 Runzhi An1†
Runzhi An1† Yongyong Cao
Yongyong Cao- 1College of Biological, Chemical Science and Engineering, Jiaxing University, Jiaxing, Zhejiang, China
- 2College of Chemical Engineering, State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, Zhejiang University of Technology, Hangzhou, China
Electrocatalytic carbon dioxide reduction (CO2RR) is a relatively feasible method to reduce the atmospheric concentration of CO2. Although a series of metal-based catalysts have gained interest for CO2RR, understanding the structure–activity relationship for Cu-based catalysts remains a great challenge. Herein, three Cu-based catalysts with different sizes and compositions (Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs) were designed to explore this relationship by density functional theory (DFT). The calculation results show a higher degree of CO2 molecule activation on CuNi3@CNTs compared to that on Cu@CNTs and Cu4@CNTs. The methane (CH4) molecule is produced on both Cu@CNTs and CuNi3@CNTs, while carbon monoxide (CO) is synthesized on Cu4@CNTs. The Cu@CNTs showed higher activity for CH4 production with a low overpotential value of 0.36 V compared to CuNi3@CNTs (0.60 V), with *CHO formation considered the potential-determining step (PDS). The overpotential value was only 0.02 V for *CO formation on the Cu4@CNTs, and *COOH formation was the PDS. The limiting potential difference analysis with the hydrogen evolution reaction (HER) indicated that the Cu@CNTs exhibited the highest selectivity of CH4 among the three catalysts. Therefore, the sizes and compositions of Cu-based catalysts greatly influence CO2RR activity and selectivity. This study provides an innovative insight into the theoretical explanation of the origin of the size and composition effects to inform the design of highly efficient electrocatalysts.
1 Introduction
With the rapid development of the global society, the increasing emissions of carbon dioxide (CO2) from the burning of fossil fuels have led to serious environmental and energy crises (Jiao, 2019; Cao et al., 2021; Wang et al., 2022). The electrocatalytic CO2 reduction reaction (CO2RR) provides a viable alternative to convert CO2 into value-added chemical products such as carbon monoxide (CO), methane (CH4), methanol (CH3OH), ethylene (CH2CH2), and ethanol (CH3CH2OH) (Xue et al., 2020; Zhang, 2022; Zhou et al., 2022). Therefore, this is a promising approach to alleviate the global demand for fossil fuels and greenhouse effects. In addition, the advantages of CO2RR include 1) the feasibility of controlling the process by adjusting the potential and related conditions; 2) electricity consumption from sustainable sources such as the Sun and wind; 3) electrochemical reaction systems that are compact, modular, and easily scaled-up (Gong et al., 2019; Jiao et al., 2019; Bai et al., 2020; Han et al., 2020; He et al., 2020). However, there remain several challenges to the practical application of CO2RR, such as slow reaction kinetics, poor efficiency, and low product selectivity due to the strong C=O double bond (806 kJ.mol-1) in CO2 molecules (Alvarez et al., 2017). Therefore, highly active electrocatalysts must be designed to improve the sluggish kinetics and energy conversion efficiency.
A recent study demonstrated the high activity and selectivity of transition metal catalysts in the conversion of CO2 to CO, CH4, or CH3OH. Among these, Cu-based catalysts have been widely applied to CO2RR due to their excellent conductivity and availability (Li et al., 2017). The catalyst size and composition structure are usually regulated to tune the catalytic activity and selectivity of metal nanoparticles. Varying particle size has size effects in CO2RR. Studies have reported on the CO2RR trends over spherical Cu nanoparticles ranging in size from 2 to 15 nm (Shen et al., 2021). A decrease in Cu nanoparticle size leads to a significant increase in low coordination sites, resulting in strong CO2 adsorption and hydrogenation. A series of different sizes of Ni-based catalysts (Ni@CNT, Ni4@CNT, and Ni (110)) were also designed to study their size effects in CO2RR. Density functional theory (DFT) calculations revealed that the Ni@CNT catalyst is much more conducive to the conversion of CO2 to CO, while CH4 molecules were produced on Ni@CNT and Ni (100) with increasing Ni size (Cao et al., 2020a). Therefore, the catalyst size may affect CO2 activation and final products.
The alloy effect of the metal catalyst is another key factor in catalytic reactions. The electronic structure of metal catalysts can be controlled by alloying with other metal atoms, which is an obvious characteristic of Cu-based catalysts (Weng et al., 2018; Meng et al., 2022). A Cu–Ni alloy catalyst embedded in the N-doped carbon framework (CuNi/NC) synthesized for CO2RR showed excellent activity and selectivity for electrocatalytic CO2RR to CO, with a high Faradaic efficiency of 99.7% and a potential of −0.6 V (Zhang, 2019; Zhong et al., 2020). Additionally, a non-noble metal CuSn alloy with a different Cu–Sn composition demonstrated a completely different distribution of products for CO2RR, including CO and formate (Li et al., 2020a). Tuning the surface composition of Cu-based catalysts via alloying with other metals could directly modify the absorbed state of the intermediate species on their active sites and determine the final product during the CO2RR. Therefore, the introduction of other metals is an effective strategy to adjust the electronic structure and improve CO2RR activity and selectivity.
This study designed different sizes and compositions of Cu-based catalysts (Cu@CNT, Cu4@CNT, and CuNi3@CNT) to explore the relationship between the structure of catalysts and activity for CO2RR based on DFT calculations. CO2 molecule adsorption and activation are essential for the following hydrogenation steps. Therefore, different CO2 adsorption configurations were optimized to explore the nature of CO2 activation on the three catalysts by analyzing their geometric and electronic properties. The mechanisms of CO2RR on different Cu-based catalysts were also assessed, and a plot was constructed to compare the selectivity of CO2RR among Cu@CNT, Cu4@CNT, and CuNi3@CNT.
2 Computational models and methods
All calculations were performed using the Vienna Ab initio Simulation Package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmiiller, 1996), including the Perdew–Burke–Ernzerhof (PBE) functions in the generalized gradient approximation (GGA) to describe electron exchange and correction effect (Blochl, 1994; Perdew et al., 1996; Kresse and Joubert, 1999). The cutoff energy for the plane wave basis was set to 520.0 eV. The Brillouin zone was sampled at 4 × 4 × 1 k points for all calculation models using the Monkhorst–Pack grid. The convergence criteria for energy for geometry optimization were set to 1.0 × 10−5 eV. The vacuum thickness was 15.0 Å to prevent interlayer interactions. van der Waals correction was implemented using the Grimme’s DFT-D3 method (Krieg, 2010).
The adsorption energy (
where
The binding energy (
where
The computational hydrogen electrode (CHE) mode was proposed by Nørskov et al (Nadolska et al., 2021), in which the changes in Gibbs free energies of CO2RR are calculated for every elementary step. The reference electrode is the reversible hydrogen electrode (RHE). Therefore, the chemical potential (μ) of the proton–electron pair is defined as follows:
The change in Gibbs free energy (
where
where e is the fundamental charge transfer, n is the proton–electron logarithm transferred, and U is the electrode applied potential relative to the RHE.
The overpotential (η) of CO2RR is defined as follows:
where
Carbon nanotubes (CNTs) are promising substitutes that have been widely applied in electrocatalysts for their high electrical conductivity, thermal stability, and abundant catalytic active sites (Grimme et al., 2010; Xiong et al., 2019; Bulmer et al., 2021; Cai et al., 2021). As an N-doped CNT (Figure 1A) may further increase interaction between CNT and the active center (Park et al., 2020; Nguyen and Shim, 2021), a single Cu atom, Cu4 cluster, and CuNi3 cluster were doped on the N-doped CNTs through the carbon vacancy defect to explore the effects on the CO2RR sizes and composition (Figures 1B–D). As shown in Figure 1B, the single Cu atom is connected to two N atoms and one C atom to form a pyramid-like structure with Eb values of −3.21 eV. The bond lengths of Cu–N and Cu–C are 2.11 Å and 1.80 Å, respectively. When the Cu4 cluster is doped on the N-doped carbon nanotubes, the three Cu atoms at the bottom interact with two N atoms and two C atoms (Figure 1C), resulting in a strong interaction with Eb values of −4.87 eV. The Cu–N, Cu–C, and Cu–Cu bond lengths are 2.05 Å, 2.03 Å, and 2.42 Å, respectively. In addition, CuNi3@CNTs are used to study the compositional effect of Cu-based catalysts for CO2RR by replacing three Cu atoms in Cu4@CNTs with three Ni atoms connected to the adjacent N atoms and C atoms on the bottom of the tetrahedron, respectively (Figure 1D). The binding energy of CuNi3@CNTs is −5.12 eV. The large binding energies indicate that the three different Cu-based catalysts all possess high thermodynamic stability, ensuring their long-term use.
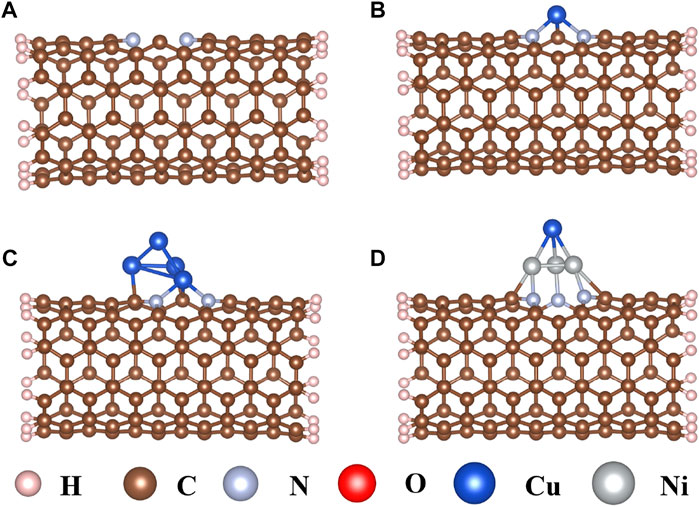
FIGURE 1. Optimized structures of (A) nitrogen-doped carbon nanotubes, (B) single Cu atom (Cu@CNTs), (C) Cu4 cluster (Cu4@CNTs), and (D) CuNi3 (CuNi3@CNTs) supported on the nitrogen-doped carbon nanotubes.
3 Results and discussion
3.1 CO2 activation on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs
CO2 adsorption and activation are crucial steps for subsequent hydrogenation in CO2RR (Jiao et al., 2019). To investigate the behaviors of CO2 adsorption, we explored the geometric and electronic structures of the adsorbed CO2 molecules. As shown in Figure 2A, the CO2 molecule is adsorbed on the single Cu atom site with an Eads value of −0.50 eV. The C=O bond length in CO2 is also slightly elongated from 1.18 to 1.25 Å. With increasing Cu active center size (from a single Cu atom to the Cu4 cluster), the Eads value of CO2 decreases to −0.19 eV. The CO2 prefers to adsorb on the edge site of the Cu4 cluster (Figure 2B) compared to the top site, with an Eads value of 0.15 eV. The adsorption site and configuration of CO2 on the CuNi3@CNTs (Figure 2C) are the same as that on the Cu4@CNTs, although the Eads value increases to −0.82 eV. The C=O bond length also increases to 1.26 Å. The top adsorption site of CuNi3@CNTs has an adsorption energy of 0.35 eV (Supplementary Figure S1). Thus, the top adsorption site was used for CO2 molecule adsorption and activation on both the Cu4@CNTs and CuNi3@CNTs. The bond angles of O−C−O in the CO2 molecule changed from 180.00° to 146.74°, 140.77°, and 139.23° on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs, respectively (Table 1). Thus, Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs all significantly activate CO2, and the size and composition effect greatly influence the degree of CO2 activation, which may further affect the activity and selectivity of CO2 hydrogenation.
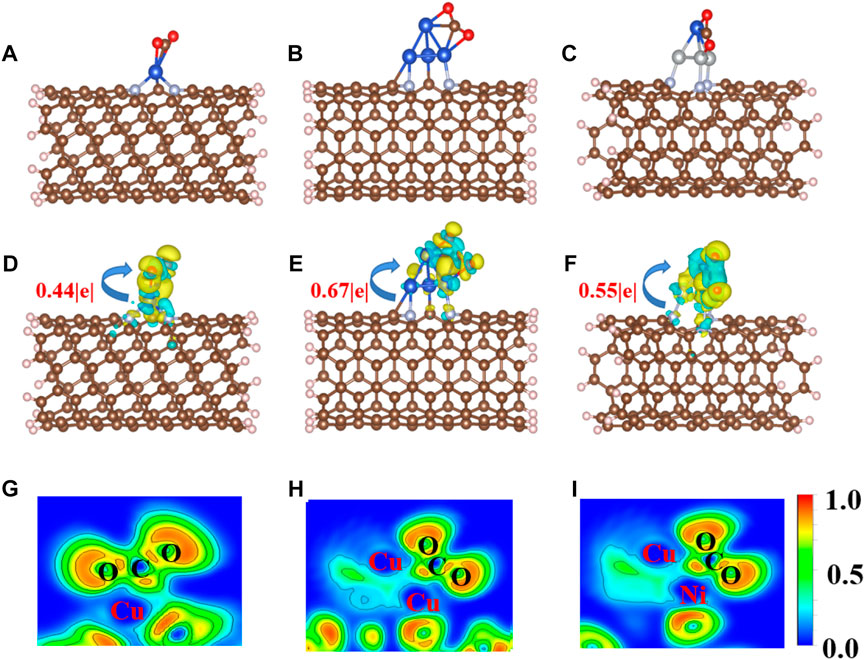
FIGURE 2. (A–C) Optimized adsorption configurations of CO2 on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs. (D–F) Calculated charge density difference (CDD) and (G–I) the electronic local function diagram (ELF) of CO2 adsorption. The charge depletion and accumulation are depicted in blue and yellow, respectively. The equivalent face value is 0.005 e/Å–3.
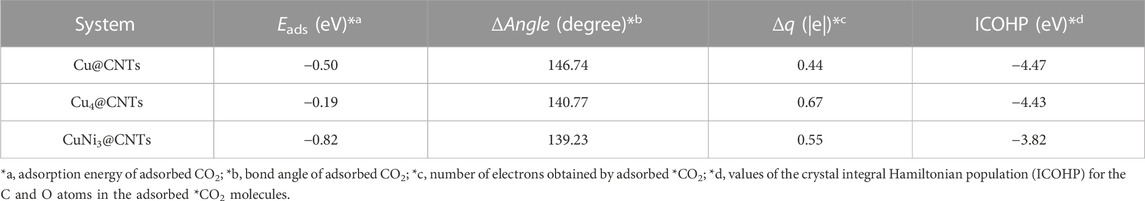
TABLE 1. Geometric and electronic structure parameters of adsorbed *CO2 on the three Cu-based catalysts.
To further investigate the activation mechanism of the CO2 molecules, the Bader charge, charge density difference (CDD), electronic local function diagram (ELF), projected density of state (PDOS), and integrated crystal orbital Hamilton population (ICOHP) with CO2 adsorption were investigated. As shown in Figures 2D–F, significant charge transfers occur between C=O double bonds and metal Cu and Ni atoms connected to CO2. In detail, the electron densities of the O atom, and between the C, O atoms and Cu, Ni atoms increase, while the electron density of the C=O double bond decreases significantly. In addition, the ELF map is used to further verify the bonding properties of C=O double bonds (Peterson et al., 2010; Reske et al., 2014; Wang et al., 2022). The results of the ELF calculation (Figures 2G–I) showed that the C=O double bond is weak, consistent with the results of the CDD analysis. The local electron densities of the Cu-C and Cu-O bonds range from 0.5 to 1.0 in the whole ELF map, which further verifies the metal coordination bond formation between Cu and Ni atoms and between the C and O atoms. The adsorbed *CO2 molecule gains 0.44|e|, 0.67|e|, and 0.55|e| from the Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs, respectively, based on the Bader charge analysis, resulting in the bending of the bond angles of the adsorbed CO2 molecules.
The PDOS of CO2 adsorption are analyzed to further reveal interactions between Cu atoms and CO2 (Figures 3A–C). The Cu-3d and Ni-3d orbitals play key roles in CO2 adsorption and activation. The C-2p, O-2p, and Cu-3d orbitals have significant peak overlaps near the Fermi level, indicating strong interactions between C, O, and Cu atoms during CO2 adsorption on Cu@CNTs and Cu4@CNTs (Figures 3A,B). With the introduction of the Ni atom, the Cu-3d and Ni-3d orbitals show hybridization, illustrating the strong interaction between the Cu and Ni atoms. Meanwhile, the broadly overlapped regions by C-2p, O-2p, Cu-3d, and Ni-3d hybrids also appear near-Fermi level, with strong peak intensities (Figure 3C). Based on the CDD and PDOS results, the mechanism of CO2 activation can be summarized as follows: anti-bonding orbitals of CO2 accept 3d-electrons from the Cu-3d and Ni-3d orbitals; meanwhile, the π electrons from the CO2 bonding orbitals revert to empty Cu-3d and Ni-3d orbitals. The activation strength of the C=O double bond of the adsorbed CO2 molecule is quantitatively studied by calculating the ICOHP values, in which lower values indicate stronger bond strength (Li et al., 2021; Song et al., 2021). As seen in Figures 3D–F, the C=O double bond anti-bonding states move down relative to the Fermi level after CO2 adsorption. Therefore, the strength of the C=O double bond significantly decreases after CO2 adsorption. The ICOHP values of the C=O double bond are −4.47, −4.43, and −3.82 eV on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs, respectively. The weaker the C=O double bond, the more efficient the CO2 activation on the catalyst surface. This result demonstrates the higher degree of CO2 activation on the CuNi3@CNTs compared to that of the Cu@CNTs and Cu4@CNTs, consistent with the results of Bader charge, CDD, and adsorption energy calculations.
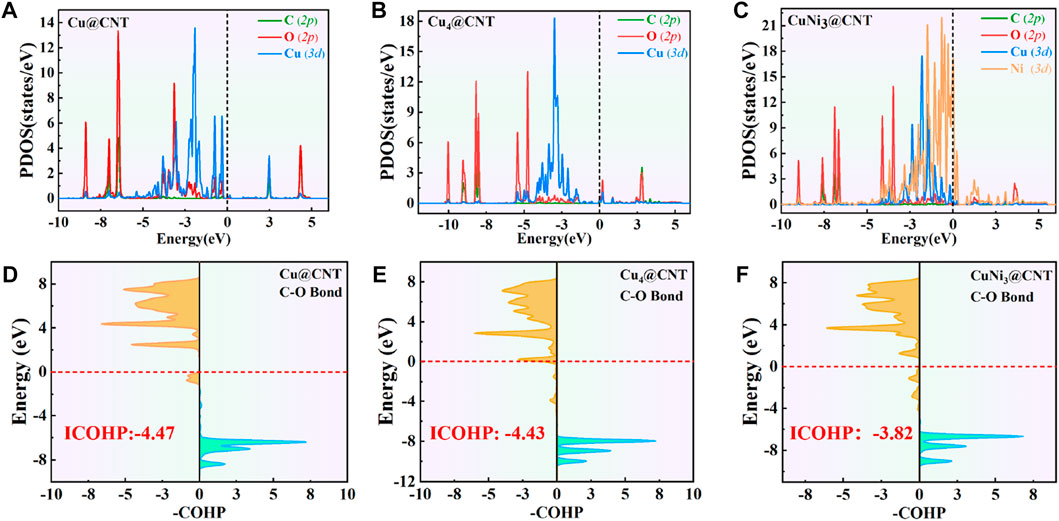
FIGURE 3. (A–C) Projected density of state (PDOS) plots. (D–F) Projected crystal orbital Hamilton population (pCOHP) of CO2 adsorption on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs. The dotted line represents the Fermi energy level.
3.2 Mechanisms of CO2RR on Cu-based catalysts
CO2RR is a complex reaction involving multi-electron (two, six, or eight-electron) reaction pathways (Jiwanti and Einaga, 2019; Niu et al., 2021; Yang et al., 2022). The overall reaction equation can be expressed as follows:
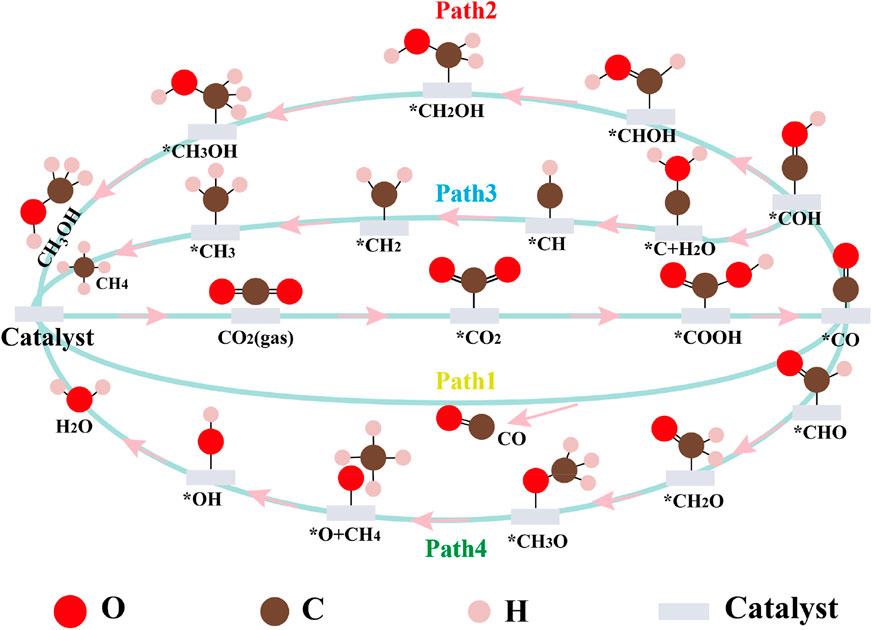
SCHEME 1. Proposed mechanisms of CO2RR to C1 products. *represents the catalytic surface.
3.3 Reaction mechanism for CO2RR to CH4 products on Cu@CNTs
To illustrate the catalytic activity and selectivity of CO2RR on Cu@CNTs, the free energy barrier diagram and corresponding intermediate configurations are shown in Figures 4, 5, respectively. Figure 4 shows that the absorbed CO2 molecule is hydrogenated to form stable *COOH with a change of the free energy value (ΔG) of 0.47 eV. In the second hydrogenation step, *COOH can react with the (H+/e−) pair to form *CO and H2O, with an exothermic energy release of –0.27 eV. The adsorbed *CO is further protonated to form *CHO (ΔG = 0.61 eV) and *COH (ΔG = 1.49 eV) intermediates, or may be directly desorbed with a ΔG value of 0.91 eV. Obviously, the formation of *CHO is more favorable than that of *CHO and *CO desorption. When the *COH species is formed, the C atom of *COH accepts three H atoms to form *CH3OH with downhill free energies of −0.47, −1.18, and −0.08 eV. *COH is also directly dissociated into *C species (ΔG = 0.60 eV), and *CH4 is formed by four hydrogenation steps of the *C intermediate. The formation of *CH and *CH2 is slightly thermodynamically favorable, with ΔG values of −1.22 and −1.83 eV, respectively. However, the formation of *CH3 has an endothermic energy of 0.78 eV. It is difficult to form the CH4 molecule in Path 3 due to the high hydrogenation barrier of the formation of *COH species for the whole reaction. In Path 4, *CHO intermediate accepts an (H+/e−) pair to form *CH2O with a ΔG value of −1.15 eV. Then, the hydrogenation of *CH2O to *CH3O is uphill by 0.26 eV. *CH3O is further hydrogenated to form *O and a CH4 molecule with a ΔG value of −0.75 eV. The leaving *O atom is continuously hydrogenated to form H2O. Thus, the conversion of CO2 to CH4 following Path 4 with an overpotential value of 0.37 V is easier than that of CO and CH3OH in all four paths, and the potential-determining step (PDS) is the *CHO formation on the Cu@CNTs. Wang et al. prepared three Cu-based catalysts for CO2RR (Weng et al., 2018). Among them, the single-atom Cu phthalocyanine catalyst exhibited the highest activity to produce CH4 with a Faradaic efficiency of 66% and a partial current density of 13 mA cm−2 at a potential of −1.06 V versus RHE, findings that are consistent with the calculation results of CO2RR to CH4 on the Cu-based single-atom catalysts in the present study.
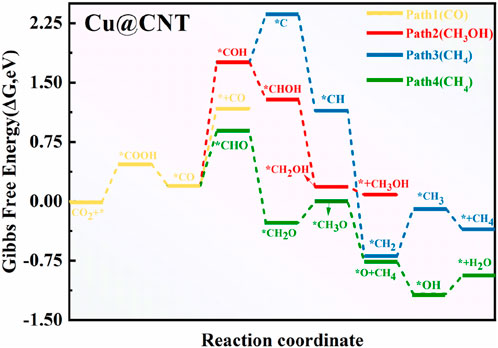
FIGURE 4. Free energy diagram of CO2RR on Cu@CNTs.
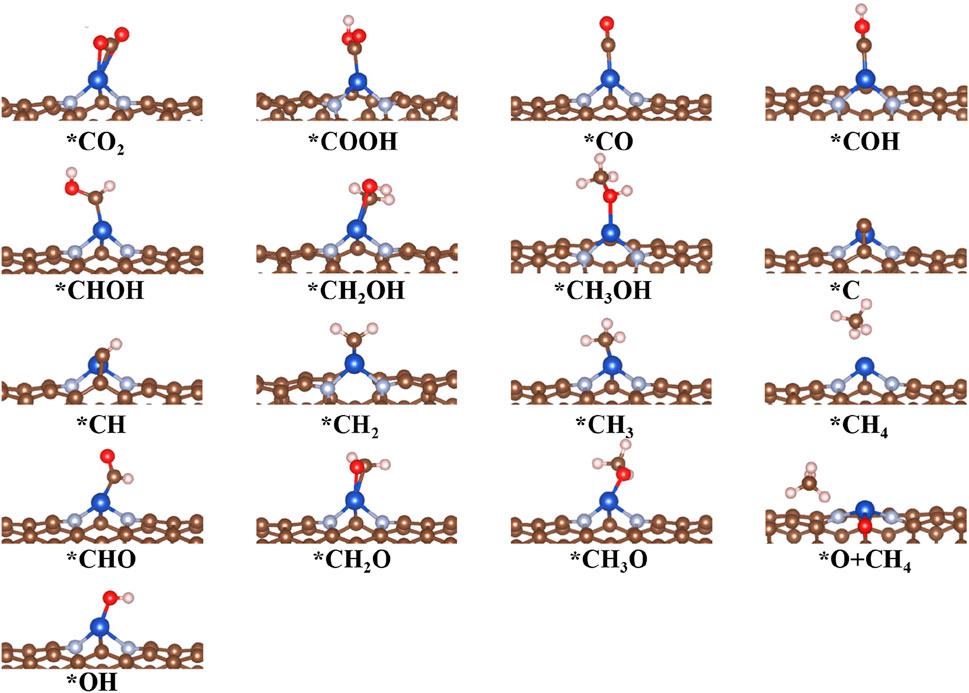
FIGURE 5. Optimized structures of intermediates for CO2RR on Cu@CNT surfaces.
3.4 Reaction mechanism for CO2RR to CO products on Cu4@CNTs
To explore the different size effects on CO2RR activity on Cu-based catalysts, the free energy profiles and optimized geometric structures for CO2RR on the Cu4@CNTs are shown in Figures 6, 7, respectively. The Cu4 clusters embedded in the CNTs catalyst provided multiple active sites for the activation of CO2 molecules compared to Cu@CNTs and further promoted hydrogenation. The adsorbed *CO2 accepts two (H+/e−) pairs to successively convert the key intermediates *COOH and *CO with an entirely endothermic process of 0.54 eV and 0.16 eV, respectively. CO desorption is a thermodynamically favorable process with a low energy of −0.01 eV compared to *CO hydrogenation to *COH (ΔG = 1.32 eV) and *CHO (ΔG = 0.62 eV). In the subsequent hydrogenation steps, the (H+/e−) pair consecutively attacks the C atom of the *CO intermediates to form *CHO, *CH2O, *CH3O, and *O+CH4 species, with PDS, the process of *O+ CH4 intermediate formation, with ΔG values of 1.32 eV in Path 4. After *COH formation, *COH hydrogen dissociates to form *C with an uphill energy of 0.72 eV. Then, the *C species is further hydrogenated to form *CH, *CH2, *CH3, and *CH4 (Path 3). The corresponding ΔG values are −0.21, −0.74, −1.11, and −0.44 eV, respectively. In Path 2, *COH is hydrogenated to form *CHOH, *CH2OH, and *CH3OH species, with ΔG values of −0.17, −0.43, and −0.86 eV. Comparison of the ΔG values of PDS in all four paths shows that the reactions are more likely to follow Path 1 and CO is produced on the Cu4@CNTs with an extremely low overpotential value of 0.02 V. More importantly, the desorption energy of CO is greatly reduced with increasing Cu size, which is conducive to the production of CO and avoids poisoning the active sites on the catalyst surface. In addition, the reported atom-pair catalyst with stable Cu10–Cu1x+ pair structures also shows high selectivity and activity for CO2RR to CO (Li et al., 2020b). Therefore, our calculation results of CO2RR on cluster catalysts are consistent with experimental results reported previously. These results demonstrate that CO2RR reduction product changes from CH4 to CO when the size of Cu–based catalysts changes from a single atom to a Cu4 cluster. Therefore, designing multiple active sites to change the final reduction products may be a universal strategy for catalyst development.
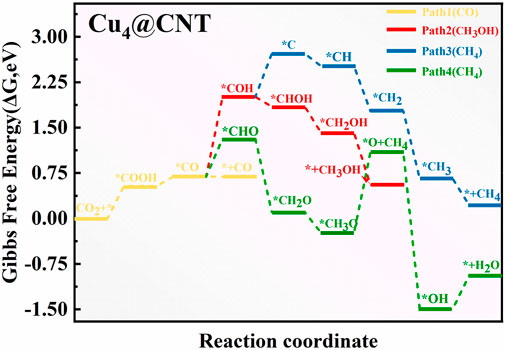
FIGURE 6. ree energy diagram of CO2RR on Cu4@CNTs.
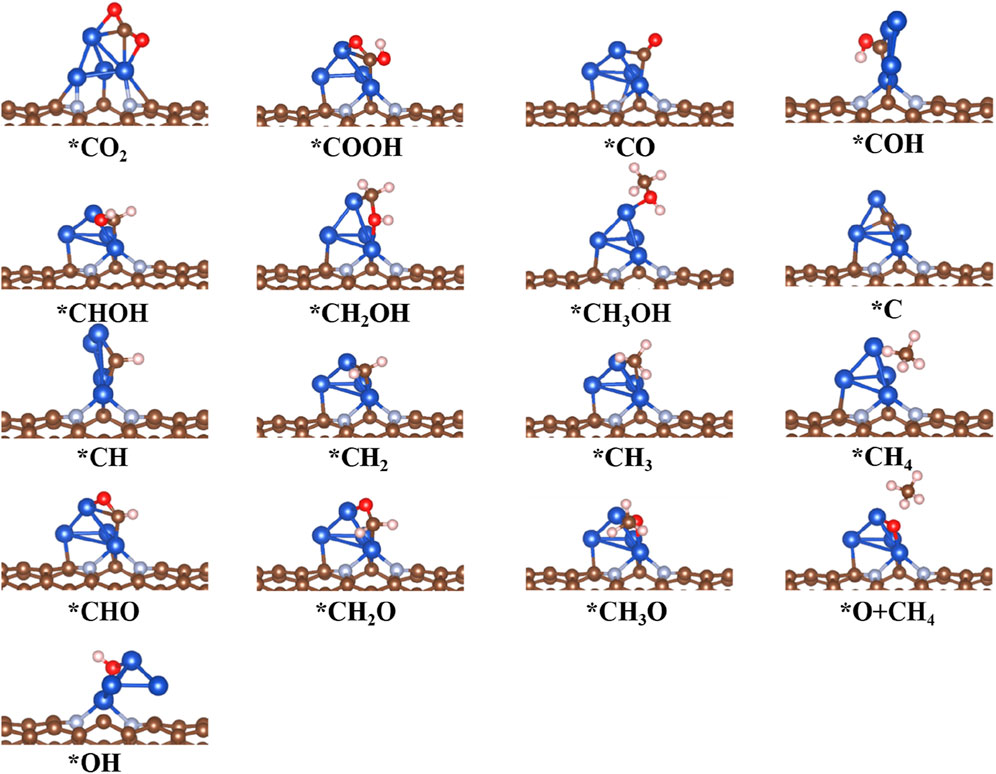
FIGURE 7. Optimized structure of intermediates for CO2RR on Cu4@CNT surfaces.
3.5 Reaction mechanism for CO2RR to CH4 products on CuNi3@CNTs
To explore the composition effect on the mechanism of CO2RR, the synthesis paths of CO2 into CH4, CO, and CH3OH products were determined on CuNi3@CNTs. The free energy and geometric structure diagrams are shown in Figures 8, 9, respectively. The *COOH intermediate is obtained in the first step of hydrogenation of the adsorbed *CO2 molecule, with a downhill energy of −0.44 eV on the CuNi3@CNTs catalyst, which differs from that on Cu@CNTs and Cu4@CNTs. According to the aforementioned charge transfer analysis, the adsorbed *CO2 molecule obtains more electrons from CuNi3@CNTs compared to Cu@CNTs and Cu4@CNTs, and the electrons of Ni-3d orbitals are partly transferred to the π anti-bonding orbitals of CO2. These strong electron transfers lead to the spontaneous formation of a *COOH intermediate. The production of *CO occurs through *COOH hydrogenation to *CO and H2O molecules. However, *CO desorption requires a large desorption energy of 1.57 eV (Path 1). Thus, *CO is further hydrogenated to *COH (ΔG = 1.83 eV) or *CHO (ΔG = 0.84 eV). In the subsequent hydrogenation steps, *CHO is hydrogenated in two steps to form *CH2O and *CH3O in Path 4. Then, the (H+/e−) pair attacks the C atom in *CH3O, resulting in CH4 formation. After release of the CH4 molecule, one *O atom remains above the bridge site of the Cu–Ni atoms (Figure 9) and is subsequently further hydrogenated into H2O molecules and released. The reactions are more likely to follow Path 2 to form CH3OH than Path 3 to form CH4 based on a comparison of the ΔG values of producing possible species (*CHOH, *CH2OH; *C, *CH2, and *CH3). As discussed previously, CO2RR is more likely to follow Path 4 to form CH4 on CuNi3@CNTs, with an overpotential value of 0.60 V and *COH formation being the PDS. For the Cu–Ni alloy catalysts, the major products of CO2RR change from ethylene (C2H4) and C2H5OH to CH4 with increasing Ni content (Park et al., 2021). These selectivity challenges are attributed to the increased adsorption energies of *CO on the Ni surface based on the results of pressure-dependent experiments and DFT calculations. We observed similar findings in our calculations. The Eads values of *CO on the CuNi3@CNTs are much higher than those on the Cu@CNTs and Cu4@CNTs. Thus, the introduction of Ni atoms in the CuNi3@CNTs changes the selectivity of CO2RR to produce CH4 compared to that on the Cu4@CNTs, which produce CO.
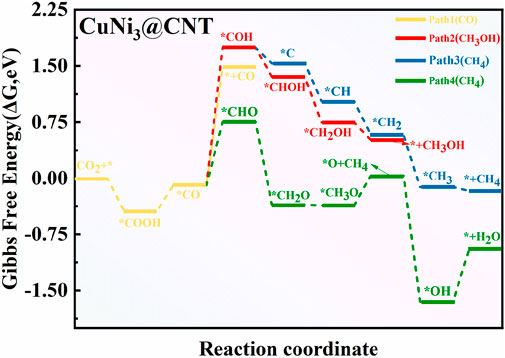
FIGURE 8. Free energy diagram of CO2RR on CuNi3@CNTs.
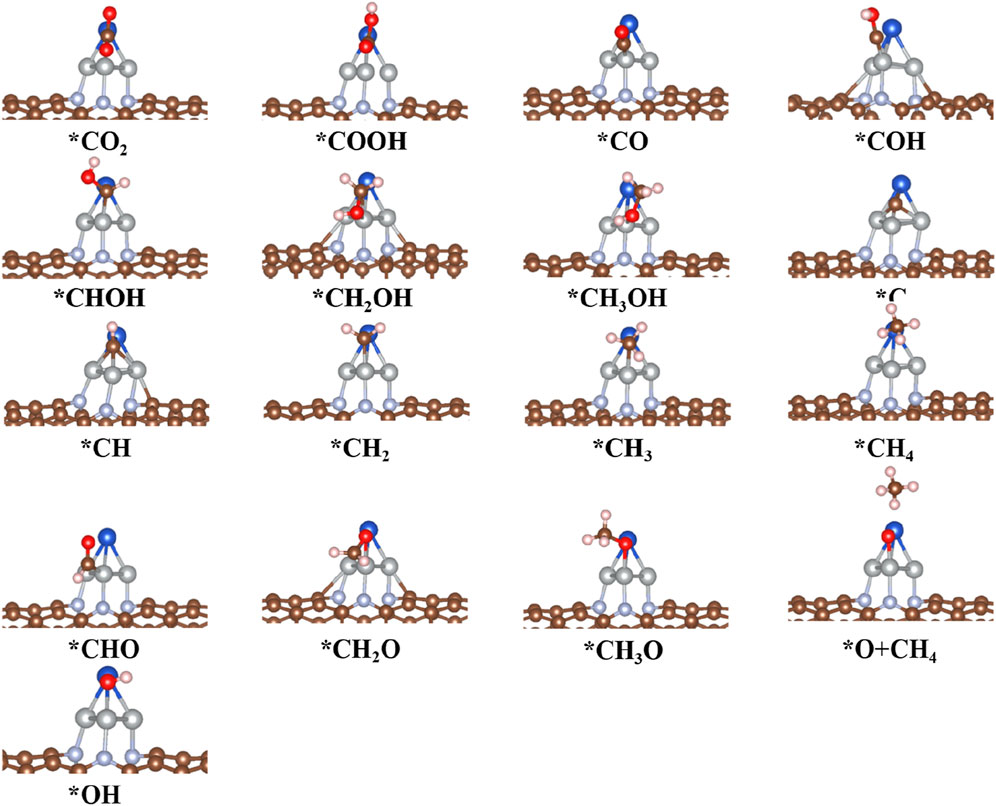
FIGURE 9. Optimized structure of intermediates for CO2RR on the CuNi3@CNT surfaces.
3.6 Competitive relationship of CO2RR vs. hydrogen evolution reaction (HER)
The hydrogen evolution reaction (HER) is usually considered a competitive reaction during CO2RR. Therefore, the selectivity and activity of CO2RR are tightly related to HER performance (Xue et al., 2020; Zhao and Liu, 2020). Figure 10A shows the free energies of HER for Cu@CNTs, Cu4@CNTs, and CuNi3@CNs. HER occurs on three catalysts with ΔG values of −1.12, 0.24, and 0.57 eV, respectively. The difference in limiting potentials for CO2RR and HER on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs were also calculated to further understand the selectivity of the catalyst for CH4 or CO formation. The optimized configurations of *H adsorption are shown in Supplementary Figure S3. The higher the UL (CO2)–UL (H2) difference, the higher is the selectivity for CO2RR over HER (Shen et al., 2021; Wei et al., 2021). As shown in Figure 10B, the selectivity toward CO2RR decreases in the order of Cu@CNTs (CH4 product) > CuNi3@CNTs (CH4 product) > Cu4@CNTs (CO product). The Cu@CNTs have only one adsorption site for the intermediates, while the CuNi3@CNTs and Cu4@CNTs have multiple adsorption sites (top, edge, and hollow sites). Thus, smaller-sized Cu@CNTs show the highest CH4 selectivity among the three catalyst types. However, the large size of the bulk Ni (110) facet shows the highest selectivity for CH4 compared to Ni@CNTs and Ni4@CNTs among the Ni-based catalysts (Cao, 2020b). In addition, the selectivity of CH4 on the CuNi3@CNTs is higher than that for CO on the Cu4@ CNTs. Therefore, the sizes and composition of Cu-based catalysts have a significant role in CO2RR selectivity.
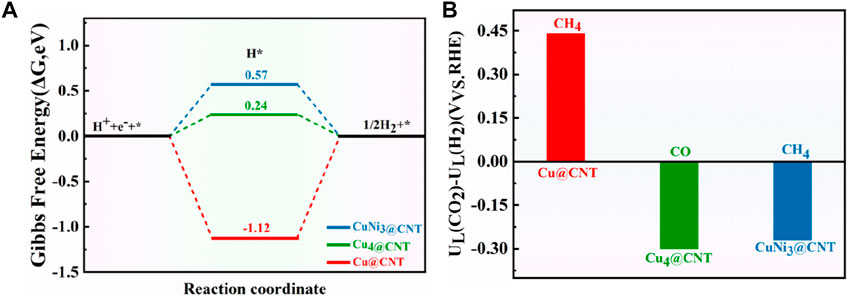
FIGURE 10. (A) Free energy diagram of the HER on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs. (B) Differences in limiting potentials for CO2RR and HER on Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs.
4 Conclusion
In summary, the size and composition effect for the activity and selectivity of CO2RR were explored using DFT. Three types of Cu-based catalysts (Cu@CNTs, Cu4@CNTs, and CuNi3@CNTs) were designed to achieve highly efficient CO2 reduction. The CuNi3@CNTs show the largest CO2 adsorption energy (−0.82 eV) due to CO2 molecule adsorption on the edge site between the Ni and Cu atoms. This promotes higher charge transfer to CO2 molecules and weakens the C=O double bond of CO2. With a size increase from single-atom Cu to the Cu4 cluster, the CO2RR product changes from CH4 to CO. More importantly, Cu4@CNTs show an extremely low overpotential of 0.02 V for CO formation, while Cu@CNTs exhibit the highest selectivity for CH4 formation among the three catalysts. When the Ni atoms are introduced in the Cu4 cluster, the CO2RR selectivity is improved compared to HER. The activity and selectivity of the Cu-based catalysts for the CO2RR depended strongly on the size and composition. Our results provide new insight into understanding the size and alloy effect of CO2RR on Cu-based catalysts.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work is financially supported by the National Natural Science Foundation of China (grant no. 22108093), the National Key Research and Development Program of China (grant no. 2018YFB1502900), the Natural Science Foundation of Zhejiang Province (grant nos. LY19B060006 and LQ20E030016), and the Innovation Jiaxxing Elite Leadership Plan and Technology Development Project of Jiaxing University (70518040).
Acknowledgments
The authors also acknowledge support from the G60 STI Valley Industry & Innovation Institute, Jiaxing University, and appreciate the help provided by J. H. Chen
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1141453/full#supplementary-material
References
Alvarez, A., Borges, M., Corral-Perez, J. J., Olcina, J. G., Urakawa, A., Cornu, D., et al. (2017). CO2 activation over catalytic surfaces. Chemphyschem 18, 3135–3141. doi:10.1002/cphc.201700782
Bai, X., Li, Q., Shi, L., Niu, X., Ling, C., and Wang, J. (2020). Hybrid Cu(0) and Cu(x) as atomic interfaces promote high-selectivity conversion of CO2 to C2H5OH at low potential. Small 16, e1901981. doi:10.1002/smll.201901981
Blochl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Bulmer, J. S., Kaniyoor, A., and Elliott, J. A. (2021). A meta-analysis of conductive and strong carbon nanotube materials. Adv. Mater 33, e2008432. doi:10.1002/adma.202008432
Cai, Y., Fu, J., Zhou, Y., Chang, Y. C., Min, Q., Zhu, J. J., et al. (2021). Insights on forming N,O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 12, 586. doi:10.1038/s41467-020-20769-x
Cao, C., Zhao, C., Fang, Q., ZhongWang, J., Zhuang, G., Deng, S., et al. (2020b). Hydrogen peroxide electrochemical synthesis on hybrid double-atoms (Pd-Cu) doped N vacancy g-C3N4: A novel design strategy for electrocatalysts screening. J. Mat. Chem. A 8, 2672–2683. doi:10.1039/C9TA12468C
Cao, C., Ma, D. D., Gu, J. F., Xie, X., Zeng, G., Li, X., et al. (2020a). Metal-organic layers leading to atomically thin bismuthene for efficient carbon dioxide electroreduction to liquid fuel. Angew. Chem. Int. Ed. Engl. 59, 15014–15020. doi:10.1002/anie.202005577
Cao, Y., Zhao, J., Zhong, X., Yao, Z., Zhuang, G., Wang, J., et al. (2021). Building highly active hybrid double–atom sites in C2N for enhanced electrocatalytic hydrogen peroxide synthesis. Green Energy Environ. 6, 846–857. doi:10.1016/j.gee.2020.12.006
Gong, Q., Ding, P., Xu, M., Zhou, W., Li, Y., Deng, J., et al. (2019). Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction. Nat. Commun. 10, 2807. doi:10.1038/s41467-019-10819-4
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Han, L., Song, S., Liu, M., Wu, Q., Luo, J., Xin, H. L., et al. (2020). Stable and efficient single-atom Zn catalyst for CO2 reduction to CH4. J. Am. Chem. Soc. 142, 12563–12567. doi:10.1021/jacs.9b12111
He, T., Zhang, L., Kour, G., and Du, A. (2020). Electrochemical reduction of carbon dioxide on precise number of Fe atoms anchored graphdiyne. J. CO2 Util. 37, 272–277. doi:10.1016/j.jcou.2019.12.025
Jiao, J. Q., Lin, R., Liu, S. J., Cheong, W. C., Li, Y. D., Chen, Z., et al. (2019). Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat. Chem. 11, 222–228. doi:10.1038/s41557-018-0201-x
Jiwanti, P. K., and Einaga, Y. (2019). Electrochemical reduction of CO2 using palladium modified boron-doped diamond electrodes: Enhancing the production of CO. Phys. Chem. Chem. Phys. 21, 15297–15301. doi:10.1039/C9CP01409H
Kresse, G., and Furthmiiller, J. (1996). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. doi:10.1103/PhysRevB.47.558
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775. doi:10.1103/PhysRevB.59.1758
Li, Q., Ouyang, Y., Lu, S., BaiZhang, X. Y., Shi, L., Ling, C., et al. (2020a). Perspective on theoretical methods and modeling relating to electro-catalysis processes. Chem. Commun. 56, 9937–9949. doi:10.1039/D0CC02998J
Li, Q., Wang, H., Luo, W., Sherrell, P. C., Chen, J., and Yang, J. (2020b). Heterogeneous single-atom catalysts for electrochemical CO2 reduction reaction. Adv. Mater 32, e2001848. doi:10.1002/adma.202001848
Li, S., Nagarajan, A. V., Alfonso, D. R., Sun, M., Kauffman, D. R., Mpourmpakis, G., et al. (2021). Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters. Angew. Chem. Int. Ed. Engl. 60, 6351–6356. doi:10.1002/anie.202016129
Li, X., Bi, W., Chen, M., Sun, Y., Ju, H., Yan, W., et al. (2017). Exclusive Ni-N4 sites realize near-unity CO selectivity for electrochemical CO2 reduction. J. Am. Chem. Soc. 139, 14889–14892. doi:10.1021/jacs.7b09074
Meng, Y., Xu, Z., Shen, Z., Xia, Q., Cao, Y., Wang, Y., et al. (2022). Understanding the water molecule effect in metalfree B-based electrocatalysts for electrochemical CO2 reduction. J. Mat. Chem. A 10, 6508–6522. doi:10.1039/D1TA10127G
Nadolska, M., Przesniak-Welenc, M., Lapinski, M., and Sadowska, K. (2021). Synthesis of phosphonated carbon nanotubes: New insight into carbon nanotubes functionalization. Materials 14, 2726. doi:10.3390/ma14112726
Nguyen, A. T. N., and Shim, J. H. (2021). All carbon hybrid N-doped carbon dots/carbon nanotube structures as an efficient catalyst for the oxygen reduction reaction. RSC Adv. 11, 12520–12530. doi:10.1039/D1RA01197A
Niu, H., Zhang, Z., Wang, X., Wan, X., Kuai, C., and Guo, Y. (2021). A feasible strategy for identifying single-atom catalysts toward electrochemical NO-to-NH3 conversion. Small 17, e2102396. doi:10.1002/smll.202102396
Park, B. J., Kim, J., and Park, H. S. (2020). Bifunctional electrocatalysts based on hierarchical graphene/iron hybrid architectures branched by N-doped CNT. J. Alloys Compd. 846, 156244. doi:10.1016/j.jallcom.2020.156244
Park, J., Cho, M., Rhee, Y. M., and Jung, Y. (2021). Theoretical study on the degree of CO2 activation in CO2-coordinated Ni(0) complexes. ACS Omega 6, 7646–7654. doi:10.1021/acsomega.0c06257
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Peterson, A. A., Pedersen, F. A., Studt, F., Rossmeisl, J., and Nørskov, J. K. (2010). How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315. doi:10.1039/c0ee00071j
Reske, R., Mistry, H., Behafarid, F., Cuenya, B. R., and Strasser, P. (2014). Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 136, 6978–6986. doi:10.1021/ja500328k
Shen, X., Liu, X., Wang, S., Chen, T., Zhang, W., Cao, L., et al. (2021). Synergistic modulation at atomically dispersed Fe/Au interface for selective CO2 electroreduction. Nano Lett. 21, 686–692. doi:10.1021/acs.nanolett.0c04291
Song, H., Tan, Y. C., Kim, B., Ringe, S., and Oh, J. (2021). Tunable product selectivity in electrochemical CO2 reduction on well-mixed Ni−Cu alloys. ACS Appl. Mat. Interfaces. 13, 55272–55280. doi:10.1021/acsami.1c19224
Wang, F., Meng, Y., Chen, X., Zhang, L., Li, G., Shen, Z., et al. (2022). Effect of nickel-based electrocatalyst size on electrochemical carbon dioxide reduction: A density functional theory study. J. Colloid Interface Sci. 615, 587–596. doi:10.1016/j.jcis.2022.02.032
Wei, N., Tian, Y., Liao, Y., Komatsu, N., Kauppinen, E. I., Lyuleeva-Husemann, A., et al. (2021). Colors of single-wall carbon nanotubes. Adv. Mater 33, e2006395. doi:10.1002/adma.202006395
Weng, Z., Wu, Y. S., Wang, M. Y., Jiang, J. B., Wang, H. L., Huo, S., et al. (2018). Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 9, 415. doi:10.1038/s41467-018-02819-7
Xiong, W., Yang, J., Shuai, L., Qiu, M., Li, X., Hou, Y., et al. (2019). CuSn alloy nanoparticles on nitrogen-doped graphene for electrocatalytic CO2 reduction. ChemElectroChem 6, 5951–5957. doi:10.1002/celc.201901381
Xue, D., Xia, H., Yan, W., Zhang, J., and Mu, S. (2020). Defect engineering on carbon-based catalysts for electrocatalytic CO2 reduction. Nanomicro Lett. 13, 5. doi:10.1007/s40820-020-00538-7
Yang, T., Tian, N., Liu, X. H., and Zhang, X. (2022). CuNi alloy nanoparticles embedded in N-doped carbon framework for electrocatalytic reduction of CO2 to CO. J. Alloys Compd. 904, 164042. doi:10.1016/j.jallcom.2022.164042
Zhang, E., Wang, T., D., Zhang, J., Li, Y., Chen, W., Li, A., et al. (2019). Bismuth single atoms resulting from transformation of metal-organic frameworks and their use as electrocatalysts for CO2 reduction. J. Am. Chem. Soc. 141, 16569–16573. doi:10.1021/jacs.9b08259
Zhang, Y., Yang, R., Li, H., and Zeng, Z. (2022). Boosting electrocatalytic reduction of CO2 to HCOOH on Ni single atom anchored WTe2 monolayer. Small 18, e2203759. doi:10.1002/smll.202203759
Zhao, X., and Liu, Y. (2020). Unveiling the active structure of single nickel atom catalysis: Critical roles of charge capacity and hydrogen bonding. J. Am. Chem. Soc. 142, 5773–5777. doi:10.1021/jacs.9b13872
Zhong, M., Tran, K., Min, Y., Wang, C., Wang, Z., Ulissi, Z., et al. (2020). Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183. doi:10.1038/s41586-020-2242-8
Keywords: electrochemical CO2 reduction reaction (CO2RR), Cu-based catalysts, density functional theory (DFT), size effect, compositional effect
Citation: An R, Chen X, Fang Q, Meng Y, Li X and Cao Y (2023) Structure–activity relationship of Cu-based catalysts for the highly efficient CO2 electrochemical reduction reaction. Front. Chem. 11:1141453. doi: 10.3389/fchem.2023.1141453
Received: 10 January 2023; Accepted: 30 January 2023;
Published: 09 February 2023.
Edited by:
Huayang Zhang, University of Adelaide, AustraliaReviewed by:
Shengwei Deng, Zhejiang University of Technology, ChinaYun-Xiang Pan, Shanghai Jiao Tong University, China
Copyright © 2023 An, Chen, Fang, Meng, Li and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Li, xili21@mail.zjxu.edu.cn; Yongyong Cao, cyy@zjxu.edu.cn
†These authors have contributed equally to this work