 Wanja Brüchle1
Wanja Brüchle1 Caroline Schwarzer2Christina Berns1Sebastian Scho1Jessica Schneefeld1
Caroline Schwarzer2Christina Berns1Sebastian Scho1Jessica Schneefeld1 Dirk Koester2,3
Dirk Koester2,3 Thomas Schack2Udo Schneider1
Thomas Schack2Udo Schneider1 Karin Rosenkranz1*
Karin Rosenkranz1*- 1Faculty of Medicine, University Clinic of Psychiatry and Psychotherapie Luebbecke, Ruhr University Bochum, Bochum, Germany
- 2Neurocognition and Action Group, Faculty of Psychology and Sports Sciences, Bielefeld University, Bielefeld, Germany
- 3Department of Business Psychology, Faculty Business and Management, BSP Business School Berlin, Berlin, Germany
Major depressive disorder (MDD) is the most common mental disorder and deficits in neuroplasticity are discussed as one pathophysiological mechanism. Physical activity (PA) enhances neuroplasticity in healthy subjects and improves clinical symptoms of MDD. However, it is unclear whether this clinical effect of PA is due to restoring deficient neuroplasticity in MDD. We investigated the effect of a 3-week PA program applied on clinical symptoms, motor excitability and plasticity, and on cognition in patients with MDD (N = 23), in comparison to a control intervention (CI; N = 18). Before and after the interventions, the clinical symptom severity was tested using self- (BDI-II) and investigator- (HAMD-17) rated scales, transcranial magnetic stimulation (TMS) protocols were used to test motor excitability and paired-associative stimulation (PAS) to test long-term-potentiation (LTP)-like plasticity. Additionally, cognitive functions such as attention, working memory and executive functions were tested. After the interventions, the BDI-II and HAMD-17 decreased significantly in both groups, but the decrease in HAMD-17 was significantly stronger in the PA group. Cognition did not change notably in either group. Motor excitability did not differ between the groups and remained unchanged by either intervention. Baseline levels of LTP-like plasticity in the motor cortex were low in both groups (PA: 113.40 ± 2.55%; CI: 116.83 ± 3.70%) and increased significantly after PA (155.06 ± 10.48%) but not after CI (122.01 ± 4.1%). Higher baseline BDI-II scores were correlated with lower levels of neuroplasticity. Importantly, the more the BDI-II score decreased during the interventions, the stronger did neuroplasticity increase. The latter effect was particularly strong after PA (r = −0.835; p < 0.001). The level of neuroplasticity related specifically to the psychological/affective items, which are tested predominantly in the BDI-II. However, the significant clinical difference in the intervention effects was shown in the HAMD-17 which focuses more on somatic/neurovegetative items known to improve earlier in the course of MDD. In summary, PA improved symptoms of MDD and restored the deficient neuroplasticity. Importantly, both changes were strongly related on the individual patients' level, highlighting the key role of neuroplasticity in the pathophysiology and the clinical relevance of neuroplasticity-enhancing interventions for the treatment of MDD.
Introduction
Major depressive disorder (MDD) is a common illness worldwide, with more than 264 million people affected (1). The pathophysiology of MDD is complex and likely due to different, possibly interacting mechanisms. Several preclinical and clinical studies described altered neuroplasticity in MDD (2–4), such as lower synaptic density in the brain which is associated with the severity of depressive symptoms (5).
Reduced LTP-like plasticity in the motor cortex has been described in MDD in studies using paired associative stimulation (PAS) as a specific transcranial magnetic stimulation (TMS) protocol (2, 6), which tests synaptic plasticity in the human brain (7). As this reduction showed some association with the symptom severity measured in clinical scales, developing interventions that aim at enhancing synaptic plasticity might be of crucial relevance in the treatment of MDD.
Physical activity (PA) is associated with higher levels of neuroplasticity in healthy subjects (8, 9), and has been identified as a protective factor against the onset of depression (10–13). The effect of PA or sports programs have been widely studied in MDD and the clinical benefit and therapeutic relevance has been shown (14–17) even of short-term interventions (18–22). Furthermore, PA seems to influence cognitive symptoms in MDD, such as deficits in attention, concentration, memory and executive functions (23, 24).
Most of these studies focused on measuring the effect of PA interventions using clinical outcome parameters, such as self- or investigator-rated depression scores, and did not investigate which neurobiological parameters, e.g., neuronal excitability or plasticity in the brain, might be associated with these clinical changes.
Our study investigated the effect of a PA program applied over a period of 3 weeks on clinical symptoms, neural excitability and PAS-induced plasticity in the motor cortex, as well as on cognitive performance in in-patients with an acute episode of MDD during their stay on the psychiatric ward. As most patients followed a more sedentary lifestyle before admission to hospital, we designed the PA program to be of moderate intensity, including elements of endurance, strength and coordination exercises that required interaction and teamwork of the participants, in order to avoid competition and the risk of perceived performance failure (25, 26). The effect of the PA program was compared to a control intervention (CI) administered over the same period of time that controlled for investigator-related effects, experience of group cohesion and social interaction, while the patients abstained from additional physical activity.
We expected—similar to previous studies—the level of PAS-induced plasticity to be low in MDD, and PA to lead to an improvement of clinical symptoms—at least of those known to be first indicators of symptom reduction, such as psychomotor retardation or loss of energy. Leading on from that, we asked (i) whether measures of PAS-induced plasticity and clinical symptoms are related on an individual subject's level, (ii) whether PA reverses neuroplasticity deficits in MDD, and (iii) whether PAS-induced plasticity could act as a marker to predict the clinical outcome of neuroplasticity enhancing interventions, such as PA.
Materials and Methods
The study was approved by the Ethics committee of the Ruhr University Bochum, medical faculty in Bad Oeynhausen (Germany) and conducted in accordance with the Declaration of Helsinki.
Subjects
After giving written informed consent, 50 in-patients meeting the clinical criteria of MDD as defined by the international classification of disease (ICD-10: F32, F33) were recruited (see Table 1 for details). The inclusion criteria were: (i) Age between 18 and 65 years, (ii) current depressive episode (BDI-II score ≥ 10 points; Hamilton depression score (HAMD-17) ≥ nine points), (iii) no concurrent brain stimulation treatment, (iv) no severe cardiovascular disease and body mass index <30 kg/m2, (v) no structural brain alteration as shown in brain imaging, and (vi) in case of concurrent medication: no major changes to antidepressant medication during the study; no medication with anticonvulsive medication or lithium; medication with benzodiazepine <1 mg/day lorazepam equivalent; no medication with antipsychotics in dosages known to alter brain excitability (27). Medication (e.g., with antidepressants) was continued as long as it complied with the inclusion criteria (see above) and left unchanged during the interventions.
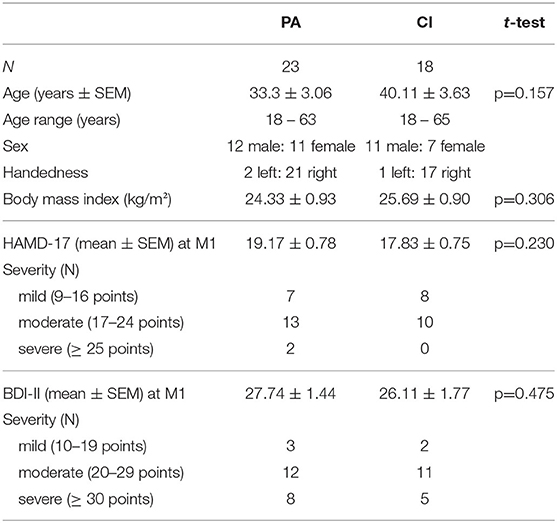
Table 1. Patients' characteristics.
Study Design
The study was performed within the setting of a primary care psychiatric university hospital on in-patients and ran for 18 months in total. Patients were screened according to the above mentioned criteria. In case of consent, they were recruited for participation in the study around 7–14 days after their admission to hospital, in order to allow for completion of diagnostics and for amelioration of severe symptoms of depression, such as suicidal ideation or agitation. They were informed that the purpose of the study was to compare the effect of two different interventions on their mood, their cognition and on neurophysiological parameters without implying a superior effect of one intervention to the other. As the recruitment rate of patients who fulfilled the inclusion criteria was expected to be low, the patients were successively recruited (rather than randomly assigned) in one of the two intervention groups and the investigators were not blinded. The first 25 patients were recruited into the PA group, and the succeeding 25 patients into the CI group. This approach ensured that 6–8 patients were participating at a time, which enabled interaction amongst participants during the sessions. Each patient participated for 3 weeks in one of the interventions. As neither the PA nor the CI sessions build up on each other, participants could join at any date.
Before the start (measurement 1; M1) and within 2–3 days after the end of the intervention period (measurement 2; M2) neurophysiological and cognitive parameters were tested (see Figure 1: experimental design), and clinical assessments by use of self- and investigator-related scales were performed.
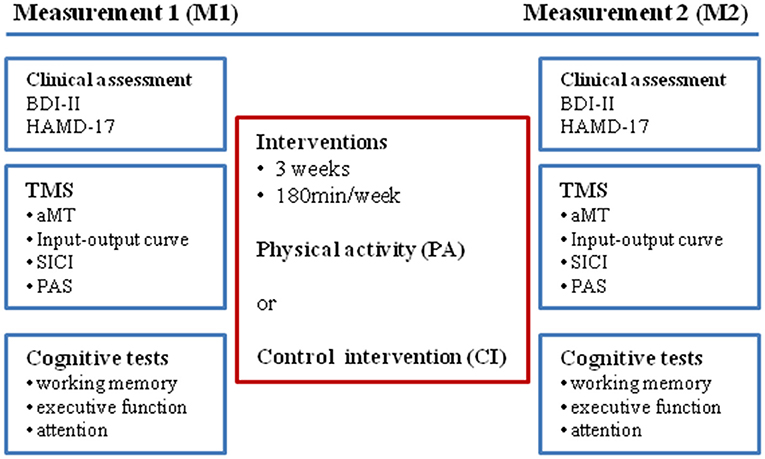
Figure 1. Experimental design.
Interventions and Assessment of Physiological Parameters
Each intervention ran for 3 weeks. For each intervention the total duration was 180 min/week; thus the total intervention time was 540 min in 3 weeks.
The PA was performed on 3 days a week (Monday, Wednesday, Friday) and was guided by an instructor. Each session lasted 60 min (without breaks) and focused on one out of three exercise types once a week, either coordination, endurance or strength training. These three exercise sessions were repeated every week. The PA program aimed to increase motivation for and to induce a positive affective response towards physical and sportive activity. It mainly consisted of interactive games, which the patients had to perform within the group or with one patient as partner. This approach reduced competitiveness among participants which could trigger a sense of underachievement or failure, or could invoke negative prior experiences (e.g., with PA at school). In addition, each PA session started with a 10-min warm-up which combined physical and cognitive tasks, by coding certain movements (e.g., circling of arms, lifting knees up) with colors (colored cards held up by the instructor). During the warm-up participants walked briskly through the room and had to perform the required movement (once) when the instructor showed the respective colored card. The color-movement associations were changed randomly in every PA session.
The CI consisted of two sessions of 90 min each per week and was guided by an instructor. While the participants remained seated on chairs, they performed different games (logical puzzles, “black stories,” card games), which required them to interact and to cooperate with each other. In the logical puzzles pieces of information were given and required the participants to put them into a matrix and find out the missing pieces of information by deduction (e.g., Who owned the zebra?). The “black stories” were mysterious stories that required the participants to reconstruct what has happened by asking and guessing. The card games prompted the participants to cooperate and to exchange cards in order to play and win together against the rule of the game (e.g., The Game—Spiel… solange Du kannst!).
The CI controlled for the presence of and attention given by an instructor and the feeling of group cohesion. The participants were cognitively engaged (solving logical problems, memorizing facts, and forming strategies). Importantly, the participants of the CI group were not included in other PA programs of the psychiatric clinic, and were instructed not to engage in any physical or sportive activity beyond their routine activity (such as walking in the hospital and hospital garden). The clinical and nursing staff was informed about the study participation and monitored the participants activity accordingly.
During the PA and CI sessions the heart rate was measured using a pulse tracker fixed to the upper arm with an elastic band (OH1, Polar Electro Oy, Kempele, Finland). The mean heart rate at rest (before the start of the session) and during the session were calculated for each participant.
Clinical Assessment
The Becks Depression Inventory II (BDI-II) was used for patients' self-assessment (28) and the Hamilton depression scale with 17 items (HAMD-17) was used for investigator-based assessment of clinical symptoms (29–31) once before and after the 3 weeks intervention period.
Neurophysiology
Transcranial Magnetic Stimulation (TMS)
TMS was performed using a Magstim 200 stimulator connected to a figure-of-eight-shaped coil with an internal wing diameter of 70 mm (Magstim Company Ltd, Whitland, UK). At the start of the experimental sessions M1 and M2 the motor “hot spot” of the abductor pollicis brevis (ABP) muscle was determined. The coil was held with the handle pointing backwards and laterally 45° to the interhemispheric line to evoke anteriorly directed current in the brain and was optimally positioned to obtain motor evoked potentials (MEP) in the APB of the dominant hand (“hot spot”). The subjects were wearing a tight fitting cotton wool cap on which the coil position was marked using a soft tip pen in order to ensure that the coil was held in a constant position during the experimental session.
Stimulation intensities are quoted as percentage of maximal stimulator output (mean ± SEM).
EMG Recording
Surface electromyographic (EMG) recordings in a belly-to-tendon montage were made from the APB and the first dorsal interosseus (FDI) muscles of the dominant hand. The raw signal was amplified and filtered with a bandpass filter of 30 Hz to 1 kHz (Digitimer D360; Welwyn Garden City, UK). Signals were digitized at 2 kHz (CED Power1401, Cambridge Electronic Design, UK) and stored on a laboratory computer for offline analysis. Online EMG was used to control for muscle relaxation during data recording and trials showing voluntary muscle activation were discarded from the analysis (<1% of trials). The recordings of the FDI were only displayed on screen during the experiments in order to support the experimenter in holding the coil in a constant position and were not analyzed further.
Motor Excitability
At the start of each measurement (M1 and M2), the active motor threshold (aMT) and the stimulus intensity (SI) needed to evoke a MEP of ~1 mV peak-to-peak amplitude (SI1mV) were defined in the APB. To determine the aMT, the EMG pattern (rectified amplitude) under maximum voluntary contraction (MVC) was displayed on the screen and a marker line was set to determine 30% of this amplitude. The subjects were instructed to activate their APB (pressing the thumb down while their hand lies in a pronated position on a cushion placed on their lap) so that the EMG amplitude was as close to that marker line as possible. The TMS measurements were always performed by two experimenters. In the measurement of the aMT, one experimenter controlled the participants APB activation level to be constant and of defined strength. The aMT was defined as the minimum intensity needed to evoke a MEP of ≥200 μV in five out of 10 trials.
The SI1mV was determined while the subjects were at rest with their hand muscles relaxed (as controlled by online EMG). Single TMS pulses were given (interstimulus interval 7 s) to determine the SI that gives a MEP of 1 mV peak-to-peak amplitude in five consecutive trials.
The input-output relationship of MEP amplitude to SI (IOcurve) was measured. For each SI of the IOcurve [50, 70, 80, 90, 100 (equal to SI1mV), 110, 120 130, and 150% of SI1mV] five consecutive TMS single pulses were applied with an interstimulus interval of 7 s and the MEPs recorded. The mean MEP amplitude per SI was calculated for each subject. Furthermore, the steepness of the IOcurve slopes defined as the steepness of the linear regression line through the given data points between 80 and 120% of SI1mV (IOslope) were calculated.
Short Interval Intracortical Inhibition (SICI)
The short-interval intracortical inhibition [SICI curve; (32, 33)] was measured using subthreshold conditioning stimulus intensities of 70, 80, and 90% of active motor threshold (aMT) and two magnetic stimulators (MagStim 200) connected via a BiStim module (Magstim Company Ltd, Whitland, UK). The conditioning stimulus preceded the suprathreshold test stimulus (intensity set at SI1mV) by 3 ms (34).
Three blocks consisting of 30 trials each were performed. Each block examined one conditioning pulse intensity and consisted of 15 MEPs elicited by the test stimulus alone (test MEPs) and 15 conditioned MEPs presented in pseudorandom order (intertrial interval 7 s). The peak-to-peak amplitude of the conditioned and test MEPs was measured for each single trial to calculate the mean amplitude and percentage SICI (conditioned MEP/ test MEP; in %) for the three different conditioning stimulus intensities. This approach allowed us to measure the level of SICI at a single conditioning intensity as well as the recruitment of SICI (SICI curve) defined as the increase of SICI with increasing intensities of the conditioning stimulus.
Plasticity in the Motor Cortex as Assessed by Paired-Associative Stimulation (PAS)
PAS consisted of 200 electrical stimuli of the median nerve at the wrist of the relaxed dominant hand paired with a single TMS pulse (at SI1mV) over the contralateral hand motor cortex with a rate of 0.25 Hz. TMS single pulses were delivered through a figure-of-eight shaped coil (diameter of each wing 70 mm) connected to a Magstim 200 stimulator and was held in the same position as described above. Electrical stimulation (Digitimer DS7A) was applied through a bipolar electrode (cathode proximal), using square-wave pulses (duration 0.2 ms) at an intensity of three times the perceptual threshold.
The electrical stimuli preceded the TMS pulses by 25 ms (PAS25). PAS25 has been shown previously to induce a long-lasting MEP increase (7, 35, 36). Subjects were instructed to look at their stimulated hand and count the peripheral electrical stimuli they perceived; they were asked the actual count by the experimenter about three to four times during the application of PAS (37). During PAS, the MEPs evoked in the APB and FDI were displayed on-line on the computer screen to control for the correct coil position and stored for off-line analysis.
Before and 10 min after the end of the PAS-intervention 20 TMS pulses were delivered (intertrial interval 7 s) using SI1mV and the MEPs recorded. Their mean amplitude was calculated. The effect of PAS was defined in each subject as change of the MEP amplitude in the APB (PASeffect = MEP after PAS / MEP before PAS; in %). In addition, PASchange refers to the change of PASeffects (PASchange = PASeffect after / before the intervention; in %).
Cognition
A PC-based test battery (Vienna test system, Schuhfried®, Austria) was used to measure different aspects of cognitive performance and executive function. Attention was tested using the work performance series which required subjects to perform additions and subtractions of single-digit numbers as fast and accurate as possible for 7 min. The Trail making test (part A and B) was used to assess the visuomotor processing speed and cognitive flexibility (38). The Response Inhibition (RI) task was used to assess voluntary control over responses within a changing context and required subjects to press a button as quickly as possible in reaction to a “Go” - signal (triangles) and to inhibit this reaction to an intermittently presented “NoGo” - signal (circle) (39, 40).
The Tower of London (TOL) assessed the planning abilities on the basis of clear rules and required the subjects to rearrange colored balls in a minimum number of moves (41, 42).
The STROOP interference test (color/word interference) was used to investigate the subjects' ability to control cognitive interference (43, 44).
Working memory performance was tested with the N-back verbal test [NBV; (45, 46)], a continuous performance measure (47, 48). A series of consonants was presented successively and subjects were required to press a button when the consonant displayed was identical to the one shown two places back (2-back paradigm) (49). To minimize the effect of familiarity, parallel versions of the tests were used in M2.
Data Analysis and Statistics
All data was tested for normal distribution by use of the Kolmogorov-Smirnov test. In case of not normally distributed data, non-parametric tests were used. All ANOVAs were tested for sphericity using Mauchly's test. In case of non-sphericity, Greenhouse-Geisser corrections were performed. Effect sizes (η2; r) were calculated for significant interactions. All data are given as mean ± SEM. Significance levels for the statistical tests are set to p ≤ 0.05; in case of multiple comparisons (results of cognitive tests) the significance level was adjusted to p ≤ 0.01.
The TMS parameters (aMT and SI1mV) of M1 and M2 were compared within groups by paired t-tests and between groups by unpaired t-tests. The IOcurve and SICI data were analyzed using ANOVA with the factor group (PA/CI) and the within-group factors intervention (before/after), stimulus intensity (IOcurve) or conditioning pulse intensity (SICI). The MEPs measured in M1 and M2 before PAS were compared by means of paired t-tests (within group) and unpaired t-tests (between groups) in order to control for correct adjustment of MEP size to 1 mV peak-to-peak amplitude. ANOVAs were performed on the raw data of MEPs with the factors group, intervention and MEP amplitude before/after PAS. For further analysis, the MEP raw data were normalized and expressed as percentage of MEPs (MEPs after PAS/ MEPs before PAS; PASeffect).
Correlations between neurophysiological data and clinical outcome (BDI-II and HAMD-17 scores) were calculated and significant results are reported giving Pearson's r for normally distributed and Kendall's tau for non-normally distributed data.
The raw data of cognitive tests was transformed into T-scores (mean = 50; SD = 10); with T-scores >50 indicating higher, and T-scores <50 indicating lower performance in comparison to a representative population (matched for age, sex and level of education) as given by the Vienna Test System (Schufried®, Austria). Similar to the analysis of the neurophysiological data, ANOVAs were performed with the between group factor group and the factors intervention as within-group factor. Post-hoc tests were performed when necessary and the significance level was adjusted to correct for multiple comparisons (see above).
Results
Subjects' and Clinical Data
Out of 50 recruited patients, 23 (of 25) in the PA group and 18 (of 25) in the CI group finished the study. Two patients of the PA group and three patients of the CI group were discharged from hospital before the M2 measurements could be taken. In addition, in the CI group two patients required emergency treatment and two patients discontinued their participation. Only results of patients who participated in both measurements (M1 and M2) and in all intervention sessions during the 3 weeks intervention period are reported. The patients in the PA and CI groups were comparable with regard to age, body mass index, BDI-II and HAMD-17 scores at M1 (see Table 1 for details).
Clinical Assessment
The subjects in the PA and CI groups did not differ in their BDI-II and HAMD-17 scores before the start of the intervention (unpaired t-tests, n.s.; see Table 1 and Figure 2). After the intervention, the BDI-II and HAMD-17 scores decreased significantly in both groups (paired t-tests; p < 0.009; see Figure 2) while the decrease in HAMD-17 score (HAMD-17 after—before intervention) was significantly stronger in the PA group (unpaired t-test; p < 0.001), there was no between-group difference in the decrease of the BDI-II score. A two-way ANOVA with the factors group (PA/CI) and intervention (before/after) showed for the HAMD-17 scores a significant interaction [ANOVA; F(1,39) = 16.52; p < 0.001], and significant main effects of the factor group [ANOVA; F(1,9) = 4.41; p = 0.042] and the factor intervention [ANOVA; F(1,39) = 112.94; p < 0.001]. For the BDI-II score data, there was no significant interaction, but a main effect of intervention [ANOVA; F(1,39) = 27.19; p < 0.001].
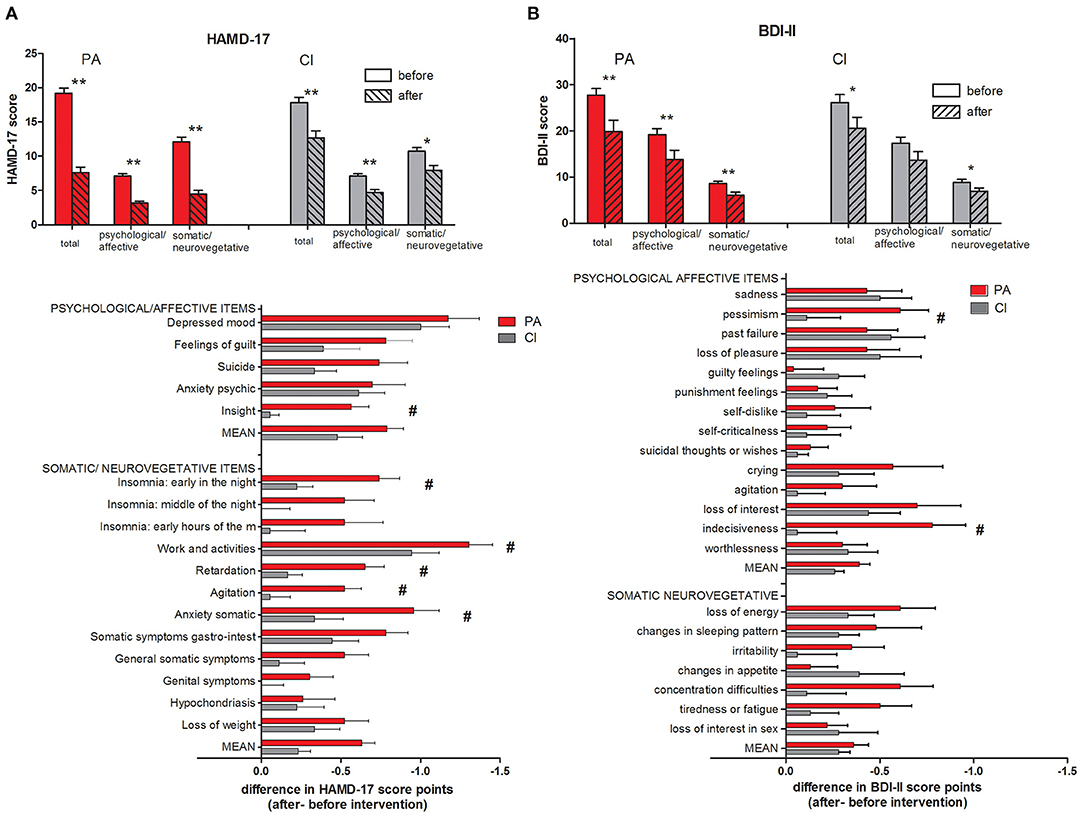
Figure 2. HAMD-17 (A) and BDI-II (B) scores with subscales. The mean scores of all items (total) as well as subscores for psychological/affective and somatic/neurovegetative items are given (mean ± SEM; t-test: **p < 0.001; *p < 0.01). Below the score change (after – before intervention) is given for each single item of the questionnaire (t-test; #p < 0.05).
A detailed analysis of the single items of the HAMD-17 scores (see Figure 2A) showed that the decrease in the items “insight,” “insomnia: early in the night,” “work and activities,” “retardation,” “agitation,” and “anxiety somatic” was significantly stronger in the PA group than in the CI group. For the BDI-II scores (see Figure 2B), the score for the items “pessimism” and “indecisiveness” decreased significantly stronger in the PA group than in the CI group.
The BDI-II and HAMD-17 scores measured before and after the interventions did not show a significant correlation in either the PA or the CI group.
Physiological Parameters
Before and during the interventions, the subjects' heart rate was continuously monitored. At rest, the mean heart rates were not different in the PA [69.00 ± 2.03 beats per minute (bpm)] and the CI group (71.39 ± 3.19 bpm; t-test: n.s.). During PA, the mean heart rate increased to 126.84 ± 2.83 bpm, showing that the patients were exercising with moderate intensity. In the CI group, the heart rate increased slightly to 83.02 ± 3.08, but this increase was not significantly different from their baseline mean heart rate (paired t-test; n.s.). The mean heart rates were not correlated to any neurophysiological or clinical parameters in either group.
Motor Cortical Excitability and Short-Interval Intracortical Inhibition
The aMT and SI1mV were not different between the two groups (see Table 2). The IOcurves (see Figures 3A,B) showed an increase of MEP amplitudes with increasing TMS intensity. The IOcurves measured before and after the interventions were not different in either the PA or the CI group (ANOVA, main effect of stimulus intensity p < 0.001; no effect of intervention). Furthermore, there were no group differences between the IOcurves (ANOVA; main effect group: p = 0.96; no significant interaction). The SICI (see Figures 3C,D) was stronger with increasing conditioning stimulus intensities. Again, there was no difference in SICI before and after the intervention within each group (ANOVAs; main effect of conditioning stimulus intensity: p < 0.001; effect of intervention: n.s.), nor was there any difference between the groups (ANOVA, main effect of factor group: p = 0.775; no significant interactions).
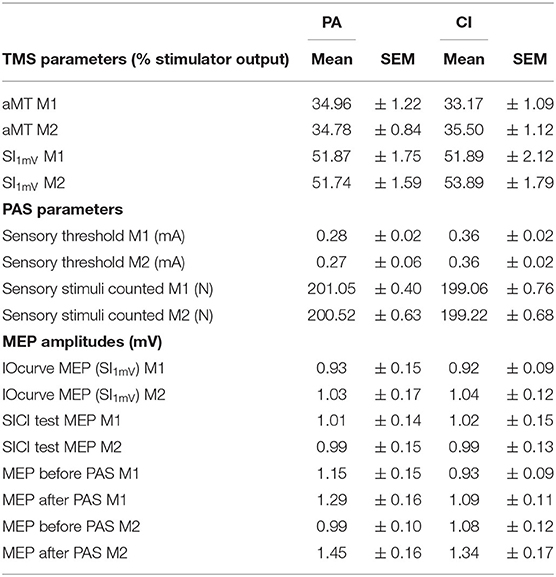
Table 2. TMS and PAS parameters and MEP amplitudes.
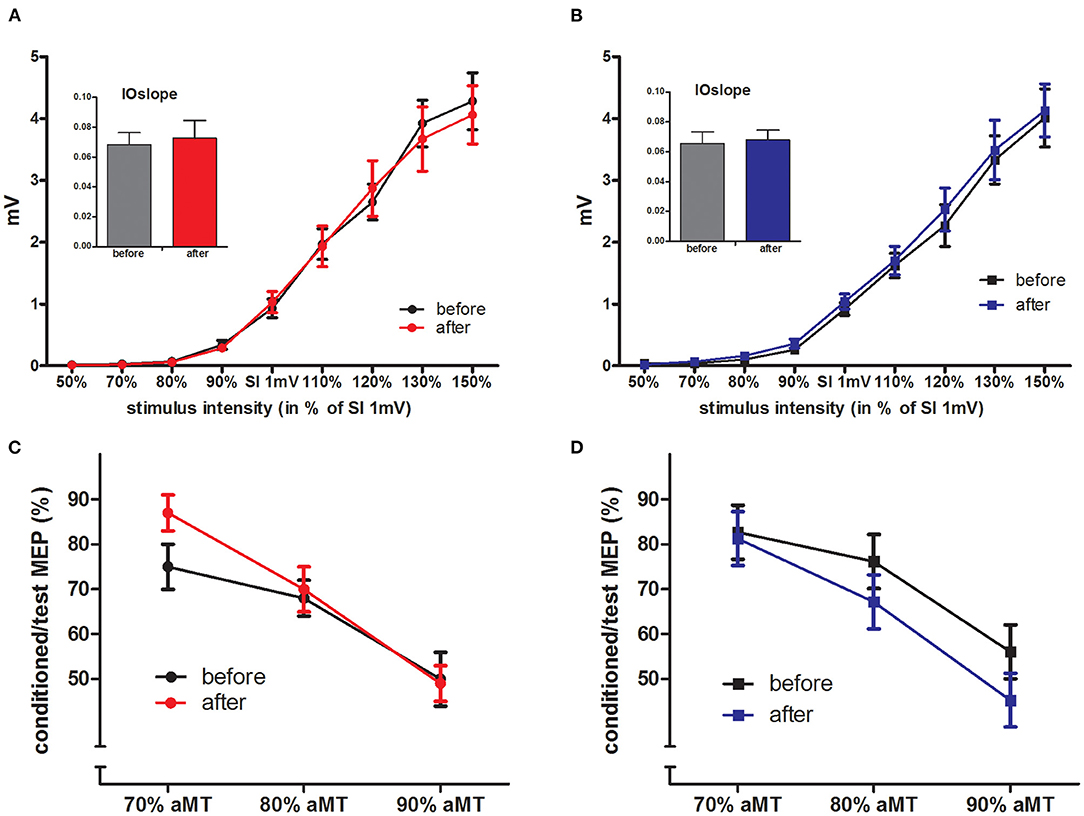
Figure 3. IOcurves and SICI. (A,B) displays the IOcurves measured in PA (A) and CI (B) groups before and after the intervention, with the IOslope given in the inserted diagram (mean ± SEM). IOcurves and IOslopes were not changed by the interventions within the groups, and there were no between-group differences. (C,D) displays the SICI results for PA (C) and CI (D) groups, again there were no differences within the groups (before/after intervention) nor between the groups.
Motor Cortical Plasticity - PAS
The mean MEP amplitudes measured before PAS were not different within each group when tested either before or after the intervention (paired t-tests, n.s.), and not different between the groups (unpaired t-tests: n.s.); thus ensuring comparability between the groups. In the PA group, PAS induced a significant increase (paired t-tests; p < 0.001) of the mean MEP amplitude before and after the intervention. The PASeffect (MEP after/before PAS, in %) was significantly stronger after PA (before: 113.40 ± 2.55%; after: 155.06 ± 10.48; paired t-test; p < 0.001; see Figure 4A).
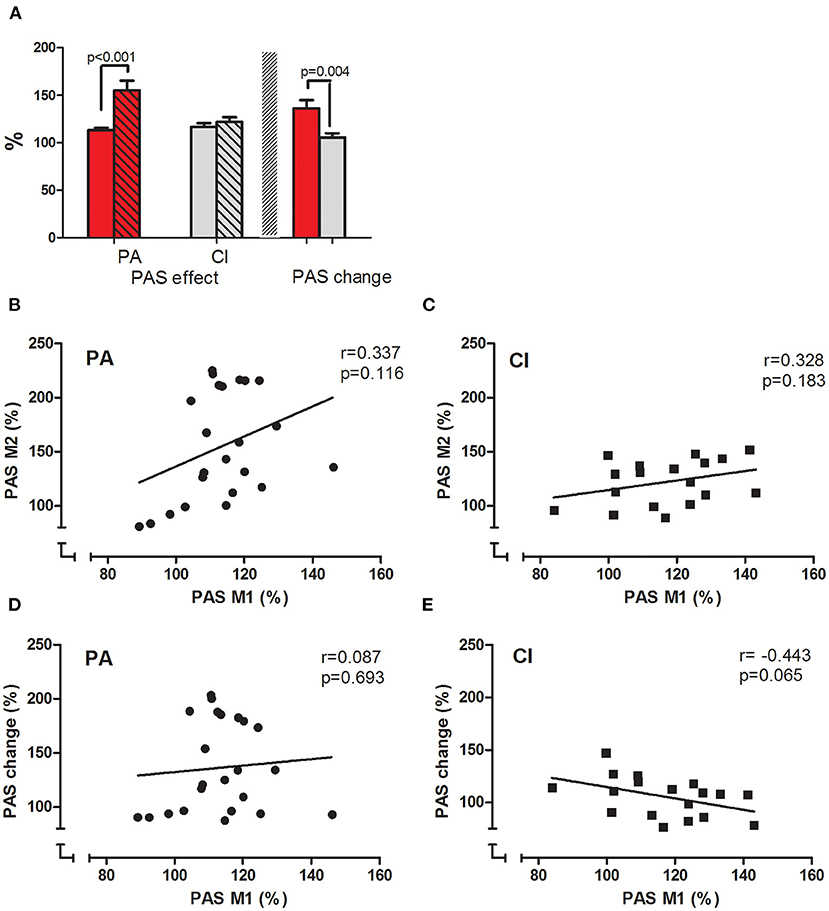
Figure 4. PASeffect and correlations. (A) Displays the PASeffects measured before (clear bars) and after (hatched bars) the interventions in PA (red bars) and CI (gray bars) groups. The PASeffect increased significantly after the intervention in the PA group, but not in the CI group (t-test results are given). (B,C) display the correlation between the PASeffects measured before and after the intervention in PA (B) and CI (C) groups; (D,E) display the correlation between the PASeffect measured before the intervention and PASchange for each group [PA: (D) and CI: (E)]. The results of Pearson's correlation are given.
In the CI group, PAS also significantly increased the mean MEP amplitude before and after the intervention (paired t-tests; p < 0.001). However, there was no difference in the PASeffects measured before and after the CI (before: 116.83 ± 3.70%; after: 122.01 ± 4.91%; paired t-test; n.s.; see Figure 4A).
The interaction of the factor intervention (before/after) and group (PA/CI) was significant [ANOVA; F(1, 39) = 9.09; p = 0.005; η2 = 0.40; r = 0.63], as were the main effects of intervention [F(1, 39) = 14.98; p < 0.001] and group [ANOVA; F(1, 39) = 4.15; p = 0.048]. The PASchange was significantly higher in the PA than in the CI group (unpaired t-test; p = 0.004).
The PASeffects measured before and after the intervention were only weakly correlated in each group (PA: Pearson's r = 0.337, p = 0.116; CI: Pearson's r = 0.328, p = 0.183; see Figures 4B,C). While the PASeffect at baseline and the PASchange showed no correlation in the PA group (Pearons's r = 0.087, p =0.693, see Figure 4D), there was a weak but non-significant correlation in the control group (Pearson's r = −0.443, p = 0.065; see Figure 4E). Thus, the baseline PASeffect did neither predict the PASeffect after the intervention, nor PASchange.
Correlations Between Clinical Scales and PAS
In both groups, there was a significant negative correlation between the BDI-II scores and the PASeffect measured before the interventions (see Figures 5A,B; PA: Pearson's r = −0.71, p < 0.001; CI: Pearson's r = −0.68, p = 0.002; for both groups together: Pearson's r = −0.695, p < 0.001): with increasing BDI-II scores the PASeffect was less strong. Furthermore, the change of the BDI-II scores and the change of the PASeffect by the intervention were significantly correlated in each groups (see Figures 5E,F; PA: Pearson's r = −0.835; p < 0.001; CI: −0.663, p = 0.003): the stronger the BDI-II score decreased, the stronger did the PASeffect increase. The latter effect was more prominent in the PA group (see Figure 5E).
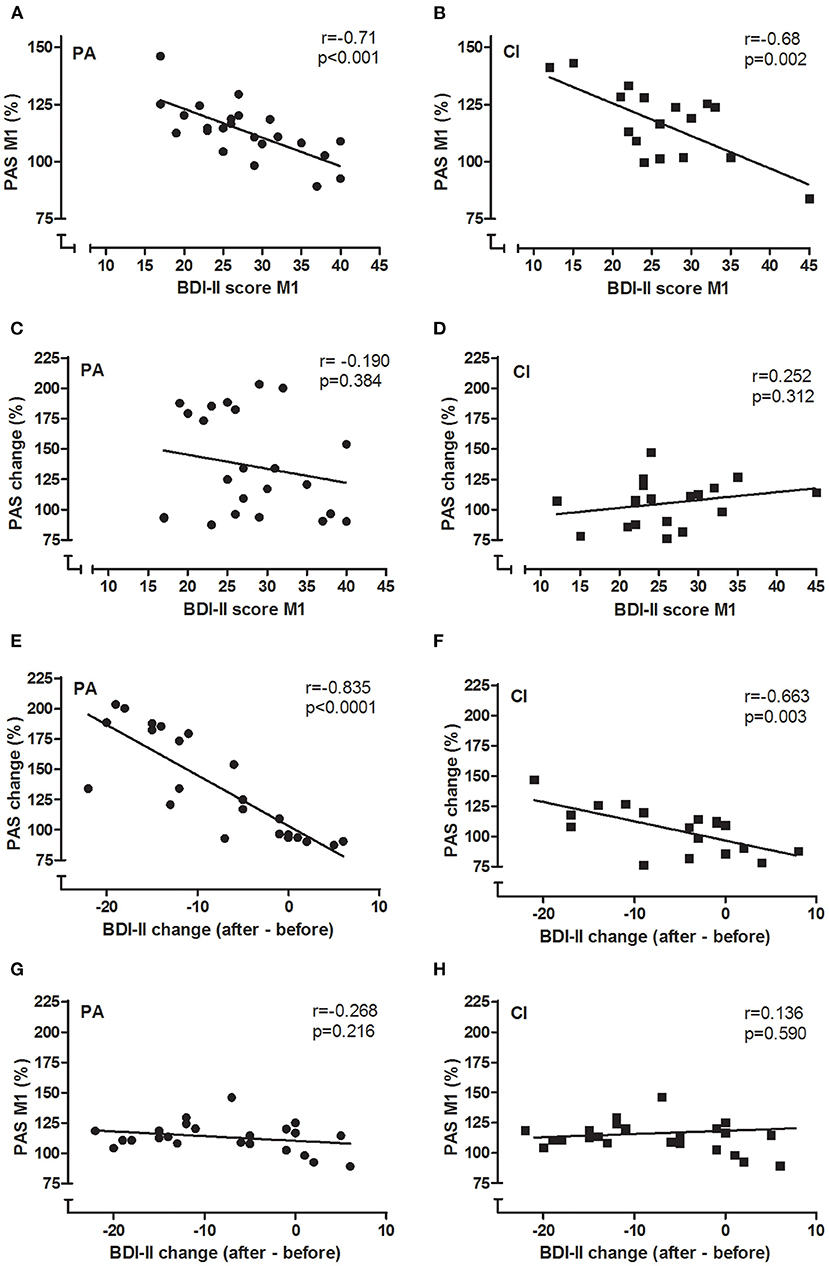
Figure 5. Correlations between BDI-II and PAS. The correlations of BDI-II and PAS are given for PA (A,C,E,G) and CI (B,D,F,H) groups. Pearson's r and p-values are given in each figure. BDI-II and PASeffect measured before the intervention (M1) were negatively correlated in each group (A,B): the higher the BDI-II score, the smaller was the PASeffect. Similarly, the change of BDI-II and PASeffects by the intervention were negatively correlated in each group (E,F). The BDI-II at baseline did not predict the amount of PASchange that could be induced by the interventions (C,D), neither did the PASeffect at baseline (M1) predict the amount of BDI-II change (G,H).
The BDI-II scores at baseline were not correlated to the PASchange in either of the groups (Figures 5C,D; PA: r = −0.19; p = 0.384; CI: r = 0.252; p = 0.312); thus, the baseline BDI-II did not predict the PASchange. Furthermore, the change of BDI-II scores were not correlated to the PASeffect at M1 in either group (Figures 5G,H; PA: r = −0.268; p = 0.216; CI: r = 0.136; p = 0.59), showing that the PASeffect at baseline did not predict the clinical effect of the interventions as shown in reduction of BDI-II scores.
There were no correlations between the HAMD-17 scores and the PAS data in either group; and no correlations between clinical scales and other neurophysiological parameters.
Cognition
Table 3 shows the results of the cognitive test performed before and after the intervention in each group (mean T-scores ± SEM). After the intervention, there was a significant increase of T-scores of some parameters within groups that indicated an improved performance (see results of paired t-tests in Table 3). There were no between-group differences in cognitive test before or after intervention, apart from a significant difference in the STROOP test (Δ after-before: baseline reading), which is of minor relevance in the absence of differences in the interference conditions.
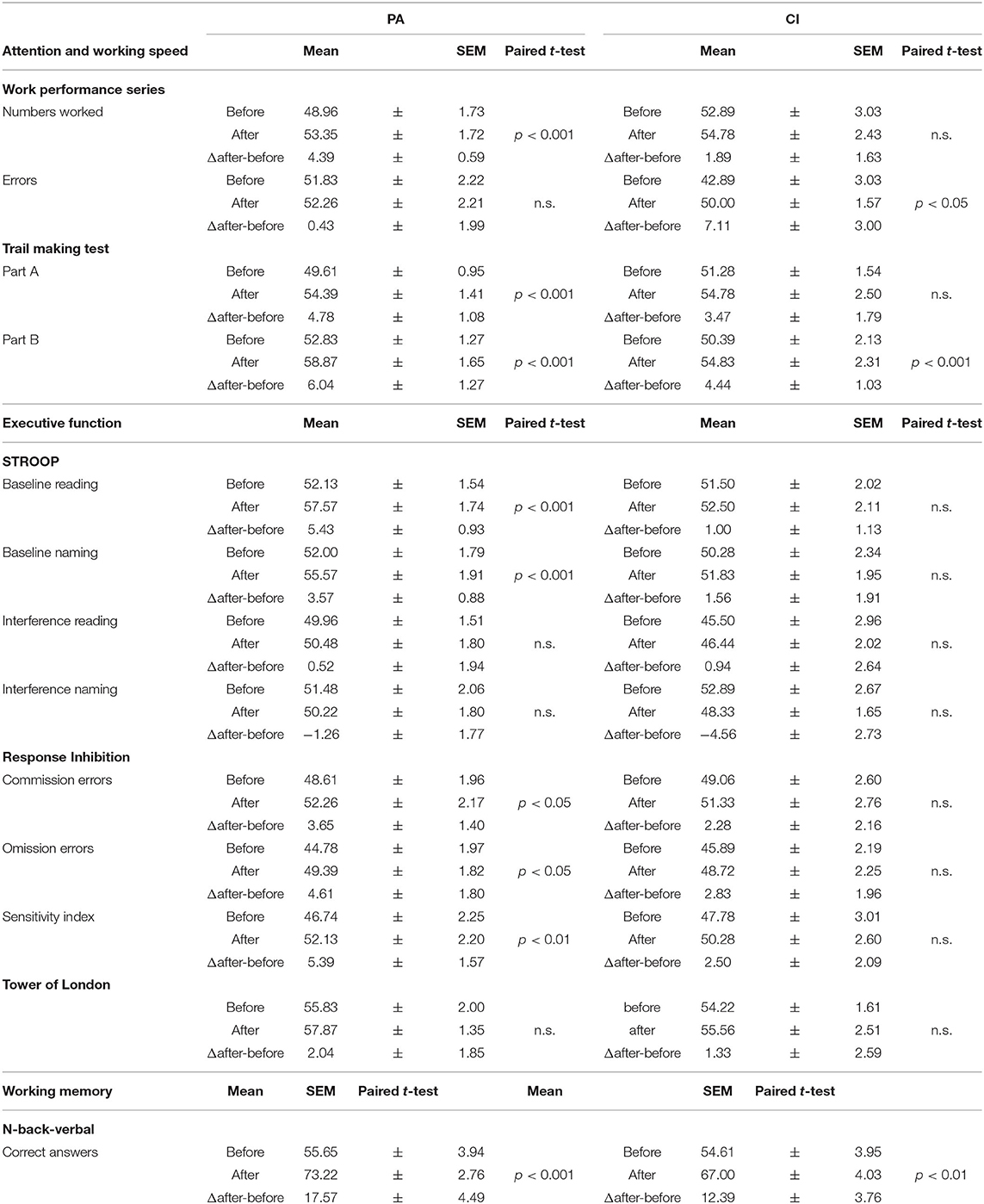
Table 3. Results of cognitive tests (T-scores).
Although there were no significant differences on how the performance was influenced by the intervention between the two groups, T-scores tended to increase stronger in the PA group: here the stronger increase of T-scores in Trail making and N-back tasks might indicate a stronger improvement of attention/ working speed and of working memory, respectively.
Discussion
Our study investigated the effect of PA—in comparison to a CI—on neuronal excitability and plasticity, as well as on clinical and cognitive symptoms in MDD. Confirming previous studies, we showed that (i) PA had a beneficial clinical effect as such as it reduced the severity of symptoms, such as psychomotor retardation and loss of energy as assessed by HAMD-17 and known to improve early in the course of MDD; and that (ii) the baseline level of motor cortical LTP-like plasticity is low in MDD.
Our study now crucially expands these findings by showing, that (iii) the severity of psychological/affective symptoms of MDD, as monitored with the BDI-II is highly correlated to the amount of LTP-like plasticity, and (iv) that PA as intervention can normalize deficient neuroplasticity which—in turn—is correlated to the reduction of clinical symptoms. In addition, (v) working memory performance (N-back verbal test), executive functions (Response Inhibition) and cognitive working speed (Trail making test) tended to improve stronger after PA than after the CI.
The reduction of PAS-induced LTP-like plasticity in the motor cortex in MDD before the interventions confirms findings of previous studies (2, 6), and different mechanisms may account for this. First, changes in structural and functional synaptic plasticity, such as reduced synaptic density in dorsolateral prefrontal cortex (DLPFC), the anterior corpus callosum (ACC), and the hippocampus (5, 50, 51) are described in MDD, likely leading to reduced functional connectivity within and between networks underlying mood and cognition. The motor cortex is an important node in the brain and processes information from various inputs (52, 53) as it is strongly interconnected with numerous brain areas. Several key structures of the cognitive network, such as the DLPFC (54), the posterior parietal cortex (55), as well as frontal areas (56) have been shown to be connected to the motor cortex, as several double-pulse TMS studies (57–60) as well as cortico-cortical paired associative stimulation studies have shown (61–64). Therefore, the PASeffect measured in the motor cortex represents a valid surrogate marker for plasticity in the networks that play a key role in the pathophysiology of MDD.
Second, the induction of LTP-like plasticity depends on postsynaptic activation of NMDA-receptors (35), and the alterations in the glutamatergic system described in MDD (65) are likely to contribute to a reduction of PASeffects.
Lastly, the presence of hallmark symptoms of MDD, such as anhedonia, loss of interest and of motivation, and psychomotor retardation, might further contribute to a reduction of synaptic plasticity. A lack of physical (66) and cognitive activity, and of social interaction, deprives the brain of important stimuli, which consequently might contribute to the downscaling or loss of synapses, which are necessary to keep the brain susceptible to plastic changes (67). Enhancing neuroplasticity is therefore a promising treatment approach, and using PA as an intervention has been proven to be clinically beneficial in MDD.
There is good evidence that PA modifies structural and functional brain plasticity (68–70). PAS-induced plasticity is higher in physically active healthy subjects compared to those with a sedentary lifestyle (8, 71). PA has been shown to increase metabolism and oxygenation, to modulate neurotransmitters and the release of neurochemical and neurotropic factors in the brain (72–77), and by these mechanisms likely contributes to the enhancement of plasticity.
After the 3 weeks of intervention, the PASeffect increased stronger in patients of the PA than of the CI group. Since parameters of neural excitability, the IOcurve and SICI, did not change, this is likely to be due to enhanced LTP-like plasticity after PA, rather than stronger motorneuronal recruitment or a reduction of GABAergic inhibition. Importantly, the PASeffect measured before the interventions was not related to PASchange, which precludes a saturation effect (“ceiling effect”).
We monitored the intensity of the PA program by measuring the heart rate, and its rise to moderate levels during the PA session indicated a moderate level of physical strain for the patients; while there was no notable change of heart rate in patients of the CI group. Given the moderate intensity and short duration of the PA program, the strength of its effect on PAS-induced plasticity in MDD was surprising. In the context of synaptic density and activity being reduced in MDD (5), it might point toward enhanced susceptibility to undergo LTP-like plasticity induction, in terms of a homeostatic mechanism. Similar to increased plasticity after sensory deprivation (78), the PA program might have re-activated synaptic connections and consequently “boosted” the efficiency of the PAS protocol to induce LTP-like plasticity (79).
Previous studies using PAS (6) and measures of synaptic density (5) have described an association between the severity of depressive symptoms and neuroplasticity in MDD. We extended these findings in our study by showing that the amount of LTP-like plasticity strongly correlates with the BDI-II scores in both groups at baseline; and further, that the amount of BDI-II score reduction and the increase of PAS plasticity seen after the interventions were correlated in both groups, though stronger in the PA. However, as the baseline PASeffect was not correlated to the BDI-II score change, and the baseline BDI-II was not correlated to the PASchange, the value of the PASeffect or the BDI-II score measured before the interventions is limited with regard to predicting either the clinical outcome or the amount of neuroplastic change.
There were no such correlations of the HAMD-17 and PASeffects, nor of the HAMD-17 and BDI-II scores. However, the difference in clinical outcome was shown in the HAMD-17, which decreased significantly stronger in the PA than in the CI group.
Several studies in MDD have already shown that self- and observer-rated scales are only moderately correlated, if at all (80, 81). Compared to the BDI-II (82), the HAMD-17 (83) has a higher sensitivity to depict changes (81, 84). The scores are quite different with regard to their item structure. The BDI-II focuses more on psychological/affective and the HAMD-17 more on somatic/neurovegetative items. As a self-rated score, the BDI is dependent on the patients' self-perception which—in turn—is often compromised by the symptoms of depression themselves. As it is focussing more on cognitive symptoms, the BDI-II is likely to be less sensitive to change because cognitive symptoms are more persistent than somatic symptoms in the course of treatment (85–87). Thus, the BDI-II has probably not been sensitive enough to depict early symptom changes that might have evolved during the 3 weeks observational period. The HAMD-17 is investigator-rated, focuses stronger on somatic/neurovegetative symptoms that are more likely to remit at an earlier phase in the course of treatment, and therefore is more perceptive to change over shorter observation periods (81). Furthermore, an observer could be more likely to see improvement in depressive symptoms than a patient affected by a cognitive bias (86, 87).
Thus, if there are first signs of symptom remittance, they are more likely to present as a reduction in HAMD-17 than in BDI-II; as was the case for the patients of the PA group. The correlation of the BDI-II and PAS-induced neuroplasticity might indicate, that factors like loss of interest, indecisiveness and pessimism, which were most strongly reduced in the PA group, might be more closely associated to deficient neuroplasticity and therefore sensitive to plasticity enhancing interventions.
The patients' attention, working memory and executive functions as tested by the cognitive test battery were not notably different from the age- and education-matched healthy control group (implemented in the test battery). Similar to other studies our findings might indicate that patients with MDD achieve this level of performance by a compensatory higher level of brain network activation, and thus their cognitive capacity is compromised by recruiting more brain resources as healthy controls (88).
Though there were no statistically significant differences, the cognitive performance tended to increase more in the PA than CI groups after the interventions, hinting toward an increased cognitive capacity by PA, as similarly described in physically active healthy subjects (89–91).
The study was performed on patients during their stay on the psychiatric ward and thus the duration of the interventions was limited to 3 weeks which might have been too short a period to induce differentiated effects on cognitive symptoms. Furthermore, we tested neuroplasticity, clinical and cognitive symptoms directly after the end of the interventions. Future studies need to address how long the PA-induced changes might last for and how they might be used to facilitate standard treatment of MDD.
In summary, we showed that a PA intervention supports the remission of clinical symptoms and normalizes deficient LTP-induced neuroplasticity in MDD, and that these two observations are highly correlated. Our study therefore further highlights the role of neuroplasticity in the pathophysiology of MDD and of PA in its treatment by showing that this intervention directly targets the deficient neuroplasticity as an underlying pathophysiological mechanism. Further research is needed to explore whether the effect of therapeutic interventions, might be predicted by clinical or neurophysiological parameters, as this would support the development of individualized treatments strategies.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics committee of the Ruhr University Bochum in Bad Oeynhausen/ Germany. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
WB, DK, and KR contributed to the experimental design. WB, CS, CB, SS, JS, and KR performed the experiments and contributed to the data acquisition. WB and KR analyzed and interpreted the data and wrote the manuscript. DK, CS, TS, and US contributed to the interpretation of the results. All authors gave their final approval of the version to be published.
Funding
This project was supported by a project grant from the Ruhr University Bochum awarded to the University Clinic of Psychiatry and Psychotherapy, Lübbecke, and to Bielefeld University, Faculty of Psychology and Sport Science, Neurocognition and Action Group (Forschungsfond für den Aufbau transdisziplinärer, medizinrelevanter Forschungskooperationen in der Region OWL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients for their participation in the study. We thank Mrs Ulrike Bökenheide for her support in the laboratory, and Dr. Elisabeth Wilking and Dr. Angelika Böhm for their support with the recruitment of patients.
References
1. James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
2. Player MJ, Taylor JL, Weickert CS, Alonzo A, Sachdev P, Martin D, et al. Neuroplasticity in depressed individuals compared with healthy controls. Neuropsychopharmacology. (2013) 38:2101–8. doi: 10.1038/npp.2013.126
3. Malykhin NV, Coupland NJ. Hippocampal neuroplasticity in major depressive disorder. Neuroscience. (2015) 309:200–13. doi: 10.1016/j.neuroscience.2015.04.047
4. Noda Y, Zomorrodi R, Vila-Rodriguez F, Downar J, Farzan F, Cash RF, et al. Impaired neuroplasticity in the prefrontal cortex in depression indexed through paired associative stimulation. Depress Anxiety. (2018) 35:448–56. doi: 10.1002/da.22738
5. Holmes SE, Scheinost D, Finnema SJ, Naganawa M, Davis MT, DellaGioia N, et al. Lower synaptic density is associated with depression severity and network alterations. Nat Commun. (2019) 10:1529. doi: 10.1038/s41467-019-09562-7
6. Kuhn M, Mainberger F, Feige B, Maier JG, Mall V, Jung NH, et al. State-dependent partial occlusion of cortical LTP-like plasticity in major depression. Neuropsychopharmacology. (2016) 41:2794. doi: 10.1038/npp.2016.97
7. Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. (2000) 123:572–84. doi: 10.1093/brain/123.3.572
8. Ridding MC, Ziemann U. Determinants of the induction of cortical plasticity by non-invasive brain stimulation in healthy subjects. J Physiol. (2010) 588:2291–304. doi: 10.1113/jphysiol.2010.190314
9. Mellow ML, Goldsworthy MR, Coussens S, Smith AE. Acute aerobic exercise and neuroplasticity of the motor cortex: a systematic review. J Sci Med Sport. (2020) 23:408–14. doi: 10.1016/j.jsams.2019.10.015
10. Teychenne M, Ball K, Salmon J. Physical activity and likelihood of depression in adults: a review. Prev Med. (2008) 46:397–411. doi: 10.1016/j.ypmed.2008.01.009
11. Mammen G, Faulkner G. Physical activity and the prevention of depression: a systematic review of prospective studies. Am J Prev Med. (2013) 45:649–57. doi: 10.1016/j.amepre.2013.08.001
12. Choi KW, Chen C-Y, Stein MB, Klimentidis YC, Wang M-J, Koenen KC, et al. Assessment of bidirectional relationships between physical activity and depression among adults: a 2-sample Mendelian randomization study. JAMA Psychiatry. (2019) 76:399–408. doi: 10.1001/jamapsychiatry.2018.4175
13. Kim S-Y, Park J-H, Lee MY, Oh K-S, Shin D-W, Shin Y-C. Physical activity and the prevention of depression: a cohort study. General Hospital Psychiatry. (2019) 60:90–7. doi: 10.1016/j.genhosppsych.2019.07.010
14. Kvam S, Kleppe CL, Nordhus IH, Hovland A. Exercise as a treatment for depression: a meta-analysis. J Affect Disord. (2016) 202:67–86. doi: 10.1016/j.jad.2016.03.063
15. Schuch FB, Vancampfort D, Richards J, Rosenbaum S, Ward PB, Stubbs B. Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res. (2016) 77:42–51. doi: 10.1016/j.jpsychires.2016.02.023
16. Schuch FB, Vancampfort D, Firth J, Rosenbaum S, Ward PB, Silva ES, et al. Physical activity and incident depression: a meta-analysis of prospective cohort studies. Am J Psychiatry. (2018) 175:631–48. doi: 10.1176/appi.ajp.2018.17111194
17. Ledochowski L, Stark R, Ruedl G, Kopp M. Körperliche Aktivität als therapeutische Intervention bei Depression. Nervenarzt. (2017) 88:765–78. doi: 10.1007/s00115-016-0222-x
18. Martinsen EW, Medhus A, Sandvik L. Effects of aerobic exercise on depression: a controlled study. Br Med J. (1985) 291:109. doi: 10.1136/bmj.291.6488.109
19. Knubben K, Reischies FM, Adli M, Schlattmann P, Bauer M, Dimeo F. A randomised, controlled study on the effects of a short-term endurance training programme in patients with major depression. Br J Sports Med. (2007) 41:29–33. doi: 10.1136/bjsm.2006.030130
20. Schuch FB, Vasconcelos-Moreno MP, Borowsky C, Fleck MP. Exercise and severe depression: preliminary results of an add-on study. J Affect Disord. (2011) 133:615–8. doi: 10.1016/j.jad.2011.04.030
21. Schuch FB, Vasconcelos-Moreno MP, Borowsky C, Zimmermann AB, Rocha NS, Fleck MP. Exercise and severe major depression: effect on symptom severity and quality of life at discharge in an inpatient cohort. J Psychiatr Res. (2015) 61:25–32. doi: 10.1016/j.jpsychires.2014.11.005
22. Ho CW, Chan SC, Wong JS, Cheung WT, Chung DW, Lau TF. Effect of aerobic exercise training on Chinese population with mild to moderate depression in Hong Kong. Rehabil Res Pract. (2014) 2014:627376. doi: 10.1155/2014/627376
23. McDermott LM, Ebmeier KP. A meta-analysis of depression severity and cognitive function. J Affect Disord. (2009) 119:1–8. doi: 10.1016/j.jad.2009.04.022
24. McClintock SM, Husain MM, Greer TL, Cullum CM. Association between depression severity and neurocognitive function in major depressive disorder: a review and synthesis. Neuropsychology. (2010) 24:9–34. doi: 10.1037/a0017336
25. Rhodes RE, Kates A. Can the affective response to exercise predict future motives and physical activity behavior? a systematic review of published evidence. Ann Behav Med. (2015) 49:715–31. doi: 10.1007/s12160-015-9704-5
26. Williams DM, Dunsiger S, Ciccolo JT, Lewis BA, Albrecht AE, Marcus BH. Acute affective response to a moderate-intensity exercise stimulus predicts physical activity participation 6 and 12 months later. Psychol Sport Exerc. (2008) 9:231–45. doi: 10.1016/j.psychsport.2007.04.002
27. Ziemann U, Reis J, Schwenkreis P, Rosanova M, Strafella A, Badawy R, et al. TMS and drugs revisited 2014. Clin Neurophysiol. (2015) 126:1847–68. doi: 10.1016/j.clinph.2014.08.028
28. Beck AT, Steer RA, Brown GK. Manual for Beck Depression Inventory II (BDI-II). San Antonio, TX: Psychological Corp (1996). doi: 10.1037/t00742-000
29. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. (1960) 23:56–62. doi: 10.1136/jnnp.23.1.56
30. Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. (1967) 6:278–96. doi: 10.1111/j.2044-8260.1967.tb00530.x
31. Williams JB. Standardizing the Hamilton depression rating scale: past, present, and future. Eur Archiv Psychiatry Clin Neurosci. (2001) 251(Suppl.2):II6–12. doi: 10.1007/BF03035120
32. Orth M, Snijders A, Rothwell J. The variability of intracortical inhibition and facilitation. Clin Neurophysiol. (2003) 114:2362–9. doi: 10.1016/S1388-2457(03)00243-8
33. Rosenkranz K, Williamon A, Rothwell JC. Motorcortical excitability and synaptic plasticity is enhanced in professional musicians. J Neurosci. (2007) 27:5200–6. doi: 10.1523/JNEUROSCI.0836-07.2007
34. Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, et al. Corticocortical inhibition in human motor cortex. J Physiol. (1993) 471:501–19. doi: 10.1113/jphysiol.1993.sp019912
35. Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol. (2002) 543:699–708. doi: 10.1113/jphysiol.2002.023317
36. Ziemann U, Ilić TV, Iliać TV, Pauli C, Meintzschel F, Ruge D. Learning modifies subsequent induction of long-term potentiation-like and long-term depression-like plasticity in human motor cortex. J Neurosci. (2004) 24:1666–72. doi: 10.1523/JNEUROSCI.5016-03.2004
37. Stefan K, Wycislo M, Classen J. Modulation of associative human motor cortical plasticity by attention. J Neurophysiol. (2004) 92:66–72. doi: 10.1152/jn.00383.2003
38. Rodewald K, Weisbrod M, Aschenbrenner S. Trail Making Test - Langensteinbacher Version. Mödling: Schuhfried GmbH (2012). p. 36.
39. Kaiser S, Mundt C, Weisbrod M. Exekutive Kontrollfunktionen und neuropsychiatrische Erkrankungen - Perspektiven für Forschung und Klinik. Fortschr Neurol Psychiatr. (2005) 73:438–50. doi: 10.1055/s-2004-830303
40. Kaiser S, Aschenbrenner S, Pfüller U, Roesch-Ely D, Weisbrod M. Response Inhibiton. Mödling: Schuhfried GmbH (2010).
41. Shallice T. Specific impairments of planning. Philos Trans R Soc Lond B, Biol Sci. (1982) 298:199–209. doi: 10.1098/rstb.1982.0082
42. Kaller CP, Unterrainer JM, Kaiser S, Weisbrod M, Aschenbrenner S. Tower of London - Freiburger Version. Mödling: Schuhfried GmbH (2011).
43. Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol. (1935) 18:643–62. doi: 10.1037/h0054651
45. Gevins AS, Custillo BC. Neuroelectric evidence for distributed processing in human working memory. Electroencephalogr Clin Neurophysiol. (1993) 87:128–43. doi: 10.1016/0013-4694(93)90119-G
46. Cohen JD, Perlstein WM, Braver TS, Nystrom LE, Noll DC, Jonides J, et al. Temporal dynamics of brain activation during a working memory task. Nature. (1997) 386:604–8. doi: 10.1038/386604a0
47. Kirchner WK. Age differences in short-term retention of rapidly changing information. J Exp Psychol. (1958) 55:352–8. doi: 10.1037/h0043688
48. Soveri A, Antfolk J, Karlsson L, Salo B, Laine M. Working memory training revisited: a multi-level meta-analysis of n-back training studies. Psychon Bull Rev. (2017) 24:1077–96. doi: 10.3758/s13423-016-1217-0
50. Czéh B, Lucassen PJ. What causes the hippocampal volume decrease in depression? Are neurogenesis, glial changes and apoptosis implicated? Eur Archiv Psychiatry Clin Neurosci. (2007) 257:250–60. doi: 10.1007/s00406-007-0728-0
51. Bennett MR. The prefrontal-limbic network in depression: a core pathology of synapse regression. Progr Neurobiol. (2011) 93:457–67. doi: 10.1016/j.pneurobio.2011.01.001
52. Tomasi D, Volkow ND. Association between functional connectivity hubs and brain networks. Cereb Cortex. (2011) 21:2003–13. doi: 10.1093/cercor/bhq268
53. Berns C, Brüchle W, Scho S, Schneefeld J, Schneider U, Rosenkranz K. Intensity dependent effect of cognitive training on motor cortical plasticity and cognitive performance in humans. Exp Brain Res. (2020) 238:2805–18. doi: 10.1007/s00221-020-05933-5
54. Takeuchi H, Taki Y, Nouchi R, Hashizume H, Sekiguchi A, Kotozaki Y, et al. Effects of multitasking-training on gray matter structure and resting state neural mechanisms. Hum Brain Mapp. (2014) 35:3646–60. doi: 10.1002/hbm.22427
55. Draganski B, May A. Training-induced structural changes in the adult human brain. Behav Brain Res. (2008) 192:137–42. doi: 10.1016/j.bbr.2008.02.015
56. Ceccarelli A, Rocca MA, Pagani E, Falini A, Comi G, Filippi M. Cognitive learning is associated with gray matter changes in healthy human individuals: a tensor-based morphometry study. Neuroimage. (2009) 48:585–9. doi: 10.1016/j.neuroimage.2009.07.009
57. Civardi C, Cantello R, Asselman P, Rothwell JC. Transcranial magnetic stimulation can be used to test connections to primary motor areas from frontal and medial cortex in humans. Neuroimage. (2001) 14:1444–53. doi: 10.1006/nimg.2001.0918
58. Koch G, Fernandez Del Olmo M, Cheeran B, Ruge D, Schippling S, Caltagirone C, et al. Focal stimulation of the posterior parietal cortex increases the excitability of the ipsilateral motor cortex. J Neurosci. (2007) 27:6815–22. doi: 10.1523/JNEUROSCI.0598-07.2007
59. Davare M, Lemon R, Olivier E. Selective modulation of interactions between ventral premotor cortex and primary motor cortex during precision grasping in humans. J Physiol. (2008) 586:2735–42. doi: 10.1113/jphysiol.2008.152603
60. Hasan A, Galea JM, Casula EP, Falkai P, Bestmann S, Rothwell JC. Muscle and timing-specific functional connectivity between the dorsolateral prefrontal cortex and the primary motor cortex. J Cogn Neurosci. (2013) 25:558–70. doi: 10.1162/jocn_a_00338
61. Koch G, Ponzo V, Di Lorenzo F, Caltagirone C, Veniero D. Hebbian and anti-Hebbian spike-timing-dependent plasticity of human cortico-cortical connections. J Neurosci. (2013) 33:9725–33. doi: 10.1523/JNEUROSCI.4988-12.2013
62. Veniero D, Ponzo V, Koch G. Paired associative stimulation enforces the communication between interconnected areas. J Neurosci. (2013) 33:13773–83. doi: 10.1523/JNEUROSCI.1777-13.2013
63. Chao C-C, Karabanov AN, Paine R, Carolina de Campos A, Kukke SN, Wu T, et al. Induction of motor associative plasticity in the posterior parietal cortex-primary motor network. Cereb Cortex. (2015) 25:365–73. doi: 10.1093/cercor/bht230
64. Kohl S, Hannah R, Rocchi L, Nord CL, Rothwell J, Voon V. Cortical paired associative stimulation influences response inhibition: cortico-cortical and cortico-subcortical networks. Biol Psychiatry. (2019) 85:355–63. doi: 10.1016/j.biopsych.2018.03.009
65. Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. (2008) 7:426–37. doi: 10.1038/nrd2462
66. Difrancesco S, Lamers F, Riese H, Merikangas KR, Beekman AT, van Hemert AM, et al. Sleep, circadian rhythm, and physical activity patterns in depressive and anxiety disorders: a 2-week ambulatory assessment study. Depress Anxiety. (2019) 36:975–86. doi: 10.1002/da.22949
67. Raichlen DA, Alexander GE. Adaptive capacity: an evolutionary neuroscience model linking exercise, cognition, and brain health. Trends Neurosci. (2017) 40:408–21. doi: 10.1016/j.tins.2017.05.001
68. Hötting K, Röder B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci Biobehav Rev. (2013) 37:2243–57. doi: 10.1016/j.neubiorev.2013.04.005
69. Voelcker-Rehage C, Niemann C. Structural and functional brain changes related to different types of physical activity across the life span. Neurosci Biobehav Rev. (2013) 37:2268–95. doi: 10.1016/j.neubiorev.2013.01.028
70. Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci. (2007) 11:342–8. doi: 10.1016/j.tics.2007.06.009
71. Cirillo J, Lavender AP, Ridding MC, Semmler JG. Motor cortex plasticity induced by paired associative stimulation is enhanced in physically active individuals. J Physiol. (2009) 587:5831–42. doi: 10.1113/jphysiol.2009.181834
72. Sparling PB, Giuffrida A, Piomelli D, Rosskopf L, Dietrich A. Exercise activates the endocannabinoid system. Neuroreport. (2003) 14:2209–11. doi: 10.1097/00001756-200312020-00015
73. Dietrich A, McDaniel WF. Endocannabinoids and exercise. Br J Sports Med. (2004) 38:536–41. doi: 10.1136/bjsm.2004.011718
74. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. (2006) 59:1116–27. doi: 10.1016/j.biopsych.2006.02.013
75. Dishman RK, Berthoud H-R, Booth FW, Cotman CW, Edgerton VR, Fleshner MR, et al. Neurobiology of exercise. Obesity. (2006) 14:345–56. doi: 10.1038/oby.2006.46
76. Sarbadhikari SN, Saha AK. Moderate exercise and chronic stress produce counteractive effects on different areas of the brain by acting through various neurotransmitter receptor subtypes: a hypothesis. Theoret Biol Med Model. (2006) 3:33. doi: 10.1186/1742-4682-3-33
77. Deslandes A, Moraes H, Ferreira C, Veiga H, Silveira H, Mouta R, et al. Exercise and mental health: many reasons to move. Neuropsychobiology. (2009) 59:191–8. doi: 10.1159/000223730
78. Orczyk JJ, Garraghty PE. Reconciling homeostatic and use-dependent plasticity in the context of somatosensory deprivation. Neural Plast. (2015) 2015:290819. doi: 10.1155/2015/290819
79. Desai NS. Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J Physiol Paris. (2003) 97:391–402. doi: 10.1016/j.jphysparis.2004.01.005
80. Carter JD, Frampton CM, Mulder RT, Luty SE, Joyce PR. The relationship of demographic, clinical, cognitive and personality variables to the discrepancy between self and clinician rated depression. J Affect Disord. (2010) 124:202–6. doi: 10.1016/j.jad.2009.11.011
81. Schneibel R, Brakemeier E-L, Wilbertz G, Dykierek P, Zobel I, Schramm E. Sensitivity to detect change and the correlation of clinical factors with the Hamilton Depression Rating Scale and the Beck Depression Inventory in depressed inpatients. Psychiatry Res. (2012) 198:62–7. doi: 10.1016/j.psychres.2011.11.014
82. Edwards BC, Lambert MJ, Moran PW, McCully T, Smith KC, Ellingson AG. A meta-analytic comparison of the Beck Depression Inventory and the Hamilton Rating Scale for Depression as measures of treatment outcome. Br J Clin Psychol. (1984) 23:93–9. doi: 10.1111/j.2044-8260.1984.tb00632.x
83. Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta-analytic comparison. J Consult Clin Psychol. (1986) 54:54–9. doi: 10.1037/0022-006X.54.1.54
84. Sayer NA, Sackeim HA, Moeller JR, Prudic J, Devanand DP, Coleman EA, et al. The relations between observer-rating and self-report of depressive symptomatology. Psychol Assess. (1993) 5:350–60. doi: 10.1037/1040-3590.5.3.350
85. Prusoff BA, Klerman GL, Paykel ES. Concordance between clinical assessments and patients' self-report in depression. Arch Gen Psychiatry. (1972) 26:546–52. doi: 10.1001/archpsyc.1972.01750240058009
86. Rush AJ, Hiser W, Giles DE. A comparison of self-reported versus clinician-related symptoms in depression. J Clin Psychiatry. (1987) 48:246–8.
87. Rush AJ, Trivedi MH, Carmody TJ, Ibrahim HM, Markowitz JC, Keitner GI, et al. Self-reported depressive symptom measures: sensitivity to detecting change in a randomized, controlled trial of chronically depressed, nonpsychotic outpatients. Neuropsychopharmacology. (2005) 30:405–16. doi: 10.1038/sj.npp.1300614
88. Harvey P-O, Fossati P, Pochon J-B, Levy R, Lebastard G, Lehéricy S, et al. Cognitive control and brain resources in major depression: an fMRI study using the n-back task. Neuroimage. (2005) 26:860–9. doi: 10.1016/j.neuroimage.2005.02.048
89. Dregan A, Gulliford MC. Leisure-time physical activity over the life course and cognitive functioning in late mid-adult years: a cohort-based investigation. Psychol Med. (2013) 43:2447–58. doi: 10.1017/S0033291713000305
90. Cox EP, O'Dwyer N, Cook R, Vetter M, Cheng HL, Rooney K, et al. Relationship between physical activity and cognitive functison in apparently healthy young to middle-aged adults: a systematic review. J Sci Med Sport. (2016) 19:616–28. doi: 10.1016/j.jsams.2015.09.003
Keywords: neuroplasticity and exercise, major depression, paired associative stimulation, transcranial magnetic stimulation, physical activity
Citation: Brüchle W, Schwarzer C, Berns C, Scho S, Schneefeld J, Koester D, Schack T, Schneider U and Rosenkranz K (2021) Physical Activity Reduces Clinical Symptoms and Restores Neuroplasticity in Major Depression. Front. Psychiatry 12:660642. doi: 10.3389/fpsyt.2021.660642
Received: 29 January 2021; Accepted: 18 May 2021;
Published: 09 June 2021.
Edited by:
Kai Wang, Anhui Medical University, ChinaReviewed by:
Jennifer L. Vande Voort, Mayo Clinic, United StatesMing-Kuei Lu, China Medical University, Taiwan
Copyright © 2021 Brüchle, Schwarzer, Berns, Scho, Schneefeld, Koester, Schack, Schneider and Rosenkranz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Rosenkranz, cm9zZW5rcmFuei5rYXJpbkBnbXguZGU=