 Agnieszka Wrobel
Agnieszka Wrobel Claudio Ottoni
Claudio Ottoni Jack C. Leo
Jack C. Leo Dirk Linke
Dirk Linke- 1Department of Biosciences, University of Oslo, Oslo, Norway
- 2Centre for Ecological and Evolutionary Synthesis, University of Oslo, Oslo, Norway
Enteric redmouth disease caused by the pathogen Yersinia ruckeri is a significant problem for fish farming around the world. Despite its importance, only a few virulence factors of Y. ruckeri have been identified and studied in detail. Here, we report and analyze the complete DNA sequence of pYR4, a plasmid from a highly pathogenic Norwegian Y. ruckeri isolate, sequenced using PacBio SMRT technology. Like the well-known pYV plasmid of human pathogenic Yersiniae, pYR4 is a member of the IncFII family. Thirty-one percent of the pYR4 sequence is unique compared to other Y. ruckeri plasmids. The unique regions contain, among others genes, a large number of mobile genetic elements and two partitioning systems. The G+C content of pYR4 is higher than that of the Y. ruckeri NVH_3758 genome, indicating its relatively recent horizontal acquisition. pYR4, as well as the related plasmid pYR3, comprises operons that encode for type IV pili and for a conjugation system (tra). In contrast to other Yersinia plasmids, pYR4 cannot be cured at elevated temperatures. Our study highlights the power of PacBio sequencing technology for identifying mis-assembled segments of genomic sequences. Comparative analysis of pYR4 and other Y. ruckeri plasmids and genomes, which were sequenced by second and the third generation sequencing technologies, showed errors in second generation sequencing assemblies. Specifically, in the Y. ruckeri 150 and Y. ruckeri ATCC29473 genome assemblies, we mapped the entire pYR3 plasmid sequence. Placing plasmid sequences on the chromosome can result in erroneous biological conclusions. Thus, PacBio sequencing or similar long-read methods should always be preferred for de novo genome sequencing. As the tra operons of pYR3, although misplaced on the chromosome during the genome assembly process, were demonstrated to have an effect on virulence, and type IV pili are virulence factors in many bacteria, we suggest that pYR4 directly contributes to Y. ruckeri virulence.
Introduction
The genus Yersinia consists of 17 different species (Reuter et al., 2014; Savin et al., 2014). Although the human pathogens within the genus are closely related to each other, they cause diverse diseases. Y. pestis, the causative agent of bubonic and pneumonic plague, is one of the most virulent organisms known (Chauhan et al., 2016). In addition, this genus includes Y. enterocolitica and Y. pseudotuberculosis, well-known human enteropathogens. Y. pestis spreads through fleabites or aerosols, whereas Y. enterocolitica and Y. pseudotuberculosis are transmitted via ingestion of contaminated food or water (Bottone, 1997; Perry and Fetherston, 1997; Jalava et al., 2006). Y. enterocolitica and Y. pseudotuberculosis are responsible for a broad range of diseases ranging from mild gastroenteritis to life-threatening septicemia (Bottone, 1997).
Y. ruckeri is a fish pathogen causing enteric redmouth disease (ERM), mainly in salmonids (Bullock et al., 1978; Busch, 1978). This bacterium contributes to enormous economic losses in aquaculture throughout the world. Y. ruckeri is mostly transmitted through contact with carrier fish (Busch, 1978; Stevenson and Airdrie, 1984). Despite the availability of vaccines, yersiniosis outbreaks still occur in fish farms (Ormsby et al., 2016). The majority of the ERM outbreaks are caused by the highly pathogenic Y. ruckeri serotype 1 belonging to biotype 1, characterized as motile with phospholipase activity (Romalde and Toranzo, 1993). For a long time, ERM has played a minor role in Norway, with only a few outbreaks per year (Hjeltnes et al., 2017). The first report of a disease outbreak caused by Y. ruckeri among Atlantic salmon was described in Norway in 1985, and this was successfully treated with antibiotics (Sparboe et al., 1986). In recent years, the number of outbreaks in the farmed Atlantic salmon population has substantially increased. The reasons for the most recent outbreaks remain unclear, and Y. ruckeri infections are nowadays a major challenge facing the Norwegian aquaculture industry, similar to other countries such as Australia (Barnes et al., 2016), Chile (Avendaño-Herrera et al., 2017), and Scotland (Ormsby et al., 2016).
Each of the human Yersinia pathogens harbors chromosomally and plasmid-encoded virulence determinants (Chauhan et al., 2016). Y. pestis usually carries two species-specific plasmids, pPCP1 and pMT1, and one highly conserved plasmid shared among the three human pathogenic Yersiniae, pYV (also called pCD1) (Ben-Gurion and Shafferman, 1981; Ferber and Brubaker, 1981; Haiko et al., 2009). This large 70-kb plasmid carries a type III secretion system (T3SS), Ysc. T3SS system encodes structural proteins, chaperones as well as effector proteins called Yops (Yersinia outer proteins) required for Yersinia extracellular survival. The effector proteins and the machinery for their delivery are required for infection and manipulation of host responses to overcome the action of phagocytes (Cornelis et al., 1998). Moreover, the plasmid encodes a major virulence factor, the Yersinia adhesin A (YadA) (Mühlenkamp et al., 2015).
Despite the economic importance, the pathogenicity of Y. ruckeri has not been studied in detail. Only few virulence factors are known, and to date all of these are encoded on the chromosome. These include bacterial adhesins important in establishing a successful colonization (Romalde and Toranzo, 1993). In particular, the chromosomally encoded adhesins YrInv and YrIlm might play a role in virulence (Wrobel et al., 2017). They belong to the intimin-invasin family of adhesins, which includes also InvA, the adhesin responsible for the initial bacterial attachment and colonization of host tissues in Y. enterocolitica and Y. pseudotuberculosis (Isberg and Leong, 1990; Wrobel et al., 2017). Other virulence factors described in Y. ruckeri include cytotoxins and haemolysins (Romalde and Toranzo, 1993), the metalloprotease Yrp1 (Secades and Guijarro, 1999), the haemolysin/cytolysin YhlA (Fernández et al., 2007), the iron uptake system ruckerbactin (Fernández et al., 2004), and a chromosomal T3SS (Liu et al., 2016). Recently, a large proteomic study of Y. ruckeri strains was performed under standard (Kumar et al., 2017) and iron-limited conditions (Kumar et al., 2016). In total, 1395 proteins were identified in the whole cell lysate of Y. ruckeri under standard culture conditions. Among them, several proteins were predicted to be virulence factors, including, among others, HtrA protease, TolB, the lipoprotein NlpD and a LuxR family transcriptional regulator. This global proteomic analysis will help in understanding the biology of the pathogen, as well as in development of new effective treatments against the ERM disease (Kumar et al., 2017).
Plasmid-borne virulence factors have been found in other fish pathogens, including Vibrio anguillarium (Crosa, 1980) and Edwardsiella tarda (Yu et al., 2012), but not in Y. ruckeri. Plasmids in Y. ruckeri strains were studied previously due to their possible involvement in virulence in analogy to the human pathogenic Yersiniae (De Grandis and Stevenson, 1982). Many authors expected to find the same virulence traits as those described for the human pathogens, such as the plasmid-encoded T3SS. However, none of the plasmid-associated virulence factors of the human-enteropathogenic Yersiniae were found in these plasmids. In general, Y. ruckeri plasmids have not yet been properly characterized and further research is required to understand their role in bacterial virulence. A study including 183 Y. ruckeri strains from different geographical locations reported 8 different plasmid profiles (Garcia et al., 1998). In this study, the most virulent sorbitol-negative Y. ruckeri strains of serotype O1 contained a large 75 MDa plasmid (~113 kb), in agreement with earlier studies (Guilvout et al., 1988; Romalde et al., 1993). In addition, smaller plasmids (12.7 MDa; ~19 kb) have been found in most of the strains (Garcia et al., 1998).
More recent studies showed that multidrug resistance plasmids in Y. ruckeri strains are a serious aquaculture concern (Toranzo et al., 1983; De Grandis and Stevenson, 1985; Carattoli et al., 2012; Huang et al., 2014). Welch et al. (2007) showed that Y. ruckeri strain YR71 carries a multidrug resistance plasmid called pYR1, which has 99% nucleotide identity with the IncA/C (incompatibility A/C) plasmid backbone of the Y. pestis isolate IP275, plasmid pIP1202. The IncA/C group comprises a large, low-copy number, multidrug resistance plasmid family within Enterobacteriaceae such as Escherichia coli, Salmonella enterica, Y. pestis, and Klebsiella pneumoniae, as well as more distantly related species such as Vibrio cholerae. Plasmids of this family are unique with regard to their structure and gene content. They contain putative transfer regions [type IV secretion system (T4SS)], regions involved in integration of mobile genetic elements, as well as regions involved in transcription (Johnson and Lang, 2012). T4SSs are widely distributed in prokaryotes as well as in some archaea. T4SSs are large macromolecular complexes typically composed of a cell-envelope spanning mating channel and an extracellular pilus structure. T4SSs are classified into two major groups type IVA (T4ASSs) and type IVB (T4BSSs). T4ASS resemble the VirB/VirD system of Agrobacterium tumefaciens while T4BSSs are related to the conjugation system of IncI plasmids. Typical examples of T4ASSs are found on conjugative plasmids, such as F, RP4 and pKM101, as well as the prototypical VirB system of A. tumefaciens. These T4ASSs export nucleoprotein complexes during conjugation. T4BSS is represented by the Legionella pneumophila icm/dot system involved in protein translocation into host cells thus allowing the pathogen to replicate intracellularly (Wallden et al., 2010).
In this work, we sequenced a plasmid—which we named pYR4—from the highly pathogenic Norwegian Y. ruckeri isolate NVH_3758 from the 1987 outbreak and performed a comparative bioinformatics analysis of the available Y. ruckeri plasmid sequences to evaluate their role in virulence.
Materials and Methods
Plasmid DNA Sequencing Technology
Genomic DNA as well as plasmid DNA was extracted from a locally important, highly pathogenic Norwegian Y. ruckeri isolate NVH_3758 (biotype 1, serotype 1) recovered from an outbreak of clinical yersiniosis in farmed Atlantic salmon, kindly provided by Prof. Duncan Colquhoun at the Norwegian Veterinary Institute in Oslo, Norway (Gulla et al., 2018). Whole genome sequencing of Y. ruckeri NVH_3758 was performed by the Norwegian Sequencing Centre (Oslo, Norway) using the Single Molecule Real Time (SMRT) sequencing technology of Pacific Biosciences. Sample preparation, reads assembly and consensus polishing were done as previously described (Wrobel et al., 2017). The final assembly yielded two contigs of circularized length of ~3.8 Mb, representing the chromosomal genome (Wrobel et al., 2017), and ~81 kb, corresponding to a new plasmid that we named pYR4. The DNA sequence of pYR4 has been deposited in the National Centre for Biotechnology Information (NCBI) database under the accession number CP032236.
Plasmid Annotation
The FASTA consensus of pYR4 from Y. ruckeri NVH_3758 was uploaded to RAST (Rapid Annotation using Subsystem Technology) for automatic annotation (Aziz et al., 2008; Seemann, 2014). After initial annotations, all open reading frames (ORFs) with initial annotations were checked using the interactive server HHpred available at the Max Planck Institute for Developmental Biology Toolkit (Söding et al., 2005) against two databases, the PDB and PFAM (Sonnhammer et al., 1998; Sussman et al., 1999). The functional annotations obtained from the HHpred server and RAST were compared and in some cases were corrected manually. Many uncharacterized proteins which were previously labeled as hypothetical by RAST were annotated based on similarity to characterized proteins. The protein sequences were uploaded into Geneious (Kearse et al., 2012). A circular representation of pYR4 showing the annotated features, the GC content and the GC skew within 50 bp-long genomic regions, was generated with Circos (Krzywinski et al., 2009). The identification of the promoter sequences of the pil and tra operons was performed with the online server BPROM (Solovyev and Salamov, 2011) as well as bTSSfinder (Shahmuradov et al., 2017) (see Table 1 and Supplementary Figure 1). The mfold Web server was used for RNA secondary structure prediction (Zuker, 2003).
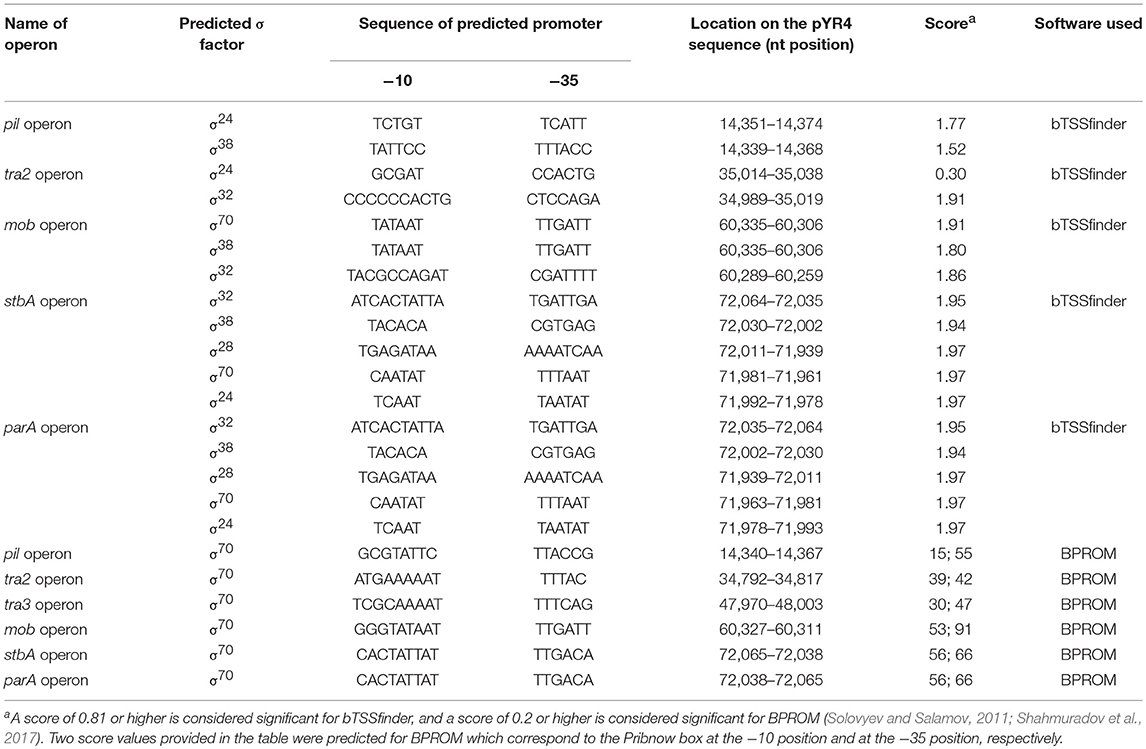
Table 1. pYR4 promoter predictions by bTSSfinder (Shahmuradov et al., 2017) and BPROM (Solovyev and Salamov, 2011) used in the present study.
RepA Phylogeny
The RepA protein sequence of pYR4 was annotated by RAST as “hypothetical.” After the initial annotation, the protein was identified as RepA using HHpred. The protein sequence was then subjected to a search using BLASTP (Altschul et al., 1997). The BLASTP search returned 100 hits, from which the first 29 RepA protein sequences were selected, after excluding sequences of hypothetical proteins and multispecies proteins, and aligned using MUSCLE (Edgar, 2004). In the final alignment, we included RepA protein sequences from pYR1, pYR3, pYR4 in addition to eight RepA protein sequences belonging to the IncA/C plasmid family. The final alignment was then used to construct the phylogenetic tree using MEGA X software by applying the Maximum Likelihood method on the Poisson correction model (Zuckerkandl and Pauling, 1965; Felsenstein, 1985; Kumar et al., 2018) (see Supplementary Figure 2).
Plasmid Comparative Analysis
We compared the nucleotide sequence of Y. ruckeri NVH_3758 plasmid pYR4 with the nucleotide sequences of the plasmids of Y. ruckeri strains YR71 (pYR1), CSF007-82 (pYR2, pYR3) and SC09 (pLT, pWKY) (Table 2) deposited in the NCBI database. To keep the same annotation system as for the pYR4 plasmid, the nucleotide sequences of the Y. ruckeri plasmids were re-annotated with RAST (see also Supplementary Figure 3 for pYR4, Supplementary Figure 4). Details of the annotation can be found in Supplementary Tables 1 and 2.
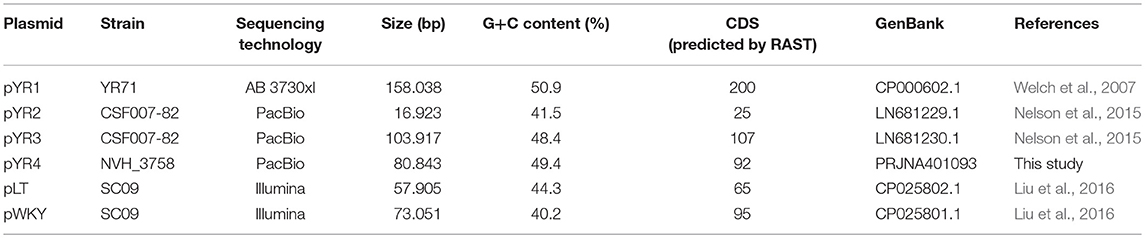
Table 2. Y. ruckeri plasmid sequences deposited in GenBank used in the present study.
Plasmid comparisons were also done with sequences of other species containing the tra and pil operons described in literature, including Erwina amylovora (pEL60, pEA68, pEA72, pEA78), Serratia entomophila (pADAP), Citrobacter freundii (pCTX-M3), and Salmonella enterica subsp. enterica serovar Typhimurium (R64) (see Supplementary Figure 5). The nucleotide sequence of Y. ruckeri NVH_3758 pYR4 plasmid was also compared to PacBio-sequenced genomic data of Y. ruckeri CSF007-82, Big Creek 74, QMA0440, SC09, and Illumina-sequenced genomes of Y. ruckeri ATCC29473 and YRB (see Supplementary Figure 5). In the comparative survey, we also included the ~57 kb-long scaffold 20 of the Y. ruckeri 150 assembly, which contains the tra and pil operons.
Finally, pYR4 was compared to the Illumina-sequenced genomes of human pathogens Y. pestis CO92, Y. pseudotuberculosis YPIII, and Y. enterocolitica 8081 (see Supplementary Figure 5). Pairwise comparisons were performed with Progressive Mauve (Darling et al., 2010) using default options and the “seed family” option to increase sensitivity. The output backbone file was then used to plot the Locally Collinear Blocks (LCB) in a circular representation with Circos.
Results
pYR4 Is a Novel Plasmid Isolated From Y. ruckeri NHV3758
In order to define the relationship between the plasmid sequenced in this study and those described in literature, we performed a comparative analysis of pYR4 with plasmid sequences generated previously by Illumina and PacBio sequencing technologies. The comparative survey with Mauve indicated no obvious similarity between pYR4 and the Y. ruckeri plasmids pYR1, pYR2, pLT, and pWKY, as no LCBs (locally collinear blocks) were detected (data not shown). On the other hand, a ~55 kb-long LCB (sequence identity >99%) that included the pil and the tra operons was present in pYR4 (from nucleotide position 9,100 to 64,005) and in the PacBio-sequenced plasmid pYR3 (Figures 1, 3, Table 2). By re-annotating pYR3 and comparing it with the higher-resolution annotation of pYR4 obtained through HHPred, we could provide a more in depth characterization of the plasmids under analysis (Nelson et al., 2015) (see Materials and Methods section, Supplementary Figure 3 for pYR4 and Supplementary Figure 4 for pYR3).
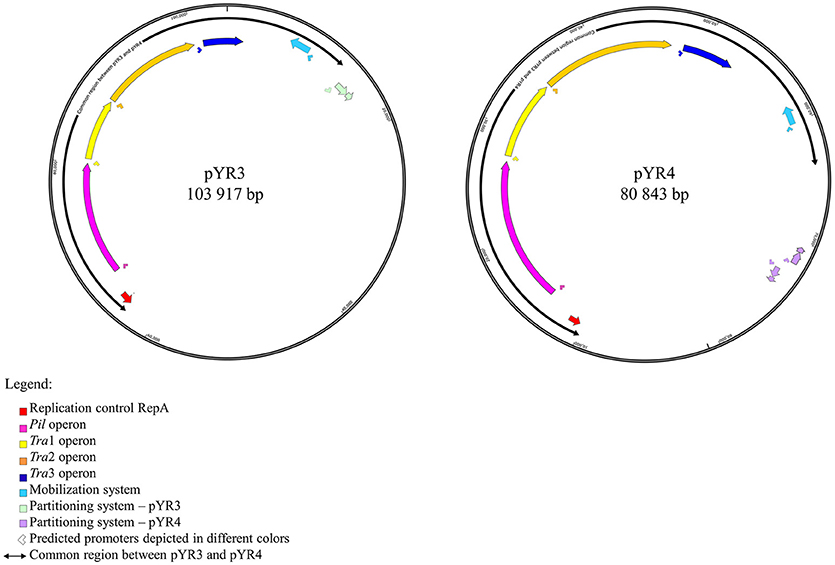
Figure 1. Schematic representation of plasmid maps of pYR3 and pYR4. Gene clusters are depicted in different colors: light blue (mobilization gene cluster), yellow (tra1 gene cluster), orange (tra2 gene cluster), dark blue (tra3 gene cluster), magenta (pil gene cluster), purple (partitioning gene cluster). Approximate locations of the promoters based on the prediction of sigma factors with BPROM and bTSSfinder is reported as arrows below each operon. Replication initiation protein is indicated in red. Common regions for pYR3 and pYR4 are represented by bidirectional arrows. Partitioning system for pYR3 and pYR4 are depicted in light green and light purple, respectively. pYR4 contains 2 partitioning systems oriented in two different directions.
The remaining portion of the plasmid sequence (>25 kb, 31% of the sequence length) appears to be unique, as no LCB was found in any of the plasmids of Y. ruckeri deposited in GenBank so far. This region contains mostly hypothetical proteins (n = 10) and mobile genetic elements (n = 10). In addition, we found a partial toxin-antitoxin system, a restriction system including both a type I restriction enzyme and a corresponding ArdA-like anti-restriction protein, and a small cluster of genes coding for two alcohol dehydrogenase enzymes and a transcription factor with high similarity to FrmR from Salmonella, a formaldehyde-sensitive regulator (Supplementary Figure 3). pYR4 contains two potential partitioning systems (ParAG and StbAB) (Figure 1). These two partitioning systems are represented by two operons oriented in opposite directions. No obvious sequence similarity with the partitioning system of pYR3 was found. Interestingly, the presence of alternative partitioning systems have already been described before for pYV from Yersinia species (Pilla and Tang, 2018). The high abundance of mobile elements may suggest that this plasmid is likely subject to structural rearrangements and that the unique ~25 kb region of pYR4 may be the result of recent horizontal gene transfer. This is also supported by the difference in G+C content between the ~55 kb region (50.4%) and the remaining portion of the plasmid (47.1%). It is worth noting that in pYR3, this complete region is replaced by a different ~45 kb region (Figure 1). These major differences suggest that Y. ruckeri NHV3758 contains a plasmid with significant differences to pYR3, which we named pYR4.
Sequence Analysis of pYR4
The nucleotide sequence of the circular pYR4 plasmid contains 80,842 base pairs (~53 MDa). The G+C content of pYR4 is 49.4%, which is around 2% less than the G+C content of the Y. ruckeri NVH_3758 genome (47.6%), suggesting acquisition by horizontal gene transfer (Figure 2) (Nishida, 2012; Hayek, 2013). The annotation of pYR4 with RAST showed 92 putative coding sequences along the entire plasmid sequence. The RAST server could annotate functions for 52 ORFs and we were able to expand this list to 71 ORFs manually, using the HHpred server (Söding et al., 2005) (see Supplementary Figure 3), leaving 21 ORFs without putative function. Fifty-five genes are encoded on the positive strand while the remaining 37 are encoded on the negative strand. The entire plasmid sequence can be divided into several gene clusters, including clusters for partitioning (parA, parG, and stbAB), a T4SS (tra), and a type IV pilus (TFP) gene cluster (pil).
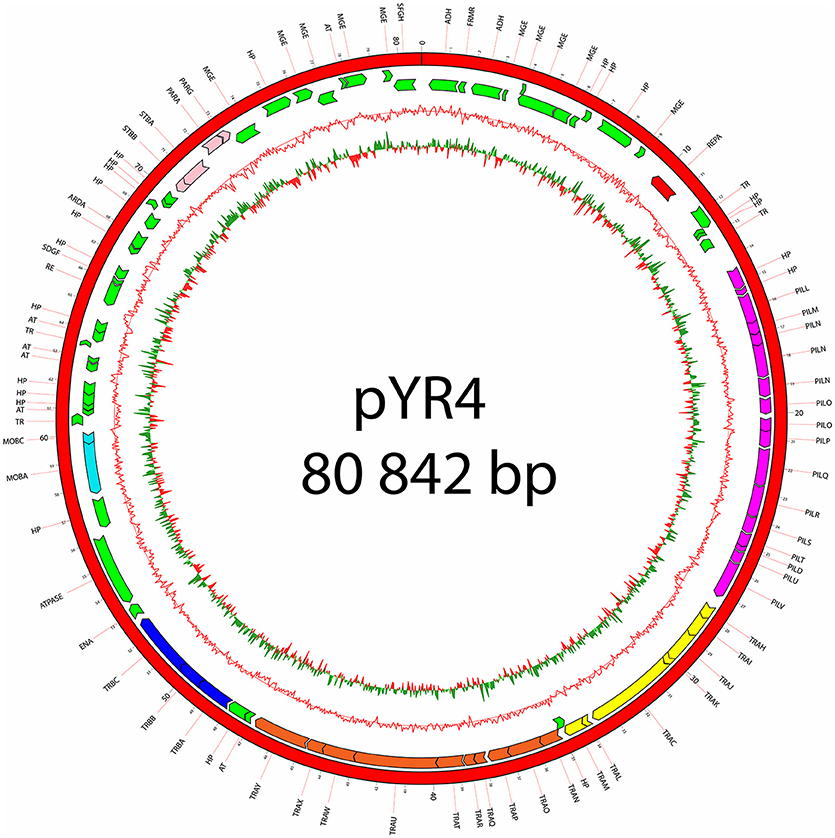
Figure 2. Plasmid map of pYR4 isolated from Y. ruckeri NVH_3758. The sequence annotations were generated with RAST (Rapid Annotation using Subsystem Technology) and further analyzed with HHpred. The rings show from inside to outside (1) the GC skew, (2) the G+C content, (3) the position of predicted ORFs in the reverse strand, and (4) the position of ORFs in the forward strand. The gene annotations are positioned in the middle of each gene on the plasmid map. Gene clusters are indicated in different colors: light blue (mobilization gene cluster), yellow (tra1 gene cluster), orange (tra2 gene cluster), dark blue (tra3 gene cluster), magenta (pil gene cluster), pink (partitioning gene cluster). Replication initiation protein is indicated in red. Hypothetical proteins (HP) and mobile genetic elements are indicated in green.
In order to understand the pYR4 plasmid physiology as well as to follow its evolution and spread, we classified pYR4 into a plasmid family. A classical method of plasmid classification is based on incompatibility (Inc) groups. In general, plasmids with the same replication system are incompatible while plasmids with different replication system are compatible. In other words, two plasmids of the same Inc group cannot be propagated in the same bacterial cell (Couturier et al., 1988). A set of 30 RepA protein sequences from IncFII plasmid family, together with 9 RepA protein sequences previously characterized as belonging to IncA/C, were aligned in order to classify pYR4. Evaluation of the RepA phylogeny showed that the pYR4 RepA protein is closely related to IncFII plasmids found in other Yersiniae species (see Supplementary Figure 2). A BLASTP search of pYR4 RepA protein returned over 100 hits of homologs found in different species. The closest RepA homologs were found in Illumina-sequenced genomes of Y. frederiksenii and Y. enterocolitica with 91 and 90% similarity over the whole protein sequence, respectively. These RepA homologs presumably are part of unnamed plasmids that were incorrectly assigned to chromosomes, since they share only 19% similarity to RepA of the well-described virulence plasmid pYV from Y. enterocolitica 8081 (Table 3). Thus, pYR4 was classified as a member of the IncFII plasmid family, in contrast to pYR1 which belongs to the IncA/C family and is represented as an outgroup in Supplementary Figure 2 (Carattoli, 2009). The IncFII plasmid family includes low-copy number plasmids mostly related to virulence, such as pYV, as well as to the dissemination of antimicrobial resistance determinants. Plasmids from this family usually carry the FII replicon alone or in association with extra replicons such as repFIA and repFIB (Carattoli, 2009; Yang et al., 2015) and are common in Yersiniae (Villa et al., 2010).
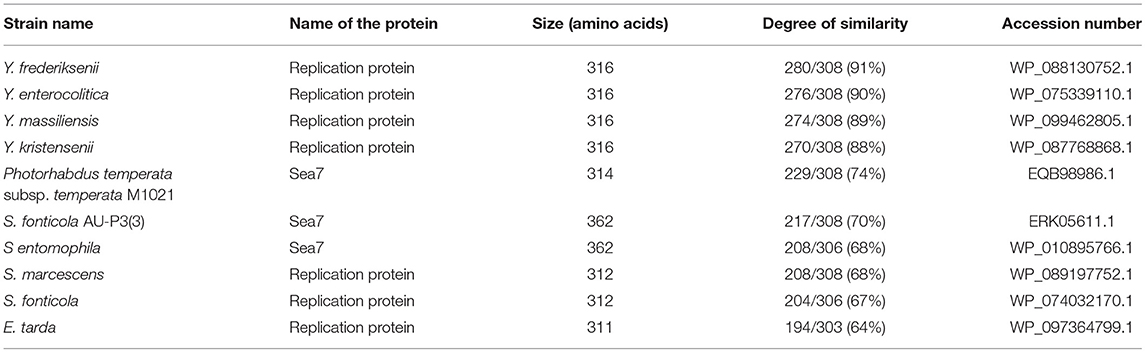
Table 3. pYR4 RepA homologs detected using BLASTP.
Comparative Analysis of Y. ruckeri Plasmid Sequences Demonstrates Errors in Assemblies of Second Generation Genome Sequencing
Our comparative survey showed that no significant LCBs were found between pYR4 and the chromosomal genomes of Y. ruckeri CSF007-82, Big Creek 74, QMA0440, SC09, or YRB (data not shown). However, we found that the ~100 kb-long scaffold 2 of the Y. ruckeri ATCC29473 Illumina assembly matched pYR3, except for a mobile element of pYR3 (Figure 3 and Supplementary Figures 4–6). Furthermore, the higher quality of PacBio sequenced plasmids (pYR3 of CSF007-82 and pYR4 of NHV-3758) made it possible in our comparative survey to place scaffolds of previous Y. ruckeri assemblies into plasmid locations. In fact, the ~57 kb-long Y. ruckeri 150 scaffold 20 that contained the tra and the pil operons could be mapped entirely to pYR3 (see Supplementary Figure 7). This scaffold included the ~55 kb-long region containing the pil and the tra operons detected in pYR4 and pYR3. Furthermore, by aligning other unplaced scaffolds of the Y. ruckeri 150 assembly, we found that four more scaffolds (23, 31, 32, 34) could be placed within pYR3 (Figure 3 and Supplementary Figures 5, 6). (An unplaced scaffold is a sequence found in an assembly that is not associated with any chromosome). Taken together, the evidence presented here suggests that the pil and tra operons are localized on plasmids pYR3 and pYR4 and that Y. ruckeri 150 and Y. ruckeri ATCC29473 contain the plasmid pYR3. In Y. ruckeri 150, the presence of plasmid- and chromosomally-borne tra clusters has been suggested based on Southern blot hybridization evidence (Méndez et al., 2009). When searching for tra genes in the assembly, we could not find copies of tra genes other than those matching pYR3 in the scaffold 20 of Y. ruckeri 150. Based on our data, chromosomal localization of the tra cluster seems very unlikely. However, resequencing or a higher quality assembly of the genome of Y. ruckeri 150 could clarify this unambiguously in the future.
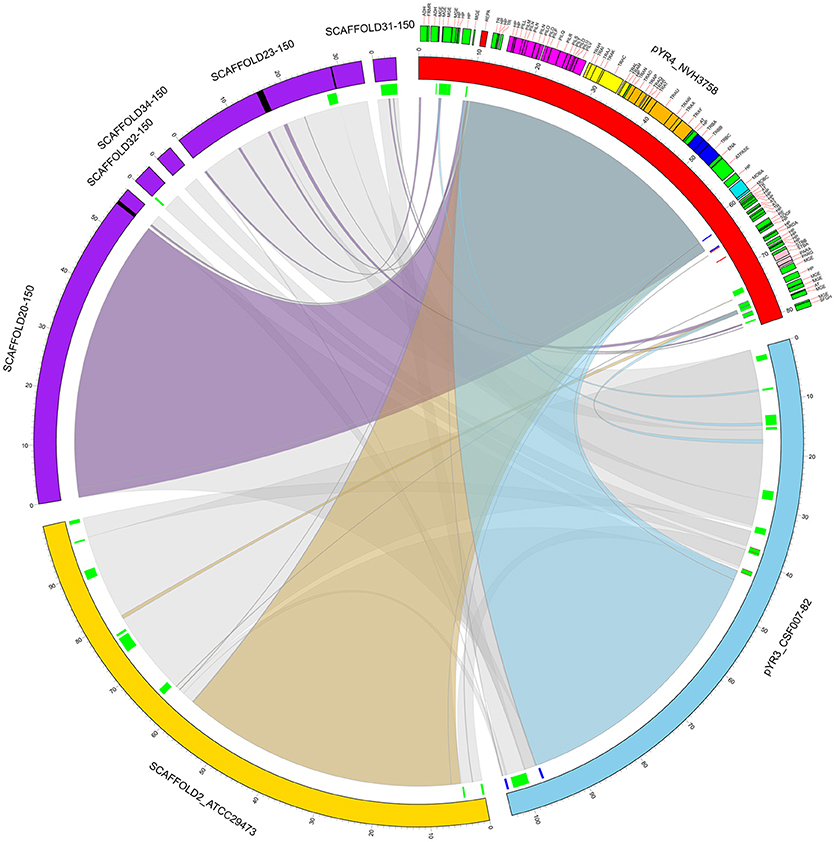
Figure 3. Circular representation of a comparative analysis of pYR4 from Y. ruckeri NVH_3758 to pYR3 and scaffolds of genomic assemblies from Y. ruckeri ATCC29473 (scaffold 2) and Y. ruckeri 150 (scaffolds 20, 23, 31, 32, 34). The pYR4 plasmid is depicted in red, pYR3 in blue, Y. ruckeri ATCC29473 scaffold 2 in yellow, and Y. ruckeri 150 scaffolds in purple. Sequence gaps (stretches of Ns) within ATCC29473 scaffolds are reported in black. Annotated features in pYR4 are shown and colored as in Figure 2. Repetitive DNA regions in pYR3 and pYR4 are colored red and blue, and mobile genetic elements are colored in green (lines in inner ring). Pairwise Locally Collinear Blocks (LCBs) as found in Mauve DNA alignments, are represented as ribbon links colored as follows: pYR4-pYR3 in light blue, pYR4-ATCC29473 in yellow, pYR4-150 in purple, pYR3-ATCC29473, and pYR3-150 in gray. Ribbon links of the ATCC29473 and 150 scaffolds with pYR3 and pYR4 are represented separately in Supplementary Figures 6, 7.
Finally, no significant LCBs were found when comparing pYR4 or pYR3 to Yersinia human pathogenic species, including the well-studied pYV plasmid from Y. enterocolitica. This suggests a very different strategy for host infection in Y. ruckeri, as the pYV plasmid is essential for virulence in Y. enterocolitica.
Sequence Analysis of pil Operon and Its Potential Involvement in Virulence
Analysis of the pYR4 nucleotide sequence of a putative pil operon showed that this region spans a 12.6 kb locus containing 17 ORFs that encode a TFP. TFPs, not to be confused with T4SSs, are surface appendages expressed by many Gram-negative bacterial species. TFPs span both bacterial membranes and they are evolutionary and structurally related to type II secretion systems. TFPs are involved in bacterial adhesion, biofilm formation, horizontal gene transfer, and pathogenesis, and in addition they mediate cell movement such as gliding motility in Myxococcus xanthus and twitching motility in Pseudomonas and Neisseria species (Shi and Sun, 2002). In the enteropathogenic Y. pseudotuberculosis, the TFP gene cluster is composed of 11 open reading frames and contributes to Y. pseudotuberculosis pathogenicity (Collyn et al., 2002). The arrangement of the pYR4 pil cluster (Figure 2; see also Table 1 and Supplementary Figure 1) resembles the pil cluster from the plasmid pADAP, which was described in S. entomophila (Hurst et al., 2011), and in plasmid R64 from S. enterica serovar Typhimurium (Kim and Komano, 1997). By analogy with the pil operon from S. entomophila, we adopted the same names for the putative proteins as described there, and designated them as PilLMNNNOOPQRSTDUV [(with the exception of the first two hypothetical proteins, designated as HP (H- for hypothetical and P- for proteins)]. It is worth mentioning that the pil operon in S. enterica includes only 14 genes (pilIJKLMNOPQRSTUV) in contrast to pYR4 (17 genes). The number of genes for pil clusters can vary, as described by Zhang et al. for S. enterica serovar Typhi, where the pil operon lacks the pilI, pilJ, and pilK genes (Zhang et al., 2000) of S. entomophila. The overall G+C content of the pil cluster is 51.7%, and thus higher than that of the Y. ruckeri NVH_3758 genome (47.6%) and the average of pYR4 (49.4%). The G+C content of the individual genes in the pil locus varies between 55.1% (pilP) and 46.8% (pilN). The ORFs of the pil cluster are encoded on the same strand and the length of the intercistronic region between each ORF ranges from 20 to 205 bp. In the region up to 333 bp upstream of the first hypothetical protein of the pil cluster, we could identify binding sites for three sigma factors (σ70, σ24, and σ38) using BPROM and bTSSfinder, indicating the presence of putative promoters sequences in that region. In fact, no putative promoter sequence was identified between the pil genes suggesting that this region may function as an operon (Figure 2) (see Table 1 and Supplementary Figure 1). Additionally, analysis of the downstream region of pilV showed the presence of a palindromic sequence (5′CTAGACAGAATAGCCTAGTCAATATTATCTATGGCATTAAGATTCTGTCAG-3′) that could serve as a transcription terminator. The analysis of the secondary structure of this region showed a steam-loop like fold with the ΔG = −15 kcal/mol using the mfold Web server (Zuker, 2003).
The comparison of the protein sequences encoded by the pil operon, for example PilO and PilT, of pYR4 with database sequences using BLASTP showed from 64 to 86% identity to PilO and PilT proteins found in the IncI1 plasmid family from Serratia species. Accession numbers for the proteins, together with their functions, are given in Table 4.
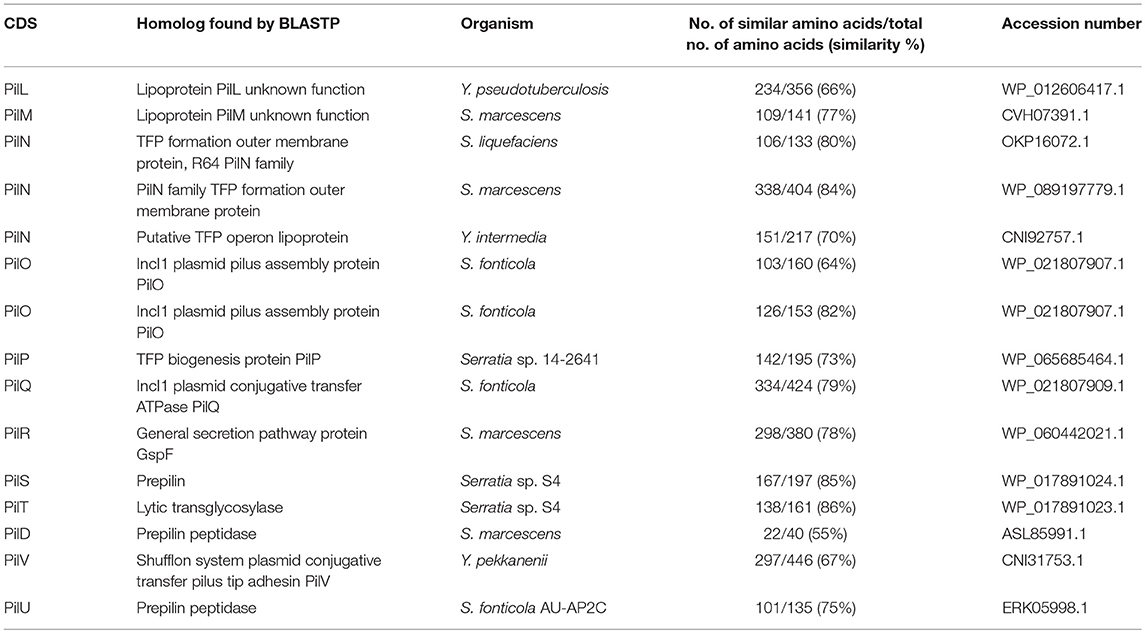
Table 4. The main characteristic features of in pil operon identified in the pYR4 plasmid from Y. ruckeri NVH_3758.
The biogenesis of TFPs involves a number of proteins. These are all present in the pil operon of pYR4, suggesting that the locus is intact and functional. The pYR4 PilS protein encodes a major pilin, which is synthesized as a prePilS. In the prePilS protein sequence we identified a hydrophilic signal peptide comprising 15 residues. A predicted cleavage site lies between the 15th (glycine) and 16th (tryptophan) residues of prePilS, which is recognized by the specific peptidase PilD (Kim and Komano, 1997). The mature PilS contains an N-terminal hydrophobic region (first 23 residues in the mature protein sequence), while the C-terminal region is rich in cysteine residues, a common feature of TFP pilins (Hurst et al., 2011). Beside the prepilins, we identified two copies of PilO and three copies of PilN, which seems to be a unique feature among plasmids from the same family. PilO and PilN are integral membrane proteins and usually exist only in one copy. An ATPase required for the assembly (PilQ) and an inner membrane protein (PilR) that we identified are also necessary for the assembly of the pili on the bacterial surface.
Sequence Analysis of the tra Regions in pYR4
Annotations of pYR4 by HHpred (Söding et al., 2005) showed the presence of the tra region that we presume encodes a T4SS and is involved in conjugation. In fact, the presence of a chromosomally-borne tra clusters in Y. ruckeri 150 was previously described (Méndez et al., 2009). In our analysis, in addition to the tra cluster identified by Méndez et al. (tra1) that is probably also plasmid-borne (see above), we could identify another two tra clusters, which we named tra2 and tra3. The tra2 cluster comprises 10 genes with the gene order TraNOPQRTUWXY and an average G+C content of 53.5%, while the tra3 cluster is composed of 3 genes with a G+C content of 52.6%. These two tra clusters are preceded by two putative promoter sequences with one located upstream from the traN gene while another one is located upstream from the trbA gene. The identification of the putative promoters sequences were based on the prediction of sigma factor binding sites using BPROM and bTSSfinder (see Table 1 and Supplementary Figure 1). The presence of the two identified putative promoter sequences and the small intercistronic region between the genes suggests that these genes might function as two operons, in addition to the tra1 operon (Figures 1, 2). The genetic organization of the tra2 and tra3 operons resembles the gene order of the tra operon found in the pADAP plasmid of S. entomophila, the R64 plasmid of S. enterica serovar Typhimurium, pCTX-M3 of C. freundii, pEL60, pEA68, pEA72, and pEA78 of E. amylovora (see Supplementary Figure 5), as previously suggested for the tra1 operon (Méndez et al., 2009).
The G+C content of the tra2 and tra3 operons (around 53%) differs from the G+C content of the chromosomes of Y. ruckeri NVH_3758 (47.6%), Y. ruckeri Big Creek 74 (47.6%), and Y. ruckeri CSF007-82 (47.5%). Additionally, the tra region was is not present in the chromosome of the Y. ruckeri strains mentioned above, indicating that the tra region may originate from another species. Méndez et al. suggested that S. entomophila, the causative agent of amber disease of the New Zealand grass grub, could be the source of the tra1 region. The G+C content of the S. entomophila pADAP tra region (tra1, tra2, tra3) is around 52%, which is close to the G+C content of pYR4. In addition, the gene order of that region is very similar. We suggest that the whole tra region encompassing tra1, tra2, and tra3 could have been acquired from this or a closely related Serratia species.
The amino acid sequences of TraH, TraI, TraJ, and TraK showed 29–34% similarity to the L. pneumophila T4BSS proteins such as DotD (for “defect in organelle trafficking”), DotC, DotB, and IcmT (for “intracellular multiplication”) (Wallden et al., 2010). The icm/dot genes are required for virulence, including intracellular growth and host cell killing (Sadosky et al., 1993; Swanson and Isberg, 1996). The pYR4 Tra proteins and their Tra homologs from L. pneumophila are similar in size, ranging from 87 residues (for TraK) to 385 residues (for TraJ). Interestingly, it has been shown before that a traI mutant strain of Y. ruckeri 150 was attenuated in an in vivo assay in rainbow trout and showed difficulty growing inside the fish (Méndez et al., 2009).
Plasmid Curing Study of pYR4 in Yersinia ruckeri NVH_3758
In order to understand the function of pYR4 and its involvement in pathogenesis, it is desirable to obtain a plasmid-cured strain. There is a wide number of plasmid curing procedures which have been successfully used to remove plasmids in Yersiniae. They for example include treatments with high temperatures or introduction of an incompatible plasmid (Sheridan et al., 1998; Ni et al., 2008). The genetic stability of the pYR4 plasmid was tested by treating Y. ruckeri cells with high temperature. Y. ruckeri NVH_3758 was grown in LB medium for 10 consecutive days at 37°C with dilutions each day (Trevors, 1986). After 10 days, colony PCR was performed on 10 clones using pil primers that could only bind to the plasmid sequence. All tested clones were PCR-positive for the pYR4 plasmid (data not shown). Based on our high quality genomic assembly, the pil operon is found only on the pYR4 plasmid and is absent from the chromosome. The results obtained from this analysis indicate that pYR4 is a very stable plasmid. The conjugative ability of the plasmid may maintain it in the population and determine its stability, so that even if individual cells lose it, they get it back from their neighbors. This suggests that the plasmid cannot be cured easily in a short time frame such, as the one that we tested. As we were unable to cure pYR4 within a reasonable amount of time, this precluded performing virulence assays to check the involvement of the plasmid in pathogenesis.
Discussion
Higher Quality of pYR4 Assembly and Identification of Unplaced Scaffolds
Comparative analysis of the pil region of pYR4 plasmid with other Y. ruckeri plasmids revealed that the pil region is not a common feature among Y. ruckeri strains. Even though we could identify different pil regions in some of the Y. ruckeri plasmids such as pWKY, we did not detect any significant sequence similarity with the pil region described here. Literature suggests that the pil operon can be encoded both on plasmids (pADAP of S. entomophila) (Hurst et al., 2011) and the chromosome (in Y. pseudotuberculosis 32777) (Collyn et al., 2002). Our data shows that in Y. ruckeri NVH_3758, the TFP gene cluster is plasmid-encoded and no other TFP gene cluster was detected on the chromosome. In fact, our genomic comparative survey indicates that previously deposited Illumina-sequenced genomic assemblies containing the tra and the pil operons—five scaffolds from a Y. ruckeri 150 assembly and a scaffold from Y. ruckeri ATCC29743—correspond to the plasmid pYR3. In particular for the 150 strain, failure in placing the genomic scaffolds may have occurred due to the presence of repetitive DNA sequences and of mobile elements in the flanking regions, e.g., the repetitive mobile element at positions 37,633–38,328 and 41,885–42,850 of pYR3 (Figure 3). This might also explain the presence of sequence gaps in the scaffolds 20 and 23 of ATCC29743 (Figure 3). These results show the power of the PacBio SMRT technology in producing higher-quality genomic assemblies, thanks to the longer average read lengths available.
Our study highlights the problem related to incorrect assemblies when using second generation sequencing (SGS) technologies. Short-read sequencing is often not enough to properly assemble plasmid sequences. Plasmids often contain many mobile repeat structures whose DNA length exceeds that provided by limitations of SGS technology (ranging 100–600 bp), thus generating unplaced scaffolds and mis-assemblies. The longer average read length provided by the PacBio SMRT sequencing can address some of the limitations of the SGS technologies, making it possible to correctly place genomic scaffolds even when containing repetitive regions and obtain higher quality assemblies. Sequencing the NVH_3758 genome using the PacBio technology yielded two contigs representing the chromosomal genome of ~3.8 Mb and the ~81 kb plasmid pYR4. Thanks to the PacBio platform, we could correctly determine the plasmid location of both the tra and the pil operons. The presence of the tra operon on the pYR4 plasmid, not on the chromosome, is reasonable as the tra operon encodes genes involved in bacterial conjugation and DNA transfer. We re-emphasize that these two systems, despite having similar names, are very distinct structurally and mechanistically. The T4SS is a secretion system that translocates nucleoprotein complexes or effector proteins into target cells, whereas TFP are contractile appendages mainly mediating adhesion and certain types of motility (Shi and Sun, 2002).
TFPs in pYR4 And Their Potential Role in Virulence and Conjugation
In the human pathogenic Yersiniae, pathogenicity is mainly related to the presence of the 70-kb virulence plasmid pYV. This plasmid encodes the Yop proteins and T3SS, which enable the bacteria to survive and multiply in the host tissues (Viboud and Bliska, 2005). Plasmids described so far in Y. ruckeri have recently gained more and more attention due to their potential association with virulence, although in-depth knowledge regarding their function is lacking.
Annotations of pYR4 showed 92 open reading frames. We could identify three different functional regions responsible for plasmid partitioning, a T4SS and a TFP. The present study strongly suggests that the pYR4 pil cluster belongs to the TFP family. In addition to attachment and motility, TFPs can be involved in DNA uptake as shown in N. gonorrhoeae (Wolfgang et al., 1998). Interestingly, there are examples of TFPs being involved in bacterial conjugation. For example, the PAPI-1 pathogenicity island of P. aeruginosa can be transferred to a recipient strain lacking this island. The mobilization of PAPI-1 was dependent on a TFP (Carter et al., 2010). The fact that the pil operon clusters together with the tra operon suggests that the two are functionally coupled and that the TFPs are involved in conjugative transfer of pYR4. However, it is also possible that the TFP is a virulence factor. We speculate that the pil proteins in pYR4, apart from being involved in virulence, can also be responsible for thin pilus formation required for liquid mating (Kim and Komano, 1997). Experiments carried out by Collyn et al. showed that the TFP gene cluster present in Y. pseudotuberculosis is not only involved in synthesis of TFPs, but also contributes to its virulence (Collyn et al., 2002). Based on these findings, we speculate that the pil operon present in Y. ruckeri could play a similarly important role in fish disease.
The difference between the G+C content of the pil operon (51.7%) and the average G+C content of the pYR4 plasmid (49.4%) suggests that the pil operon could have been acquired relatively recently by horizontal gene transfer. Likewise, the difference in G+C content compared to the Y. ruckeri NVH_3758 chromosome (47.6%) suggests that the plasmid has been acquired from a different species. Previous studies show that for a plasmid to be horizontally transferred, the difference in the G+C composition between the genome and the plasmid should be in the range from 1 to 5% (Hurst et al., 2011). The G+C content of the plasmid pADAP of S. entomophila is 53%. Most of the pil proteins from pYR4 (PilL, PilN, PilO, PilP, PilQ, PilR, PilS, PilT, PilD) are highly similar to pil proteins found in TFPs in Serratia species, whereas others (PilL, PilU, PilN) are more similar to those found in other Yersiniae. These findings, in addition to the same gene organization of the pil cluster, suggest that the TFP locus in pYR4 may have been acquired from S. entomophila, which occupies a similar aquatic environment as Y. ruckeri (Grimont et al., 1988).
T4SSs and the tra Operon
Analysis of the pYR4 protein encoding sequences allowed us to identify a complete T4SS, which we named tra. The corresponding coding region consists of three operons. The genetic organization of the tra operons (tra1, tra2, tra3) is very similar to that found in pADAP of S. entomophila, pCTX-M3 of C. freundii and pEL60, pEA67, pEA72, and pEA78 of E. amylovora, as previously described for tra1 (Méndez et al., 2009). Comparative analysis of the pYR4 tra operons to the pYR3 tra operons from Y. ruckeri CSF007-82 showed 99.9% nucleotide sequence similarity, in contrast to tra operons from other Y. ruckeri strains, where we did not detect any strong sequence similarity. In 2007, Welch et al. showed the presence of multidrug resistance of the plasmid pYR1 in Y. ruckeri YR71. pYR1 also contains a T4SS for conjugative transfer. However, the T4SS of pYR1 does not show high levels of sequence similarity—and no LCB was found in the Mauve alignment—to the tra operons described here (Welch et al., 2007).
Interestingly, similarities between the pYR4 Tra and L. pneumophila Icm/Dot proteins implicate a role in Y. ruckeri virulence. L. pneumophila is the causative agent of Legionnaires' disease. As an intracellular pathogen, L. pneumophila is able to grow and multiply within human macrophages, leading to their killing. In L. pneumophila, many virulence genes located in icm (“for intracellular multiplication”) and dot (“defect in organelle trafficking”) locus have been identified. Some of the Dot/Icm proteins are homologous to the Tra proteins found in plasmid R64 (Komano et al., 2000). pYR4 Tra proteins display amino acid sequence similarity to Icm/Dot proteins ranging from 29 to 34%. They are similar in size and their predicted functions are similar. However, as we could not identify any genes encoding putative effector proteins in pYR4 of NVH_3758, it is possible that the tra locus is purely conjugative. Nevertheless, we cannot conclusively rule out a role for the tra locus in virulence. Interestingly, Méndez et al. generated the traI mutant strain of Y. ruckeri 150 in which the traI gene was disrupted. In the in vivo competition assays in rainbow trout, the virulence of the traI mutant strain was reported to be attenuated when compared to the WT strain, suggesting that TraI is involved in virulence of this bacterium.
Conclusions
In conclusion, the results presented here suggest that pYR4 from the 1987 outbreak strain Y. ruckeri NVH_3758 is a conjugative plasmid that encodes a T4SS and a TFP that might contribute to Y. ruckeri virulence. The 55 kDa plasmid backbone is identical to that of pYR3 and has presumably been acquired by horizontal gene transfer through conjugation from a Serratia species that occupies the same biological niche, as previously suggested (Méndez et al., 2009). In addition, pYR4 contains a a previously undescribed ~25 kDa region with a partitioning system completely different from that of pYR3. This region contains a set of mobile elements, several hydrogenase enzymes with a corresponding transcription factor, and additional hypothetical genes that need further investigation.
The plasmid could contribute to the dissemination of Y. ruckeri virulence by spreading the TFP-encoding pil locus among non-virulent strains. Further experiments are required to elucidate the function of the pil and tra regions and to clarify their role in virulence. However, we would like to highlight that based on 100% sequence similarity between TraI of pYR4 and TraI of Y. ruckeri 150 that we now know is located on pYR3 based on our re-assembly of the Y. ruckeri 150 genome, we assume that at least the tra region is directly involved in virulence. traI deletion in Y. ruckeri 150 lead to a significant decrease in virulence (Méndez et al., 2009). Additionally, our study demonstrates the power of PacBio SMRT sequencing technology in producing assemblies of high quality and accuracy, compared to sequencing technologies based on shorter read lengths such as Illumina and 454. The latter methods failed to show the plasmid localization of the pil and tra regions in multiple cases.
Data Availability
The sequence of pYR4 described herein has been deposited in GenBank with the accession number CP032236.
Author Contributions
AW and CO conceived, planned, and carried out the experiments. AW, CO, JL, and DL contributed to the discussion and interpretation of the results, and wrote the manuscript. All authors provided critical feedback and contributed to the final shape of the manuscript.
Funding
This work by supported by Departmental funds of the Department of Biosciences, University of Oslo (COMPI), by Research Council of Norway Young Investigator grant 249793 (to JL), and by Research Council of Norway FriMedBio grant 240483 (to DL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HL and handling Editor declared their shared affiliation.
Acknowledgments
We would like to thank the sequencing service provided by the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo and supported by the Functional Genomics and Infrastructure programs of the Research Council of Norway and the Southeastern Regional Health Authorities. We express gratitude to Prof. Duncan Colquhoun (Norwegian Veterinary Institute) for providing Y. ruckeri NVH_3758.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00373/full#supplementary-material
References
Altschul, S., Madden, T., Schäffer, A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Avendaño-Herrera, R., Tapia-Cammas, D., Aedo, A., Saldivia, P., Ortega, C., and Irgang, R. (2017). Disease caused by Yersinia ruckeri serotype O2b found in chilean-farmed coho salmon, Oncorhynchus kisutch. J. Fish Dis. 40, 279–285. doi: 10.1111/jfd.12502
Aziz, R., Bartels, D., Best, A., DeJongh, M., Disz, T., Edwards, R., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Barnes, A., Delamare-Deboutteville, J., Gudkovs, N., Brosnahan, C., Morrison, R., and Carson, J. (2016). Whole genome analysis of Yersinia ruckeri isolated over 27 years in Australia and New Zealand reveals geographical endemism over multiple lineages and recent evolution under host selection. Microb. Genomics 2:e000095. doi: 10.1099/mgen.0.000095
Ben-Gurion, R., and Shafferman, A. (1981). Essential virulence determinants of different Yersinia are carried on a common plasmid. Plasmid 5, 183–187. doi: 10.1016/0147-619X(81)90019-6
Bottone, E. (1997). Yersinia enterocolitica: the charisma continues. Clin. Microbiol. Rev. 10, 257–276.
Bullock, G., Stuckey, H., and Shotts, J. R. E. (1978). Enteric redmouth bacterium: comparison of isolates from different geographic areas. J. Fish Dis. 1, 351–356. doi: 10.1111/j.1365-2761.1978.tb00039.x
Carattoli, A. (2009). Resistance plasmid families in Enterobacteriaceae. Antimicrob. Agents Chemother. 53, 2227–2238. doi: 10.1128/AAC.01707-08
Carattoli, A., Villa, L., Poirel, L., Bonnin, R., and Nordmann, P. (2012). Evolution of IncA /C bla CMY-2 -carrying plasmids by acquisition of the bla NDM-1 carbapenemase gene. Antimicrob. Agents Chemother. 56, 783–786. doi: 10.1128/AAC.05116-11
Carter, M., Chen, J., and Lory, S. (2010). The Pseudomonas aeruginosa pathogenicity island PAPI-1 is transferred via a novel type IV pilus. J. Bacteriol. 192, 3249–3258. doi: 10.1128/JB.00041-10
Chauhan, N., Wrobel, A., Skurnik, M., and Leo, J. (2016). Yersinia adhesins: an arsenal for infection. Proteomics-Clin. Appl. 10, 949–963. doi: 10.1002/prca.201600012
Collyn, F., Léty, M., Nair, S., Escuyer, V., Younes, A., Simonet, M., et al. (2002). Yersinia pseudotuberculosis harbors a type IV pilus gene cluster that contributes to pathogenicity. Infect. Immun. 70, 6196–6205. doi: 10.1128/IAI.70.11.6196-6205.2002
Cornelis, G., Boland, A., Boyd, A., Geuijen, C., Iriarte, M., Neyt, C., et al. (1998). The virulence plasmid of Yersinia, an antihost genome. Microbiol. Mol. Biol. Rev. 62, 1315–1352.
Couturier, M., Bex, F., Bergquist, P., and Maas, W. (1988). Identification and classification of bacterial plasmids. Microbiol. Rev. 52, 375–395.
Crosa, J. (1980). A plasmid associated with virulence in the marine fish pathogen Vibrio anguillarum specifies an iron sequestering system. Nature 284, 566–568. doi: 10.1038/284566a0
Darling, A., Mau, B., and Perna, N. (2010). Progressive mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
De Grandis, S., and Stevenson, R. (1982). Variations in plasmid profiles and growth characteristics of Yersinia ruckeri strains. FEMS Microbiol. Lett. 15, 199–202. doi: 10.1111/j.1574-6968.1982.tb00067.x
De Grandis, S., and Stevenson, R. (1985). Antimicrobial susceptibility patterns and R plasmid-mediated resistance of the fish pathogen Yersinia ruckeri. Antimicrob. Agents Chemother. 27, 938–942. doi: 10.1128/AAC.27.6.938
Edgar, R. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Fernández, L., Marquez, I., and Guijarro, J. (2004). Identification of specific in vivo-induced (ivi) genes in Yersinia ruckeri and analysis of ruckerbactin, a catecholate siderophore iron acquisition system. Appl. Environ. Microbiol. 70, 5199–5207. doi: 10.1128/AEM.70.9.5199-5207.2004
Fernández, L., Prieto, M., and Guijaro, J. (2007). The iron- and temperature-regulated haemolysin YhlA is a virulence factor of Yersinia ruckeri. Microbiology 153, 483–489. doi: 10.1099/mic.0.29284-0
Garcia, J., Dominguez, L., Larsen, J., and Pedersen, K. (1998). Ribotyping and plasmid profiling of Yersinia ruckeri. J. Appl. Microbiol. 85, 949–955. doi: 10.1111/j.1365-2672.1998.tb05258.x
Grimont, P., Jackson, T., Ageron, E., and Noonan, M. (1988). Serratia entomophila sp. nov. associated with amber disease in the New Zealand grass grub Costelytra zealandica. Int. J. Syst. Bacteriol. 38, 1–6. doi: 10.1099/00207713-38-1-1
Guilvout, I., Quilici, M., Rabot, S., Lesel, R., and Mazigh, D. (1988). BamHI restriction endonuclease analysis of Yersinia ruckeri plasmids and their relatedness to the genus Yersinia 42- to 47-megadalton plasmid. Appl. Environ. Microbiol. 54, 2594–2597.
Gulla, S., Barnes, A., Welch, T., Romalde, J., Ryder, D., Ormsby, M., et al. (2018). Multi-locus variable number of tandem repeat Analysis (MLVA) of Yersinia ruckeri confirms the existence of host-specificity, geographic endemism and anthropogenic dissemination of virulent clones. Appl. Environ. Microbiol. 84:e00730–18. doi: 10.1128/AEM.00730-18
Haiko, J., Suomalainen, M., Ojala, T., Lähteenmäki, K., and Korhonen, T. (2009). Invited review: breaking barriers – attack on innate immune defences by omptin surface proteases of enterobacterial pathogens. Innate Immun. 15, 67–80. doi: 10.1177/1753425909102559
Hayek, N. (2013). Lateral transfer and GC content of bacterial resistance genes. Front. Microbiol. 4:41. doi: 10.3389/fmicb.2013.00041
Hjeltnes, B., Bornø, G., Jansen, M., Haukaas, A., and Walde, C. (2017). The Health Situation in Norwegian Aquaculture 2016. Norwegian Veterinary Institute 2017.
Huang, Y., Michael, G., Becker, R., Kaspar, H., Mankertz, J., Schwarz, S., et al. (2014). Pheno- and genotypic analysis of antimicrobial resistance properties of Yersinia ruckeri from fish. Vet. Microbiol. 171, 406–412. doi: 10.1016/j.vetmic.2013.10.026
Hurst, M., Becher, S., and O'Callaghan, M. (2011). Nucleotide sequence of the Serratia entomophila plasmid pADAP and the Serratia proteamaculans pU143 plasmid virulence associated region. Plasmid 65, 32–41. doi: 10.1016/j.plasmid.2010.10.001
Isberg, R., and Leong, J. (1990). Multiple β1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60, 861–871. doi: 10.1016/0092-8674(90)90099-Z
Jalava, K., Hakkinen, M., Valkonen, M., Nakari, U., Palo, T., Hallanvuo, S., et al. (2006). An outbreak of gastrointestinal illness and erythema nodosum from grated carrots contaminated with Yersinia pseudotuberculosis. J. Infect. Dis. 194, 1209–1216. doi: 10.1086/508191
Johnson, T., and Lang, K. (2012). IncA/C plasmids: an emerging threat to human and animal health? Mob. Genet. Elements 2, 55–58. doi: 10.4161/mge.19626
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, S., and Komano, T. (1997). The plasmid R64 thin pilus identified as a type IV pilus. J. Bacteriol. 179, 3594–3603. doi: 10.1128/jb.179.11.3594-3603.1997
Komano, T., Yoshida, T., Narahara, K., and Furuya, N. (2000). The transfer region of IncI1 plasmid R64: similarities between R64 tra and Legionella icm/dot genes. Mol. Microbiol. 35, 1348–1359. doi: 10.1046/j.1365-2958.2000.01769.x
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 18, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, G., Hummel, K., Ahrens, M., Menanteau-Ledouble, S., Welch, T., Eisenacher, M., et al. (2016). Shotgun proteomic analysis of Yersinia ruckeri strains under normal and iron-limited conditions. Vet. Res. 47:100. doi: 10.1186/s13567-016-0384-3
Kumar, G., Hummel, K., Welch, T., Razzazi-Fazeli, E., and El-Matbouli, M. (2017). Global proteomic profiling of Yersinia ruckeri strains. Vet. Res. 48:55. doi: 10.1186/s13567-017-0460-3
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Liu, T., Wang, K., Wang, J., Chen, D., Huang, X., Ouyang, P., et al. (2016). Genome sequence of the fish pathogen Yersinia ruckeri SC09 provides insights into niche adaptation and pathogenic mechanism. Int. J. Mol. Sci. 17:557. doi: 10.3390/ijms17040557
Méndez, J., Fernández, L., Ménendez, A., Reimundo, P., Pérez-Pascual, D., Navais, R., et al. (2009). A chromosomally located traHIJKCLMN operon encoding a putative type IV secretion system is involved in the virulence of Yersinia ruckeri. Appl. Environ. Microbiol. 75, 937–945. doi: 10.1128/AEM.01377-08
Mühlenkamp, M., Oberhettinger, P., Leo, J., Linke, D., and Schütz, M. (2015). Yersinia adhesin A (YadA) – Beauty & beast. Int. J. Med. Microbiol. 305, 252–258. doi: 10.1016/j.ijmm.2014.12.008
Nelson, M., Lapatra, S., Welch, T. (2015). Complete genome sequence of Yersinia ruckeri strain CSF007-82, etiologic agent of red mouth disease in salmonid fish. Genome Announc. 3:e01491–14. doi: 10.1128/genomeA.01491-14
Ni, B., Du, Z., Guo, Z., Zhang, Y., and Yang, R. (2008). Curing of four different plasmids in Yersinia pestis using plasmid incompatibility. Lett. Appl. Microbiol. 47, 235–240. doi: 10.1111/j.1472-765X.2008.02426.x
Nishida, H. (2012). Comparative analyses of base compositions, DNA sizes, and dinucleotide frequency profiles in archaeal and bacterial chromosomes and plasmids. Int. J. Evol. Biol. 2012:342482. doi: 10.1155/2012/342482
Ormsby, M., Caws, T., Burchmore, R., Wallis, T., Verner-Jeffreys, D., and Davies, R. (2016). Yersinia ruckeri isolates recovered from diseased atlantic salmon (Salmo salar) in scotland are more diverse than those from rainbow trout (Oncorhynchus mykiss) and represent distinct subpopulations. Appl. Environ. Microbiol. 82, 5785–5794. doi: 10.1128/AEM.01173-16
Perry, R., and Fetherston, J. (1997). Yersinia pestis — etiologic agent of plague. Clin. Microbiol. Rev. 10, 35–66.
Pilla, G., and Tang, C. M. (2018). Going around in circles: virulence plasmids in enteric pathogens. Nat. Rev. Microbiol. 16, 484–95. doi: 10.1038/s41579-018-0031-2
Reuter, S., Connor, T., Barquist, L., Walker, D., Feltwell, T., Harris, S., et al. (2014). Parallel independent evolution of pathogenicity within the genus Yersinia. Proc. Natl. Acad. Sci. U.S.A. 111, 6768–6773. doi: 10.1073/pnas.1317161111
Romalde, J., MagariÑos, B., Barja, J., and Toranzo, A. (1993). Antigenic and molecular characterization of Yersinia ruckeri proposal for a new intraspecies classification. Syst. Appl. Microbiol. 16, 411–419. doi: 10.1016/S0723-2020(11)80274-2
Romalde, J. L., and Toranzo, A. E. (1993). Pathological activities of Yersinia ruckeri, the enteric redmouth (ERM) bacterium. FEMS Microbiol. Lett. 112, 291–299. doi: 10.1111/j.1574-6968.1993.tb06465.x
Sadosky, A. B., Wiater, L. A., and Shuman, H. A. (1993). Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61, 5361–5373.
Savin, C., Martin, L., Bouchier, C., Filali, S., Chenau, J., Zhou, Z., et al. (2014). The Yersinia pseudotuberculosis complex: characterization and delineation of a new species, Yersinia wautersii. Int. J. Med. Microbiol. 304, 452–463. doi: 10.1016/j.ijmm.2014.02.002
Secades, P., and Guijarro, J. A. (1999). Purification and characterization of an extracellular protease from the fish pathogen Yersinia ruckeri and effect of culture condition on production. Appl. Environ. Microbiol. 65, 3969–3975.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shahmuradov, I., Mohamad Razali, R., Bougouffa, S., Radovanovic, A., and Bajic, V. (2017). bTSSfinder: a novel tool for the prediction of promoters in cyanobacteria and Escherichia coli. Bioinformatics 33, 334–340. doi: 10.1093/bioinformatics/btw629
Sheridan, J. J., Logue, C. M., McDowell, D. A., Blair, I. S., and Hegarty, T. (1998). A study of the growth kinetics of Yersinia enterocolitica serotype O:3 in pure and meat culture systems. J. Appl. Microbiol. 85, 293–301. doi: 10.1046/j.1365-2672.1998.00504.x
Shi, W., and Sun, H. (2002). Type IV pilus-dependent motility and its possible role in bacterial pathogenesis. Infect. Immun. 70, 1–4. doi: 10.1128/IAI.70.1.1-4.2002
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, 244–248. doi: 10.1093/nar/gki408
Solovyev, V., and Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, ed R. W. Li (New York, NY: Nova Science Publishers), 61–78.
Sonnhammer, E., Eddy, S., Birney, E., Bateman, A., and Durbin, R. (1998). Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26, 320–322. doi: 10.1093/nar/26.1.320
Sparboe, O., Koren, C., Hastein, T., Poppe, T., and Stenwig, H. (1986). The first isolation of Yersinia ruckeri from farmed Norwegian salmon. Bull. Eur. Assoc. Fish Pathol. 6, 41–42.
Stevenson, R., and Airdrie, R. (1984). Serological variation among Yersinia ruckeri strains. J. Fish Dis. 7, 247–254. doi: 10.1111/j.1365-2761.1984.tb00930.x
Sussman, J. L., Abola, E. E., Lin, D., Jiang, J., Manning, N. O., and Prilusky, J. (1999). The protein data bank. Bridging the gap between the sequence and 3D structure world. Genetica 1062, 149–158. doi: 10.1023/A:1003753517358
Swanson, M. S., and Isberg, R. R. (1996). Identification of Legionella pneumophila mutants that have aberrant intracellular fates. Infect. Immun. 64, 2585–2594.
Toranzo, A. E., Barja, J. L., Colwell, R. R., and Hetrick, F. M. (1983). Characterization of plasmids in bacterial fish pathogens. Infect. Immun. 39, 184–192.
Trevors, J. T. (1986). Plasmid curing in bacteria. FEMS Microbiol. Lett. 32, 149–157. doi: 10.1111/j.1574-6968.1986.tb01189.x
Viboud, G. I., and Bliska, J. B. (2005). Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89. doi: 10.1146/annurev.micro.59.030804.121320
Villa, L., García-Fernández, A., Fortini, D., and Carattoli, A. (2010). Replicon sequence typing of IncF plasmids carrying virulence and resistance determinants. J. Antimicrob. Chemother. 65, 2518–2529. doi: 10.1093/jac/dkq347
Wallden, K., Rivera-Calzada, A., and Waksman, G. (2010). Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12, 1203–1212. doi: 10.1111/j.1462-5822.2010.01499.x
Welch, T. J., Fricke, W. F., Mcdermott, P. F., White, D. G., Rosso, M., Rasko, D. A., et al. (2007). Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS ONE 2:e309. doi: 10.1371/journal.pone.0000309
Wolfgang, M., Lauer, P., Park, H., Brossay, L., Hebert, J., and Koomey, M. (1998). PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol. Microbiol. 29, 321–330. doi: 10.1046/j.1365-2958.1998.00935.x
Wrobel, A., Ottoni, C., Leo, J. C., Gulla, S., and Linke, D. (2017). The repeat structure of two paralogous genes, Yersinia ruckeri Invasin (yrInv) and a “Y. ruckeri Invasin-like molecule”, (yrIlm) sheds light on the evolution of adhesive capacities of a fish pathogen. J. Struct. Biol. 201, 171–183. doi: 10.1016/j.jsb.2017.08.008
Yang, Q., Sun, J., Li, L., Deng, H., Liu, B., Fang, L., et al. (2015). IncF plasmid diversity in multi-drug resistant Escherichia coli strains from animals in China. Front. Microbiol. 6:964. doi: 10.3389/fmicb.2015.00964
Yu, J., Cho, M., Kim, J., and Kang, H. (2012). Large antibiotic-resistance plasmid of Edwardsiella tarda contributes to virulence in fish. Microb. Pathog. 52, 259–266. doi: 10.1016/j.micpath.2012.01.006
Zhang, X.-L., Tsui, I. S. M., Yip, C. M. C., Fung, A. W. Y., Wong, D. K.-H., Dai, X., et al. (2000). Salmonella enterica Serovar typhi uses type IVB Pili to enter human intestinal epithelial cells. Infect. Immun. 68, 3067–3073. doi: 10.1128/IAI.68.6.3067-3073.2000
Zuckerkandl, E., and Pauling, L. (1965). “Evolutionary divergence and convergence in proteins,” in Evolving Genes and Proteins, eds V. Bryson and H. J. Vogel (New York, NY: Academic Press), 97–166.
Keywords: tra operon, pil operon, conjugative plasmid, Yersinia ruckeri, type IV secretion system
Citation: Wrobel A, Ottoni C, Leo JC and Linke D (2018) pYR4 From a Norwegian Isolate of Yersinia ruckeri Is a Putative Virulence Plasmid Encoding Both a Type IV Pilus and a Type IV Secretion System. Front. Cell. Infect. Microbiol. 8:373. doi: 10.3389/fcimb.2018.00373
Received: 06 July 2018; Accepted: 04 October 2018;
Published: 30 October 2018.
Edited by:
Victoria Auerbuch, University of California, Santa Cruz, United StatesReviewed by:
Hanh N. Lam, University of California, Santa Cruz, United StatesGokhlesh Kumar, Veterinärmedizinische Universität Wien, Austria
Copyright © 2018 Wrobel, Ottoni, Leo and Linke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Linke, ZGlyay5saW5rZUBpYnYudWlvLm5v