 Alan Anticevic1,2,3,4,5,6*
Alan Anticevic1,2,3,4,5,6* Michael W. Cole7
Michael W. Cole7 Grega Repovs8
Grega Repovs8 Aleksandar Savic9
Aleksandar Savic9 Naomi R. Driesen1Genevieve Yang1,4
Naomi R. Driesen1Genevieve Yang1,4 Youngsun T. Cho1
Youngsun T. Cho1 John D. Murray10
John D. Murray10 David C. Glahn1,5
David C. Glahn1,5 Xiao-Jing Wang10
Xiao-Jing Wang10 John H. Krystal1,2,3,11
John H. Krystal1,2,3,11
- 1Department of Psychiatry, Yale University School of Medicine, New Haven, CT, USA
- 2NIAAA Center for the Translational Neuroscience of Alcoholism, New Haven, CT, USA
- 3Abraham Ribicoff Research Facilities, Connecticut Mental Health Center, New Haven, CT, USA
- 4Interdepartmental Neuroscience Program, Yale University, New Haven, CT, USA
- 5Olin Neuropsychiatry Research Center, Institute of Living, Hartford Hospital, Hartford, CT, USA
- 6Department of Psychology, Yale University, New Haven, CT, USA
- 7Department of Psychology, Washington University in St. Louis, St. Louis, MO, USA
- 8Department of Psychology, University of Ljubljana, Ljubljana, Slovenia
- 9Department of Psychiatry, University of Zagreb School of Medicine, Zagreb, Croatia
- 10Center for Neural Science, New York University, New York, NY, USA
- 11Department of Neurobiology, Yale University School of Medicine, New Haven, CT, USA
Neuropsychiatric diseases such as schizophrenia and bipolar illness alter the structure and function of distributed neural networks. Functional neuroimaging tools have evolved sufficiently to reliably detect system-level disturbances in neural networks. This review focuses on recent findings in schizophrenia and bipolar illness using resting-state neuroimaging, an advantageous approach for biomarker development given its ease of data collection and lack of task-based confounds. These benefits notwithstanding, neuroimaging does not yet allow the evaluation of individual neurons within local circuits, where pharmacological treatments ultimately exert their effects. This limitation constitutes an important obstacle in translating findings from animal research to humans and from healthy humans to patient populations. Integrating new neuroscientific tools may help to bridge some of these gaps. We specifically discuss two complementary approaches. The first is pharmacological manipulations in healthy volunteers, which transiently mimic some cardinal features of psychiatric conditions. We specifically focus on recent neuroimaging studies using the NMDA receptor antagonist, ketamine, to probe glutamate synaptic dysfunction associated with schizophrenia. Second, we discuss the combination of human pharmacological imaging with biophysically informed computational models developed to guide the interpretation of functional imaging studies and to inform the development of pathophysiologic hypotheses. To illustrate this approach, we review clinical investigations in addition to recent findings of how computational modeling has guided inferences drawn from our studies involving ketamine administration to healthy subjects. Thus, this review asserts that linking experimental studies in humans with computational models will advance to effort to bridge cellular, systems, and clinical neuroscience approaches to psychiatric disorders.
Introduction
The human brain is a complex, dynamic system with computations occurring at several levels of organization, from individual synapses to networks that span multiple brain regions. These large-scale neural systems ultimately produce complex behaviors that are profoundly altered in the context of psychotropic drug administration or neuropsychiatric disease.
One example is schizophrenia – a common, multi-faceted, and heterogeneous neuropsychiatric syndrome (1) associated with disturbances in perception (2), belief (3), emotion (4), and cognition (5). A number of theoretical models of schizophrenia suggest that the clinical features of this disorder emerge from disturbances in neural connectivity and deficits in synaptic plasticity (6). Progress in explicating the pathophysiology of schizophrenia has been slowed by our limited understanding of the neurobiology of schizophrenia, shortcomings of animal models for this disorder, and the challenge of translating basic and clinical research approaches to this disorder. This knowledge gap has constrained our ability to develop new and more effective pharmacotherapies for schizophrenia (7), accounting for little change in the public health impact of this disease over the past two decades (8).
Disturbances in the structural and functional connectivity of the cerebral cortex are thought to be central to the neurobiology of schizophrenia (6) and are thought to impair the function of large-scale neural systems (9–13). Efforts have been made to reconcile these system-level observations with the cellular neuropathology of schizophrenia. One leading mechanistic model proposes that glutamate synaptic abnormalities associated with schizophrenia, mimicked in part by the effects of drugs that block the N-methyl-d-aspartate glutamate receptor (NMDAR) (7), disturb the local balance of excitation and inhibition and thereby contribute to alterations in large-scale neural system functional connectivity (14). This influential hypothesis is based on a key observation: sub-anesthetic doses of non-competitive NMDAR antagonists such as ketamine produce symptoms resembling those of schizophrenia in healthy humans (15). There is also growing evidence from pre-clinical (16), post-mortem (17), neuroimaging (18), and pharmacological experiments (15) illustrating that abnormal NMDA receptor function may be one pathophysiological mechanism occurring in people with schizophrenia. Alterations in synaptic function of gamma-aminobutyric acid (GABA) (19, 20) and dopamine (21) have also been implicated in schizophrenia, which likely affect local circuit computations (22). Collectively, this work provides insight into how disturbances in synaptic glutamate signaling (among other neurotransmitters) could contribute to producing schizophrenia symptoms (7). Nevertheless, there remains an important explanatory gap between mechanistic cellular-level hypotheses of schizophrenia and non-invasive neuroimaging studies that characterize the function of neural systems.
Functional neuroimaging has consistently revealed both region-specific and network alterations in schizophrenia across a number of cognitive measures (23). For instance, there is now substantial evidence suggesting profound alterations in networks supporting complex cognition in schizophrenia [e.g., working memory (WM) (24–26)]. This work has been complemented by a parallel focus on characterizing functional connectivity alterations in schizophrenia (27). Resting-state functional connectivity is based on the analysis of low-frequency fluctuations present in the blood-oxygenation-level-dependent (BOLD) signal (28, 29). These low-frequency fluctuations have been shown to be temporally correlated within spatially distinct but functionally related networks (30), establishing an intrinsic functional architecture (31) across species (32). The functional networks identified at rest also are correlated with other measures of structural and functional connectivity in healthy populations (33) and allow for characterization of distributed circuit abnormalities in neuropsychiatric illness (34, 35). Such approaches have been successfully applied to the study of schizophrenia and have increasingly revealed neuroimaging markers of this complex illness (12, 27, 36–38), which may be consistent with established theoretical models of this disease (39).
This review first focuses on resting-state functional connectivity MRI (rs-fcMRI) studies of schizophrenia that are providing distinct insights into cortical dysfunction associated with this disorder (37, 38). We highlight emerging connectivity strategies that deal with the complexity and heterogeneity of schizophrenia anatomy, physiology, and behavior (40). For instance, we articulate how data-driven tools that capture distributed connectivity abnormalities – such as global brain connectivity (GBC) – have the potential to avoid biases and identify network disturbances that traditional seed-based approaches may fail to detect. We next detail recent specific efforts to assay thalamo-cortical dysfunction in schizophrenia (38), long thought to be important to the clinical features of this disorder (41–43). We also discuss the potential utility of such data-driven approaches to discover variations in large-scale systems that may be altered across diagnostic categories (e.g., bipolar illness with psychosis and schizophrenia) and that may inform symptom-based or circuit-based diagnostic systems, such as the NIMH Research Domain Criteria (RDoC) (44–46).
The encouraging progress in understanding functional connectivity alterations in schizophrenia creates new opportunity to reconcile system-level findings with hypotheses emerging from molecular and cellular studies of this disorder. Failure to integrate these multiple levels of research undermines our understanding of the pathophysiology of this disorder and it will impede medications development. To this end, we next turn to studies using pharmacological models (47), such as the NMDAR antagonist ketamine, to test hypotheses related to the causes of circuit dysfunction in schizophrenia (18). Here we review a focused set of pharmacological neuroimaging (ph-fMRI) studies that utilized NMDAR antagonists to alter behavior and connectivity in healthy volunteers as a way to better understand schizophrenia (18, 48–54). These ph-fMRI investigations provide insight into how specific manipulations of synaptic function have effects that scale to produce both behavioral and system-level alterations that may be observed in patients. We articulate a set of directions for future studies that can capitalize on ph-fMRI as a tool to provide insight into specific illness-related mechanisms, especially when combined with advanced functional connectivity approaches.
Finally, we briefly articulate the utility of neuroscience theory and computational models for iteratively guiding our pharmacological and clinical experiments (55, 56). We focus on one type of computational modeling that may hold promise in this regard – namely, biophysically realistic computational models that contain cellular-level detail necessary to characterize specific synaptic disturbances that may occur in disease states (56, 57). This level of biophysical detail can provide a vital opportunity to test both hypothesized synaptic alterations in schizophrenia (55) and also possible pharmacotherapies that may attenuate such disturbances (58). Despite possible advantages, we note some key limitations of these modeling approaches, pertaining to the constrained behavioral repertoire and neural architectures that are currently effectively modeled in this way, largely owing to gaps in our basic understanding of neurobiology that can constrain such models. Therefore, we articulate a key objective for the future of schizophrenia research: biophysically realistic computational models need to be systematically developed and scaled to the level of neural systems (59), to inform fMRI-level observations in schizophrenia as well as those observed following pharmacological manipulations in healthy humans (Figure 1). We argue that this approach could be especially productive for the study of functional connectivity in psychiatric conditions, with the ultimate aim of developing mechanistically derived biomarker predictions.
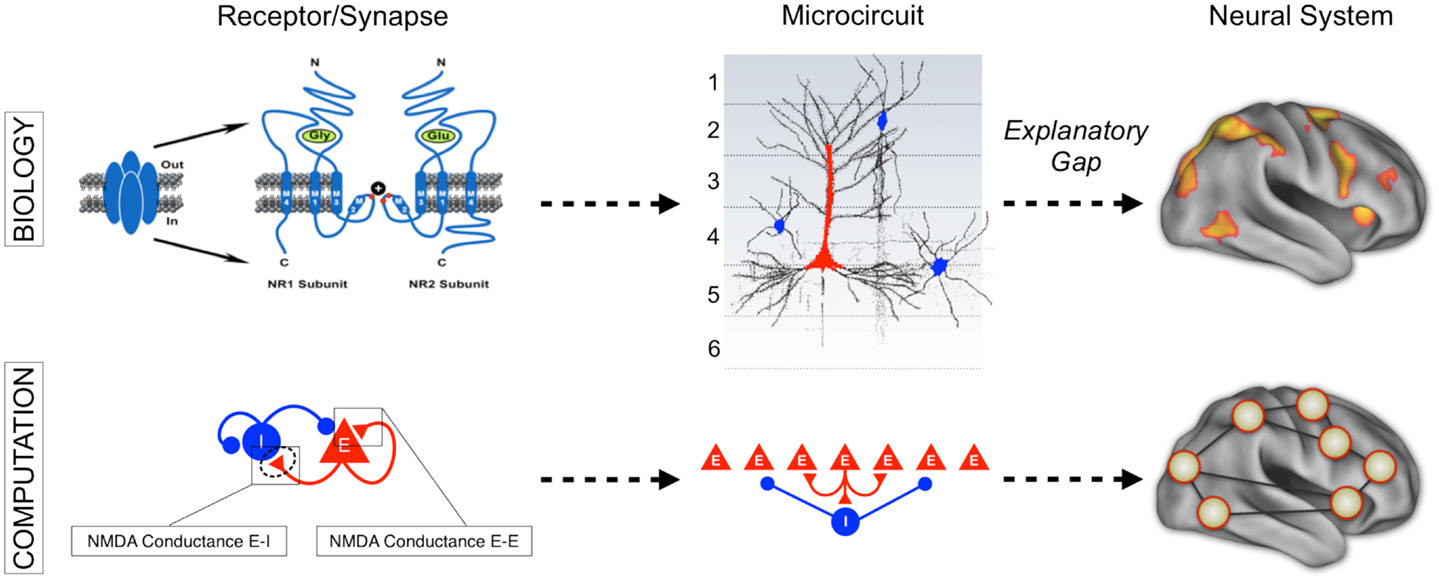
Figure 1. Understanding complex mental illness from synapses to circuits to neural systems. A major challenge facing the field of clinical neuroscience is building the links across levels of inquiry, from the level of receptors and cells, to microcircuits, and ultimately scale to the level of neural systems and behavior. At present, there is a vast explanatory gap across these levels in our understanding of psychiatric symptoms (top panel). Bridging this gap represents a major effort in explaining how alterations of specific mechanisms across neural systems may produce complex behavioral alterations seen in serious mental illness. This challenge is also exemplified by the National Institute of Mental Health Research Domain Criteria (RDoC) initiative (45) in order to map the biology across levels of inquiry onto behavior in a more systematic and data-driven way. We argue that computational modeling approaches (55) combined with additional experimental tools such as functional neuroimaging and pharmacology (47) offer one possible path toward this objective (bottom panel). We detail emerging efforts in functional connectivity work that may present a unique opportunity in this regard (59). Note: top left receptor figure was adapted with permission from Kotermanski and Johnson (60).
Building on these insights, we emphasize the need for efforts to translate our basic discoveries in neuroscience and cellular-level hypotheses in schizophrenia with system-level observations that may directly relate to the complex behavioral abnormalities observed in this illness. We highlight multiple complementary neuroscientific approaches, including recent clinical studies (37), pharmacological neuroimaging experiments (54), and theoretical/computational neuroscience approaches (18, 58) that can be synergistically harnessed to inform our understanding of underlying mechanisms and guide development of better pharmacotherapies for schizophrenia.
Developments in Functional Connectivity Applications to Neuropsychiatric Disease
Nearly two decades ago Biswal and colleagues (61) demonstrated that coherent fluctuations in the BOLD signal exist across time and space, demarcating a functional network architecture in the human brain. Such early studies highlighted this phenomenon by focusing on the fluctuations between the left and right motor cortex. This key property of the BOLD signal has in turn generated a paradigm shift in non-invasive human neuroimaging and allowed for characterization of distributed neural networks across species in the absence of task-mediated effects (28–30, 32, 62). Importantly, a close correspondence between resting-state networks and task-based networks has been established within individual subjects and using meta-analytic techniques (31). The value of this approach has been enhanced by the subsequent emergence of analytic techniques that have provided insights into the component structure and regulation of distributed cortical networks (62). For instance, recent advances in graph-theoretical approaches have shown that cortical and subcortical networks can be segregated into unique community structures, providing a comprehensive data-driven mapping of human functional networks (63). Another set of studies has demonstrated the temporal non-stationary properties of the large-scale functional networks, delineating temporal functional modes of the human brain (64). In other words, there seem to be distinct and independent patterns of connectivity over both space and time. Although a comprehensive review of connectivity method developments is beyond the scope of this manuscript, we refer the reader to recent detailed reports on this topic (62). Here we specifically focus on select clinical applications in schizophrenia and bipolar illness.
Resting-state functional connectivity MRI approaches have been increasingly applied to neuropsychiatric illness (65). Use of this technique is built upon the hypothesis that specific neuropsychiatric conditions are brain disorders that affect computations across large-scale networks of regions or specific circuits in such a way that these alterations can be identified with functional connectivity measures. This hypothesis suggests that such disturbances in neural network function may reflect alterations in more basic cellular-level mechanisms. Thus, disturbances in molecular signaling or synaptic function would be hypothesized to scale and produce disturbances in large-scale neural systems (66). Such network-level disturbances in a given functional system may then reflect specific psychiatric symptoms (12, 27). This framework has guided the application of functional connectivity with the objective of identifying putative biomarkers via BOLD fMRI that could reveal neural system-level disturbances in neuropsychiatric conditions even in the absence of specific tasks. Such an approach may hold promise if its sensitivity and specificity is ultimately refined to the point of diagnostic utility.
Specific advantages of rs-fcMRI include: (i) speed, as the functional network architecture can be reliably assayed within 10 min of data acquisition (67), and perhaps even faster with recent advances in multi-band imaging technology (68, 69); (ii) possible cost-effectiveness. Task-based studies may require longer time for data acquisition, depending on the precise paradigm being imaged. (iii) Lack of performance confounds, which likely affects nearly every cognitive activation task when applied to a psychiatric population (70). (iv) In addition, when coupled with techniques such as GBC or independent component analyses (ICA), rs-fcMRI studies allow the unbiased, simultaneous examination of all neural networks (see below for more discussion). All of these features suggest that rs-fcMRI may be a feasible method for eventually guiding and tracking symptoms, illness progression and possibly response to pharmacotherapies and/or behavioral treatments.
The approach itself, however, is not without limitations. For instance, the size of the correlation coefficient is often used to examine the strength of coupling between different network nodes (71). While this is an analysis-level consideration, the majority of published studies use this technique. This approach may not be ideal for examining shared versus non-shared variance between regions and may obscure more complex differences that could occur in psychiatric conditions (71). For instance, it may be important to uniquely identify functional connectivity alterations that truly reflect altered neuronal communication between two network nodes, as opposed to the influence of a third region on both (a correlation-based approach could not disambiguate these possibilities). It is also important to emphasize that functional connectivity, as measured at rest, is purely correlational and therefore can only be used to make tentative causal inferences. That is, if there is evidence for an alteration in a given circuit, this alteration could be predictive of disease onset or could be a consequence of some upstream physiological alteration. Moreover, there are additional complications involved in the study of neuropsychiatric illness concerning what is a primary deficit, as opposed to a compensation or treatment effects. These potential confounds underscore the complementary value of causal experimental designs involving pharmacologic, neuro-stimulation, or cognitive manipulations of circuit activity. Finally, there are still notable differences in the methods used to analyze rs-fcMRI data. For instance, one ongoing controversial issue relates to removal of the global signal (i.e., global signal regression, GSR) from functional connectivity data (72, 73). This step effectively regresses out variance associated with global brain fluctuations, which can be large in magnitude and can perhaps obscure certain real functional relationships (74). Nonetheless, this step effectively shifts the mean of the connectivity distribution, resulting in a portion of correlations being moved into the negative range – thus spuriously inducing at least some anti-correlations. There is convincing evidence from electrophysiology experiments using animals that anti-correlations indeed exist (75, 76). However, GSR could still complicate some aspects of between-group comparisons and the interpretations of clinical studies (73), especially when examining system-level relationships across groups. Therefore, future studies should carefully continue to consider the possible impact of this step on between-group differences in clinical rs-fcMRI studies.
Another methodological issue pertains to how networks are identified. Many early functional connectivity studies both in healthy adults and in clinical populations used a seed-based approach, whereby the correlation from a given region of interest is examined with all other voxels in the brain. This approach has also been complemented by more complex graph theoretical and network-based methods (77). Seed-based approaches inherently assume a consistent difference across a set of regions in a clinical condition. However, the pattern of dysconnectivity for a single region could indeed be more variable both across subjects and within regions. Such spatial variability in functional networks across subjects may present a limitation of seed-based approaches when applied in studies of clinical populations (11, 40). This may be the case in complex mental illnesses such as schizophrenia. Therefore, detecting more complex patterns of dysconnectivity requires new approaches taking into account these confounds (78–80).
New methods are needed to detect more complex patterns of disorder-related disturbances in connectivity (80), illustrated by the GBC approach (11, 35, 40, 81). The GBC technique is specifically designed to consider connectivity from a given voxel or (area) to all other voxels (or areas) simultaneously by computing either average connectivity strength (weighted GBC) or by counting the number of connections above a given connection strength (un-weighted GBC). Thus, this approach is data-driven and unbiased as to the location of connectivity disruption. That is, unlike typical seed-based approaches, GBC requires only the seed region to be relatively consistently located across subjects, while the target regions can vary substantially across subjects (Figure 2). For example, if a given area is perturbed in its functional connectivity consistently, irrespective of the overall network spatial configuration where the perturbation is, GBC will remain sensitive to this alteration. Further, unlike typical seed-based approaches, GBC involves one statistical test per voxel (or ROI) rather than one test per voxel-to-voxel pairing – substantially reducing multiple comparisons (e.g., 30,000 rather than ~900 million tests). These two improvements over typical seed-based approaches can dramatically increase the chance of identifying group differences in connectivity, or individual differences in connectivity correlated with behavioral symptoms (11, 35, 40, 81). Indeed, consistent with these proposed advantages, GBC has now been applied to identifying regions with large-scale disruptions in functional connectivity across a variety of mental illnesses, such as schizophrenia (11), bipolar disorder (35), and obsessive-compulsive disorder (OCD) (82). GBC is explicitly designed to address questions about brain connectivity that are qualitatively distinct from traditional seed-based functional connectivity analyses. For instance, areas of high GBC are “hubs” of connectivity in the brain that are maximally functionally connected with other areas and may play a role in coordinating large-scale patterns of brain activity (40). In that sense, a group difference in GBC may reflect areas and networks in which the large-scale coordination of information processing is affected in the disease state. For instance, decreased GBC may reflect decreased participation of a brain region in broader networks. Conversely, increased GBC may suggest a pathological broadening or synchronization of functional networks. Related to this point, we have now published a manuscript examining distributed networks in OCD using GBC where specific striatal and orbitofrontal circuits have been implicated. Yet, GBC remains sensitive to alterations in neuropsychiatric conditions that may actually be associated with more focal deficits (which may be the case in OCD versus schizophrenia). In that sense, GBC is not necessarily more useful for “global” versus “restricted” deficits, but rather dysconnectivity to a given “hub” region that may be affected in its participation in wide-spread neural networks. These properties of GBC – detection of regions with large-scale functional connectivity disruptions along with tolerance for individual differences – makes this method particularly powerful for identifying regions with consistently large and distributed disruptions that have functional consequences reflected in individual differences in psychiatric symptoms. Continued refinement of graph-theoretical data-driven metrics such as GBC may allow a powerful path forward for delineating biomarkers within and across diagnostic categories.
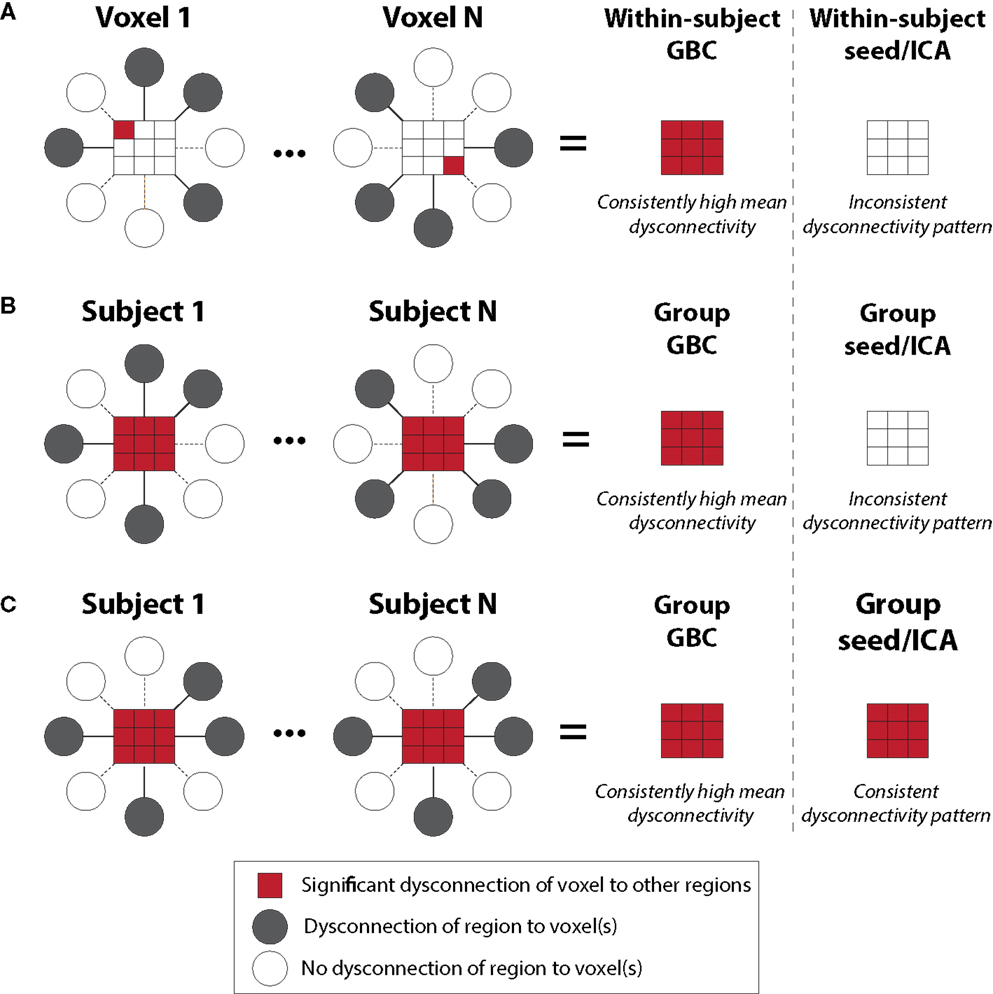
Figure 2. Global brain connectivity identifies globally dysconnected regions despite substantial within or between subject variability. We hypothesized that some brain regions may have global variable disruptions in functional connectivity, which may contribute to individual differences in symptom severity in brain disorders. GBC can be used as a data-driven approach to search for such regions. GBC is computed as the average connectivity of each voxel to all others. Gray circles indicate regions with altered connectivity to the depicted red voxels. (A) GBC will identify a region with consistent global dysconnectivity even when there are substantial differences in connectivity patterns within the region. Seed or ICA approaches would be unable to identify dysconnectivity in such a region because of inconsistent dysconnectivity patterns across neighboring voxels. (B) GBC, unlike seed or ICA approaches, will identify a region with consistent global dysconnectivity even when there are substantial individual differences in connectivity across subjects. This allows the identification of regions with many individual differences in connectivity, which might correlate with individual differences in symptoms. (C) GBC is also sensitive to consistent group differences in connectivity that might be identified using seed or ICA approaches, though GBC might be more sensitive due to pooling of results for both consistent and inconsistent dysconnectivity. GBC, global brain connectivity; ICA, independent component analysis. Note: figure adapted with permission from Cole and colleagues (11).
Another data-driven method that has increasing applications in psychiatric neuroimaging involves ICA of BOLD signal fluctuations at rest. Here ICA provides a tool to identify spatially (78, 83) or temporally (64) independent modes of brain function. ICA has been successfully used to identify distributed network abnormalities in both schizophrenia and bipolar illness (84). In particular, temporal ICA (64) may provide a novel and powerful method to assay the non-stationary properties of distributed neural systems across different clinical conditions. It remains to be systematically tested if recently defined temporal functional modes exhibit alterations in neuropsychiatric illness. One possibility, given wide-spread neurotransmitter disruptions in schizophrenia, is that the temporal functional modes are substantially more non-stationary in this illness. Powerful new acquisition sequences [e.g., multi-band imaging (68)], that allow much finer temporal sampling, will provide critical technological innovations at the level of neuroimaging acquisition with direct clinical applications, in particular when using ICA-type measures.
Collectively, the continued success of these methods suggests that ongoing refinement of tools to examine functional connectivity remains vital for development of biomarkers for psychiatric illness. Next, we turn to specific findings in the field of schizophrenia and bipolar research. We detail recent methodological advances in connectivity work that have been directly applied to better understand network disruption in these psychiatric conditions.
Emerging Evidence Suggests Large-Scale Dysconnectivity in Schizophrenia
There are now a number of influential hypotheses suggesting that, at its core, schizophrenia may be a disorder of large-scale neural connectivity (6, 85), i.e., “dysconnectivity.” Here we refer to dysconnectivity as an alteration in neural communication that specifically produces behavioral pathology, as opposed to a simple alteration in functional connectivity that deviates from the norm. The models proposing such dysconnectivity range from theoretical to formal computational hypotheses (42, 43, 66, 86–88). This work has inspired the search for connectivity biomarkers for schizophrenia and it broadly includes: (i) studies that use rs-fcMRI, that is investigations that study the intrinsic properties of BOLD signal fluctuations to delineate system-level alterations in psychiatric illness (27); (ii) studies that examine task-based dysconnectivity, that is alterations in connectivity during specific task contexts; (iii) pharmacological studies of individuals at rest or performing tasks, which we discuss in the upcoming section. While the literature on task-based connectivity has offered important clues regarding network alterations during specific cognitive processes (e.g., WM) (27, 89, 90), here we specifically detail emerging efforts to use rs-fcMRI to define biomarkers for schizophrenia. This is not to say that characterizing functional connectivity during cognitive processing is not important. In fact, such studies represent a vital effort to move our understanding beyond activation-based hypotheses regarding cognitive deficits in schizophrenia or regional hypotheses of specific symptoms. In the future, task-based functional connectivity studies will ultimately aid our understanding of network dysfunction with respect to specific symptoms or specific cognitive processes known to be profoundly affected in schizophrenia (e.g., WM) (26). Additionally, task-based studies of functional connectivity provide an important assurance that observed differences in resting-state functional connectivity do not merely reflect differences in the dynamics and content of mental processes subjects might engage in during rest, but rather stable disruptions in functional connectivity that persist across mental states (90). However, rs-fcMRI can be used to identify neural-system alterations even more broadly. With that in mind, we argue that rs-fcMRI offers a unique tool to identify distributed neural system alterations, extending beyond a given task context. Moreover, as noted above, this approach in particular bypasses task performance confounds which plague the task-based activation literature (70) and such concerns will likely extend to task-based connectivity work.
Despite its promise, a major obstacle in delineating successful neural markers for schizophrenia (or any other severe neuropsychiatric disease) using connectivity (or any other approach for that matter) has been the complexity of this illness and the vast range of behaviors it affects (91), as well as the complex temporal dynamics of its progression. That is, the apparent clinical complexity of schizophrenia is a major obstacle to biomarker development that may apply to all patients. In addition, the associated clinical heterogeneity (i.e., differences in symptoms across patients), the presence of numerous comorbidities and environmental modifiers, the ubiquitous confound of long-term antipsychotic treatment, and the wide range of affected behaviors collectively reduce the likelihood that a single biomarker would be applicable to the entire syndrome. Moreover, many researchers and clinicians would argue that schizophrenia is a theoretical construct used to label co-occurring symptoms whereby it may be challenging (or perhaps impossible) to map out a reliable and replicable marker of the entire syndrome in a parsimonious way [although theoretical models of the disease often consider it as one entity (66)]. We argue that functional connectivity tools offer a useful path forward in this regard by providing methods to test specific large-scale dysconnectivity patterns in relation to this heterogeneity that may be otherwise difficult to capture.
Certain studies using rs-fcMRI have, however, attempted to deal with clinical heterogeneity by focusing on the relationship between altered functional connectivity in specific pathways and linking those pathways to particular features of schizophrenia. For instance, studies by Hoffmann and colleagues have focused on better understanding alterations in connectivity specifically associated with auditory hallucinations (92), which many patients experience (1). They found compelling evidence for disruptions between regions associated with auditory processing. Specifically, they found that when they seeded Wernicke’s area, there was significantly greater functional connectivity to Brodmann’s areas 45/46 among hallucinating patients compared with non-hallucinating patients. In a subsequent analysis, they reported that the functional connectivity within a functional loop including the Wernicke’s area, inferior frontal gyrus, and putamen was robustly greater for hallucinating patients compared with non-hallucinating patients. Vercammen and colleagues also found that patients with schizophrenia evidenced attenuated functional connectivity between the left TPJ (temporo-parietal junction) and the right Broca’s area (93). These are examples where targeted seed-based approaches may identify alterations in circumscribed circuits associated with specific symptoms.
Another set of emerging studies have studied the “salience” and “control” systems, focused on striatal and insular dysconnectivity in schizophrenia (94, 95), particularly in relation to the “aberrant salience” hypothesis (96, 97). Briefly, the “aberrant salience” hypothesis has been linked to abnormal striatal dopamine function, suggesting that during psychotic states patients have a higher likelihood of forming inappropriate associations and respond excessively to random neutral events. Related to this issue a study by Tu and colleagues examined whether schizophrenia is associated with functional connectivity alterations within the cingulo-opercular (CO) network specifically (98). They identified significantly reduced functional connectivity in the bilateral putamen for patients with schizophrenia, which was related to cognitive performance in patients. The authors concluded that schizophrenia is associated with disconnection within cortico-striatal circuits. A complementary study by Moran and colleagues (99) focused on the anterior insula as a key node involved in modulation of distributed neural systems (as part of the CO system). The authors tested for disruptions in the functional relationships between the insula and control/default networks in patients with schizophrenia. Similarly, they examined a relationship between these network disturbances and cognitive deficits. Consistent with a priori predictions, Moran and colleagues found strong support for the disrupted relationship between the anterior insula and the control-executive and default-mode networks in schizophrenia, which again was predictive of cognitive performance. Most recently, a study by Palaniyappan and colleagues (100) also focused on the relationship between the salience (insular) system and the executive [lateral prefrontal cortex (PFC)] networks in schizophrenia. They explicitly tested for evidence of disrupted directional influence across the networks. In other words, similar to Moran and colleagues, they used an auto-regressive technique (i.e., Granger causality) to examine both feed-forward and reciprocal connectivity between the aforementioned networks. The authors reported significant differences in patients with regard to time-lagged functional relationships between executive and insular systems. The authors conclude that this “breakdown” in directional functional relationships may reflect aberrant processing of novel salient information. These studies provide compelling emerging evidence that there may indeed exist causal breakdowns across functional networks in schizophrenia. Nevertheless, it is important to note that lag-based causality measures in the context of BOLD signal analyses have not been without controversy (101–103), primarily because of systematic difference in the hemodynamic response function lags across areas. Given these concerns, the use of Granger causality in rs-fcMRI studies needs to be interpreted with caution [see Ref. (104) for a more detailed treatment of using auto-regression techniques with the BOLD signal]. Future studies using concurrent electrophysiology/fMRI protocols could provide convergent evidence to address issues regarding temporal dependencies across cortical networks when analyzing the BOLD signal.
We discussed studies that focused on either specific regions or networks that may be abnormal in schizophrenia. An alternative tactic, precisely because of the complexity of this illness, is to use data-driven methods to study large-scale connectivity alterations that may be difficult to pinpoint a priori. A number of prominent and well-established models of schizophrenia neuropathology implicate profound disruptions in PFC function (105), likely stemming from a confluence of glutamate, GABA, and dopamine alterations that could jointly affect PFC function (7, 19, 106–108) (see Box 1). One area that has been repeatedly implicated in schizophrenia neuropathology is the dorso-lateral PFC (23). However, this evidence has largely been marshaled through task-based studies such as those examining WM deficits in this illness (70). A deficit in task-evoked computations of a given region does not necessarily guarantee that the same node will show other forms of functional “dysconnectivity.” Moreover, such evidence does not guarantee that this same area may be disrupted in its functional connectivity in the absence of a task (i.e., during resting-state). Nevertheless, PFC functional deficits have been considered a hallmark feature of the illness. Therefore, one promising approach is to examine global PFC dysconnectivity in a data-driven way.
Box 1. Glutamate versus dopamine: upstream versus down-stream mechanisms in schizophrenia.
It is increasingly acknowledged that schizophrenia is associated with disturbances in multiple neurotransmitter systems, including alterations at the cortical microcircuit level in glutamate and γ-aminobutyric acid (GABA), as well as disturbances in dopaminergic neurotransmission along striatal-thalamic-cortical pathways (7, 106, 109). Additional studies have also implicated the glial system as possibly compromised in schizophrenia (110, 111) and other neuropsychiatric conditions (112). However, it remains unknown whether dopamine or glutamate alterations are the upstream causes or down-stream consequences of the disease process (109). Addressing this question has important implications for appropriately constraining computational models, both at the microcircuit level (58) and expanding those mechanisms to the system-level (18). There are two broad possibilities to consider: (i) there may be dissociable groups of patients each associated with a primary abnormality in one of the broad neurotransmitter systems, but as a consequence there may be secondary abnormalities in the other system due to shared pathways and functional loops (43, 88). (ii) One alternative possibility is that there are always primary alterations in only one of the two systems, followed by alterations in the other. Disambiguating between these causal possibilities can have important implications for developing optimized targeted pharmacotherapies for specific patient groups, which is a likely possibility given the heterogeneity of the illness. By extension, resolving these issues may have implications for optimized pharmacotherapies for a given phase of illness [if initial stages of schizophrenia are primary associated with hyper-glutamatergic neurotransmission (113)]. Also, these two possibilities have important implications for the utility of pharmacological models of psychosis (e.g., amphetamine versus the NMDAR antagonist challenge studies in healthy volunteers).
Here it is important to consider some key differences between the dopamine and glutamate hypotheses in schizophrenia in relation to pharmacological findings and their therapeutic effects: (i) dopaminergic medication has not proved successful at ameliorating the full range of impairing symptoms in schizophrenia, particularly cognitive deficits (26); (ii) pharmacological models of schizophrenia targeting the dopamine system (e.g., amphetamine challenge) have typically produced a clinical profile marked by acute psychosis, as opposed to a broader range of symptoms produced by pharmacological agents targeting the glutamate system (15, 114, 115). Therefore, while it is certainly important to acknowledge dopamine as a key component of disrupted neurotransmission in schizophrenia (21, 116, 117), it remains to be determined if dopamine is indeed a down-stream cause or a consequence of primary disruptions in glutamate (118). Relatedly, irrespective of cause or consequence arguments it will vital to consider the diversity of DA receptors (D1 versus D2) and their respective sites of influence in cortex (22, 119, 120) versus striatum (108), which in turn generates important constraints for computational modeling studies that incorporate dopaminergic and glutamatergic signaling mechanisms.
As articulated above, the GBC functional connectivity approach is specifically designed to test the hypothesis that a given functional brain region has altered coupling with the rest of the brain (or a large anatomical portion of the brain, such as the PFC). To test the efficacy of this approach, Cole and colleagues (11) used a restricted GBC (rGBC) approach focused on PFC in particular in a sample of patients with chronic schizophrenia relative to demographically matched healthy comparison subjects. Cole and colleagues reported that bilateral regions centered on the lateral PFC showed reductions in their PFC rGBC, suggesting that these regions have profound alterations in their connectivity patterns with the rest of PFC. To further test the relationship between identified PFC rGBC alterations and cognitive deficits, Cole and colleagues quantified the relationship between IQ and the PFC global connectivity alterations. The authors found that greater connectivity between the identified lateral PFC and the rest of PFC predicted better cognitive performance, suggesting that a lower index of global PFC coupling may in part relate to cognitive deficits in schizophrenia. Moreover, the authors additionally examined the pattern of whole-brain coupling of the discovered right lateral PFC region using seed-based techniques. The authors found that patients diagnosed with schizophrenia showed increased coupling with sensory cortices (Figure 3B, posterior regions, shown in yellow-red). Patients also showed reduced connectivity with prefrontal and other higher-order temporal regions (Figure 3B, shown in blue). Collectively, this initial report demonstrates that data-driven functional connectivity approaches could identify regions previously implicated in schizophrenia using task-based methods. Moreover, these identified areas showed robust and complex alterations of connectivity with the rest of the brain and importantly, related to observed symptoms and other cognitive measures.
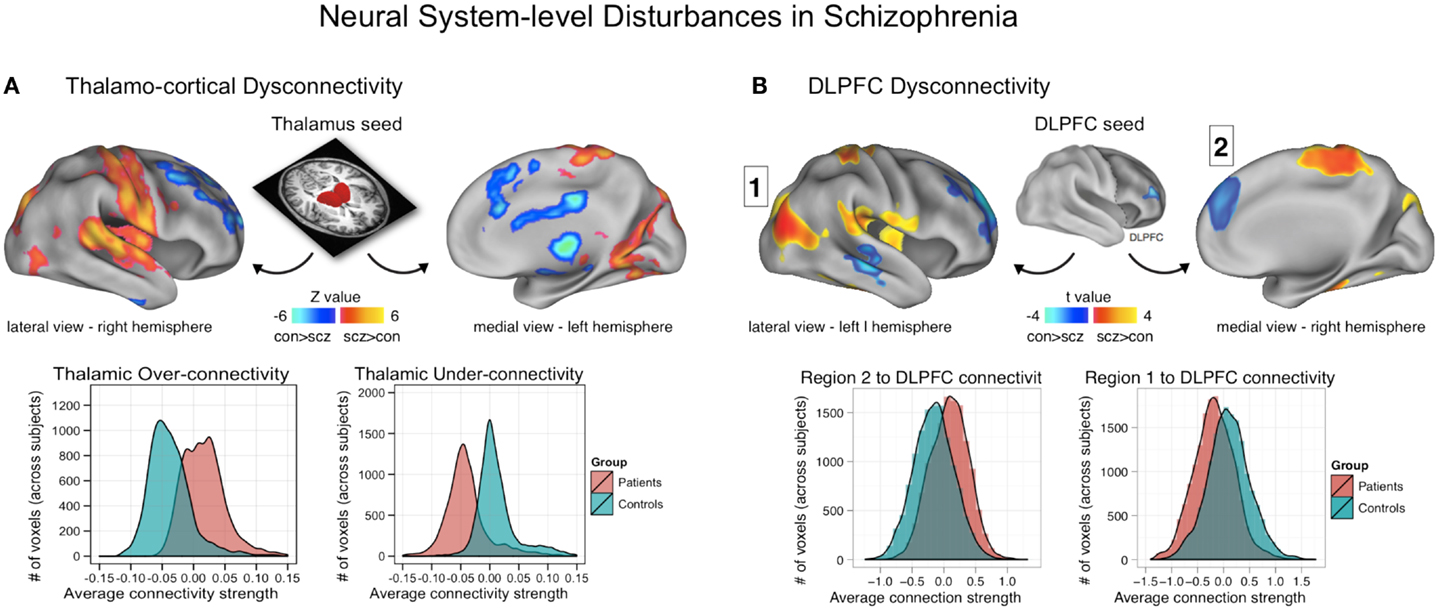
Figure 3. Neural system-level dysconnectivity in schizophrenia – emerging biomarkers. (A) Results from a recent large connectivity investigation examining thalamo-cortical connectivity alterations in 90 patients diagnosed with schizophrenia relative to 90 matched healthy controls (38). Anticevic and colleagues found robust alterations in thalamo-cortical information flow in schizophrenia, whereby sensory-motor cortical regions showed over-connectivity in schizophrenia (regions shown in yellow-red), but prefrontal-striatal-cerebellar regions showed under connectivity in schizophrenia relative to controls (regions shown in blue). Anticevic and colleagues fully replicated this pattern in an independent sample. Woodward and colleagues, using complementary approaches, found highly comparable effects (37). (B) A similar pattern of over/under connectivity was identified in patients with schizophrenia when using a DLPFC seed region identified via GBC; as with the thalamic seed, there was increased coupling with sensory (posterior regions, shown in yellow-red) but reduced connectivity with prefrontal and other higher-order temporal regions (shown in blue). This over/under pattern recapitulated qualitatively the observations found for the thalamic analysis in (A), as shown in the distribution plots on the bottom of each panel. Note: figures adapted with permission from Anticevic and colleagues (38) and Cole and colleagues (11).
Similar data-driven efforts have been applied to study global connectivity alterations in other psychiatric conditions. Specifically, Anticevic and colleagues extended the approach to study bipolar patients with and without a history of psychotic symptoms (35). While examining neural system-level disturbances in schizophrenia is a vital objective, psychosis occurs across a number of diagnostic categories. For instance, many bipolar patients experience frank psychosis (121). Given this clinical observation, some studies divide bipolar patients based on the presence or absence of psychotic symptoms (122, 123). In that sense, there may be somewhat limited utility in predefined diagnostic boundaries for understanding variation in neural circuits that could possibly be affected across neuropsychiatric conditions (45). Recognizing this limitation, there are emerging efforts to map common aspects of neural dysfunction across diagnostic categories.
Focusing on bipolar illness, Anticevic and colleagues examined the possibility that there are similar abnormalities in bipolar illness with a history of psychosis to those found in schizophrenia. Consistent with this hypothesis, Anticevic and colleagues found that bipolar illness is indeed associated with altered PFC rGBC specifically in a medial prefrontal/ventral cingulate region implicated in regulation of emotion (35). Importantly, this effect was primarily driven by a presence of psychosis history, and the magnitude of the observed PFC rGBC disruption was correlated with the severity of prior psychosis. This effect, however, was obtained in euthymic bipolar individuals (as opposed to symptomatic schizophrenia patients above), which suggests that at least some alterations in global connectivity may be a stable (trait) feature of the illness and may not necessarily only manifest in overtly symptomatic individuals. In that sense, data-driven functional connectivity may provide a tool to examine individual variability in both current and/or lifetime symptoms. Such data-driven approaches can also be used to establish alterations in functional neural architecture over time – namely, whether the observed disruptions in schizophrenia and bipolar illness change longitudinally as the illness progresses. This possibility is consistent with prominent neurodevelopmental models of severe psychiatric illness (14, 124–126), which suggest that there may be profound functional changes along the illness progression, to which these tools may be sensitive.
While promising, a key challenge facing data-driven approaches will be to establish whether identified effects consistently replicate across sites and samples. It may be possible that illness sample heterogeneity, illness stage, symptom severity, anatomical heterogeneity, or other factors will critically impact the patterns of data-driven effects across studies and samples. That is, it may be possible that different foci are detected as showing disruptions in different samples. Therefore, it will be critical to determine which of the identified data-driven global connectivity alterations are stable and replicable (perhaps capturing some core disturbances) and which alter as a function of other variables (perhaps capturing state effects). Such ongoing data-driven efforts should also continue to capitalize on recent advances in graph theory (80) as well as neuroimaging acquisition-level improvements (64, 69, 127) which could jointly improve the sensitivity of resting-state connectivity-derived metrics.
As described above, the PFC has been repeatedly implicated in schizophrenia neuropathology. However, despite data-driven efforts to map PFC dysfunction, the PFC remains a challenge in the study of schizophrenia, owing largely to the complex individual differences in function and anatomy of the PFC. Therefore, to ultimately establish a viable large-scale, brain-wide marker of neural alterations in schizophrenia, an alternative approach can be taken. One possible path is to start from the thalamus as a key region of interest (42). That is, while the PFC has classically been implicated in schizophrenia neuropathology, recent studies highlight disturbances in additional neural foci. In particular, the thalamus has emerged as an important disrupted locus in schizophrenia. While we presented some arguments against using seed-based approaches, the thalamus may present a unique opportunity in this case. Here we argue for the utility of specifying a seed when there is a specific theoretically guided hypothesis implicating a given area in the disease process. In this case, examining thalamic connectivity using rs-fcMRI in schizophrenia capitalizes on several key aspects of this subcortical region: (i) the thalamus is topographically connected to the entire cortex (128, 129), and may therefore represent a node particularly sensitive to network-level disturbances (42); and (ii) the thalamus contains anatomically and functionally segregated nuclei readily identifiable via neuroimaging (130) thus providing a lens for examining parallel yet distributed large-scale connectivity disturbances in neuropsychiatric disease.
Consistent with this view, a large body of evidence implicates significant thalamo-cortical communication disturbances in schizophrenia neuropathology (41, 42, 131–134). In fact, a fundamental aspect of large-scale brain organization preserved across mammalian species is thalamo-cortico-striatal sub-circuits that are thought to integrate various functions such as emotion processing and motor output (135–138). Such circuits may become profoundly dysregulated in schizophrenia, and the thalamus, as an organized hub of cortical and subcortical connections, may be especially sensitive to such dysregulation. Essentially, the thalamus serves as a nexus for parallel circuits through which diverse cortical and subcortical functions are integrated and distributed throughout the cortical mantle (128, 139, 140). These thalamic circuits have been implicated in schizophrenia pathophysiology on the basis of neuropathology studies (88, 141–143), pre-clinical lesion models (144, 145), structural imaging studies (146, 147), and computational models (43). Moreover, thalamic abnormalities are repeatedly implicated in sensory gating (148, 149) and filtering disruptions (150, 151) associated with this disorder (42). Indeed, one prominent model of schizophrenia neuropathology is centered on the thalamus as a key hub of disrupted computations in this illness. This “cognitive dysmetria” hypothesis articulates a distributed disruption in information processing across widespread cortical and subcortical nodes (39). In their seminal theoretical piece, Andreasen and colleagues argue that in order to explain the full range of schizophrenia symptoms, the field has to move away from region-specific models, but rather consider a distributed processing deficit, which could also parsimoniously explain a computational abnormality in a given node of a distributed complex system. In that sense, the thalamus is a uniquely positioned set of nuclei that communicates with virtually every cortical territory and is likely to be profoundly affected in schizophrenia.
Building on theoretical, pre-clinical, and anatomical work, recent studies of functional connectivity have begun to map thalamo-cortical alterations in schizophrenia. The first study to do so, by Welsh and colleagues (152), focused on the medio-dorsal nucleus of the thalamus as a seed region. This focused approach is justified given that specific thalamic nuclei in schizophrenia may show particularly profound functional connectivity disruptions. The medio-dorsal nucleus projects heavily to PFC regions (153, 154), and is thought to be compromised in schizophrenia (142, 144, 155). Welsh and colleagues found lower connectivity between the medio-dorsal nucleus and the PFC in patients with schizophrenia relative to healthy comparison subjects. However, this early investigation was based on a very small sample and could therefore be limited in its ability to provide conclusions regarding more subtle disruptions elsewhere. Moreover, Welsh and colleagues could not provide information regarding additional thalamic nuclei given the explicit focus on the medio-dorsal nucleus. In a subsequent study, Woodward and colleagues employed a substantially more powered sample (N = 62) and extended the approach to other thalamic nuclei (37). The authors used a parcellation scheme of thalamo-cortical connections at rest provided by Zhang and colleagues (129). In the study by Zhang and colleagues, cortical areas were clustered into subdivisions that exhibited unique functional connectivity with distinct thalamic nuclei based on the similarities in resting-state BOLD signal. Woodward and colleagues harnessed this segmentation scheme to test the hypothesis that unique thalamo-cortical circuits may show different patterns of disturbances in schizophrenia. Strikingly, Woodward and colleagues found that compared to healthy controls, the thalamic segmentations associated with the PFC showed reduced connectivity in schizophrenia. In contrast, cortical territories centered on sensory-motor regions showed increases in thalamic coupling in schizophrenia. This evidence suggests that there exists a profound alteration in thalamo-cortical information flow in schizophrenia but one that seems to follow an anatomical dissociation between sensory and higher order association regions. Building on this robust evidence for thalamo-cortical alterations in schizophrenia it is important to provide information about the specific connections being affected. Specifically, the cortical parcellation scheme, while a powerful initial demonstration, did not allow for examination of the cerebellum for instance. Cerebellum is a structure that, like the striatum, has projections to the cortex by way of the thalamus (156), and has been implicated in schizophrenia pathophysiology (39). Lastly, perhaps due to restricted power, the authors could not examine subtle relationships between symptoms and identified dysconnectivity.
A subsequent report by Anticevic and colleagues examined thalamo-cortical dysconnectivity in 90 patients diagnosed with schizophrenia relative to 90 matched healthy comparison subjects (38). The key objective of this investigation was to determine if thalamo-cortical disturbances span across diagnostic boundaries that share similar symptoms. This cross-diagnostic extension directly informs the objectives articulated by the NIMH RDoC initiative, which aims to develop biomarker-driven diagnostic systems (45). First, the authors replicated the core findings by Woodward and colleagues, demonstrating that schizophrenia is associated with increased coupling between the thalamus and all sensory-motor cortices. In contrast, frontal-striatal-cerebellar nodes showed reduced coupling with the thalamus in schizophrenia relative to healthy comparison subjects (Figure 3A). Both patterns were fully replicated in an independent and smaller sample of patients. Critically, further analyses demonstrated that these two sources of disturbance were functionally related. That is, those patients with the highest sensory-motor-thalamic coupling also showed the lowest prefrontal-striatal-cerebellar-thalamic coupling. This effect was most prominent for thalamic clusters centered on the medio-dorsal nucleus with known dense projections to the PFC, ruling out the possibility of pan-thalamic dysconnectivity that is uniform. Furthermore, the magnitude of the sensory-motor-thalamic over-connectivity was correlated with PANSS symptom severity across patients, confirming its functional relevance. The magnitude of this correlation, however, was small (r = 0.23) – indicating that the observed pattern explains only a small portion of the variance in symptom variation across subjects. An alternative possibility, given that the majority of patients were quite symptomatic, is that the small magnitude reflects a restricted range in symptoms whereby there was little variability in symptom severity across the patient sample.
The identified thalamic dysconnectivity was successfully used for diagnostic classification via multivariate pattern analysis (MVPA) (157) with high levels of sensitivity and specificity across both the discovery and replication samples. This implies that the identified dysconnectivity patterns, while not yet qualifying for a robust biomarker, may be refined and used to predict risk and assess treatment response. In particular, there are major ongoing improvements in neuroimaging acquisition (127) and processing (69) technology as a direct consequence of the Human Connectome Project (158) that can enable future studies to iteratively refine neuroimaging approaches. Such studies can focus on the identified patterns to ultimately improve methods and fine-tune the identified patterns for biomarker use.
Lastly, the authors found that the bipolar illness sample exhibited an “intermediate” pattern of disturbance such that the patterns of thalamo-cortical connectivity were “shifted” relative to healthy comparison subjects but not as severely altered as those identified in schizophrenia. This finding in particular offers promise for using neuroimaging markers to inform our understanding of shared disturbances in the underlying biology that cut across diagnostic categories (123). The next step will be to understand how such shared neural system-level “endophenotypes” (159, 160) map onto co-occurring behavioral disturbances (e.g., psychosis) as well as onto possibly shared alterations at the level of cortical microcircuits (which we discuss in the last section).
Most recently, Klingner and colleagues provided convergent results from a sample of 22 patients diagnosed with schizophrenia and 22 matched comparison subjects (161). In their study they separately examined the left and right thalamic seeds. They found robust evidence for increased thalamic connectivity with bilateral sensory-motor and auditory cortices. The authors conclude that their results suggest a possible “lack of thalamic control on motor/sensory information processing resulting in increased (and less filtered) forwarding of information to the prefrontal cortex.” This hypothesis is consistent with findings from the two earlier and larger studies (37, 38). Interestingly, Klingner and colleagues did not observe notable reductions in thalamic coupling with frontal-striatal-cerebellar nodes in schizophrenia, reported by both aforementioned groups. There are at least two possible interpretations for this difference: (i) the sample size in the Klingner study was much smaller and possibly underpowered to find both sets of patterns (although the effect size analysis and replication analyses by Anticevic and colleagues argues against this possibility); and (ii) Klingner and colleagues may have employed techniques, such as using GSR as a preprocessing step, that could have led to different results (73). It remains to be systematically determined what the true contribution of the global signal is in these analyses, especially in the patient groups. One possibility is that the global signal carries biologically meaningful information regarding cortical-thalamic disruptions in schizophrenia that needs to be carefully characterized. Methodological issues notwithstanding, this emerging body of work strongly and consistently implicates disruptions in thalamo-cortical information flow as a neural system marker in schizophrenia.
While this initial progress in mapping thalamo-cortical disturbances in schizophrenia represents a promising advance in psychiatric neuroimaging research, there are still fundamental gaps in our understanding of how such findings relate to neuropathological mechanisms of this illness. There are a number of future directions that the field should pursue to better understand these observations. For instance, it remains unknown why there are dissociable disturbances across thalamic nuclei in schizophrenia. Future studies focused more exclusively on the medio-dorsal nucleus versus, say, the pulvinar (known to be more involved in visual processing) could begin to explain mechanism behind these differences. Although the original study by Welsh and colleagues provides clues here, follow-up studies with more power that focus on the medio-dorsal nucleus could provide finer-grained information regarding its patterns of dysconnectivity with the PFC.
Another complex issue that is not adequately resolved by any of the noted investigations relates to medication effects. For instance, there are considerations of medication dose, type of medications (given that patients are often treated with multiple drugs from different medication classes), and possible systematic differences in the medications received by patients carrying the diagnoses of schizophrenia, schizoaffective, and bipolar (e.g., mood stabilizers and anti-depressants). While all studies address this issue statistically to a certain extent (by computing chlorpromazine equivalents and then co-varying for the medication dose), future studies in un-medicated patients are needed. It is possible, however, that medication may not necessarily be a confound in this case – instead, antipsychotic medication could actually stabilize thalamo-cortical dysconnectivity. Studies explicitly aimed at testing medication effects on connectivity could provide more detailed insight into this issue. It also remains unknown if the identified patterns of large-scale thalamo-cortical dysconnectivity are characteristic only of chronic stages of schizophrenia or whether they already appear in the prodromal or early stages of the illness. Establishing the link between identified thalamo-cortical dysconnectivity and illness progression remains a vital effort to inform the viability of this marker for predicting and/or tracking risk and progression of the disease. While one of the studies noted above provides a functional link between sensory-motor-thalamic over-connectivity and PANSS symptoms, it remains unknown if these patterns relate to cognitive and executive functional deficits characteristic of the schizophrenia syndrome (26). It may be possible that alterations in thalamo-cortical function (especially the PFC component) in part relate to cognitive deficits observed in this illness.
Additional questions still exist pertaining to the cross-diagnostic relevance of these observations. As noted above, it was shown by Anticevic and colleagues that qualitatively similar patterns of thalamic dysconnectivity are apparent in bipolar illness (although smaller in magnitude than those found in schizophrenia). It remains to be determined if those bipolar patients with a history of co-occurring psychosis are quantitatively more similar to alterations identified in schizophrenia (123). Such finer-grained cross-diagnostic investigations have further potential to inform and refine the clinical relevance of the identified marker. These studies should be complemented with targeted efforts to improve the classification provided by Anticevic and colleagues and allow for even more precision in harnessing this putative biomarker. Recently Fox and Greicius discussed progress in neuropsychiatric studies using resting-state connectivity. They appropriately concluded at the time that in schizophrenia there has been remarkably little progress in producing replicable results (34), perhaps owing to the complexity and heterogeneity of this neuropsychiatric illness noted above. These recent studies reviewed here, which collectively focused on identifying patterns of thalamo-cortical disruption in schizophrenia, may be converging on a parsimonious final common pathway of this complex disease, at least at the neural systems level (43). In that sense, these effects may be one of the better-replicated findings in the schizophrenia connectivity literature to date, offering promise for biomarker development and refinement.
A longer-term goal will entail bridging this neural system-level marker of schizophrenia with evolving cellular-level hypotheses of schizophrenia neuropathology. It remains unknown how the identified neural system-level markers relate to hypotheses that propose disruptions at the cellular level in schizophrenia. For instance, Anticevic and colleagues articulate a possible role of the disruption in cortical excitation (E) and inhibition (I) balance within the cortical microcircuit in producing system-wide disruptions, which may occur in schizophrenia (see next section) and in turn affect cognition (162). It remains unknown, however, how such alterations can scale to produce the presently observed pattern of aberrant thalamo-cortical connectivity. A corollary of this hypothesis relates to observations in bipolar illness, which was associated with an intermediate pattern of thalamo-cortical alterations. In bipolar illness different cellular-level hypotheses have been proposed from those hypothesized to occur in schizophrenia (112, 163, 164) [although some authors have articulated shared disturbances in GABA interneuron function (165)]. Therefore, either distinct mechanisms operate in these different neuropsychiatric conditions that converge on the same alterations or there may be, at least in part, a shared alteration in some of the same mechanisms across the two conditions. That is, it is possible that some patients with bipolar illness share some of the features of cellular neuropathology that affect patients with schizophrenia (165), especially those bipolar patients who present with co-occurring psychosis (166). Moreover, alterations along a number of distinct neurotransmitter pathways, involving a confluence of glutamate (7, 167), GABA (19), and dopamine disturbance (106), could jointly converge on a profound disturbance in thalamo-cortical function. In the upcoming sections, we discuss additional neuroscientific tools, namely pharmacological neuroimaging and computational modeling, which can be combined to help elucidate the role of specific cellular and synaptic mechanisms in observed system-level disruptions that occur in schizophrenia.
Pharmacological Neuroimaging – Toward a Mechanistic Understanding of System-Level Disruptions in Psychiatric Illness
We presented evidence, supported by several emerging studies, for profound alterations in thalamo-cortical information flow in schizophrenia, as well as evidence for alterations in PFC connectivity. Yet, the mechanisms that could inform rationally guided pharmacotherapy for these disturbances in schizophrenia remain unknown. One leading mechanistic hypothesis proposes possible disruptions in the E/I balance in the cortical micro-circuitry resulting from hypo-function of the NMDAR (7), which might affect cortical computations, leading to large-scale dysconnectivity (14). A way to link such pharmacological mechanisms to neural system-level observations is to directly compare clinical patient studies and results following pharmacological manipulations, or to separately test the effects of pharmacological manipulations in healthy volunteers.
A powerful candidate approach is to use ketamine, a non-competitive NMDAR antagonist and a leading schizophrenia pharmacological model, which transiently, reversibly, and safely induces characteristic schizophrenia symptoms in healthy volunteers (7). Here pharmacological manipulations provide a method with which researchers can test the effects of a given neurotransmitter perturbation in a constrained, causal, and hypothesis-driven way. Moreover, such synaptic hypotheses can be implemented directly into computational models to generate experimental predictions (discussed in the last section). A prevailing hypothesis regarding ketamine’s effects on cortical micro-circuitry proposes preferential antagonism of interneurons with subsequent disinhibition of pyramidal cells (168), a mechanism we implemented in a recent computational modeling investigation (58) (see below for a discussion). As an extension of this hypothesis, a cortex-wide disruption in E/I balance might de-stabilize thalamo-cortical information flow in ways observed in schizophrenia (or other neuropsychiatric conditions). It should be noted that it still remains unclear what the relative contribution is of NMDARs on pyramidal cells (169) versus interneurons (58) may be in relation to the hypothesized alterations in E/I balance. Understanding the relative contribution of such cell-specific receptor alterations remains an important future direction, which could inform targeted pharmacotherapies. Such detailed studies of how cellular-level alterations could give rise to thalamo-cortical alterations following pharmacological manipulations remain to be done. Nevertheless, there is emerging evidence from a few focused investigations detailing the effects of ketamine on large-scale cortical connectivity. These investigations provide preliminary clues for how ketamine’s effects on large-scale systems may resemble effects seen in schizophrenia.
For instance, a recent resting-state connectivity study by Driesen and colleagues investigated the effects of acute ketamine administration to healthy volunteers on large-scale global cortical connectivity (Figure 4A). The authors use a resting-state connectivity approach similar to that applied by Cole and colleagues in chronic schizophrenia (40), extended to include the entire brain (i.e., all voxels without imposing a PFC restriction). A number of studies have demonstrated that NMDAR antagonist administration is associated with excessive pyramidal cell activity, increases extracellular glutamate levels (16), and increases in perfusion and cortical metabolism (48–51, 53). A logical extension of this hypothesis is that administration of ketamine may profoundly affect large-scale cortical connectivity. Consistent with pre-clinical studies, Driesen and colleagues found that NMDAR antagonist administration resulted in a global elevation of functional connectivity (i.e., everywhere in the brain). This observation is broadly consistent with cellular-level hypotheses of ketamine’s effects on glutamate release, which may increase coupling of cortical circuits at rest by increasing the E/I ratio (i.e., decreasing cortical microcircuit inhibition). It is, however, critical to point out that chronic schizophrenia has typically been associated with reductions in cortical connectivity (170) and activation (70), especially in the PFC (11), as described above. Therefore, there are at least some evident discrepancies between the effects of pharmacological models such as ketamine and the actual illness (54). This important discrepancy between ketamine’s effects on PFC circuits and observations in schizophrenia should be reconciled in prospective studies that directly compare pharmacological and clinical effects using resting-state connectivity measures. One possible factor that could explain differences between NMDAR antagonist effects and chronic schizophrenia is that such pharmacological models may be relevant only to specific patient subgroups or illness stages. One possibility is that the increased connectivity under ketamine is similar to the early stages of psychotic illness. This hypothesis is consistent with elevated glutamate levels reported early in the illness course (113, 171). It is also consistent with the observation that ketamine tends to produce symptoms associated with incipient illness stages, rather than auditory hallucinations that occur in frank psychosis and chronic schizophrenia (15, 49, 172–174). Moreover, significant functional dynamical changes may occur during schizophrenia progression (46) that could profoundly affect PFC function, structure, and integrity. This hypothesis is supported by recent meta-analytic findings reporting decreases in glutamate across the illness progression (113). Whether such alterations across the schizophrenia illness course are reflected in PFC connectivity changes remains unknown, as does ketamine’s impact on PFC functional network architecture. Future pharmacological studies as well as cross-sectional and longitudinal clinical investigation are needed to test this hypothesis. In addition, careful pre-clinical experiments could possibly inform such hypotheses (118).
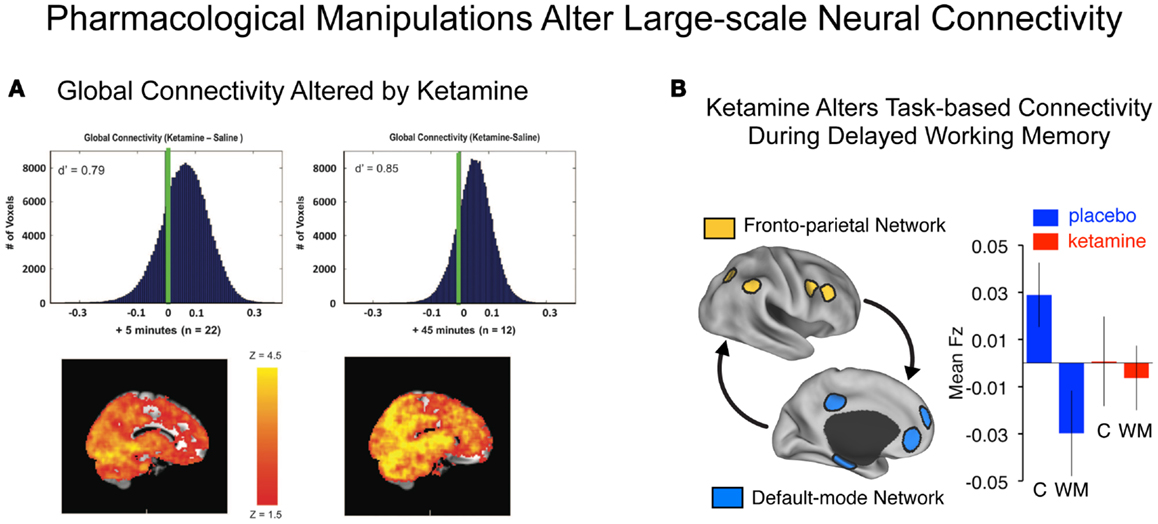
Figure 4. Characterizing connectivity alterations using pharmacological neuroimaging. While functional connectivity neuroimaging alone has been a powerful tool for characterizing neural system-level alterations in schizophrenia, it is ultimately a correlational tool. That is, we are examining alterations in the associations as a function of illness presence or absence. To move toward understanding the possible role of specific neurotransmitter mechanisms in schizophrenia, however, neuroimaging studies can be combined with pharmacological manipulations (47, 175). Such causal experimental manipulations can shed light on the role of specific neurotransmitter systems in schizophrenia (15). (A) Driesen and colleagues have shown that administration of ketamine profoundly altered the global connectivity of the brain. Specifically they demonstrated that the GBC measure increased following ketamine administration, possibly reflecting a hyper-glutamatergic state or a state of cortical disinhibition (54) consistent with animal models (111, 176). Formal computational models are needed in order to provide a deeper intuition for such pharmacological effects on large-scale neural systems. (B) Anticevic and colleagues have shown that administration of an NMDAR antagonist, ketamine, alters the connectivity of large-scale anti-correlated neural systems during performance of a working memory task (18). Although this connectivity study was performed during task performance rather than rest, it illustrates a proof of concept for how a pharmacological challenge can alter connectivity in a causal way. Note: figures adapted with permission from Driesen and colleagues (54) and Anticevic and colleagues (18).
A complementary area of research has investigated the effects of ketamine on functional connectivity (177) in relation to its potential anti-depressant effects (178). While a comprehensive treatment of anti-depressant effects of ketamine is beyond the scope of this review, it is important to note that studies examining its effects on glutamateric pathways in the context of mood symptoms (178) may be highly informative for developing our understanding of its relevance to schizophrenia (111). Briefly, emerging models in this area postulate that ketamine may act as anti-depressant by promoting synaptic plasticity via intra-cellular signaling pathways, ultimately promoting brain-derived neurotrophic factor expression via synaptic potentiation (179) and in turns synaptic growth (178). In that sense, acute NMDAR antagonism may promote synaptic plasticity along specific pathways impacted in mood disorders, such as ventral medial PFC (180, 181, p. 916). Conversely, when administered to patients diagnosed with schizophrenia, NMDAR antagonists seem to worsen their symptom profile (182), perhaps by “pushing” an already aberrantly elevated glutamatergic signaling profile upward. Collectively such dissociable effects of ketamine may imply that along distinct circuits there may be an inverted-U relationship between ketamine’s effects and symptoms: depressed patients may be positioned on the low end of the inverted-U (178) and schizophrenia patents may be positioned on the higher end (183). Both task-based and resting-state functional connectivity techniques are well positioned to interrogate such system-level effects of NMDAR antagonists in humans.
As an example of such an approach, another study examining the effects of ketamine by Anticevic and colleagues focused on understanding the functional impact of NMDAR antagonism on the organization of the large-scale, anti-correlated neural systems (Figure 4B). This pharmacological neuroimaging investigation was explicitly focused on understanding ketamine effects on WM. However, in the context of this cognitive question, Anticevic and colleagues also assessed whether the task-based functional connectivity of large-scale neural systems is affected by ketamine administration. As we noted above, such task-based connectivity approaches can be useful to pinpoint how large-scale systems are affected during specific cognitive operations. The authors found that an acute administration of a low ketamine dose profoundly altered the typically observed anti-correlated structure of the large-scale neural systems – namely the task-positive fronto-parietal regions (184) and the task-negative regions (typically termed the default-mode network, DMN) (28). In line with this observed decrease in functional connectivity, Driesen and colleagues (185) also found a reduction in WM task-based functional connectivity along fronto-parietal areas (when using a DLPFC seed) following ketamine administration. The investigators reported increased functional connectivity of the DLPFC during rest using the exact same seed. This set of observations in particular sheds light on how a pharmacological manipulation such as NMDAR antagonism may have profoundly different effects in the context of a cognitive task (e.g., WM) and during rest. The mechanisms behind this observed difference are beyond the scope of this review and will be discussed in forthcoming studies.
Briefly, it may be possible that large-scale, network-level synchrony during WM is critically dependent on appropriate content-specific signals between neural subpopulations (186). In contrast, the BOLD fluctuations during rest likely relate to coupling between regions at the infra-slow-frequency ranges (187). It may be possible that a reduction in appropriate task-evoked synchrony by NMDAR antagonism reduces the ability of a network to form a coherent and optimal level of functional connectivity during WM. This may be exacerbated by an amplification of shared “noise” in the system, which is reflected in the apparent hyper-synchrony at rest (54). Collectively, therefore, an NMDAR antagonist may “disinhibit” the system, which gives rise to infra-slow, spatially distributed fluctuations across large areas of cortex that manifest in aberrant hyper-connectivity at rest. Precisely due to this elevated background noise, in combination with disrupted capacity for appropriate task-evoked synchrony, the net effect during WM may be a reduction in functional connectivity (which contrasts with observations at rest). Moreover, task-evoked activity can suppress the slow fluctuations associated with rest (188); if this suppression were weakened in schizophrenia, the “signal-to-noise” ratio would be degraded and task-based functional connectivity could be reduced. Because task-based and resting-state synchrony may be separated by timescale, EEG studies, combined with fMRI, could potentially address such hypotheses (189–191). Moreover, precisely because of such important differences between task and rest in certain contexts (e.g., ketamine manipulation) it remains important for task-based and resting-state investigations to inform one another. In the upcoming section, we explicitly discuss the recent developments in computational modeling that can provide a platform for formal integration of neuroscience theory both in the context of resting-state studies as well as formal task-based experiments.
Bridging Levels of Analysis via Computational Modeling
Above we discussed recent clinical and pharmacological neuroimaging findings that shed light on the nature of large-scale neural system alterations in schizophrenia. In particular, the pharmacological experiments provide a causal method to explicitly manipulate specific neurotransmitter mechanisms that may be involved in schizophrenia. In that sense, these studies can begin to address given neurotransmitter contributions to neural system-level and behavioral alterations observed in schizophrenia. Still, these studies cannot measure synaptic and cellular-level phenomena alone. Therefore, one possible methodological integration involves a formal link between such pharmacological/clinical experiments and computational models that contain this level of functional detail. One branch of computational model that provides a particularly productive platform involves biophysically based models that contain the relevant synaptic mechanisms (57, 192) thought to be disrupted in neuropsychiatric illness (55). Such microcircuit models have been already harnessed to make predictions in the context of ketamine experimental manipulations of WM (58) (discussed more below). There is, however, an ongoing need to scale such models to the level of neural systems to provide relevant predictions for both resting-state and task-based clinical and pharmacological studies.
Recent computational models have been developed to explicitly capture how the global pattern of resting-state functional connectivity arises through cortico-cortical interactions (193). In particular, modeling studies in this area have focused on the extent to which functional connectivity can be predicted by long-range anatomical connectivity (Figure 5A). The dynamic interactions between neural populations will also shape functional connectivity. The starting point for these models is an anatomical coupling matrix reflecting long-range connections between cortical regions, derived either from tracer studies in macaque monkeys or from diffusion tractography in humans. The activity of a local region (a node in the large-scale network) follows some dynamics and is shaped by input from other areas, propagating via the long-range connections. Ongoing activity, either due to chaos or noise, produces fluctuations in the activity across the network. The functional connectivity of the model can then be calculated and compared to functional connectivity observed in experiments. Honey and colleagues (194) studied long-range chaotic synchronization when local nodes follow oscillatory dynamics, using connectivity from human diffusion tractography. They found that the global dynamics of the network could partially explain the presence of strong functional connections between regions that lack direct anatomical connection. Cabral and colleagues (195) used a similar oscillatory model with connectivity data from healthy subjects, and parametrically varied the overall strength of long-range connections. They found that decreasing long-range connection strength altered functional connectivity patterns in a manner similar to those observed in schizophrenia (9), with reduced overall functional connectivity strength and changes in certain graph-theoretic measures of the functional connectivity matrix.
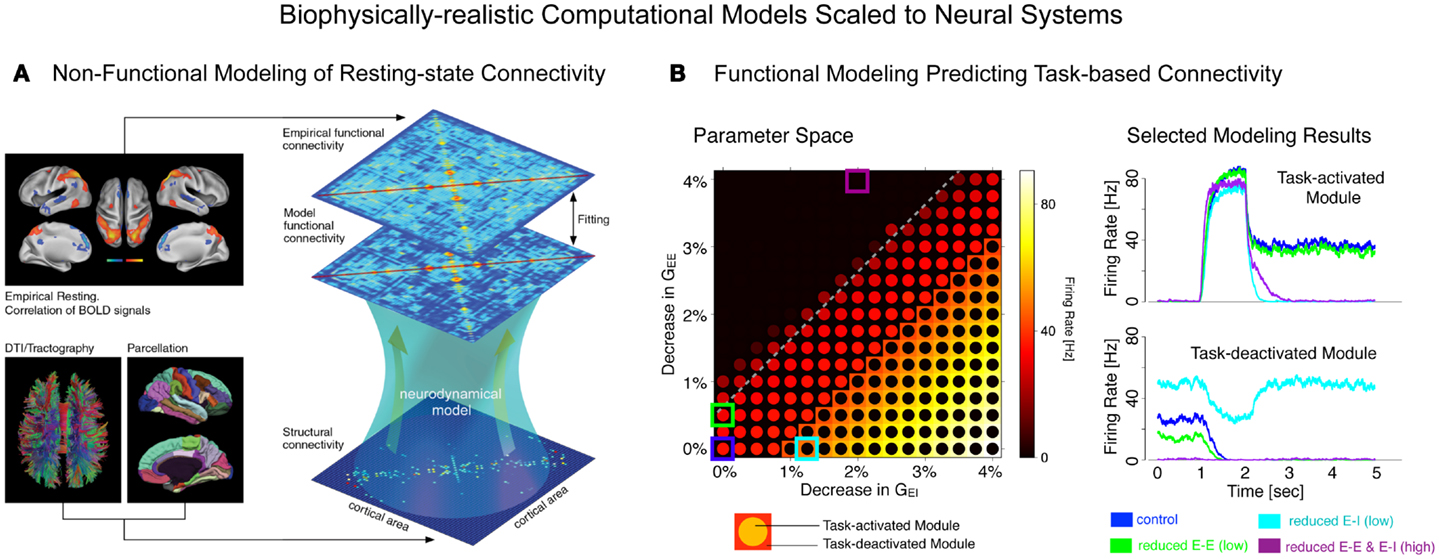
Figure 5. Computational modeling of system-level effects via biophysically realistic computational approaches. While detailed microcircuit models have made an impact on our understanding of cortical dynamics (56), the challenge remains to scale such models to incorporate dynamic interactions across large-scale neural systems, which are likely profoundly affected in schizophrenia (and other severe neuropsychiatric conditions). (A) A recently published elegant study by Deco and colleagues (59) illustrates an approach where a biophysically realistic model of cortical computations has been applied to understand the generation of slow-frequency fluctuations in the BOLD signal. The authors used diffusion-spectrum imaging to anatomically constrain the model and in turn fitted the modeling results to empirically-derived resting-state functional connectivity data. The result illustrates that coherent fluctuations in the BOLD signal (i.e., resting functional connectivity) may emerge from a system that is at the edge of chaos, allowing linear but transient departures in neuronal firing rates. (B) Anticevic and colleagues have used a functional model of working memory, a cognitive process that is profoundly affected in schizophrenia (26), to better understand the role of NMDA receptor function in the interaction of large-scale anti-correlated neural systems. Specifically they studied the functional antagonism present during a cognitive task between the task-activated (fronto-parietal module) and task-deactivated (default-mode module) networks (196). Following a complete parameter sweep (left), the authors found that a small perturbation of the NMDARs on inhibitory interneurons within each cortical microcircuit captured the firing that was observed experimentally following ketamine administration in healthy volunteers (18). Collectively, these studies offer examples for how biologically constrained modeling approaches can be applied to understand large-scale neural system physiology in both resting-state (non-functional) and task-based (functional) settings. Note: (A) of the figure was adapted with permission from Deco and colleagues (59).
Deco and colleagues (59, 193) extended this approach, deriving long-range connectivity from human diffusion tractography and implementing the local node dynamics with a biophysically based model of a cortical microcircuit. In particular, the local microcircuit incorporates recurrent excitation with realistic synaptic dynamics, and the strength of recurrent excitation enables multi-stable dynamics that can subserve cognitive computations such as WM (197, 198). Noisy background inputs to nodes induce fluctuations in activity that are shaped into correlated patterns by long-range coupling between nodes through the anatomically derived connectivity. Functional connectivity in the model is given by the pattern of these correlated fluctuations. The authors parametrically varied two global parameters with biophysical relevance: the strength of recurrent excitation within local nodes, and the strength of long-range connections between nodes. The strengths of local and long-range connections combine to provide recurrent excitation in the network. They found that the similarity between model and experimental functional connectivity patterns was maximized when the network’s baseline state is near the boundary in parameter space between stability and instability induced by excess excitation. These studies reveal that the pattern of functional connectivity in the model is sensitive to both the pattern of anatomical connectivity and the physiological parameters that scale local and long-range connections. The biophysical basis of these models makes them directly applicable to address the dynamical consequences of anatomical and physiological changes induced by disease processes or pharmacological manipulation. Changes in the strengths of local and long-range connections may induce differential effects in the patterns of functional connectivity. Therefore, fitting models to functionally connectivity in patients could distinguish among distinct synaptic alterations. This approach could also potentially reveal the effects of complex drug actions on local circuit tuning and long-range interactions.
The models described above were explicitly constructed to simulate resting-state fluctuations, rather than to implement a particular function such as WM. Nevertheless, functional models can still make predictions that can be tested using functional connectivity, especially task-based functional connectivity (81). To this end, Anticevic and colleagues extended a microcircuit model of WM to study interactions between large-scale networks and their disruption by synaptic perturbation (Figure 5B) (18). The biophysically based model consists of two modules of spiking microcircuits: one that is task-activated and capable of WM computations, and one that is task-deactivated from a high-activity baseline state (hypothesized to model the activity pattern of the default-mode network in the context of cognitive activation). The interactions between these modules are mutually suppressive, via long-range projections onto inhibitory interneurons, a feature founded on findings of anti-correlated fluctuations between task-positive and default-mode networks (30). Within the model authors specifically studied the functional impact of disinhibition via NMDA hypo-function on interneurons, to relate such microcircuit hypotheses to neural changes observed under ketamine administration. The authors found that disinhibition of the entire network results in a failure to shut off the default-mode module, impairing the pattern of activation and deactivation during WM tasks. This modeling study provides one example of how a microcircuit model of a specific cognitive process (i.e., WM) can be scaled to incorporate system-level interactions and make predictions relevant for task-based functional connectivity. In addition, one strength of computational models is the ability to systematically explore different operating regimes in the space of model parameters. For example, Anticevic and colleagues contrasted reductions in recurrent excitation onto interneurons versus pyramidal cells, generating testable predictions for elevated versus reduced excitation/inhibition balance. As a test of the model architecture and operation, the authors analyzed task-based functional connectivity between task-activated and task-deactivated networks, computed across trials during the delay period of a WM task. Under control conditions, the two networks exhibited robust negative correlation, in line with effective antagonistic interactions in the model. In contrast, under ketamine administration this negative correlation disappears (Figure 4B), in line with the model prediction that the task-positive module cannot effectively suppress the hyperactive default-mode module. Collectively, this study provides preliminary evidence that functional models can be directly related to functional connectivity predictions in the context of WM. Future computational/experimental studies should be designed to extend this framework to more complex processes and symptoms that may be disrupted in schizophrenia. We argue that such ongoing efforts for the integration of theory, pharmacological experiments and clinical work will be a vital path for the field of clinical neuroscience to provide testable and rationally guided advances for understanding disease mechanisms and putative treatments.
Concluding Remarks
Collectively, we articulated recent focused developments in three areas of clinical neuroscience of schizophrenia: (i) we reviewed methodological advances in resting-state functional connectivity that were directly translated to understand neural system-level disturbances in schizophrenia. We specifically focused on data-driven techniques that offer a promising way to detect disrupted connectivity while bypassing the likely complexity and regional heterogeneity of network alterations that are present in schizophrenia. We also discussed ongoing developments in studies of thalamo-cortical dysconnectivity in schizophrenia that are directly informed by influential theoretical models of the illness. (ii) We highlighted select pharmacological studies of the NMDARs that offer a causal way to understand neural system alterations in psychiatric illness. (iii) We articulated the developments in biophysically based computational modeling studies that provide a platform for testing specific synaptic alterations. In turn, we demonstrate that such models can potentially be scaled to the level of neural systems to make relevant predictions for both resting-state or task-based connectivity experiments. We argue that the ongoing blend of these three approaches can provide advances in the field of clinical neuroscience that create a final output that is much greater than the sum of its parts.
Conflict of Interest Statement
John H. Krystal consults for several pharmaceutical and biotechnology companies with compensation less than $10,000 per year. All other authors declare that they have no conflict of interest.
Acknowledgments
Financial support for this study was provided by NIH grant DP5OD012109-01 (PI: Alan Anticevic), NIAAA grant 2P50AA012870-11 (PI: John H. Krystal), NIH grant MH096801 (PI: Michael W. Cole), NIH grant R01 MH062349 (PI: Xiao-Jing Wang) the National Alliance for Research on Schizophrenia and Depression (PIs: Alan Anticevic), the Fulbright Foundation (Aleksandar Savic), and the Yale Center for Clinical Investigation (YCCI, PI: Alan Anticevic). We thank Sharif Youssef for assistance with this manuscript.
References
1. Walker E, Kestler L, Bollini A, Hochman KM. Schizophrenia: etiology and course. Annu Rev Psychol (2004) 55:401–30. doi:10.1146/annurev.psych.55.090902.141950
2. Yoon JH, Tamir D, Minzenberg MJ, Ragland DJ, Ursu S, Carter CS. Multivariate pattern analysis of functional magnetic resonance imaging data reveals deficits in distributed representations in schizophrenia. Biol Psychiatry (2008) 64:1035–41. doi:10.1016/j.biopsych.2008.07.025
3. Corlett PR, Honey GD, Fletcher PC. From prediction error to psychosis: ketamine as a pharmacological model of delusions. J Psychopharmacol (2007) 21:238–52. doi:10.1177/0269881107077716
4. Holt DJ, Lakshmanan B, Freudenreich O, Goff DC, Rauch SL, Kuperberg GR. Dysfunction of a cortical midline network during emotional appraisals in schizophrenia. Schizophr Bull (2011) 37:164–76. doi:10.1093/schbul/sbp067
5. Barch DM, Braver TS. Cognitive control in schizophrenia: psychological and neural mechanisms. In: Engle RW, Sedek G, von Hecker U, McIntosh AM, editors. Cognitive Limitations in Aging and Psychopathology. Cambridge: Cambridge University Press (2007). p. 122–59.
6. Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry (2006) 59:929–39. doi:10.1016/j.biopsych.2005.10.005
7. Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) (2003) 169:215–33. doi:10.1007/s00213-003-1582-z
8. Murray CJ, Lopez AD, Harvard School of Public Health; World Health Organization; World Bank. The Global Burden of Disease: A Comprehensive Assessment of Mortality and Disability from Diseases, Injuries, and Risk Factors in 1990 and Projected to 2020. Cambridge, MA: Published by the Harvard School of Public Health on Behalf of the World Health Organization and the World Bank; Distributed by Harvard University Press (1996).
9. Lynall M-E, Bassett DS, Kerwin R, McKenna PJ, Kitzbichler M, Muller U, et al. Functional connectivity and brain networks in schizophrenia. J Neurosci (2010) 30:9477–87. doi:10.1523/JNEUROSCI.0333-10.2010
10. van den Heuvel MP, Mandl RC, Stam CJ, Kahn RS, Pol HE. Aberrant frontal and temporal complex network structure in schizophrenia: a graph theoretical analysis. J Neurosci (2010) 30:15915–26. doi:10.1523/JNEUROSCI.2874-10.2010
11. Cole MW, Anticevic A, Repovs G, Barch DM. Variable global dysconnectivity and individual differences in schizophrenia. Biol Psychiatry (2011) 70:43–50. doi:10.1016/j.biopsych.2011.02.010
12. Fornito A, Yoon J, Zalesky A, Bullmore ET, Carter CS. General and specific functional connectivity disturbances in first-episode schizophrenia during cognitive control performance. Biol Psychiatry (2011) 70:64–72. doi:10.1016/j.biopsych.2011.02.019
13. Salomon R, Bleich-Cohen M, Hahamy-Dubossarsky A, Dinstien I, Weizman R, Poyurovsky M, et al. Global functional connectivity deficits in schizophrenia depend on behavioral state. J Neurosci (2011) 31:12972–81. doi:10.1523/JNEUROSCI.2987-11.2011
14. Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr Opin Neurobiol (2013) 23:283–90. doi:10.1016/j.conb.2012.11.004
15. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51:199–214. doi:10.1001/archpsyc.1994.03950030035004
16. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci (2007) 27:11496–500. doi:10.1523/JNEUROSCI.2213-07.2007
17. Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons and cortical gamma oscillations in schizophrenia. Schizophr Bull (2012) 38(5):950–7. doi:10.1093/schbul/sbs010
18. Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ, et al. NMDA receptor function in large-scale anti-correlated neural systems with implications for cognition and schizophrenia. Proc Natl Acad Sci U S A (2012) 109:16720–5. doi:10.1073/pnas.1208494109
19. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci (2005) 6:312–24. doi:10.1038/nrn1648
20. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci (2012) 35:57–67. doi:10.1016/j.tins.2011.10.004
21. Toda M, Abi-Dargham A. Dopamine hypothesis of schizophrenia: making sense of it all. Curr Psychiatry Rep (2007) 9:329–36. doi:10.1007/s11920-007-0041-7
22. Durstewitz D, Seamans JK. The computational role of dopamine D1 receptors in working memory. Neural Netw (2002) 15:561–72. doi:10.1016/S0893-6080(02)00049-7
23. Glahn DC, Ragland JD, Abramoff A, Barrett J, Laird AR, Bearden CE, et al. Beyond hypofrontality: a quantitative meta-analysis of functional neuroimaging studies of working memory in schizophrenia. Hum Brain Mapp (2005) 25:60–9. doi:10.1002/hbm.20138
24. Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatr (1994) 6:348–57.
25. Meyer-Lindenberg A, Poline JB, Kohn PD, Holt JL, Egan MF, Weinberger DR, et al. Evidence for abnormal cortical functional connectivity during working memory in schizophrenia. Am J Psychiatry (2001) 158:1809–17. doi:10.1176/appi.ajp.158.11.1809
26. Barch DM, Ceaser A. Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cogn Sci (2012) 16:27–34. doi:10.1016/j.tics.2011.11.015
27. Fornito A, Zalesky A, Pantelis C, Bullmore ET. Schizophrenia, neuroimaging and connectomics. Neuroimage (2012) 62:2296–314. doi:10.1016/j.neuroimage.2011.12.090
28. Raichle ME, Snyder AZ. A default mode of brain function: a brief history of an evolving idea. Neuroimage (2007) 37:1083–90; discussion 1097–9. doi:10.1016/j.neuroimage.2007.02.041
29. Biswal BB, Mennes M, Zuo X-N, Gohel S, Kelly C, Smith SM, et al. Toward discovery science of human brain function. Proc Natl Acad Sci U S A (2010) 107:4734–9. doi:10.1073/pnas.0911855107
30. Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci U S A (2005) 102:9673–8. doi:10.1073/pnas.0504136102
31. Smith SM, Fox PT, Miller KL, Glahn DC, Fox PM, Mackay CE, et al. Correspondence of the brain’s functional architecture during activation and rest. Proc Natl Acad Sci U S A (2009) 106:13040–5. doi:10.1073/pnas.0905267106
32. Vincent JL, Patel GH, Fox MD, Snyder AZ, Baker JT, Van Essen DC, et al. Intrinsic functional architecture in the anaesthetized monkey brain. Nature (2007) 447:83–6. doi:10.1038/nature05758
33. Greicius MD, Supekar K, Menon V, Dougherty RF. Resting-state functional connectivity reflects structural connectivity in the default mode network. Cereb Cortex (2009) 19:72–8. doi:10.1093/cercor/bhn059
34. Fox MD, Greicius M. Clinical applications of resting state functional connectivity. Front Syst Neurosci (2010) 4:19. doi:10.3389/fnsys.2010.00019
35. Anticevic A, Brumbaugh MS, Winkler AM, Lombardo LE, Barrett J, Corlett PR, et al. Global prefrontal and fronto-amygdala dysconnectivity in bipolar I disorder with psychosis history. Biol Psychiatry (2012) 73:565–73. doi:10.1016/j.biopsych.2012.07.031
36. Whitfield-Gabrieli S, Thermenos H, Milanovic S, Tsuang M, Faraone S, McCarley R, et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Natl Acad Sci U S A (2009) 106:1279–84. doi:10.1073/pnas.0809141106
37. Woodward ND, Karbasforoushan H, Heckers S. Thalamocortical dysconnectivity in schizophrenia. Am J Psychiatry (2012) 169:1092–9. doi:10.1176/appi.ajp.2012.12010056
38. Anticevic A, Cole MW, Repovs G, Murray JD, Brumbaugh MS, Winkler AM, et al. Characterizing thalamo-cortical disturbances in schizophrenia and bipolar illness. Cereb Cortex (2013). doi:10.1093/cercor/bht165
39. Andreasen NC, Paradiso S, O’Leary DS. “Cognitive dysmetria” as an integrative theory of schizophrenia: a dysfunction in cortical-subcortical-cerebellar circuitry? Schizophr Bull (1998) 24:203–18. doi:10.1093/oxfordjournals.schbul.a033321
40. Cole MW, Pathak S, Schneider W. Identifying the brain’s most globally connected regions. Neuroimage (2010) 49:3132–48. doi:10.1016/j.neuroimage.2009.11.001
41. Carlsson M, Carlsson A. Schizophrenia: a subcortical neurotransmitter imbalance syndrome? Schizophr Bull (1990) 16:425–32. doi:10.1093/schbul/16.3.425
43. Lisman JE, Pi HJ, Zhang Y, Otmakhova NA. A thalamo-hippocampal-ventral tegmental area loop may produce the positive feedback that underlies the psychotic break in schizophrenia. Biol Psychiatry (2010) 68:17–24. doi:10.1016/j.biopsych.2010.04.007
44. Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry (2009) 66:988–9. doi:10.1016/j.biopsych.2009.10.008
45. Cuthbert BN, Insel TR. Toward new approaches to psychotic disorders: the NIMH research domain criteria project. Schizophr Bull (2010) 36:1061–2. doi:10.1093/schbul/sbq108
47. Honey G, Bullmore E. Human pharmacological MRI. Trends Pharmacol Sci (2004) 25:366–74. doi:10.1016/j.tips.2004.05.009
48. Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport (1995) 6:869–72. doi:10.1097/00001756-199504190-00011
49. Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry (1997) 154:805–11.
50. Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol (1997) 7:9–24. doi:10.1016/S0924-977X(96)00039-9
51. Vollenweider FX, Vontobel P, Oye I, Hell D, Leenders KL. Effects of (S)-ketamine on striatal dopamine: a [11C]raclopride PET study of a model psychosis in humans. J Psychiatr Res (2000) 34:35–43. doi:10.1016/S0022-3956(99)00031-X
52. Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology (2001) 25:455–67. doi:10.1016/S0893-133X(01)00243-3
53. Lahti AC, Holcomb HH, Medoff DR, Weiler MA, Tamminga CA, Carpenter WT. Abnormal patterns of regional cerebral blood flow in schizophrenia with primary negative symptoms during an effortful auditory recognition task. Am J Psychiatry (2002) 158:1797–808. doi:10.1176/appi.ajp.158.11.1797
54. Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D’Souza DC, et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry (2013) 18(11):1199–204. doi:10.1038/mp.2012.194
55. Wang X-J. Toward a prefrontal microcircuit model for cognitive deficits in schizophrenia. Pharmacopsychiatry (2006) 39(Suppl 1):S80–7. doi:10.1055/s-2006-931501
56. Wang X-J. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev (2010) 90:1195–268. doi:10.1152/physrev.00035.2008
57. Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci (2001) 24:455–63. doi:10.1016/S0166-2236(00)01868-3
58. Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH. Linking microcircuit dysfunction to cognitive impairment: effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex (2012). doi:10.1093/cercor/bhs370
59. Deco G, Ponce-Alvarez A, Mantini D, Romani GL, Hagmann P, Corbetta M. Resting-state functional connectivity emerges from structurally and dynamically shaped slow linear fluctuations. J Neurosci (2013) 33:11239–52. doi:10.1523/JNEUROSCI.1091-13.2013
60. Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci (2009) 29:2774–9. doi:10.1523/JNEUROSCI.3703-08.2009
61. Biswal B, Yetkin FZ, Haughton VM, Hyde JS. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med (1995) 34:537–41. doi:10.1002/mrm.1910340409
62. Buckner RL, Krienen FM, Yeo BT. Opportunities and limitations of intrinsic functional connectivity MRI. Nat Neurosci (2013) 16:832–7. doi:10.1038/nn.3423
63. Power JD, Cohen AL, Nelson SM, Wig GS, Barnes KA, Church JA, et al. Functional network organization of the human brain. Neuron (2011) 72:665–78. doi:10.1016/j.neuron.2011.09.006
64. Smith SM, Miller KL, Moeller S, Xu J, Auerbach EJ, Woolrich MW, et al. Temporally-independent functional modes of spontaneous brain activity. Proc Natl Acad Sci U S A (2012) 109:3131–6. doi:10.1073/pnas.1121329109
65. Greicius M. Resting-state functional connectivity in neuropsychiatric disorders. Curr Opin Neurol (2008) 21:424–30. doi:10.1097/WCO.0b013e328306f2c5
66. Loh M, Rolls ET, Deco G. A dynamical systems hypothesis of schizophrenia. PLoS Comput Biol (2007) 3:e228. doi:10.1371/journal.pcbi.0030228
67. Van Dijk KR, Hedden T, Venkataraman A, Evans KC, Lazar SW, Buckner RL. Intrinsic functional connectivity as a tool for human connectomics: theory, properties, and optimization. J Neurophysiol (2010) 103:297–321. doi:10.1152/jn.00783.2009
68. Feinberg DA, Moeller S, Smith SM, Auerbach E, Ramanna S, Glasser MF, et al. Multiplexed echo planar imaging for sub-second whole brain fMRI and fast diffusion imaging. PLoS One (2010) 5:e15710. doi:10.1371/journal.pone.0015710
69. Glasser MF, Sotiropoulos SN, Wilson JA, Coalson TS, Fischl B, Andersson JL, et al. The minimal preprocessing pipelines for the human connectome project. Neuroimage (2013) 80:105–24. doi:10.1016/j.neuroimage.2013.04.127
70. Van Snellenberg JX, Torres IJ, Thornton AE. Functional neuroimaging of working memory in schizophrenia: task performance as a moderating variable. Neuropsychology (2006) 20(5):497–510. doi:10.1037/0894-4105.20.5.497
71. Friston KJ. Functional and effective connectivity: a review. Brain Connect (2011) 1:13–36. doi:10.1089/brain.2011.0008
72. Murphy K, Birn RM, Handwerker DA, Jones TB, Bandettini PA. The impact of global signal regression on resting state correlations: are anti-correlated networks introduced? Neuroimage (2009) 44:893–905. doi:10.1016/j.neuroimage.2008.09.036
73. Saad ZS, Gotts SJ, Murphy K, Chen G, Jo HJ, Martin A, et al. Trouble at rest: how correlation patterns and group differences become distorted after global signal regression. Brain Connect (2012) 2:25–32. doi:10.1089/brain.2012.0080
74. Fox M, Zhang D, Snyder A, Raichle M. The global signal and observed anticorrelated resting state brain networks. J Neurophysiol (2009) 101:3270–83. doi:10.1152/jn.90777.2008
75. Mantini D, Gerits A, Nelissen K, Durand JB, Joly O, Simone L, et al. Default mode of brain function in monkeys. J Neurosci (2011) 31:12954–62. doi:10.1523/JNEUROSCI.2318-11.2011
76. Keller CJ, Bickel S, Honey CJ, Groppe DM, Entz L, Craddock RC, et al. Neurophysiological investigation of spontaneous correlated and anticorrelated fluctuations of the BOLD signal. J Neurosci (2013) 33:6333–42. doi:10.1523/JNEUROSCI.4837-12.2013
77. Bullmore E, Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci (2009) 10:186–98. doi:10.1038/nrn2575
78. Calhoun VD, Sui J, Kiehl K, Turner J, Allen E, Pearlson G. Exploring the psychosis functional connectome: aberrant intrinsic networks in schizophrenia and bipolar disorder. Front Psychiatry (2011) 2:75. doi:10.3389/fpsyt.2011.00075
79. Khadka S, Meda SA, Stevens MC, Glahn DC, Calhoun VD, Sweeney JA, et al. Is aberrant functional connectivity a psychosis endophenotype? A resting state functional magnetic resonance imaging study. Biol Psychiatry (2013) 74(6):458–66. doi:10.1016/j.biopsych.2013.04.024
80. van den Heuvel MP, Sporns O, Collin G, Scheewe T, Mandl RC, Cahn W, et al. Abnormal rich club organization and functional brain dynamics in schizophrenia. JAMA Psychiatry (2013) 70:783–92. doi:10.1001/jamapsychiatry.2013.1328
81. Cole MW, Yarkoni T, Repovs G, Anticevic A, Braver TS. Global connectivity of prefrontal cortex predicts cognitive control and intelligence. J Neurosci (2012) 32:8988–99. doi:10.1523/JNEUROSCI.0536-12.2012
82. Anticevic A, Hu S, Zhang S, Savic A, Billingslea E, Wasylink S, et al. Global resting-state fMRI analysis identifies frontal cortex, striatal, and cerebellar dysconnectivity in obsessive-compulsive disorder. Biol Psychiatry (2013). doi:10.1016/j.biopsych.2013.10.021
83. Calhoun VD, Maciejewski PK, Pearlson GD, Kiehl KA. Temporal lobe and “default” hemodynamic brain modes discriminate between schizophrenia and bipolar disorder. Hum Brain Mapp (2008) 29:1265–75. doi:10.1002/hbm.20463
84. Meda SA, Gill A, Stevens MC, Lorenzoni RP, Glahn DC, Calhoun VD, et al. Differences in resting-state functional magnetic resonance imaging functional network connectivity between schizophrenia and psychotic bipolar probands and their unaffected first-degree relatives. Biol Psychiatry (2012) 71:881–9. doi:10.1016/j.biopsych.2012.01.025
86. Rolls ET, Loh M, Deco G, Winterer G. Computational models of schizophrenia and dopamine modulation in the prefrontal cortex. Nat Rev Neurosci (2008) 9:696–709. doi:10.1038/nrn2462
87. Rolls ET, Deco G. A computational neuroscience approach to schizophrenia and its onset. Neurosci Biobehav Rev (2010) 35(8):1644–53. doi:10.1016/j.neubiorev.2010.09.001
88. Lisman J. Excitation, inhibition, local oscillations, or large-scale loops: what causes the symptoms of schizophrenia? Curr Opin Neurobiol (2012) 22:537–44. doi:10.1016/j.conb.2011.10.018
89. Repovs G, Csernansky JG, Barch DM. Brain network connectivity in individuals with schizophrenia and their siblings. Biol Psychiatry (2011) 15:967–73. doi:10.1016/j.biopsych.2010.11.009
90. Repovs G, Barch DM. Working memory related brain network connectivity in individuals with schizophrenia and their siblings. Front Hum Neurosci (2012) 6:137. doi:10.3389/fnhum.2012.00137
91. Anticevic A, Corlett PR. Cognition-emotion dysinteraction in schizophrenia. Front Psychol (2012) 3:392. doi:10.3389/fpsyg.2012.00392
92. Hoffman RE, Fernandez T, Pittman B, Hampson M. Elevated functional connectivity along a corticostriatal loop and the mechanism of auditory/verbal hallucinations in patients with schizophrenia. Biol Psychiatry (2011) 69:407–14. doi:10.1016/j.biopsych.2010.09.050
93. Vercammen A, Knegtering H, den Boer JA, Liemburg EJ, Aleman A. Auditory hallucinations in schizophrenia are associated with reduced functional connectivity of the temporo-parietal area. Biol Psychiatry (2010) 67:912–8. doi:10.1016/j.biopsych.2009.11.017
94. Palaniyappan L. Does the salience network play a cardinal role in psychosis? An emerging hypothesis of insular dysfunction. J Psychiatry Neurosci (2012) 37:17–27. doi:10.1503/jpn.100176
95. Palaniyappan L, White TP, Liddle PF. The concept of salience network dysfunction in schizophrenia: from neuroimaging observations to therapeutic opportunities. Curr Top Med Chem (2012) 12:2324–38. doi:10.2174/156802612805289881
96. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry (2003) 160:13–23. doi:10.1176/appi.ajp.160.1.13
97. Kapur S, Mizrahi R, Li M. From dopamine to salience to psychosis-linking biology, pharmacology and phenomenology of psychosis. Schizophr Res (2005) 79(1):59–68. doi:10.1016/j.schres.2005.01.003
98. Tu PC, Hsieh JC, Li CT, Bai YM, Su TP. Cortico-striatal disconnection within the cingulo-opercular network in schizophrenia revealed by intrinsic functional connectivity analysis: a resting fMRI study. Neuroimage (2012) 59(1):238–47. doi:10.1016/j.neuroimage.2011.07.086.
99. Moran LV, Tagamets MA, Sampath H, O’Donnell A, Stein EA, Kochunov P, et al. Disruption of anterior insula modulation of large-scale brain networks in schizophrenia. Biol Psychiatry (2013) 74:467–74. doi:10.1016/j.biopsych.2013.02.029
100. Palaniyappan L, Simmonite M, White TP, Liddle EB, Liddle PF. Neural primacy of the salience processing system in schizophrenia. Neuron (2013) 79:814–28. doi:10.1016/j.neuron.2013.06.027
101. Schippers MB, Renken R, Keysers C. The effect of intra- and inter-subject variability of hemodynamic responses on group level Granger causality analyses. Neuroimage (2011) 57:22–36. doi:10.1016/j.neuroimage.2011.02.008
102. Smith SM, Bandettini PA, Miller KL, Behrens T. The danger of systematic bias in group-level FMRI-lag-based causality estimation. Neuroimage (2012) 59(2):1228–9. doi:10.1016/j.neuroimage.2011.08.015
103. Wen X, Rangarajan G, Ding M. Is Granger causality a viable technique for analyzing fMRI data? PLoS One (2013) 8:e67428. doi:10.1371/journal.pone.0067428
104. Smith SM, Miller KL, Salimi-Khorshidi G, Webster M, Beckmann CF, Nichols TE, et al. Network modelling methods for FMRI. Neuroimage (2011) 54:875–91. doi:10.1016/j.neuroimage.2010.08.063
105. Goldman-Rakic PS. Prefrontal cortical dysfunction in schizophrenia: the relevance of working memory. In: Carroll BJ, Barrett JE, editors. Psychopathology and the Brain. New York: Raven Press, Ltd. (1991). p. 1–23.
106. Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci (2003) 1003:138–58. doi:10.1196/annals.1300.063
107. Guillin O, Abi-Dargham A, Laruelle M. Neurobiology of dopamine in schizophrenia. Int Rev Neurobiol (2007) 78:1–39. doi:10.1016/S0074-7742(06)78001-1
108. Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch Gen Psychiatry (2012) 69(8):776–86. doi:10.1001/archgenpsychiatry.2012.169
109. Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol (2006) 26(4–6):365–84. doi:10.1007/s10571-006-9062-8
110. Bernstein H-G, Steiner J, Bogerts B. Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother (2009) 9:1059–71. doi:10.1586/ern.09.59
111. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2011) 37(1):4–15. doi:10.1038/npp.2011.181
112. Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A (1998) 95:13290–5. doi:10.1073/pnas.95.22.13290
113. Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull (2013) 39:120–9. doi:10.1093/schbul/sbr069
114. Krystal JH, Perry EB, Gueorguieva R, Belger A, Madonick SH, Abi-Dargham A, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry (2005) 62:985–94. doi:10.1001/archpsyc.62.9.985
115. Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry (2012) 17:664–5. doi:10.1038/mp.2011.171
116. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry (1999) 46:56–72. doi:10.1016/S0006-3223(99)00067-0
117. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry (2000) 48:627–40. doi:10.1016/S0006-3223(00)00976-8
118. Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron (2013) 10:81–93. doi:10.1016/j.neuron.2013.02.011
119. Durstewitz D, Kelc M, Gunturkun O. A neurocomputational theory of the dopaminergic modulation of working memory functions. J Neurosci (1999) 19:2807–22.
120. Durstewitz D, Seamans JK, Sejnowski TJ. Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J Neurophysiol (2000) 83(3):1733–50.
122. Pearlson GD, Wong DF, Tune LE, Ross CA, Chase GA, Links JM, et al. In vivo D2 dopamine receptor density in psychotic and non-psychotic patients with bipolar disorder. Arch Gen Psychiatry (1995) 52:471–7. doi:10.1001/archpsyc.1995.03950180057008
123. Glahn DC, Bearden CE, Barguil M, Barrett J, Reichenberg A, Bowden CL, et al. The neurocognitive signature of psychotic bipolar disorder. Biol Psychiatry (2007) 62:910–6. doi:10.1016/j.biopsych.2007.02.001
124. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry (1987) 44:660–9. doi:10.1001/archpsyc.1987.01800190080012
125. Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron (2000) 28:325–34. doi:10.1016/S0896-6273(00)00111-2
126. Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci (2010) 11:100–13. doi:10.1038/nrn2774
127. Van Essen DC, Ugurbil K, Auerbach E, Barch DM, Behrens TE, Bucholz R, et al. The human connectome project: a data acquisition perspective. Neuroimage (2012) 62:2222–31. doi:10.1016/j.neuroimage.2012.02.018
128. Behrens TE, Johansen-Berg H, Woolrich MW, Smith SM, Wheeler-Kingshott CA, Boulby PA, et al. Non-invasive mapping of connections between human thalamus and cortex using diffusion imaging. Nat Neurosci (2003) 6:750–7. doi:10.1038/nn1075
129. Zhang D, Snyder AZ, Shimony JS, Fox MD, Raichle ME. Noninvasive functional and structural connectivity mapping of the human thalamocortical system. Cereb Cortex (2010) 20:1187–94. doi:10.1093/cercor/bhp182
130. Fischl B, Salat DH, van der Kouwe AJ, Makris N, Ségonne F, Quinn BT, et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage (2004) 23(Suppl 1):S69–84. doi:10.1016/j.neuroimage.2004.07.016
131. Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia – implications for schizophrenia and Parkinson’s disease. Trends Neurosci (1990) 13:272–6. doi:10.1016/0166-2236(90)90108-M
132. Andreasen NC, Arndt S, Swayze V, Cizadlo T, Flaum M, O’Leary D, et al. Thalamic abnormalities in schizophrenia visualized through magnetic resonance image averaging. Science (1994) 266:294–8. doi:10.1126/science.7939669
133. Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol (2001) 41:237–60. doi:10.1146/annurev.pharmtox.41.1.237
134. Lewis DA, Cruz DA, Melchitzky DS, Pierri JN. Lamina-specific deficits in parvalbumin-immunoreactive varicosities in the prefrontal cortex of subjects with schizophrenia: evidence for fewer projections from the thalamus. Am J Psychiatry (2001) 158:1411–22. doi:10.1176/appi.ajp.158.9.1411
135. Alexander GE, Delong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci (1986) 9:357–81. doi:10.1146/annurev.ne.09.030186.002041
136. Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron (2002) 33:163–75. doi:10.1016/S0896-6273(01)00582-7
137. Sherman SM. The thalamus is more than just a relay. Curr Opin Neurobiol (2007) 17:417–22. doi:10.1016/j.conb.2007.07.003
138. Sherman SM. Thalamocortical interactions. Curr Opin Neurobiol (2012) 22:575–9. doi:10.1016/j.conb.2012.03.005
139. Haber S, McFarland NR. The place of the thalamus in frontal cortical-basal ganglia circuits. Neuroscientist (2001) 7:315–24. doi:10.1177/107385840100700408
140. Johansen-Berg H, Behrens TE, Sillery E, Ciccarelli O, Thompson AJ, Smith SM, et al. Functional-anatomical validation and individual variation of diffusion tractography-based segmentation of the human thalamus. Cereb Cortex (2005) 15:31–9. doi:10.1093/cercor/bhh105
141. Pakkenberg B. Pronounced reduction of total neuron number in mediodorsal thalamic nucleus and nucleus accumbens in schizophrenics. Arch Gen Psychiatry (1990) 47:1023–8. doi:10.1001/archpsyc.1990.01810230039007
142. Lewis DA. Is there a neuropathology of schizophrenia? Recent findings converge on altered thalamic-prefrontal cortical connectivity. Neuroscientist (2000) 6:208–18. doi:10.1177/107385840000600311
143. Cronenwett WJ, Csernansky J. Thalamic pathology in schizophrenia. Curr Top Behav Neurosci (2010) 4:509–28. doi:10.1007/7854_2010_55
144. Volk DW, Lewis DA. Effects of a mediodorsal thalamus lesion on prefrontal inhibitory circuitry: implications for schizophrenia. Biol Psychiatry (2003) 53:385–9. doi:10.1016/S0006-3223(02)01547-0
145. Selemon LD, Begovic A, Rakic P. Selective reduction of neuron number and volume of the mediodorsal nucleus of the thalamus in macaques following irradiation at early gestational ages. J Comp Neurol (2009) 515:454–64. doi:10.1002/cne.22078
146. Csernansky JG, Schindler MK, Splinter NR, Wang L, Gado M, Selemon LD, et al. Abnormalities of thalamic volume and shape in schizophrenia. Am J Psychiatry (2004) 161:896–902. doi:10.1176/appi.ajp.161.5.896
147. Harms MP, Wang L, Mamah D, Barch DM, Thompson PA, Csernansky JG. Thalamic shape abnormalities in individuals with schizophrenia and their nonpsychotic siblings. J Neurosci (2007) 27:13835–42. doi:10.1523/JNEUROSCI.2571-07.2007
148. Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (2001) 156:117–54. doi:10.1007/s002130100811
149. Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull (2007) 33:69–94. doi:10.1093/schbul/sbl060
150. Oltmanns TF, Neale JM. Schizophrenic performance when distractors are present: attentional deficit or differential task difficulty? J Abnorm Psychol (1975) 84:205–9. doi:10.1037/h0076721
151. Anticevic A, Repovs G, Corlett PR, Barch DM. Negative and non-emotional interference with visual working memory in schizophrenia. Biol Psychiatry (2011) 70:1159–68. doi:10.1016/j.biopsych.2011.07.010
152. Welsh RC, Chen AC, Taylor SF. Low-frequency BOLD fluctuations demonstrate altered thalamocortical connectivity in schizophrenia. Schizophr Bull (2010) 36:713–22. doi:10.1093/schbul/sbn145
153. Giguere M, Goldman-Rakic PS. Mediodorsal nucleus: areal, laminar, and tangential distribution of afferents and efferents in the frontal lobe of rhesus monkeys. J Comp Neurol (1988) 277:195–213. doi:10.1002/cne.902770204
154. Ray JP, Price JL. The organization of projections from the mediodorsal nucleus of the thalamus to orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol (1993) 337:1–31. doi:10.1002/cne.903370102
155. Parnaudeau S, O’Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, et al. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron (2013) 77:1151–62. doi:10.1016/j.neuron.2013.01.038
156. Rouiller EM, Liang F, Babalian A, Moret V, Wiesendanger M. Cerebellothalamocortical and pallidothalamocortical projections to the primary and supplementary motor cortical areas: a multiple tracing study in macaque monkeys. J Comp Neurol (1994) 345(2):185–213.
157. Norman KA, Polyn SM, Detre GJ, Haxby JV. Beyond mind-reading: multi-voxel pattern analysis of fMRI data. Trends Cogn Sci (2006) 10:424–30. doi:10.1016/j.tics.2006.07.005
158. Van Essen DC, Ugurbil K. The future of the human connectome. Neuroimage (2012) 62:1299–310. doi:10.1016/j.neuroimage.2012.01.032
159. Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry (2003) 160:636–45. doi:10.1176/appi.ajp.160.4.636
160. Meyer-Lindenberg A. From maps to mechanisms through neuroimaging of schizophrenia. Nature (2010) 468:194–202. doi:10.1038/nature09569
161. Klingner CM, Langbein K, Dietzek M, Smesny S, Witte OW, Sauer H, et al. Thalamocortical connectivity during resting state in schizophrenia. Eur Arch Psychiatry Clin Neurosci (2013). doi:10.1007/s00406-013-0417-0
162. Anticevic A, Cole MW, Murray JD, Corlett PR, Wang XJ, Krystal JH. The role of default network deactivation in cognition and disease. Trends Cogn Sci (2012) 16:584–92. doi:10.1016/j.tics.2012.10.008
163. Drevets WC, Price JL, Simpson JR Jr, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature (1997) 386:824–7. doi:10.1038/386824a0
164. Drevets WC, Savitz J, Trimble M. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr (2008) 13(8):663–81.
165. Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology (2001) 25:1–27. doi:10.1016/S0893-133X(01)00225-1
166. Keshavan MS, Clementz BA, Pearlson GD, Sweeney JA, Tamminga CA. Reimagining psychoses: an agnostic approach to diagnosis. Schizophr Res (2013) 146:10–6. doi:10.1016/j.schres.2013.02.022
167. MacDonald AW III, Chafee MV. Translational and developmental perspective on N-methyl-D-aspartate synaptic deficits in schizophrenia. Dev Psychopathol (2006) 18(3):853–76. doi:10.1017/S0954579406060421
168. Greene R. Circuit analysis of NMDAR hypofunction in the hippocampus, in vitro, and psychosis of schizophrenia. Hippocampus (2001) 11:569–77. doi:10.1002/hipo.1072
169. Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron (2013) 77:736–49. doi:10.1016/j.neuron.2012.12.032
170. Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci U S A (2006) 103:19878–83. doi:10.1073/pnas.0609440103
171. de la Fuente-Sandoval C, León-Ortiz P, Favila R, Stephano S, Mamo D, Ramírez-Bermúdez J, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology (2011) 36:1781–91. doi:10.1038/npp.2011.65
172. Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry (1999) 156(10):1646–9.
173. Thomas LE, Woods SW. The schizophrenia prodrome: a developmentally informed review and update for psychopharmacologic treatment. Child Adolesc Psychiatr Clin N Am (2006) 15(1):109–33. doi:10.1016/j.chc.2005.08.011
174. Moore JW, Cambridge VC, Morgan H, Giorlando F, Adapa R, Fletcher PC. Time, action and psychosis: using subjective time to investigate the effects of ketamine on sense of agency. Neuropsychologia (2013) 51:377–84. doi:10.1016/j.neuropsychologia.2012.07.005
175. Honey RA, Honey GD, O’Loughlin C, Sharar SR, Kumaran D, Bullmore ET, et al. Acute ketamine administration alters the brain responses to executive demands in a verbal working memory task: an FMRI study. Neuropsychopharmacology (2004) 29:1203–4. doi:10.1038/sj.npp.1300438
176. Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci (1997) 17:2921–7.
177. Scheidegger M, Walter M, Lehmann M, Metzger C. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS One (2012) 7(9):e44799. doi:10.1371/journal.pone.0044799
178. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry (2013) 73(12):1133–41. doi:10.1016/j.biopsych.2013.03.026
179. Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry (2012) 72:555–61. doi:10.1016/j.biopsych.2012.03.029
180. Nugent AC, Diazgranados N, Carlson PJ, Ibrahim L, Luckenbaugh DA, Brutsche N, et al. Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disord (2013). doi:10.1111/bdi.12118
181. Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, et al. The default mode network and self-referential processes in depression. Proc Natl Acad Sci U S A (2009) 106:1942–7. doi:10.1073/pnas.0812686106
182. Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull (2012) 38:942–9. doi:10.1093/schbul/sbs075
183. Moghaddam B. Bringing order to the glutamate chaos in schizophrenia. Neuron (2003) 40:881–4. doi:10.1016/S0896-6273(03)00757-8
184. Fornito A, Harrison BJ, Zalesky A, Simons JS. Competitive and cooperative dynamics of large-scale brain functional networks supporting recollection. Proc Natl Acad Sci U S A (2012) 109(31):12788–93. doi:10.1073/pnas.1204185109
185. Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D’Souza DC, et al. The impact of NMDA receptor blockade on human working memory-related prefrontal function and connectivity. Neuropsychopharmacology (2013) 38(13):2613–22. doi:10.1038/npp.2013.170
186. Salazar RF, Dotson NM, Bressler SL, Gray CM. Content-specific fronto-parietal synchronization during visual working memory. Science (2012) 338:1097–100. doi:10.1126/science.1224000
187. Pan WJ, Thompson GJ, Magnuson ME, Jaeger D, Keilholz S. Infraslow LFP correlates to resting-state fMRI BOLD signals. Neuroimage (2013) 74:288–97. doi:10.1016/j.neuroimage.2013.02.035
188. He BJ. Spontaneous and task-evoked brain activity negatively interact. J Neurosci (2013) 33:4672–82. doi:10.1523/JNEUROSCI.2922-12.2013
189. Musso F, Brinkmeyer J, Ecker D, London MK, Thieme G, Warbrick T, et al. Ketamine effects on brain function – simultaneous fMRI/EEG during a visual oddball task. Neuroimage (2011) 58:508–25. doi:10.1016/j.neuroimage.2011.06.045
190. Schmidt A, Bachmann R, Kometer M, Csomor PA, Stephan KE, Seifritz E, et al. Mismatch negativity encoding of prediction errors predicts S-ketamine-induced cognitive impairments. Neuropsychopharmacology (2012) 37:865–75. doi:10.1038/npp.2011.261
191. Gil-da-Costa R, Stoner GR, Fung R, Albright TD. Nonhuman primate model of schizophrenia using a noninvasive EEG method. Proc Natl Acad Sci U S A (2013) 110:15425–30. doi:10.1073/pnas.1312264110
192. Wang X-J, Tegnér J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci U S A (2004) 101:1368–73. doi:10.1073/pnas.0305337101
193. Deco G, Jirsa VK, McIntosh A. Emerging concepts for the dynamical organization of resting-state activity in the brain. Nat Rev Neurosci (2011) 12:43–56. doi:10.1038/nrn2961
194. Honey CJ, Sporns O, Cammoun L, Gigandet X, Thiran JP, Meuli R, et al. Predicting human resting-state functional connectivity from structural connectivity. Proc Natl Acad Sci U S A (2009) 106:2035–40. doi:10.1073/pnas.0811168106
195. Cabral J, Hugues E, Kringelbach ML, Deco G. Modeling the outcome of structural disconnection on resting-state functional connectivity. Neuroimage (2012) 62:1342–53. doi:10.1016/j.neuroimage.2012.06.007
196. Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci (2008) 1124:1–38. doi:10.1196/annals.1440.011
197. Brunel N, Wang XJ. Effects of neuromodulation in a cortical network model of object working memory dominated by recurrent inhibition. J Comput Neurosci (2001) 11:63–85. doi:10.1023/A:1011204814320
Keywords: schizophrenia, pharmacology, functional connectivity, computational modeling, thalamus, NMDA receptors, glutamate
Citation: Anticevic A, Cole MW, Repovs G, Savic A, Driesen NR, Yang G, Cho YT, Murray JD, Glahn DC, Wang X-J and Krystal JH (2013) Connectivity, pharmacology, and computation: toward a mechanistic understanding of neural system dysfunction in schizophrenia. Front. Psychiatry 4:169. doi: 10.3389/fpsyt.2013.00169
Received: 15 September 2013; Paper pending published: 12 October 2013;
Accepted: 04 December 2013; Published online: 24 December 2013.
Edited by:
André Schmidt, University of Basel, SwitzerlandReviewed by:
Martin Walter, Otto-von-Guericke-Universität Magdeburg, GermanyBernhard J. Mitterauer, Voltronics-Institute for Basic Research, Psychopathology and Brain Philosophy, Austria
Andrei Manoliu, Technische Universität München, Germany
Copyright: © 2013 Anticevic, Cole, Repovs, Savic, Driesen, Yang, Cho, Murray, Glahn, Wang and Krystal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan Anticevic, Department of Psychiatry, Yale University, 34 Park Street, New Haven, CT 06519, USA e-mail:YWxhbi5hbnRpY2V2aWNAeWFsZS5lZHU=