction learning of conditioned fear in isogenic 129S1 (S1) mice can be recovered by administration of an HDAC inhibitor (Whittle et al., 2013). Administration of another non-selective HDAC inhibitor sodium butyrate (NaB) has a facilitating effect on the extinction of a fear memory in mice (Itzhak et al., 2012), which might be due, at least in part, to epigenetic-related mechanisms in the hippocampus and IfL cortex (Stafford et al., 2012). Furthermore, overexpression of HDAC1 in the hippocampus has also been shown to facilitate the extinction of contextual fear memories, and this effect can be prevented by inhibition of HDAC1 (Bahari-Javan et al., 2012). Finally, inhibition of p300 (a histone acetyltransferase) in the IfL cortex can enhance extinction of fear conditioning in mice, which was suggested to result from the influence of p300 on LTP in this brain region (Marek et al., 2011).
While there have been substantially fewer studies examining the epigenetic changes that accompany the extinction of drug-seeking behavior, similar results to the fear-conditioning literature have been observed. Malvaez et al. (2010) examined the effect of HDAC inhibition on the extinction of a cocaine-induced CPP. They found that systemic administration of NaB facilitated the extinction of the cocaine memory and attenuated reinstatement of cocaine-seeking behavior. Importantly, these behavioral effects correlated with enhanced acetylation of histone H3 in the NAc. Systemic administration of the HDAC3 inhibitor RGFP966 also facilitates the extinction of a cocaine-related memory, and it was suggested that this effect was mediated by enhancement of memory consolidation during extinction learning (Malvaez et al., 2013). These effects were also associated with histone acetylation linked to gene expression in the IfL cortex, hippocampus, and NAc. Taken together, observations from the fear and addiction fields have provided intriguing insights into the possible therapeutic targets related to epigenetics that could potentially be utilized to facilitate the extinction of emotionally salient memories. While further research is needed to fully clarify the roles of these mechanisms in the extinction of drug-related memories, this is a promising area of investigation for the extinction of drug cues given the established role of epigenetic mechanisms in memory.
Extinction Versus Reconsolidation
The widely held belief that extinction learning involves the acquisition of new memories has been challenged recently with the idea that behavior typically interpreted as extinction learning may actually represent reconsolidation of previously formed memories (for reviews on this topic, see Dudai and Eisenberg, 2004; Nader and Einarsson, 2010; Sorg, 2012). During the initial coding of events, memories are labile, but subsequently consolidate into long-term storage through protein synthesis-dependent mechanisms (Quirk et al., 2010). Thus, extinction training may serve to reverse or update previously formed contingencies (Sorg, 2012). As such, exposure to extinction training shortly after reactivation of a fear memory attenuates recovery, renewal, and reinstatement of conditioned fear (Monfils et al., 2009; Quirk et al., 2010). Importantly, studies have shown that timing of the CS presentation is critical in order to temporarily activate the labile state in which updates to the CS-US association can occur. Reconsolidation typically requires short presentations of the CS (Nader and Hardt, 2009), and presentation of the CS alone within 6 h after memory reactivation results in behavioral effects that reflect unlearning as opposed to the inhibition of fear (Nader et al., 2000; Quirk et al., 2010). Theoretically, the ability to modify existing memories, as opposed to creating new inhibitory associations through the facilitation of extinction learning, could be advantageous over extinction-based exposure therapies. Studies show that while extinction learning can be facilitated pharmacologically, these effects can be context-dependent (Bouton, 2000, 2002, 2004; Milad et al., 2005; Woods and Bouton, 2006). Modification of the original memory, rather than the creation of competitive memories, might manifest a behavior that is more resistant to the influence of context (Quirk et al., 2010), an idea that has clinical support. For instance, administration of a beta-adrenergic receptor antagonist during reconsolidation removes the fear-arousing aspects of the conditioned memory (Soeter and Kindt, 2011). This effect was not specific to the initial stimuli used in the fear-conditioning paradigm and generalized to related stimuli. While there is excitement in the field that revolves around the influence of reconsolidation on extinction behavior, more research is clearly needed to fully elucidate the contributions of both processes in the inhibition of behavior.
Conclusion
In this review, we focused on studies that incorporate learning principles in extinction training with the goal of lessening the influence of these cues on addictive behavior. It has been widely recognized that drug use and relapse are strongly cue specific (Drummond and Glautier, 1994) and one of the most important factors that contributes to relapse is the impact of drug cues on drug-seeking behavior. In recent years, there has been increasing attention on the neural mechanisms that underlie extinction learning in an effort to manipulate and possibly enhance learning that occurs during inhibitory conditioning. Clinically, extinction-based behavioral therapies have generally proven ineffective for suppression of relapse to drug taking. This lack of efficacy may relate to the fact that extinction learning does not erase the original drug memory but instead involves formation of a new extinction memory that acts in competition for control of behavior with the drug memory. However, the intransigent nature of the drug-memory appears to promote subsequent relapse to drug-taking. The temporal relationship of extinction and relapse are depicted in Figure 4. While extinction training alone can initially reduce drug-seeking behavior, these effects are likely context-specific. Thus, when the addict is exposed to drug cues outside of the treatment environment, the drug memory that was suppressed but not erased during extinction training, can reinitiate drug-seeking and drug use. Although speculative, pharmacological facilitation of extinction learning may enhance formation of an inhibitory memory that is much “stronger” than the initial drug memory and may help protect against cue-induced relapse. Recent research has shed light on pharmacologically targeting glutamatergic, adrenergic, and epigenetic mechanisms to enhance inhibitory learning during extinction training. Furthermore, while the neurocircuitry of extinction likely involves a distributed network of different brain regions that include the mPFC, NAc, amydala, hippocampus, and hypothalamus, recent studies have implicated opposing roles of the PrL and IfL subregions of the PFC in the control of drug-related behavior. A model has emerged in which drug-seeking is likely a PrL cortex driven behavior while extinction learning and the resulting inhibition of drug-seeking is a IfL cortex driven behavior. One aim of future research is to elucidate the contribution of these different neural regions and mechanisms to the facilitation of extinction learning to ultimately develop more effective treatments for addiction.
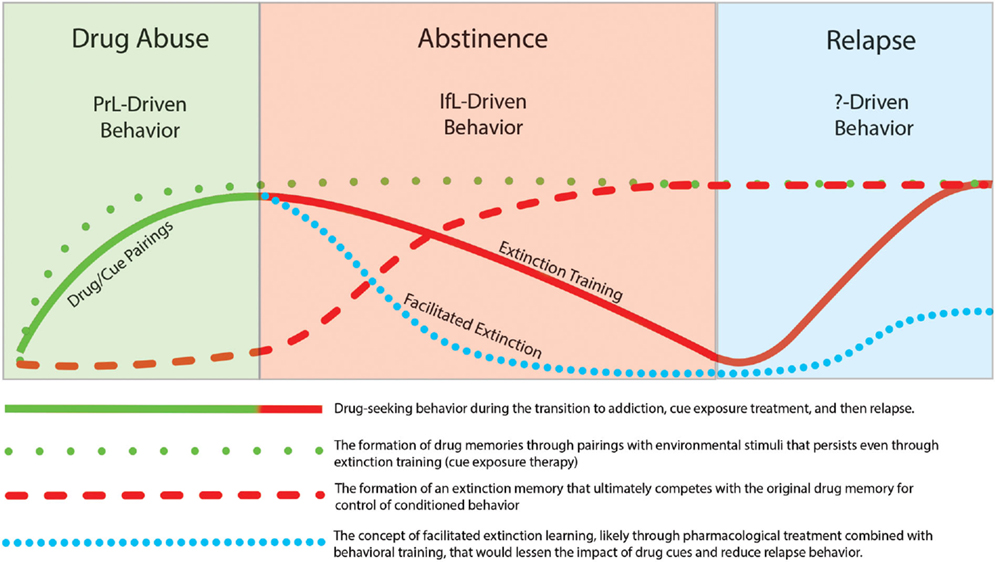
Figure 4. Illustration showing that while behavioral extinction training can reduce drug-seeking behavior, the persistence of the original drug memory can promote subsequent relapse. However, pharmacological facilitation of the extinction process may promote a stronger and more persistent extinction memory that may lead to reductions in the rate of relapse.
Although there have been substantial advances in our understanding of the neural mechanisms involved in the extinction of drug-related memories, a number of important issues need to be addressed by additional studies in the field of drug addiction. For example, while the neural circuits that mediate extinction of fear behavior do not overlap directly with those in drug-seeking behaviors, are the mechanisms that mediate extinction the same for all drugs of abuse? There is strong evidence for involvement of the PrL cortex in cocaine-seeking and IfL cortex in cocaine extinction behavior. However, there are few and sometimes conflicting findings with other drugs of abuse, such as heroin (Rogers et al., 2008), methamphetamine (Rocha and Kalivas, 2010), and alcohol (Millan et al., 2010). In addition, as recent research begins to highlight the importance of other structures in the extinction of drug memories, how do they interact with the established role of the PFC in mediating extinction behavior? The identification of the specific roles of the hippocampus, amygdala, and hypothalamus and their influence on a “final common pathway” through the PFC could provide insight into possible therapeutic targets to enhance extinction learning.
The standard procedure for extinction training is repeated presentations of the CS in absence of the US. While this method has permeated the literature since the days of Pavlov, it is not clear whether this is the most effective approach. It is of interest that several studies have examined the “retrieval-extinction” approach that combines extinction training with brief drug-memory retrieval (to activate the labile state of the memory) that have produced encouraging results (Hutton-Bedbrook and McNally, 2013).
With an increased focus on the importance of consolidation in promotion of extinction learning, a particularly interesting area of future research will be to understand the effect of sleep and sleep insomnia in extinction learning. Coordinated activity in the PFC and hippocampus during sleep is critical for the consolidation of memories (Euston et al., 2007). Sleep has been shown to promote retention of fear extinction memory (Pace-Schott et al., 2009, 2012). Interestingly, while extinction training can attenuate sleep disturbances (Wellman et al., 2008), the bidirectional relationship between these two processes and how they contribute to the extinction of drug memories is largely unexplored.
Lastly, there are multiple studies showing that context is a major hurdle in using extinction-based treatment approaches, and another important area of research will be to determine whether context specificity of extinction can be prevented. Context is not limited to common environmental stimuli associated with drug use and can include factors such as drug states and the passage of time (Bouton et al., 2012). Thus, there are many types of stimuli that serve as contextual cues to promote relapse, and future research is needed in order to understand how pharmacological manipulation of extinction training could be used to minimize the influence of context.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bahari-Javan, S., Maddalena, A., Kerimoglu, C., Wittnam, J., Held, T., Bahr, M., et al. (2012). HDAC1 regulates fear extinction in mice. J. Neurosci. 32, 5062–5073. doi:10.1523/JNEUROSCI.0079-12.2012
Balleine, B. W., and Killcross, S. (2006). Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 29, 272–279. doi:10.1016/j.tins.2006.03.002
Barth, A. M., Vizi, E. S., and Lendvai, B. (2007). Noradrenergic enhancement of Ca2(responses of basal dendrites in layer 5 pyramidal neurons of the prefrontal cortex. Neurochem. Int. 51, 323–327. doi:10.1016/j.neuint.2007.05.008
Baxter, M. G., and Murray, E. A. (2002). The amygdala and reward. Nat. Rev. Neurosci. 3, 563–573. doi:10.1038/nrn875
Ben-Shahar, O., Sacramento, A. D., Miller, B. W., Webb, S. M., Wroten, M. G., Silva, H. E., et al. (2013). Deficits in ventromedial prefrontal cortex group 1 metabotropic glutamate receptor function mediate resistance to extinction during protracted withdrawal from an extensive history of cocaine self-administration. J. Neurosci. 33, 495–506a. doi:10.1523/JNEUROSCI.3710-12.2013
Berlau, D. J., and McGaugh, J. L. (2006). Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol. Learn. Mem. 86, 123–132. doi:10.1016/j.nlm.2005.12.008
Billa, S. K., Sinha, N., Rudrabhatla, S. R., and Moron, J. A. (2009). Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur. J. Neurosci. 29, 55–64. doi:10.1111/j.1460-9568.2008.06560.x
Blundell, P., Hall, G., and Killcross, S. (2001). Lesions of the basolateral amygdala disrupt selective aspects of reinforcer representation in rats. J. Neurosci. 21, 9018–9026.
Bonci, A., Bernardi, G., Grillner, P., and Mercuri, N. B. (2003). The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends Pharmacol. Sci. 24, 172–177. doi:10.1016/S0165-6147(03)00068-3
Bouton, M. E. (2000). A learning theory perspective on lapse, relapse, and the maintenance of behavior change. Health Psychol. 19, 57–63. doi:10.1037/0278-6133.19.Suppl1.57
Bouton, M. E. (2002). Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol. Psychiatry 52, 976–986. doi:10.1016/S0006-3223(02)01546-9
Bouton, M. E. (2004). Context and behavioral processes in extinction. Learn. Mem. 11, 485–494. doi:10.1101/lm.78804
Bouton, M. E., Winterbauer, N. E., and Todd, T. P. (2012). Relapse processes after the extinction of instrumental learning: renewal, resurgence, and reacquisition. Behav. Processes 90, 130–141. doi:10.1016/j.beproc.2012.03.004
Bredy, T. W., and Barad, M. (2008). The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn. Mem. 15, 39–45. doi:10.1101/lm.801108
Bredy, T. W., Wu, H., Crego, C., Zellhoefer, J., Sun, Y. E., and Barad, M. (2007). Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem. 14, 268–276. doi:10.1101/lm.500907
Brown, E. E., and Fibiger, H. C. (1993). Differential effects of excitotoxic lesions of the amygdala on cocaine-induced conditioned locomotion and conditioned place preference. Psychopharmacology (Berl.) 113, 123–130. doi:10.1007/BF02244344
Cahill, L., Babinsky, R., Markowitsch, H. J., and McGaugh, J. L. (1995). The amygdala and emotional memory. Nature 377, 295–296. doi:10.1038/377295a0
Cahill, L., McGaugh, J. L., and Weinberger, N. M. (2001). The neurobiology of learning and memory: some reminders to remember. Trends Neurosci. 24, 578–581. doi:10.1016/S0166-2236(00)01885-3
Capriles, N., Rodaros, D., Sorge, R. E., and Stewart, J. (2003). A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl.) 168, 66–74. doi:10.1007/s00213-002-1283-z
Carroll, M. E. (1998). Acquisition and reacquisition (relapse) of drug abuse: modulation by alternative reinforcers. NIDA Res. Monogr. 169, 6–25.
Chaudhri, N., Sahuque, L. L., Cone, J. J., and Janak, P. H. (2008). Reinstated ethanol-seeking in rats is modulated by environmental context and requires the nucleus accumbens core. Eur. J. Neurosci. 28, 2288–2298. doi:10.1111/j.1460-9568.2008.06517.x
Childress, A. R., Hole, A. V., Ehrman, R. N., Robbins, S. J., McLellan, A. T., and O’Brien, C. P. (1993). Cue reactivity and cue reactivity interventions in drug dependence. NIDA Res. Monogr. 137, 73–95.
Childress, A. R., McLellan, A. T., Ehrman, R., and O’Brien, C. P. (1988). Classically conditioned responses in opioid and cocaine dependence: a role in relapse? NIDA Res. Monogr. 84, 25–43.
Childress, A. R., Mozley, P. D., McElgin, W., Fitzgerald, J., Reivich, M., and O’Brien, C. P. (1999). Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 156, 11–18.
Cleva, R. M., Gass, J. T., Widholm, J. J., and Olive, M. F. (2010). Glutamatergic targets for enhancing extinction learning in drug addiction. Curr. Neuropharmacol. 8, 394–408. doi:10.2174/157015910793358169
Cleva, R. M., Hicks, M. P., Gass, J. T., Wischerath, K. C., Plasters, E. T., Widholm, J. J., et al. (2011). mGluR5 positive allosteric modulation enhances extinction learning following cocaine self-administration. Behav. Neurosci. 125, 10–19. doi:10.1037/a0022339
Conklin, C. A., and Tiffany, S. T. (2002a). Applying extinction research and theory to cue-exposure addiction treatments. Addiction 97, 155–167. doi:10.1046/j.1360-0443.2002.00014.x
Conklin, C. A., and Tiffany, S. T. (2002b). Cue-exposure treatment: time for change. Addiction 97, 1219–1221. doi:10.1046/j.1360-0443.2002.00205.x
Corcoran, K. A., Desmond, T. J., Frey, K. A., and Maren, S. (2005). Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J. Neurosci. 25, 8978–8987. doi:10.1523/JNEUROSCI.2246-05.2005
Corcoran, K. A., and Maren, S. (2001). Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J. Neurosci. 21, 1720–1726.
Corty, E. W., and Coon, B. (1995). The extinction of naturally occurring conditioned reactions in psychoactive substance users: analog studies. Addict. Behav. 20, 605–618. doi:10.1016/0306-4603(95)00020-D
Crombag, H. S., and Shaham, Y. (2002). Renewal of drug seeking by contextual cues after prolonged extinction in rats. Behav. Neurosci. 116, 169–173. doi:10.1037/0735-7044.116.1.169
Davis, A. R., Shields, A. D., Brigman, J. L., Norcross, M., McElligott, Z. A., Holmes, A., et al. (2008). Yohimbine impairs extinction of cocaine-conditioned place preference in an alpha2-adrenergic receptor independent process. Learn. Mem. 15, 667–676. doi:10.1101/lm.1079308
Davis, M., Barad, M., Otto, M., and Southwick, S. (2006). Combining pharmacotherapy with cognitive behavioral therapy: traditional and new approaches. J. Trauma. Stress 19, 571–581. doi:10.1002/jts.20149
Deschaux, O., Vendruscolo, L. F., Schlosburg, J. E., Diaz-Aguilar, L., Yuan, C. J., Sobieraj, J. C., et al. (2012). Hippocampal neurogenesis protects against cocaine-primed relapse. Addict. Biol.. [Epub ahead of print]. doi:10.1111/adb.12019
Di Chiara, G. (1999). Drug addiction as dopamine-dependent associative learning disorder. Eur. J. Pharmacol. 375, 13–30. doi:10.1016/S0014-2999(99)00372-6
Di Chiara, G., and Bassareo, V. (2007). Reward system and addiction: what dopamine does and doesn’t do. Curr. Opin. Pharmacol. 7, 69–76. doi:10.1016/j.coph.2006.11.003
Di Ciano, P., and Everitt, B. J. (2004). Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J. Neurosci. 24, 7167–7173. doi:10.1523/JNEUROSCI.1581-04.2004
Di Pietro, N. C., Black, Y. D., and Kantak, K. M. (2006). Context-dependent prefrontal cortex regulation of cocaine self-administration and reinstatement behaviors in rats. Eur. J. Neurosci. 24, 3285–3298. doi:10.1111/j.1460-9568.2006.05193.x
Drummond, D. C., and Glautier, S. (1994). A controlled trial of cue exposure treatment in alcohol dependence. J. Consult. Clin. Psychol. 62, 809–817. doi:10.1037/0022-006X.62.4.809
Dudai, Y., and Eisenberg, M. (2004). Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron 44, 93–100. doi:10.1016/j.neuron.2004.09.003
Epstein, D. H., Preston, K. L., Stewart, J., and Shaham, Y. (2006). Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl.) 189, 1–16. doi:10.1007/s00213-006-0529-6
Euston, D. R., Tatsuno, M., and McNaughton, B. L. (2007). Fast-forward playback of recent memory sequences in prefrontal cortex during sleep. Science 318, 1147–1150. doi:10.1126/science.1148979
Everitt, B. J., Cardinal, R. N., Parkinson, J. A., and Robbins, T. W. (2003). Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann. N. Y. Acad. Sci. 985, 233–250. doi:10.1111/j.1749-6632.2003.tb07085.x
Ferrante, M., Migliore, M., and Ascoli, G. A. (2009). Feed-forward inhibition as a buffer of the neuronal input-output relation. Proc. Natl. Acad. Sci. U.S.A. 106, 18004–18009. doi:10.1073/pnas.0904784106
Fontanez-Nuin, D. E., Santini, E., Quirk, G. J., and Porter, J. T. (2011). Memory for fear extinction requires mGluR5-mediated activation of infralimbic neurons. Cereb. Cortex 21, 727–735. doi:10.1093/cercor/bhq147
Fuchs, R. A., Branham, R. K., and See, R. E. (2006). Different neural substrates mediate cocaine seeking after abstinence versus extinction training: a critical role for the dorsolateral caudate-putamen. J. Neurosci. 26, 3584–3588. doi:10.1523/JNEUROSCI.5146-05.2006
Fujita, Y., Morinobu, S., Takei, S., Fuchikami, M., Matsumoto, T., Yamamoto, S., et al. (2012). Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J. Psychiatr. Res. 46, 635–643. doi:10.1016/j.jpsychires.2012.01.026
Gass, J. T., and Olive, M. F. (2009). Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol. Psychiatry 65, 717–720. doi:10.1016/j.biopsych.2008.11.001
Goldstein, R. Z., and Volkow, N. D. (2011). Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 12, 652–669. doi:10.1038/nrn3119
Graff, J., and Tsai, L. H. (2013). The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 53, 311–330. doi:10.1146/annurev-pharmtox-011112-140216
Grasing, K., He, S., and Li, N. (2005). Selegiline modifies the extinction of responding following morphine self-administration, but does not alter cue-induced reinstatement, reacquisition of morphine reinforcement, or precipitated withdrawal. Pharmacol. Res. 51, 69–78. doi:10.1016/j.phrs.2004.07.004
Hammersley, R. (1992). Cue exposure and learning theory. Addict. Behav. 17, 297–300. doi:10.1016/0306-4603(92)90035-T
Hatfield, T., Han, J. S., Conley, M., Gallagher, M., and Holland, P. (1996). Neurotoxic lesions of basolateral, but not central, amygdala interfere with Pavlovian second-order conditioning and reinforcer devaluation effects. J. Neurosci. 16, 5256–5265.
Heidbreder, C. A., and Groenewegen, H. J. (2003). The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci. Biobehav. Rev. 27, 555–579. doi:10.1016/j.neubiorev.2003.09.003
Hiroi, N., and White, N. M. (1991). The lateral nucleus of the amygdala mediates expression of the amphetamine-produced conditioned place preference. J. Neurosci. 11, 2107–2116.
Hutton-Bedbrook, K., and McNally, G. P. (2013). The promises and pitfalls of retrieval-extinction procedures in preventing relapse to drug seeking. Front. Psychiatry 4:14. doi:10.3389/fpsyt.2013.00014
Hyman, S. E. (2005). Addiction: a disease of learning and memory. Am. J. Psychiatry 162, 1414–1422. doi:10.1176/appi.ajp.162.8.1414
Itzhak, Y., Anderson, K. L., Kelley, J. B., and Petkov, M. (2012). Histone acetylation rescues contextual fear conditioning in nNOS KO mice and accelerates extinction of cued fear conditioning in wild type mice. Neurobiol. Learn. Mem. 97, 409–417. doi:10.1016/j.nlm.2012.03.005
Izquierdo, I., Bevilaqua, L. R., Rossato, J. I., Bonini, J. S., Medina, J. H., and Cammarota, M. (2006). Different molecular cascades in different sites of the brain control memory consolidation. Trends Neurosci. 29, 496–505. doi:10.1016/j.tins.2006.07.005
Ji, G., and Neugebauer, V. (2012). Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain 5, 36. doi:10.1186/1756-6606-5-36
Ji, J., and Maren, S. (2005). Electrolytic lesions of the dorsal hippocampus disrupt renewal of conditional fear after extinction. Learn. Mem. 12, 270–276. doi:10.1101/lm.91705
Kalivas, P. W., Peters, J., and Knackstedt, L. (2006). Animal models and brain circuits in drug addiction. Mol. Interv. 6, 339–344. doi:10.1124/mi.6.6.7
Kalivas, P. W., and Volkow, N. D. (2005). The neural basis of addiction: a pathology of motivation and choice. Am. J. Psychiatry 162, 1403–1413. doi:10.1176/appi.ajp.162.8.1403
Kaplan, G. B., and Moore, K. A. (2011). The use of cognitive enhancers in animal models of fear extinction. Pharmacol. Biochem. Behav. 99, 217–228. doi:10.1016/j.pbb.2011.01.009
Kelley, A. E. (2004). Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44, 161–179. doi:10.1016/j.neuron.2004.09.016
Knackstedt, L. A., Moussawi, K., LaLumiere, R., Schwendt, M., Klugmann, M., and Kalivas, P. W. (2010). Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J. Neurosci. 30, 7984–7992. doi:10.1523/JNEUROSCI.1244-10.2010
Koya, E., Uejima, J. L., Wihbey, K. A., Bossert, J. M., Hope, B. T., and Shaham, Y. (2009). Role of ventral medial prefrontal cortex in incubation of cocaine craving. Neuropharmacology 56(Suppl. 1), 177–185. doi:10.1016/j.neuropharm.2008.04.022
Kupferschmidt, D. A., Tribe, E., and Erb, S. (2009). Effects of repeated yohimbine on the extinction and reinstatement of cocaine seeking. Pharmacol. Biochem. Behav. 91, 473–480. doi:10.1016/j.pbb.2008.08.026
LaBar, K. S. (2003). Emotional memory functions of the human amygdala. Curr. Neurol. Neurosci. Rep. 3, 363–364. doi:10.1007/s11910-003-0015-z
LaBar, K. S., and Cabeza, R. (2006). Cognitive neuroscience of emotional memory. Nat. Rev. Neurosci. 7, 54–64. doi:10.1038/nrn1825
LaLumiere, R. T., and Kalivas, P. W. (2008). Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 28, 3170–3177. doi:10.1523/JNEUROSCI.5129-07.2008
LaLumiere, R. T., Niehoff, K. E., and Kalivas, P. W. (2010). The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn. Mem. 17, 168–175. doi:10.1101/lm.1576810
LaLumiere, R. T., Smith, K. C., and Kalivas, P. W. (2012). Neural circuit competition in cocaine-seeking: roles of the infralimbic cortex and nucleus accumbens shell. Eur. J. Neurosci. 35, 614–622. doi:10.1111/j.1460-9568.2012.07991.x
Lattal, K. M., Barrett, R. M., and Wood, M. A. (2007). Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav. Neurosci. 121, 1125–1131. doi:10.1037/0735-7044.121.5.1125
Lattal, K. M., and Lattal, K. A. (2012). Facets of Pavlovian and operant extinction. Behav. Processes 90, 1–8. doi:10.1016/j.beproc.2012.03.009
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559. doi:10.1074/jbc.M402229200
Levenson, J. M., and Sweatt, J. D. (2005). Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6, 108–118. doi:10.1038/nrn1604
Likhtik, E., Pelletier, J. G., Paz, R., and Pare, D. (2005). Prefrontal control of the amygdala. J. Neurosci. 25, 7429–7437. doi:10.1523/JNEUROSCI.2314-05.2005
Malvaez, M., McQuown, S. C., Rogge, G. A., Astarabadi, M., Jacques, V., Carreiro, S., et al. (2013). HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U.S.A. 110, 2647–2652. doi:10.1073/pnas.1213364110
Malvaez, M., Sanchis-Segura, C., Vo, D., Lattal, K. M., and Wood, M. A. (2010). Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol. Psychiatry 67, 36–43. doi:10.1016/j.biopsych.2009.07.032
Marchant, N. J., Furlong, T. M., and McNally, G. P. (2010). Medial dorsal hypothalamus mediates the inhibition of reward seeking after extinction. J. Neurosci. 30, 14102–14115. doi:10.1523/JNEUROSCI.4079-10.2010
Marchant, N. J., Millan, E. Z., and McNally, G. P. (2012). The hypothalamus and the neurobiology of drug seeking. Cell. Mol. Life Sci. 69, 581–597. doi:10.1007/s00018-011-0817-0
Marek, R., Coelho, C. M., Sullivan, R. K., Baker-Andresen, D., Li, X., Ratnu, V., et al. (2011). Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J. Neurosci. 31, 7486–7491. doi:10.1523/JNEUROSCI.0133-11.2011
Maren, S. (2005). Building and burying fear memories in the brain. Neuroscientist 11, 89–99. doi:10.1177/1073858404269232
McFarland, K., Davidge, S. B., Lapish, C. C., and Kalivas, P. W. (2004). Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J. Neurosci. 24, 1551–1560. doi:10.1523/JNEUROSCI.4177-03.2004
McFarland, K., and Kalivas, P. W. (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 21, 8655–8663.
McGaugh, J. L. (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 27, 1–28. doi:10.1146/annurev.neuro.27.070203.144157
McLaughlin, J., and See, R. E. (2003). Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl.) 168, 57–65. doi:10.1007/s00213-002-1196-x
Milad, M. R., Orr, S. P., Pitman, R. K., and Rauch, S. L. (2005). Context modulation of memory for fear extinction in humans. Psychophysiology 42, 456–464. doi:10.1111/j.1469-8986.2005.00302.x
Milad, M. R., and Quirk, G. J. (2012). Fear extinction as a model for translational neuroscience: ten years of progress. Annu. Rev. Psychol. 63, 129–151. doi:10.1146/annurev.psych.121208.131631
Millan, E. Z., Furlong, T. M., and McNally, G. P. (2010). Accumbens shell-hypothalamus interactions mediate extinction of alcohol seeking. J. Neurosci. 30, 4626–4635. doi:10.1523/JNEUROSCI.4933-09.2010
Millan, E. Z., Marchant, N. J., and McNally, G. P. (2011). Extinction of drug seeking. Behav. Brain Res. 217, 454–462. doi:10.1016/j.bbr.2010.10.037
Millan, E. Z., and McNally, G. P. (2011). Accumbens shell AMPA receptors mediate expression of extinguished reward seeking through interactions with basolateral amygdala. Learn. Mem. 18, 414–421. doi:10.1101/lm.2144411
Monfils, M. H., Cowansage, K. K., Klann, E., and Ledoux, J. E. (2009). Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 324, 951–955. doi:10.1126/science.1167975
Mueller, D., and Cahill, S. P. (2010). Noradrenergic modulation of extinction learning and exposure therapy. Behav. Brain Res. 208, 1–11. doi:10.1016/j.bbr.2009.11.025
Mueller, D., Porter, J. T., and Quirk, G. J. (2008). Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J. Neurosci. 28, 369–375. doi:10.1523/JNEUROSCI.3248-07.2008
Myers, K. M., and Carlezon, W. A. Jr. (2012). D-cycloserine effects on extinction of conditioned responses to drug-related cues. Biol. Psychiatry 71, 947–955. doi:10.1016/j.biopsych.2012.02.030
Myers, K. M., Carlezon, W. A. Jr., and Davis, M. (2011). Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology 36, 274–293. doi:10.1038/npp.2010.88
Myers, K. M., and Davis, M. (2002). Behavioral and neural analysis of extinction. Neuron 36, 567–584. doi:10.1016/S0896-6273(02)01064-4
Myers, K. M., and Davis, M. (2007). Mechanisms of fear extinction. Mol. Psychiatry 12, 120–150. doi:10.1038/sj.mp.4001939
Nader, K., and Einarsson, E. O. (2010). Memory reconsolidation: an update. Ann. N. Y. Acad. Sci. 1191, 27–41. doi:10.1111/j.1749-6632
Nader, K., and Hardt, O. (2009). A single standard for memory: the case for reconsolidation. Nat. Rev. Neurosci. 10, 224–234. doi:10.1038/nrn2590
Nader, K., Schafe, G. E., and Le Doux, J. E. (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406, 722–726. doi:10.1038/35021052
Neisewander, J. L., Baker, D. A., Fuchs, R. A., Tran-Nguyen, L. T., Palmer, A., and Marshall, J. F. (2000). Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J. Neurosci. 20, 798–805.
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75. doi:10.1038/nrn2303
Nic Dhonnchadha, B. A., Lin, A., Leite-Morris, K. A., Kaplan, G. B., Man, H. Y., and Kantak, K. M. (2013). Alterations in expression and phosphorylation of GluA1 receptors following cocaine-cue extinction learning. Behav. Brain Res. 238, 119–123. doi:10.1016/j.bbr.2012.10.012
Noonan, M. A., Bulin, S. E., Fuller, D. C., and Eisch, A. J. (2010). Reduction of adult hippocampal neurogenesis confers vulnerability in an animal model of cocaine addiction. J. Neurosci. 30, 304–315. doi:10.1523/JNEUROSCI.4256-09.2010
O’Brien, C. P., Childress, A. R., McLellan, A. T., and Ehrman, R. (1992). A learning model of addiction. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 70, 157–177.
Olmstead, M. C. (2006). Animal models of drug addiction: where do we go from here? Q. J. Exp. Psychol. (Hove) 59, 625–653. doi:10.1080/17470210500356308
Ouyang, M., and Thomas, S. A. (2005). A requirement for memory retrieval during and after long-term extinction learning. Proc. Natl. Acad. Sci. U.S.A. 102, 9347–9352. doi:10.1073/pnas.0502315102
Ovari, J., and Leri, F. (2008). Inactivation of the ventromedial prefrontal cortex mimics re-emergence of heroin seeking caused by heroin reconditioning. Neurosci. Lett. 444, 52–55. doi:10.1016/j.neulet.2008.08.015
Pace-Schott, E. F., Milad, M. R., Orr, S. P., Rauch, S. L., Stickgold, R., and Pitman, R. K. (2009). Sleep promotes generalization of extinction of conditioned fear. Sleep 32, 19–26.
Pace-Schott, E. F., Verga, P. W., Bennett, T. S., and Spencer, R. M. (2012). Sleep promotes consolidation and generalization of extinction learning in simulated exposure therapy for spider fear. J. Psychiatr. Res. 46, 1036–1044. doi:10.1016/j.jpsychires.2012.04.015
Pedreira, M. E., and Maldonado, H. (2003). Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron 38, 863–869. doi:10.1016/S0896-6273(03)00352-0
Peters, J., Kalivas, P. W., and Quirk, G. J. (2009). Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn. Mem. 16, 279–288. doi:10.1101/lm.1041309
Peters, J., LaLumiere, R. T., and Kalivas, P. W. (2008a). Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J. Neurosci. 28, 6046–6053. doi:10.1523/JNEUROSCI.1045-08.2008
Peters, J., Vallone, J., Laurendi, K., and Kalivas, P. W. (2008b). Opposing roles for the ventral prefrontal cortex and the basolateral amygdala on the spontaneous recovery of cocaine-seeking in rats. Psychopharmacology (Berl.) 197, 319–326. doi:10.1007/s00213-007-1034-2
Phelps, E. A., Delgado, M. R., Nearing, K. I., and Ledoux, J. E. (2004). Extinction learning in humans: role of the amygdala and vmPFC. Neuron 43, 897–905. doi:10.1016/j.neuron.2004.08.042
Power, A. E., Berlau, D. J., McGaugh, J. L., and Steward, O. (2006). Anisomycin infused into the hippocampus fails to block "reconsolidation" but impairs extinction: the role of re-exposure duration. Learn. Mem. 13, 27–34. doi:10.1101/lm.91206
Quirk, G. J., Garcia, R., and Gonzalez-Lima, F. (2006). Prefrontal mechanisms in extinction of conditioned fear. Biol. Psychiatry 60, 337–343. doi:10.1016/j.biopsych.2006.03.010
Quirk, G. J., Likhtik, E., Pelletier, J. G., and Pare, D. (2003). Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J. Neurosci. 23, 8800–8807.
Quirk, G. J., and Mueller, D. (2008). Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33, 56–72. doi:10.1038/sj.npp.1301555
Quirk, G. J., Pare, D., Richardson, R., Herry, C., Monfils, M. H., Schiller, D., et al. (2010). Erasing fear memories with extinction training. J. Neurosci. 30, 14993–14997. doi:10.1523/JNEUROSCI.4268-10.2010
Rescorla, R. A., and Wagner, A. R. (1972). “A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and nonreinforcement,” in Classical Conditioning II: Current Research and Theory, eds A. H. Black, and W. K. Prokasy (New York: Appleton-Century-Crofts), 64–99.
Rizos, Z., Ovari, J., and Leri, F. (2005). Reconditioning of heroin place preference requires the basolateral amygdala. Pharmacol. Biochem. Behav. 82, 300–305. doi:10.1016/j.pbb.2005.08.019
Robbins, T. W., and Everitt, B. J. (2002). Limbic-striatal memory systems and drug addiction. Neurobiol. Learn. Mem. 78, 625–636. doi:10.1006/nlme.2002.4103
Rocha, A., and Kalivas, P. W. (2010). Role of the prefrontal cortex and nucleus accumbens in reinstating methamphetamine seeking. Eur. J. Neurosci. 31, 903–909. doi:10.1111/j.1460-9568.2010.07134.x
Rogers, J. L., Ghee, S., and See, R. E. (2008). The neural circuitry underlying reinstatement of heroin-seeking behavior in an animal model of relapse. Neuroscience 151, 579–588. doi:10.1016/j.neuroscience.2007.10.012
Schmidt, E. F., Sutton, M. A., Schad, C. A., Karanian, D. A., Brodkin, E. S., and Self, D. W. (2001). Extinction training regulates tyrosine hydroxylase during withdrawal from cocaine self-administration. J. Neurosci. 21, RC137.
Schroeder, J. P., and Packard, M. G. (2004). Facilitation of memory for extinction of drug-induced conditioned reward: role of amygdala and acetylcholine. Learn. Mem. 11, 641–647. doi:10.1101/lm.78504
See, R. E. (2005). Neural substrates of cocaine-cue associations that trigger relapse. Eur. J. Pharmacol. 526, 140–146. doi:10.1016/j.ejphar.2005.09.034
See, R. E., Fuchs, R. A., Ledford, C. C., and McLaughlin, J. (2003). Drug addiction, relapse, and the amygdala. Ann. N. Y. Acad. Sci. 985, 294–307. doi:10.1111/j.1749-6632.2003.tb07089.x
Self, D. W., and Choi, K. H. (2004). Extinction-induced neuroplasticity attenuates stress-induced cocaine seeking: a state-dependent learning hypothesis. Stress 7, 145–155. doi:10.1080/10253890400012677
Self, D. W., Choi, K. H., Simmons, D., Walker, J. R., and Smagula, C. S. (2004). Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn. Mem. 11, 648–657. doi:10.1101/lm.81404
Shalev, U., Grimm, J. W., and Shaham, Y. (2002). Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol. Rev. 54, 1–42. doi:10.1124/pr.54.1.1
Shidara, M., and Richmond, B. J. (2002). Anterior cingulate: single neuronal signals related to degree of reward expectancy. Science 296, 1709–1711. doi:10.1126/science.1069504
Sierra-Mercado, D., Padilla-Coreano, N., and Quirk, G. J. (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36, 529–538. doi:10.1038/npp.2010.184
Sinha, R. (2001). How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl.) 158, 343–359. doi:10.1007/s002130100917
Sinha, R., Fuse, T., Aubin, L. R., and O’Malley, S. S. (2000). Psychological stress, drug-related cues and cocaine craving. Psychopharmacology (Berl.) 152, 140–148. doi:10.1007/s002130000499
Soeter, M., and Kindt, M. (2011). Disrupting reconsolidation: pharmacological and behavioral manipulations. Learn. Mem. 18, 357–366. doi:10.1101/lm.2148511
Sorg, B. A. (2012). Reconsolidation of drug memories. Neurosci. Biobehav. Rev. 36, 1400–1417. doi:10.1016/j.neubiorev.2012.02.004
Stafford, J. M., Raybuck, J. D., Ryabinin, A. E., and Lattal, K. M. (2012). Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol. Psychiatry 72, 25–33. doi:10.1016/j.biopsych.2011.12.012
Stewart, J. (2000). Pathways to relapse: the neurobiology of drug- and stress-induced relapse to drug-taking. J. Psychiatry Neurosci. 25, 125–136.
Stewart, J. (2003). Stress and relapse to drug seeking: studies in laboratory animals shed light on mechanisms and sources of long-term vulnerability. Am. J. Addict. 12, 1–17. doi:10.1111/j.1521-0391.2003.tb00535.x
Sun, N., and Laviolette, S. R. (2012). Inactivation of the basolateral amygdala during opiate reward learning disinhibits prelimbic cortical neurons and modulates associative memory extinction. Psychopharmacology (Berl.) 222, 645–661. doi:10.1007/s00213-012-2665-5
Sun, W., Akins, C. K., Mattingly, A. E., and Rebec, G. V. (2005). Ionotropic glutamate receptors in the ventral tegmental area regulate cocaine-seeking behavior in rats. Neuropsychopharmacology 30, 2073–2081. doi:10.1038/sj.npp.1300744
Sutton, M. A., Schmidt, E. F., Choi, K. H., Schad, C. A., Whisler, K., Simmons, D., et al. (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421, 70–75. doi:10.1038/nature01249
Szalay, J. J., Morin, N. D., and Kantak, K. M. (2011). Involvement of the dorsal subiculum and rostral basolateral amygdala in cocaine cue extinction learning in rats. Eur. J. Neurosci. 33, 1299–1307. doi:10.1111/j.1460-9568.2010.07581.x
Szapiro, G., Vianna, M. R., McGaugh, J. L., Medina, J. H., and Izquierdo, I. (2003). The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus 13, 53–58. doi:10.1002/hipo.10043
Thompson, R. H., and Swanson, L. W. (1998). Organization of inputs to the dorsomedial nucleus of the hypothalamus: a reexamination with Fluorogold and PHAL in the rat. Brain Res. Brain Res. Rev. 27, 89–118. doi:10.1016/S0165-0173(98)00010-1
Tobena, A., Fernandez-Teruel, A., Escorihuela, R. M., Nunez, J. F., Zapata, A., Ferre, P., et al. (1993). Limits of habituation and extinction: implications for relapse prevention programs in addictions. Drug Alcohol Depend. 32, 209–217. doi:10.1016/0376-8716(93)90085-5
Tsankova, N., Renthal, W., Kumar, A., and Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367. doi:10.1038/nrn2132
Tully, K., Li, Y., Tsvetkov, E., and Bolshakov, V. Y. (2007). Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. U.S.A. 104, 14146–14150. doi:10.1073/pnas.0704621104
Vianna, M. R., Igaz, L. M., Coitinho, A. S., Medina, J. H., and Izquierdo, I. (2003). Memory extinction requires gene expression in rat hippocampus. Neurobiol. Learn. Mem. 79, 199–203. doi:10.1016/S1074-7427(03)00003-0
Volkow, N. D., Fowler, J. S., Wang, G. J., and Goldstein, R. Z. (2002). Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol. Learn. Mem. 78, 610–624. doi:10.1006/nlme.2002.4099
Vorel, S. R., Liu, X., Hayes, R. J., Spector, J. A., and Gardner, E. L. (2001). Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292, 1175–1178. doi:10.1126/science.1058043
Weiss, F. (2005). Neurobiology of craving, conditioned reward and relapse. Curr. Opin. Pharmacol. 5, 9–19. doi:10.1016/j.coph.2004.11.001
Wellman, L. L., Yang, L., Tang, X., and Sanford, L. D. (2008). Contextual fear extinction ameliorates sleep disturbances found following fear conditioning in rats. Sleep 31, 1035–1042.
Wells, A. M., Lasseter, H. C., Xie, X., Cowhey, K. E., Reittinger, A. M., and Fuchs, R. A. (2011). Interaction between the basolateral amygdala and dorsal hippocampus is critical for cocaine memory reconsolidation and subsequent drug context-induced cocaine-seeking behavior in rats. Learn. Mem. 18, 693–702. doi:10.1101/lm.2273111
Whitelaw, R. B., Markou, A., Robbins, T. W., and Everitt, B. J. (1996). Excitotoxic lesions of the basolateral amygdala impair the acquisition of cocaine-seeking behaviour under a second-order schedule of reinforcement. Psychopharmacology (Berl.) 127, 213–224. doi:10.1007/BF02246129
Whittle, N., Schmuckermair, C., Gunduz Cinar, O., Hauschild, M., Ferraguti, F., Holmes, A., et al. (2013). Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology 64, 414–423. doi:10.1016/j.neuropharm.2012.06.001
Widholm, J. J., Gass, J. T., Cleva, R. M., and Olive, M. F. (2011). The mGluR5 positive allosteric modulator CDPPB does not alter extinction or contextual reinstatement of methamphetamine-seeking behavior in rats. J. Addict. Res. Ther. S1, ii:004.
Wise, R. A. (2004). Dopamine, learning and motivation. Nat. Rev. Neurosci. 5, 483–494. doi:10.1038/nrn1406
Woods, A. M., and Bouton, M. E. (2006). D-cycloserine facilitates extinction but does not eliminate renewal of the conditioned emotional response. Behav. Neurosci. 120, 1159–1162. doi:10.1037/0735-7044.120.5.1159
Keywords: extinction learning, prelimbic, infralimbic, prefrontal cortex, addiction
Citation: Gass JT and Chandler LJ (2013) The plasticity of extinction: contribution of the prefrontal cortex in treating addiction through inhibitory learning. Front. Psychiatry 4:46. doi: 10.3389/fpsyt.2013.00046
Received: 27 February 2013; Accepted: 16 May 2013;
Published online: 30 May 2013.
Edited by:
Nicholas W. Gilpin, Louisiana State University Health Sciences Center New Orleans, USACopyright: © 2013 Gass and Chandler. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: L. J. Chandler, Department of Neurosciences, Medical University of South Carolina, 67 President Street MSC 861, Charleston, SC 29425, USA e-mail:Y2hhbmRqQG11c2MuZWR1