 Naveen Thakur1
Naveen Thakur1 Amar Nath Sharma1
Amar Nath Sharma1 Mangesh Dattu Hade1
Mangesh Dattu Hade1 Ajay Chhaya2
Ajay Chhaya2 Ashwani Kumar1
Ashwani Kumar1 Ravinder Singh Jolly1
Ravinder Singh Jolly1 Kanak L. Dikshit1,2*
Kanak L. Dikshit1,2*- 1CSIR-Institute of Microbial Technology, Chandigarh, India
- 2Department of Biotechnology, Panjab University, Chandigarh, India
Mycobacterium tuberculosis (Mtb) produces an unconventional flavohemoglobin (MtbFHb) that carries a FAD-binding site similar to D-lactate dehydrogenases (D-LDH) and oxidizes D-lactate into pyruvate. The molecular mechanism by which MtbFHb functions in Mtb remains unknown. We discovered that the D-LDH-type FAD-binding site in MtbFHb overlaps with another FAD-binding motif similar to thioredoxin reductases and reduces DTNB in the presence of NADPH similar to trxB of Mtb. These results suggested that MtbFHb is functioning as a disulfide oxidoreductase. Interestingly, D-lactate created a conformational change in MtbFHb and attenuated its ability to oxidize NADPH. Mass spectroscopy demonstrated that MtbFHb reduces des-myo-inositol mycothiol in the presence of D-lactate unlike NADPH, indicating that D-lactate changes the specificity of MtbFHb from di-thiol to di-mycothiol. When M. smegmatis carrying deletion in the fhbII gene (encoding a homolog of MtbFHb) was complemented with the fhb gene of Mtb, it exhibited four- to fivefold reductions in lipid peroxidation and significant enhancement in the cell survival under oxidative stress. These results were corroborated by reduced lipid peroxidation and enhanced cell survival of wild-type M. smegmatis after overexpression of the fhb gene of Mtb. Since D-lactate is a by-product of lipid peroxidation and MtbFHb is a membrane-associated protein, D-lactate-mediated reduction of mycothiol disulfide by MtbFHb may uniquely equip Mtb to relieve the toxicity of D-lactate accumulation and protect the cell from oxidative damage, simultaneously balancing the redox environment under oxidative stress that may be vital for the pathogenesis of Mtb.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, continues to be a major threat to human life, causing millions of deaths annually (Bloom and Murray, 1992; Dye, 2006). The extraordinary ability of the tubercle bacillus to survive within the intracellular environment of its host, where a highly toxic environment exists due to scarcity of oxygen and high levels of toxic reactive species, depends primarily on its multi-tier defense system and metabolic flexibility. Being an obligate aerobe, Mtb continually remains exposed to endogenous reactive oxygen as a part of normal aerobic respiration and additionally gets exposed to copious amounts of host-generated toxic reactive species during intracellular infection (Chan et. al., 1995; Cook et al., 2009). To encounter these bactericidal components of the host, Mtb produces a number of scavenging enzymes for reactive oxygen and nitrogen species, such as superoxide dismutase, catalases, peroxidases (Manca et al., 1999; Voscuil et al., 2011), and truncated hemoglobins (Ouellet et al., 2002; Pathania et al., 2002). Together with antioxidants like thioredoxins, peroxiredoxins, and mycothiol, these enzymes provide highly efficient machinery to Mtb for the detoxification of toxic reactive species and balancing the redox environment of the cell (Bryk et al., 2000; Arner and Holmgren, 2000; Kumar et al., 2011; Zeida et al., 2015). Mycothiol, a functional analogue of glutathione, is the major low-molecular-mass thiol of actinomycetes and is present in high levels in Mtb (Newton et al., 1996; Reyes et al., 2018) where it acts as a major detoxification system not only against reactive oxygen and nitrogen species but also against toxins, electrophiles, and other reactive species (Buchmeier et al., 2003; Ung and Av-Gay, 2006). Under oxidative stress, mycothiol is oxidized into mycothione, which gets reduced and recycled into mycothiol by a flavoprotein, mycothione reductase, using NADPH as a co-factor (Patel and Blanchard, 1998; Patel and Blanchard, 1999). The genome of Mtb encodes a single copy of the trxR gene, encoding thioredoxin reductase (Bryk et al., 2002) and a NADPH-dependent disulfide reductase (Mtr), specific for mycothione (Patel and Blanchard, 1998). These flavoenzymes constitute crucial components of the antioxidant network of Mtb and play an important role in balancing the redox environment of the cell under different physiological conditions.
Mtb produces a unique hexa-coordinated flavohemoglobin (MtbFHb) having antioxidant properties (Gupta et al., 2012). It belongs to a new class of two domain Hbs (type II FHbs) that is distinct from conventional type I FHbs (Bonamore and Boffi, 2008) and exclusively present in actinomycetes (Gupta et al., 2011). MtbFHb carries a FAD-binding motif, similar to D-lactate dehydrogenases and oxidizes D-lactate into pyruvate in a FAD-dependent manner. The functional relevance of using D-lactate as an electron donor and the molecular mechanism by which MtbFHb operates under oxidative stress are not known at present. The D-lactate metabolizing enzyme, D-lactate dehydrogenase (D-LDH), is missing in many mycobacterial species (Pinchuk et al., 2009), and a D-LDH activity has not been demonstrated in Mtb so far. It led to the speculation that MtbFHb provides a unique ability to Mtb for eliminating the accumulation of D-lactate that is produced in the cells as a by-product of lipid peroxidation under oxidative stress. MtbFHb represents the first example of a flavin and heme containing two-domain protein that mediates electron transfer from D-lactate to the heme domain via FAD bound to the reductase domain. Interestingly, the FAD binding site, similar to D-LDH, is present in type II FHbs of only few virulent mycobacteria. This co-factor binding site is absent or mutated in most of the pathogenic and non-pathogenic mycobacteria, suggesting that D-lactate-mediated electron transfer may not be the primary function of type II FHbs and may be specifically required in some pathogenic mycobacteria. The functional relevance of D-lactate oxidation and its role in modulating the physiological function(s) of MtbFHb remain to be elucidated.
Here, we have identified a new co-factor binding site within the reductase domain of MtbFHb, which is similar to the FAD-binding site of thioredoxin reductases (Dym and Eisenberg, 2001). Interestingly, this FAD-binding site overlaps with the previously identified D-LDH-type FAD-binding site of MtbFHb, indicating that these FAD-binding sites may have cooperating functions in electron transfer. The present study unravels the functional relevance of two overlapping FAD-binding sites in MtbFHb and provides new insights into the molecular mechanism of MtbFHb function in Mtb. Here, we have demonstrated that MtbFHb carries two distinct disulfide reductase activities and acts as a NADPH-dependent disulfide reductase and a D-lactate-dependent mycothiol disulfide reductase.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
Escherichia coli JM109 and E. coli BL21DE3 were used routinely for the cloning and the expression of recombinant proteins, respectively. Cultures of E. coli were grown in Luria-Bertani (LB) or Terrific broth (containing 24 g of yeast extract, 12 g of Bacto-tryptone, 12.3 g of K2HPO4, and 2.3 g of KH2PO4) at 37°C and 180 rpm. Mycobacterial strains, M. tuberculosis H37Ra (designated as Mtb) and M. smegmatis mc2 155, were used for the experimental studies and were grown in Middlebrook 7H10 agar (Difco) or 7H9 broth, supplemented with OADC (10% bovine serum albumin fraction V, dextrose, and sodium chloride), 0.2% glycerol, and 0.05% Tween 80. When required, kanamycin (Sigma) and hygromycin B (Sigma) were added at a concentration of 50 and 200 µg/ml, respectively, for E. coli and 25 and 50 µg/ml, respectively for selective growth of mycobacteria. The plasmids, p19Kpro (Garbe et al., 1994) and pVV16 (Stover et al., 1991), were used for the expression of recombinant genes in mycobacteria.
Cloning, Expression, and Purification of Mycobacterial FHbs
Mycobacterial genes, Rv0385 and MSMEG_0719, encoding the fhb gene of Mtb and fhbII gene of M. smegmatis, respectively, were cloned and expressed as described earlier (Gupta et al., 2012; Thakur et al., 2018). Recombinant FHbs when expressed in E. coli appeared to be associated with the cell membrane and only a minor fraction of the protein came in soluble cytoplasmic fraction. The soluble protein fraction of MtbFHb and MsFHbII were purified essentially as described earlier (Gupta et al., 2012; Thakur et al., 2018). The protein and hemoglobin profiles were monitored at 280 and 414 nm, respectively. The fhb gene of Mtb and the fhbII gene of M. smegmatis were cloned on E. coli-mycobacterial expression vectors, p19Kpro, at BamHI-HindIII sites and at NdeI-BamHI sites on pVV16. Resulting plasmids, designated as p19kproMtbFHb and pVV16MtbFHb, were transformed via electroporation into M. smegmatis following standard procedure for the expression of recombinant proteins.
Site-Directed Mutagenesis of MtbFHb
Three cysteine residues (cys188, cys289, and cys360) of MtbFHb were mutated individually using overlapping extension PCR to create three cysteine mutants, MtbFHbcys188ala, MtbFHbcys289ala, and MtbFHbcys360ala, where the cysteine residue was mutated to alanine. A list of primers to create these mutants is provided in the Supplementary Materials. These mutants were cloned on an expression vector, pET9b, as described earlier for wild-type MtbFHb (Gupta et al., 2012). Mutant MtbFHbcys188ala was expressed in E. coli and protein was purified as mentioned previously (Gupta et al., 2012). No protein expression was observed for two other cysteine mutants, MtbFHbcys289ala and MtbFHbcys360ala.
Treatment of MtbFHb by Iodoacetamide
Iodoacetamide (IMA), a sulfhydryl-reactive alkylating agent, has been used to alkylate cysteines in MtbFHb to check the role of cysteine in the protein function. Alkylation of the protein was carried out using a reduction alkylation system (G-biosciences). The reaction was set up in 500 µl of Tris-Cl (pH 7.2) buffer carrying 400 µg MtbFHb mutant (MtbFHbcys188ala), 2 µl of reduction buffer, and 10 µl of reductant solution from the kit. The reaction mixture was incubated at 37°C for 30 min to reduce the protein. Thereafter, alkylation of the protein was performed by adding 2.5 µl of alkylation buffer and 25 µl of 0.4 M of freshly prepared iodoacetamide. The reaction was incubated at room temperature for 30 min protected from light. Reaction mixture was then passed through G-50 desalting column to remove salts from the protein.
Disulfide Reductase Activity Assay
Disulfide reductase activity of the protein was determined by reduction of insulin. Insulin stock solution (10 mg/ml) was prepared in 0.05 M Tris-Cl (pH 8.0) adjusting pH 2 to 3 by the addition of 1.0 M HCl and rapidly titrating the solution back to 8.0 with 1.0 M NaOH. The solution of insulin was perfectly clear and was stored at −20°C. The final reaction mixture (500 µl) contained 0.1 M potassium phosphate (pH 7.0), 2 mM EDTA, 0.13 mM insulin, and 0.33 mM dithiothreitol (DTT). The reaction was initiated by adding 2 µM protein and an increase in the absorbance at 650 nm was monitored to check the reduction of insulin. NADPH-dependent disulfide reductase activity of FHbs was checked by the reduction of DTNB [5,5’-dithiobis-(2-nitrobenzoic acid)] using NADPH as an electron donor. The assay was performed at room temperature in a buffer containing 100 mM potassium phosphate (pH 7.0), 2 mM EDTA, 5 mM DTNB, 250 µM NADPH, and 2 µM of FHb protein. Reduction of DTNB by MtbFHb and MsFHbII was monitored by the increase in absorbance at 412 nm. The kinetic constants were checked by varying the concentration of DTNB (50 µM to 500 µM) and keeping the concentration of protein constant (2 µM). DTNB reduction by both the FHbs was calculated by fitting the data to the Michaelis–Menten equation using GraphPad prism 8.
Electron Transfer and Reduction of MtbFHb Bound Co-Factors by NADPH and D-Lactate
Oxidation of NADPH and D-lactate as well as the reduction of FAD and the heme by MtbFHb and MsFHbII was checked spectrophotometrically as described previously (Singh et al., 2014). Briefly, the reaction was set up in 500 µl with 8 µM oxygenated MtbFHb or MsFHbII in 0.05 M potassium phosphate buffer (pH 7.2). Reaction was initiated after adding a specified amount of NADPH or D-lactate. Spectral changes exhibiting conversion of oxy-FHb into the ferrous form were monitored at different time intervals spectrophotometrically. Reduction of FAD was checked by the decrease in the absorbance at 460 nm, while reduction of heme was followed by the shift of Soret peak at 414 to 430 nm.
NADP/NADPH Oxidase Activity
Oxidation of NADPH or NADH was checked spectrophotometrically by monitoring spectral changes at 340 nm. The assay was performed in 50 mM phosphate buffer (pH 7.0) containing 1 mM protein (MtbFHb or MsFHbII) and 1 mM EDTA. Reaction was initiated by adding 250 μM NADH or NADPH and spectra were recorded at 340 nm at specified time intervals as mentioned previously (Thakur et al., 2014).
Circular Dichroism Spectroscopy
CD spectra of proteins were checked using a Jasco J-810 spectropolarimeter. Measurements in the far-UV region (250 to 190 nm) were carried out on protein solutions (0.4 mg/ml) employing a cell with a path length of 0.1 cm at 25°C. The mean residue ellipticity (θ) was calculated using a mean residue molecular mass of 110 Da. Each spectrum reported represents an average of 5 scans.
Assay for Lipid Peroxidation
The lipid peroxides were determined through colorimetric assay using FOX II reagent (G-Bioscience) following manufacturer instructions. This reaction sequence involves the oxidation of ferrous to ferric ions by lipid peroxide with the subsequent binding of ferric ion to the ferric sensitive dye xylenol orange, yielding an orange to purple complex (color depends on the amount of –OOH present), which is measured at 560 nm. Lipid peroxidation in the cell sample was determined following the published procedure (Cha et al., 2004). Briefly, cell pellet (having 50 µg protein) was suspended in 1 ml of 50 mM Tris-Cl (pH 7.4) containing 0.5% SDS and sonicated, followed by removal of SDS after washing with deionized water twice. Resulting pellet was then dissolved in 1 ml methanol/chloroform (2:1 v/v) solution. The suspension was vortexed at room temperature. From this, 200 µl aliquot was taken and mixed with 800 µl of FOXII reagents and incubated for 30 min. The level of lipid peroxide in the cell was monitored by measuring the absorbance at 560 nm using the standard curve prepared from H2O2.
Synthesis of Des-Myo-Inositol Disulfide Mycothione
Des-myo-inositol mycothione, a synthetic analog of mycothione, was synthesized chemically exactly following the published procedure (Patel and Blanchard, 1998). All experiments were performed at 25°C. 1NMR spectra were obtained at Jeol ECX300 MHz spectrophotometer. Details of synthesis are provided in Supplementary Material.
Mass Spectrometric Analysis of Des-Myo-Inositol Mycothione
In the absence of any spectrophotometric method to detect D-lactate-mediated reduction of mycothiol disulfide (mycothione), we used mass spectrophotometric analysis of the product after reduction of des-myo-inositol disulfide mycothione. All assays were performed in a 100-µl reaction mixture containing 50 mM HEPES (pH 7.6) and 0.1 mM EDTA. MtbFHb and MsFHbII (5 µg each) were incubated individually with 100 μM des-myo-inositol mycothione along with 250 μM NADPH or 250 μM D-lactate at 25°C for 5 min and the product was analyzed via LC-MS using C18 column, with acetonitrile and water gradient. MSH was eluted at 3.81–4.20 min.
Generation of fhb Gene Knock Out Mutants of Mycobacteria
To investigate the physiological function of mycobacterial FHbs, attempts were made to create FHb knock out strains of Mtb and M. smegmatis using plasmid vector pML 523 following the procedure described earlier (Song et al., 2009). Briefly, 1,100-bp upstream and downstream regions of the fhb gene of Mtb and fhbII gene of M. smegmatis, carrying 125-bp coding regions of amino terminus with SpeI restriction site at the N-terminus and SwaI restriction site at the C-terminus, were amplified by PCR (list of primers is provided in Supplemental Materials). Each amplified product was cloned at the SpeI and the SwaI sites of plasmid vector, pML523. Similarly, 1,100-bp downstream regions of the fhb gene of Mtb and the fhbII gene of M. smegmatis carrying 150-bp coding regions of the C-terminus was amplified individually carrying the PacI restriction site at the N-terminus and the NsiI restriction site at the C-terminus. These amplified products were cloned individually at the PacI and NsiI restriction sites of pML523 plasmid. Resulting gene knock out plasmids, pML523MtbFHb carrying upstream and downstream regions of the Mtb fhb gene, and pML523MsFHbII carrying upstream and downstream regions of MsfhbII gene were used transformed into Mtb and M. smegmatis for the fhb and fhbII gene deletion, respectively. Transformed cells were plated on Middlebrook 7H9 agar plates carrying 50 µg/ml hygromycin and transformants were screened for the expression of GFP (green fluorescent protein) and the presence of XylE after spraying 2% catechol using published procedure (Song et al., 2009). No colonies were obtained for Mtb after repeated attempts but few colonies were obtained for M. smegmatis. These colonies were screened for the gene knock out. Deletion of the fhbII gene in M. smegmatis was confirmed through multiple PCR primer sets specific for the gene and resulting strain was designated as MsΔfhbII (Figure S7).
Quantitative RT-PCR
Wild type, MsΔfhbII, and Mtb fhb gene overexpressing cells of M. smegmatis were cultured in Middlebrook 7H9 medium at 37°C at 200 rpm for 18 h. Thereafter, cells were taken out and centrifuged at 14,000 rpm. RNA was purified using TRI-Reagent (Sigma-Aldrich) as per manufacturer instructions. All the samples were treated with DNA-free DNase (Ambion) to remove any contaminating DNA. Thereafter, cDNA was synthesized using the Superscript III first strand synthesis system and qRT-PCR was performed in a Bio-Rad cycler using 2x SYBR real-time PCR pre-mix (Sso Fast™ Evagreen Supermix, Bio-Rad). Primers used for qRT-PCR are listed in Supplementary Material. The sigA gene was used as housekeeping control. Relative fold change in the transcript level of the fhbII gene and Mtb fhb gene in M. smegmatis was calculated. All qRT-PCR experiments were performed with three replicates and signals were normalized to the housekeeping sigA mRNA and quantified by the Livak (2−ΔΔCt) method (Livak and Schmittgen, 2001). Statistical analysis was performed using one-tailed t-test, and the results were plotted based on average and standard deviation taken from three replicates using GraphPad Prism version 8.4.3 for windows.
Growth Properties and Stress Responses of MsΔfhbII and Wild-Type M. smegmatis After Expression of the Mtb fhb Gene
Since the MsΔfhbII strain of M. smegmatis is resistant to hygromycin, we transformed pVVMtbFHb into wild type and MsΔfhbII strains of M. smegmatis to select kanamycin-resistant colonies expressing the Mtb fhb gene. Growth properties of recombinant strains along with its isogenic wild-type M. smegmatis were checked under aerobic and oxidative stress conditions. Overnight grown cells of wild type and MtbFHb expressing MsΔfhbII strains of M. smegmatis as well as their isogenic control cells were freshly inoculated into Middlebrook 7H9 growth medium to adjust the final cell OD600 at 0.05 nm. Hygromycin (25 µg/ml) or kanamycin (25 µg/ml) was supplemented wherever required. Growth profiles of these cells were checked periodically by taking optical density at 600 nm. H2O2, CHP, and diamide were used as oxidants to impose oxidative stress. For CFU count, Mtb cells were grown to mid-log phase and washed twice in 7H9 medium and single-cell suspension was then prepared by centrifuging the culture at 8,000 rpm for 10 min in the presence of sterilized glass beads. The OD of cells was adjusted to 0.03. Cells were then exposed to oxidants for 30 min and CFU was checked after plating and counting colonies after 2 days.
Statistical Analysis
Statistical analysis of the data was carried out using two-way ANOVA followed with a post-hoc Dunnett multiple comparison test and one-tailed t-test. Transcript level was quantified by the Livak (2−ΔΔCt) method, and results were plotted based on average and standard deviation taken from three replicates using GraphPad Prism version 8.4.3 for windows.
Results
MtbFHb Carries Two Overlapping FAD-Binding Sites
MtbFHb, encoded by Rv0385 gene of Mtb, has been identified as a novel hexa-coordinated flavo-heme protein carrying a globin and a reductase domain (Gupta et al., 2012). The reductase domain of MtbFHb has a FAD-binding motif similar to respiratory D-LDH and oxidizes D-lactate into pyruvate in a FAD-dependent manner. FAD-binding to this co-factor binding motif of MtbFHb has been validated earlier by site-directed mutagenesis (Gupta et al., 2012). Structure-based sequence alignment of MtbFHb with its homologs (type II FHbs) in mycobacteria revealed that except Mtb and few virulent mycobacteria, the D-LDH type FAD-binding site is absent or mutated in other type II FHbs of mycobacteria and other actinomycetes (Figure S1), indicating that the oxidation of D-lactate may not be the primary function of MtbFHb and its homologs. Detailed sequence analysis revealed the presence of a new FAD-binding site (D(x)8GxxP) within the reductase domain of MtbFHb, which is similar to the FAD-binding site of thioredoxin reductases (Dym and Eisenberg, 2001). This FAD-binding motif overlaps with previously identified D-LDH-type FAD-binding site in MtbFHb and appeared conserved among homologs of MtbFHb present in mycobacteria and other actinomycetes (Figures 1 and S1), suggesting that the primary function of MtbFHb and its homologs may be related to disulfide oxidoreductases.
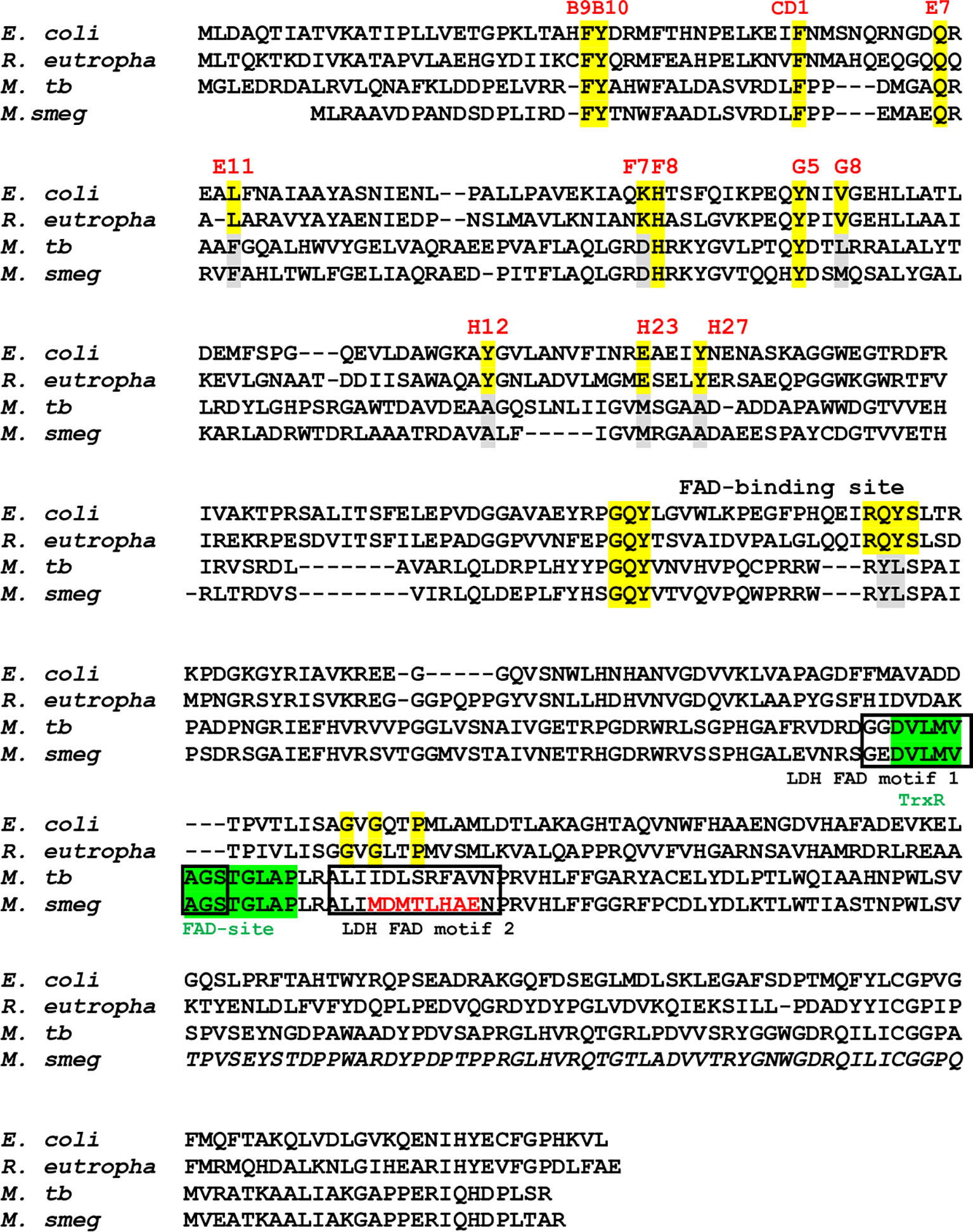
Figure 1 Sequence alignment of type I FHbs of E. coli and R. eutropha with type II FHbs of Mtb and M. smegmatis showing key residues of globin and reductase domains. The conserved residues within the globin domain are marked and highlighted in yellow and mutated residues of type II residues are shown in gray. Putative thioredoxin reductase-type FAD binding site is highlighted in green [D(x) 8GxxP] and LDH-type FAD binding sites 1 and 2 [A(x)7AxN, G(x)7 GS] are shown in black boxes. Mutation within the intergenic region of D-LDH binding site of MsFHbII is shown in red. Bacterial strain are designated as E. coli (Escherichia coli), R. eutropha (Ralstonia eutropha), M. tb (Mycobacterium tuberculosis), and M. smeg (Mycobacterium smegmatis).
MtbFHb Acts as a Disulfide Reductase
Since thioredoxin reductase-type FAD-binding motif in MtbFHb and other type II FHbs are fully conserved, we attempted to see if MtbFHb and its homologs are in fact functioning as novel heme containing disulfide reductase. To validate that we selected type II FHb (MsFHbII) of M. smegmatis, which has close sequence similarity with MtbFHb and carries similar thioredoxin reductase-like FAD-binding motif (Figure 1), we first tested whether MtbFHb and MsFHbII are able to reduce insulin, similar to thioredoxin reductases. Protein disulfide reductase activity of these FHbs, taking thioredoxin reductase (trxB) of Mtb as a reference, was tested by insulin disulfide reduction assay where the protein catalyzes the reduction of insulin in the presence of thiol reductant, dithiothreitol (DTT). Both MtbFHb and MsFHbII displayed rapid reduction of insulin in the presence of DTT (Figure 2A), very similar to trxB, indicating that MtbFHb and MsFHbII are acting as a disulfide reductase and their thioredoxin-type FAD binding site is functional. Earlier, we created an MtbFHb mutant, carrying mutations at GS residues of the conserved D-LDH type FAD binding motifs (GX7GS.AX7AXN). This mutant appeared defective in FAD binding (Gupta et al., 2012) and oxidation of D-lactate into pyruvate. We found that this mutant is unable to oxidize NADPH also unlike wild-type MtbFHb. Since GS residues of D-LDH-type FAD-binding motif also overlaps with thioredoxin reductase-type FAD-binding site of MtbFHb (Figure 1), it is quite likely that a change in the overlapping region of two FAD-binding sites has disrupted the association of MtbFHb with FAD at both of these locations.
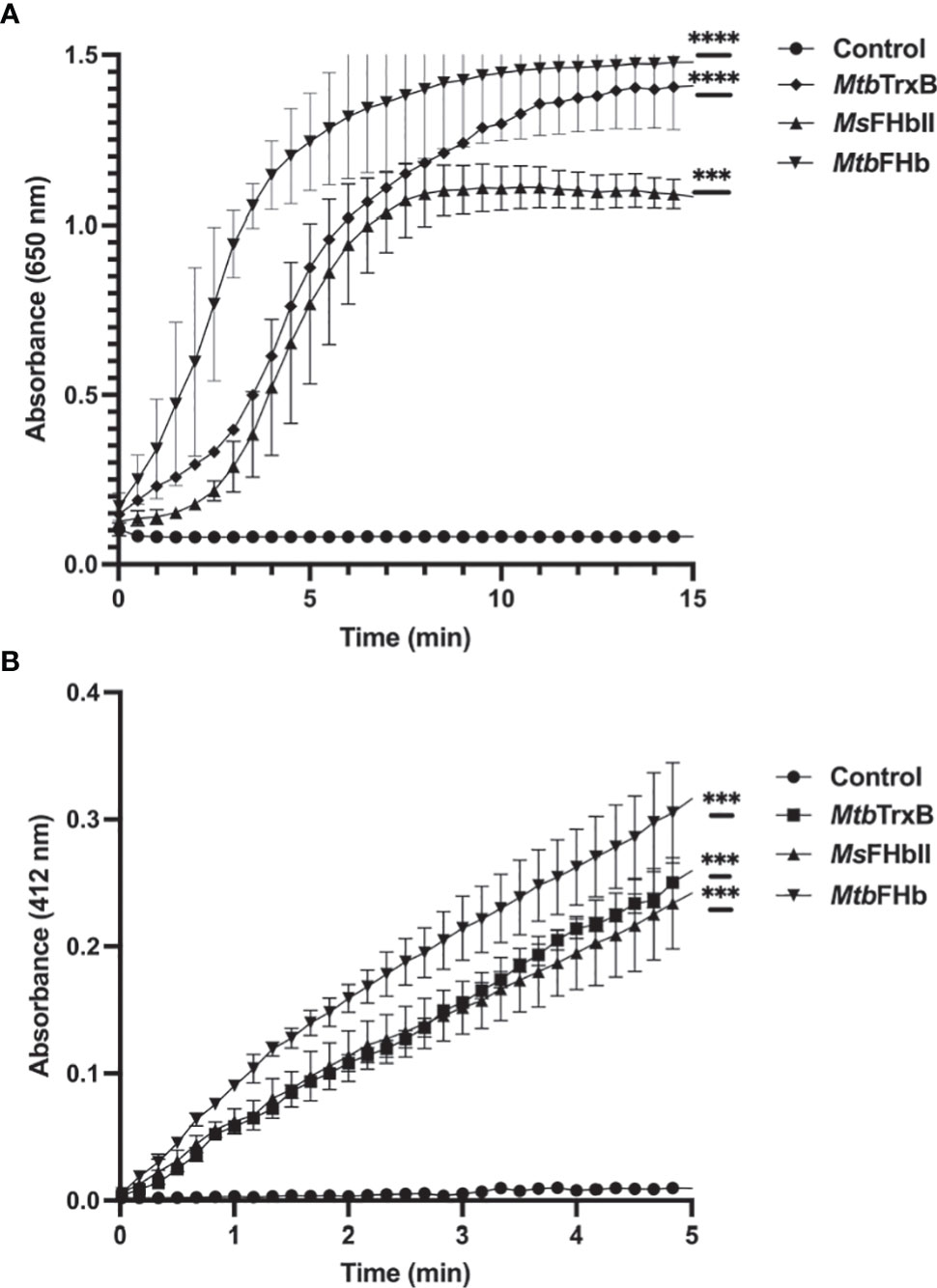
Figure 2 (A) Insulin disulfide reductase activity of MtbFHb, MsFHbII, and trxB of M. tuberculosis. Insulin reduction assay was set up in 500 µl containing 0.1 M potassium phosphate (pH 7.0), 2 mM EDTA, 0.13 mM insulin, and 0.33 mM DTT. The reaction was carried out at 25°C by adding 2 µM proteins (Mtb FHb, Ms FHbII, or Mtb trxB) and monitoring insulin reduction by the increase in optical density at 650 nm. (B) NADPH-dependent reduction of DTNB by MtbFHb, MsFHbII, and trxB. The assay was carried out at 25°C in a 500-µl reaction mixture containing 100 mM potassium phosphate (pH 7.0), 2 mM EDTA, 5 mM DTNB, and 250 µM NADPH. Reaction was carried out by adding 2 µM proteins (MtbFHb, MsFHbII, or Mtb trxB). The control reaction was set up without any protein. Reduction of DTNB was monitored by the increase in absorbance at 412 nm. Experiments were repeated at least five times. Results are presented as the mean values ± standard deviations. Two-way ANOVA was employed to analyze statistical significance. ***p < 0.001, ****p < 0.0001.
Disulfide Reductase Activity of MtbFHb Is NADPH Dependent
MtbFHb and MsFHbII lack a NADH binding site and their NADH oxidase activity is attenuated (Gupta et al., 2012; Thakur et al., 2018) unlike conventional type I FHb (MsFHbI) of M. smegmatis (Thakur et al., 2014) and other bacteria. Since thioredoxin reductases are NADPH-dependent oxidoreductases and MtbFHb and MsFHbII carry thioredoxin reductase-like FAD binding motif, we checked whether these FHbs are able to oxidize NADPH. The oxidation of NADPH appeared quite fast in the presence of MtbFHb and MsFHbII, indicating that these FHbs are using NADPH as an electron donor instead of NADH (Figure S2). We further checked disulfide reductase activities of MtbFHb and MsFHbII in the presence of a generic disulfide substrate, DTNB (5,5’-dithiobis,2-nitrobenzoic acid), using NADH, NADPH, and D-lactate to check the involvement of these electron donors in this process. Reduction of DTNB by both the proteins increased in the presence of NADPH in a time-dependent manner, similar to the trxB of Mtb (Figure 2B), but D-lactate and NADH were not utilized as electron donors for the DTNB reduction. NADPH-dependent reduction of DTNB by MtbFHb and MsFHbII appeared comparable to that of trxB of Mtb. These results demonstrated that MtbFHb and MsFHbII carry a NADPH-dependent disulfide reductase activity. MtbFHb and MsFHbII reduced DTNB with Km values of 98.5 ± 6.8 μM and 93.7 ± 7.6 μM, respectively, thereby indicating that the specificity of these FHbs is significantly higher towards DTNB as compared to Mtr of Mtb, which has 3300 ± 700 μM Km for the reduction of DTNB (Patel and Blanchard, 1999). The DTNB reduction by MtbFHb and MsFHbII was estimated as 1608 ± 66 mM/min/mg and 1079 ± 48 mM/min/mg, respectively.
Role of Cysteines in Disulfide Reductase Activity of MtbFHb
Although the reductase domain of MtbFHb carries three cysteines, it lacks a typical disulfide active site (CXXC) unlike conventional disulfide reductases. To investigate the role of cysteines in protein function, site-directed mutagenesis has been carried out to create three cysteine mutants of MtbFHb (MtbFHbcys188ala, MtbFHbcys289ala, and MtbFHbcys360ala) where cys188, cys289, and cys360 residues were mutated individually into alanine. Mutation in cys188 of MtbFHb did not affect the protein function of MtbFHb as MtbFHbcys188ala mutant exhibited NADPH-dependent reduction of DTNB appeared very similar to wild type (Figure 3). The other two MtbFHb mutants, MtbFHbcys289ala and MtbFHbcys360ala, could not be overexpressed in E. coli, possibly due to rapid protein degradation. It is likely that these cysteine residues are crucial for conformational stability and functional activity of the protein as these are conserved among all type II FHbs of mycobacteria. To overcome this problem, we selected cysteine mutant MtbFHbcys188ala that carries only two cysteines and subjected it to IMA treatment after reduction to block the formation of disulfide bond if that is created within two cysteine (cys289 and cys360) residues of the protein. NADPH-dependent reduction of DTNB by MtbFHbcys188ala was reduced significantly after IMA treatment as compared to the untreated one. These results suggested that cys289 and cys360 residues are involved in modulating the disulfide reductase activity of MtbFHb.
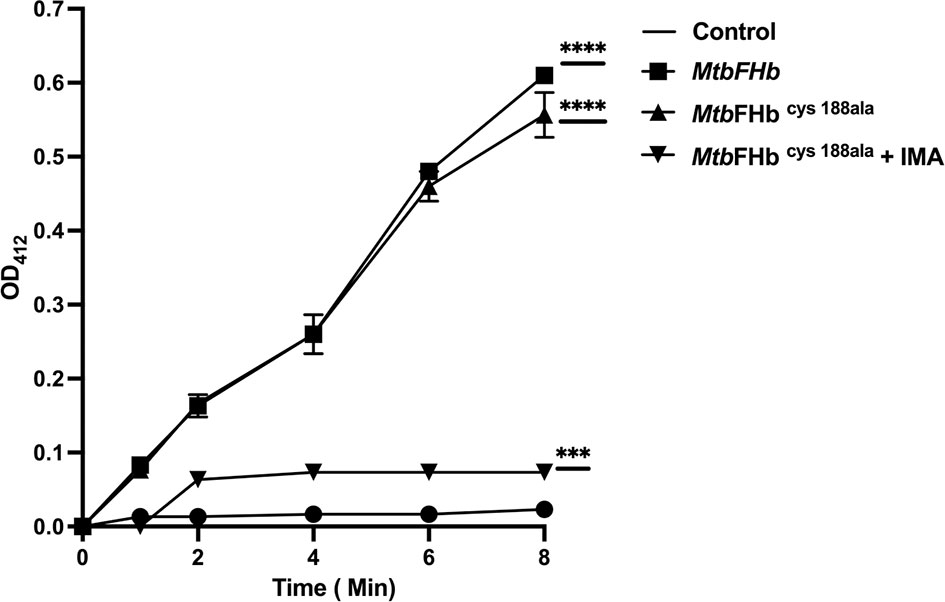
Figure 3 Reduction of DTNB by MtbFHb and mutant MtbFHbcys188ala. Iodoacetamide (IMA) treatment of MtbFHbcys188ala mutant protein was carried out first in 500 µl of Tris-Cl (pH 7.2) buffer carrying 400 µg of protein, 2 µl of reduction buffer, and 10 µl of reductant solution from the kit (G-bioscience). The reaction mixture was incubated at 37°C for 30 min to reduce the protein. Thereafter, alkylation of the protein was performed by adding 2.5 µl of alkylation buffer and 25 µl of 0. 4 M of freshly prepared IMA. The reaction was incubated at room temperature for 30 min protected from light. Reaction mixture was then passed through G-50 desalting column to remove salts from the protein. DTNB reduction assay mixture contained wild type and the mutant MtbFHb (with 50 micromole heme content), 0.5 mM NADPH, and 0.1 mM DTNB. MtbFHbcys188ala (IMA) is designated for MtbFHbcys188ala treated with IMA. The change in absorbance at 412 nm was monitored at 30°C at a specified time. Data are shown as mean ± standard deviation of five independent scans. ***p < 0.001, ****p < 0.0001.
Electron-Transfer During Oxidation of NADPH and D-Lactate by MtbFHb
To understand the mechanism by which MtbFHb participates in electron transfer using NADPH and D-lactate as electron donors, we checked the pattern of heme and FAD reduction during oxidation of NADPH and D-lactate by MtbFHb and compared it with MsFHbII that carries a D-LDH type FAD-binding motif with mutated intergenic region (Figure 1). The electron transfer during oxidation of D-lactate and NADPH occurs in a two-step process, wherein these electron donors donate electrons and reduce the FAD, which further transmits electrons to the heme iron. Therefore, we compared the efficiency of redox state changes in the FAD and the heme moieties in MtbFHb and MsFHbII and pattern of electron transfer during oxidation of D-lactate and NADPH. When MtbFHb was reduced with NADPH, a rapid decrease occurred at 460 nm and the optical spectrum of the FAD changed very fast at 250 µM NADPH, reaching to fully reduced state within 2 min and simultaneously displaying a fast increase at 430 nm, which indicated rapid reduction of the heme iron. Similar behavior was observed in the case of MsFHbII (Figures 4A, B). These data demonstrated that those two reductive steps, which include reduction of FAD by NADPH followed by reduction of heme by an intra-molecular electron transfer, occur more or less in a similar manner in the case of MtbFHb and MsFHbII. However, when D-lactate was used as an electron donor, the optical spectrum of the flavin at 460 nm changed very fast in the presence of MtbFHb and reached to fully reduced state in the presence of 250 µM D-lactate, whereas reduction of FAD by D-lactate occurred very slowly and got reduced only partially in the case of MsFHbII under identical conditions (Figure 4C). Similarly, the rate of heme reduction by D-lactate appeared nearly 4- to 5-fold faster in MtbFHb in comparison to MsFHbII (Figure 4D), indicating that the transfer of electrons to the FAD via D-lactate is not very efficient in the case of MsFHbII in comparison to MtbFHb. Thus, both FHbs appear to have similar NADPH-dependent disulfide reductase activities, but the efficiency of electron-transfer by D-lactate and reduction of FAD and the heme iron appeared extremely slow in MsFHbII in comparison to MtbFHb. These results suggested that the D-lactate and NADPH are utilized efficiently as electron donors by MtbFHb and both its FAD-binding sites are efficient in electron transfer, unlike MsFHbII, where transfer of electrons occurs very slowly in the presence of D-lactate.
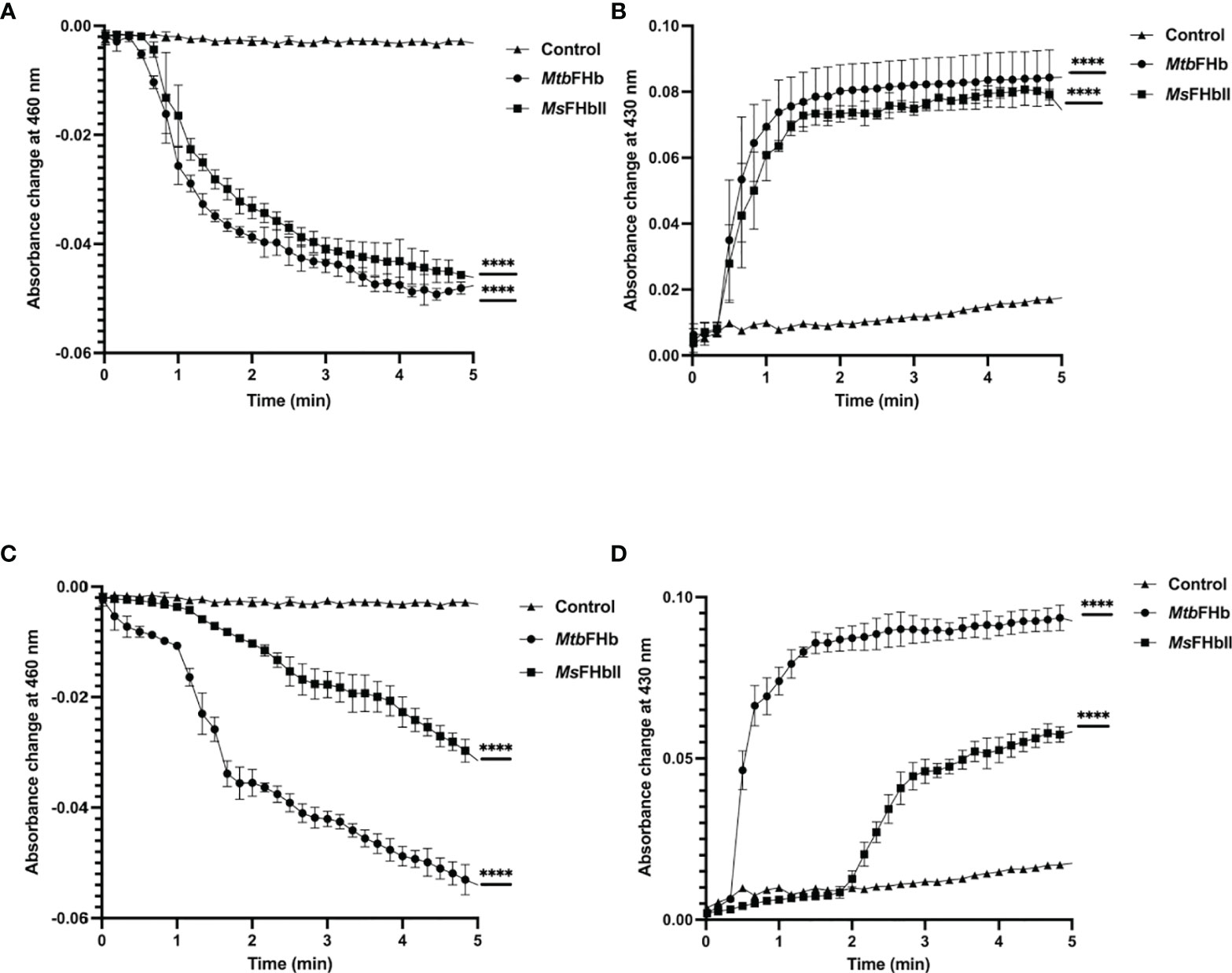
Figure 4 Profile of heme and FAD reduction during oxidation of NADPH and D-lactate by MtbFHb and MsFHbII. Reaction was set up in 500 µl with 8 µM oxygen bound MtbFHb or MsFHbII in 0.05 M potassium phosphate buffer (pH 7.2). Reaction was initiated after adding 250 μM D-lactate or 250 μM NADPH and recording the spectral changes with change in time. Heme reduction was followed by the shift in Soret peak from 414 nm to 430 nm (B, D), while reduction of FAD was checked by the decrease in absorbance at 460 nm (A, C). Experiments were repeated at least five times and data are shown as mean ± standard deviation of five independent scans. Two-way ANOVA was employed to analyze statistical significance. ****p < 0.0001.
D-Lactate Inhibits Oxidation of NADPH and FAD-Mediated Electron Transfer by MtbFHb
Since two overlapping FAD-binding sites of MtbFHb appeared fully functional in electron transfer, we checked how these two co-factors’ binding sites coordinate their function. Our earlier studies demonstrated that FAD associates with MtbFHb in 1:1 stoichiometry (Gupta et. al., 2012) and one FAD moiety remains attached with one molecule of MtbFHb; thus, only one FAD-binding site may be operative at a time with respect to its specific electron donor (NADPH or D-lactate). It led us to explore how these two overlapping FAD-binding sites in MtbFHb operate and modulate protein function in the presence of two different electron donors (NADPH and D-lactate). Therefore, we checked the reduction of FAD by NADPH alone and in combination with D-lactate by monitoring spectral changes at 460 nm and 340 nm to check the reduction of FAD and the oxidation of NADPH, respectively. The reduction of FAD occurs very fast and gets fully saturated at 250 µM when D-lactate and NADPH are used as electron donor individually, whereas at lower concentrations, it remains partially reduced (Figures 5A, B). It was further confirmed by the oxidation of NADPH by MtbFHb. (Figure 5C), which indicated that MtbFHb oxidizes NADPH very efficiently in a concentration-dependent manner (Figure 5C). As shown in Figure 5A, FAD bound with MtbFHb gets fully reduced in the presence of 250 µM NADPH but the oxidation of NADPH was inhibited completely when D-lactate was incorporated in the reaction (Figure 5D). These results suggested that electron transfer by NADPH to the FAD in MtbFHb gets restricted in the presence of D-lactate.
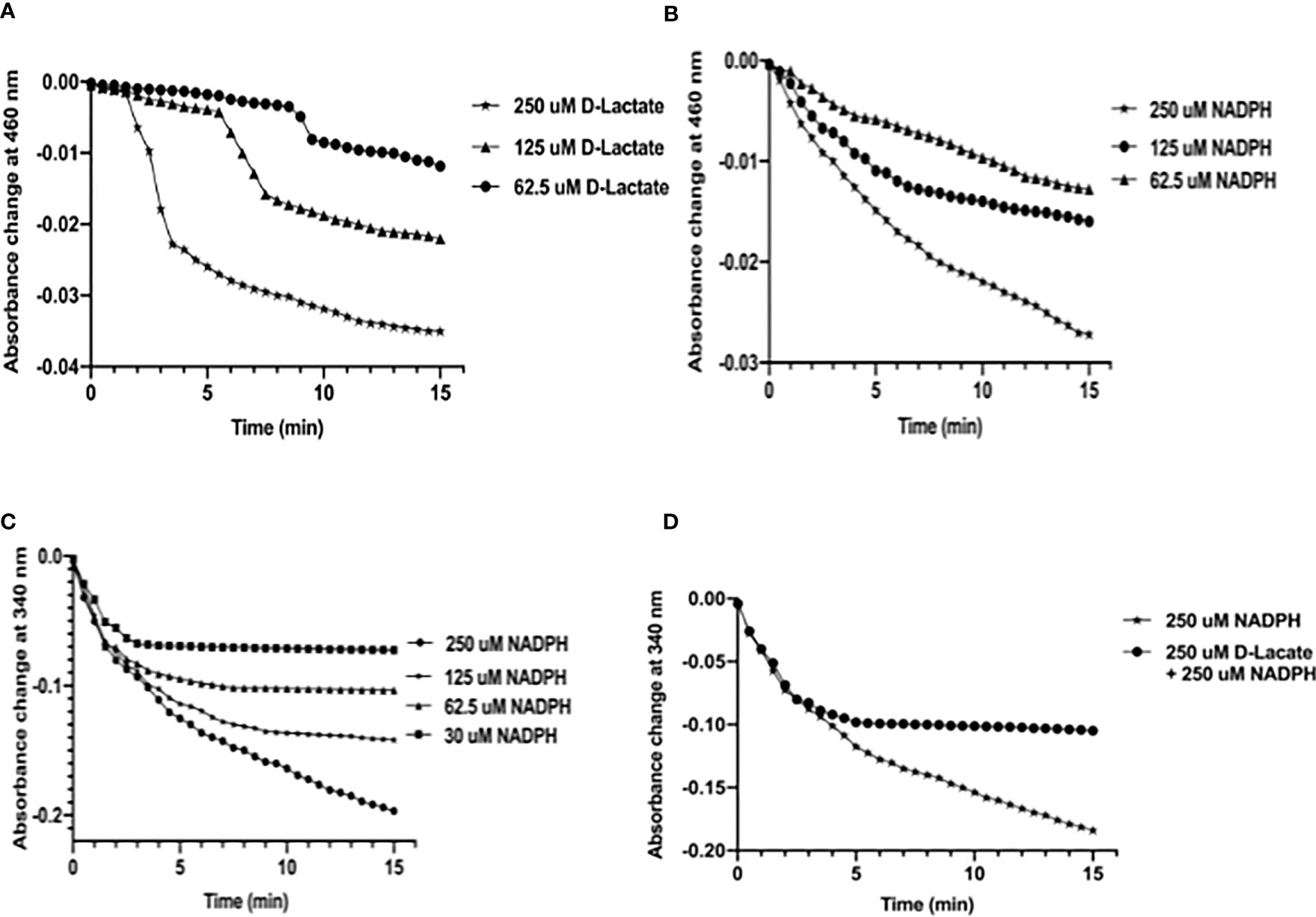
Figure 5 D-lactate inhibits oxidation of NADPH by MtbFHb. (A) Profile of FAD reduction by MtbFHb in the presence of different concentrations of D-lactate. (B) Profile of FAD reduction by MtbFHb in the presence of varying concentrations of NADPH. (C) Oxidation of NADPH by MtbFHb. (D) Inhibition of NADPH oxidation by MtbFHb in the presence of D-lactate. The reaction was set in 500 µl containing specified amount of NADPH or D-lactate in 0.5 M potassium phosphate buffer (pH 7.2) and 1 mM EDTA. Baseline level of FAD reduction or NADPH oxidation was set before addition of the protein in the reaction mixture. Reduction of heme in the presence of different concentrations of D-lactate or NADPH was monitored spectrophotometrically at 460 nm after adding MtbFHb (8 µM) in the reaction mixture. Data are shown as mean ± standard deviation of three independent experiments. Two-way ANOVA was employed to analyze statistical significance. ****p < 0.0001.
D-Lactate Mediates Conformational Changes in MtbFHb
The addition of D-lactate to the ferric MtbFHb (with Soret and visible bands at 414 and 535/570 nm) results in a species with Soret and visible bands at 426 and 529/558 nm, indicating reduction of the heme iron (Figure S3). Since D-lactate inhibited oxidation of NADPH by MtbFHb, it is quite likely that a conformational change takes place in MtbFHb in the presence of D-lactate. To test this possibility, we checked far-UV-CD spectra of the MtbFHb in the presence and the absence of D-lactate to see if D-lactate induces any structural change in MtbFHb that prevents access of NADPH towards the FAD moiety and disrupts the electron transfer. The CD spectrum of native MtbFHb exhibited two negative peaks at 208 and 222 nm (Figure 6A) that are characteristics of alpha-helical structure, as reported for trHbN of Mtb (Lama et al., 2006). In the presence of D-lactate, the negative peak at 222 nm gets more pronounced and a new peak appears at 212 nm, which indicated significant conformational change in the heme domain. No such change has been observed in MtbFHb in the presence of NADPH (Figure 6B). These results confirmed a conformational change in MtbFHb during its interaction with D-lactate.
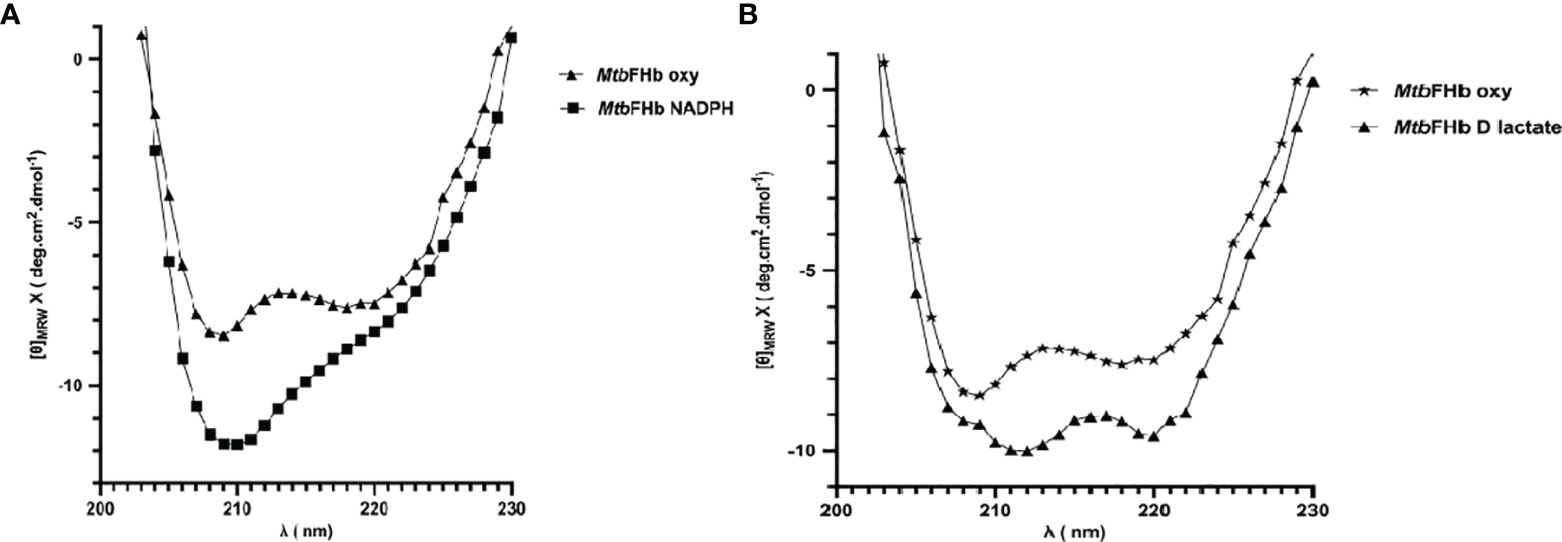
Figure 6 Secondary structure analysis of MtbFHb. The near-UV CD spectra of native MTbFHb after reduction with NADPH (A) and D-lactate (B). Protein sample (20 µM) was prepared in 50 mM Tris-Cl (pH 7.8). Spectra were recorded individually in the far-UV region (250 to 190 nm) and were carried out on protein solutions (0.4 mg/ml) employing a cell with a path length of 0.1 cm at 25°C after addition of NADPH (10 µM) or D-lactate (10 µM). Each spectrum reported represents an average of 5 scans.
D-Lactate Changes the Specificity of MtbFHb Towards Mycothiol Disulfide
In an attempt to understand the molecular mechanism by which disulfide reductase activity of MtbFHb is modulated by two different co-factors, we checked how it would respond towards mycothiol that exists as a major low-molecular-mass thiol in mycobacteria (Newton et al., 1996) and tested whether MtbFHb is able to use mycothiol disulfide as a substrate that gets accumulated under oxidative stress. Mycothiol is structurally unusual, containing an alpha 1-1 glycosidic linkage between the two monosaccharides, D-glucosamine and myo-inositol. A structural analogue of oxidized mycothiol, des-myo-inositol mycothiol, has been successfully used previously to test mycothione reductase activity of a NADPH-dependent mycothione reductase (Patel and Blanchard, 1998; Patel and Blanchard, 1999). Therefore, we synthesized this synthetic analogue of mycothione (Figure S4) using the published procedure (see Supplementary Information) and used it as a substrate to check whether MtbFHb displays any mycothione reductase activity using NADPH or D-lactate as an electron donor. Due to difficulty in checking the oxidation of D-lactate spectrophotometrically, we used a mass spectrometry-based approach to check the reduction of des-myo-inositol mycothiol. MtbFHb was incubated with D-lactate and NADPH separately and the reaction products were analyzed via LC-MS to confirm reduction of disulfide linkage of the product. When NADPH was used as an electron donor, no visible reactivity was observed on des-myo-inositol mycothiol in the presence of MtbFHb (Figure S5). Interestingly, when MtbFHb was incubated with des-myo-inositol mycothiol in the presence of D-lactate, it exhibited the appearance of a main peak corresponding to half the size of the original substrate (Figures 7A, B), indicating reduction of disulfide linkage of the substrate. These results suggested that MtbFHb is capable of reducing di-mycothiol in the presence of D-lactate but not in the presence of NADPH, indicating that the interaction of D-lactate with MtbFHb mediates a conformational change that attenuates its interactions with NADPH and changes its substrate specificity towards mycothiol disulfide.
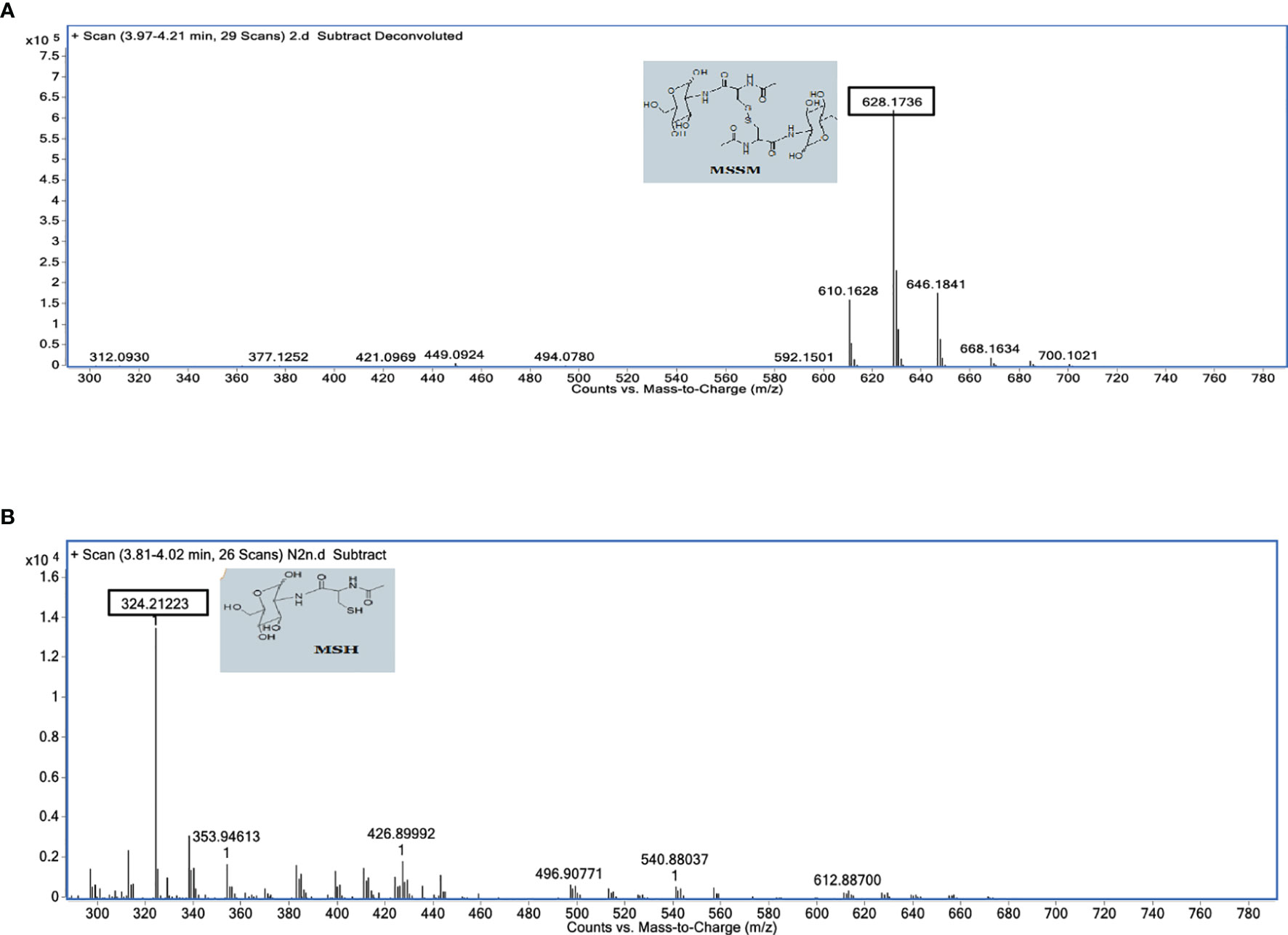
Figure 7 Mass spectrophotometric analysis of des-myo-inositol mycothiol (synthetic analog of mycothione) reduction in the absence (A) and the presence (B) of D-lactate. Assays were performed in a 100-µl reaction mixture containing 50 mM HEPES (pH 7.6) and 0.1 mM EDTA. MtbFHb and MsFHbII (5 µg each) were incubated individually with 100 μM des-myo-inositol mycothione along with 250 μM NADPH or 250 μM D-lactate at 25°C for 5 min and the product was analyzed via LC-MS using C18 column, with acetonitrile and water gradient. MSH was eluted at 3.81–4.20 min. MSH, Myo-inositol mycothiol; MSSH, des-myo-inositol mycothiol.
Deletion of the fhb Gene in Mtb and fhbII Gene in M. smegmatis
To assess the physiological function of MtbFHb, we attempted to construct an fhb gene deleted strain of Mtb. Unfortunately, no viable colonies of Mtb were obtained despite our repeated attempts to knock out fhb gene. Since MsFHbII shares close sequence similarity with MtbFHb and exhibits similar NADPH-dependent disulfide reductase activity, we constructed a fhbII gene knock out strain of M. smegmatis and used it as a model to evaluate the physiological function of MtbFHb. Disruption of the fhbII gene in M. smegmatis was confirmed through PCR analysis of the genome of wild type and the mutant strain (Figure S6). The selected mutant strain, thereafter, was designated as MsΔfhbII. The growth of MsΔfhbII appeared slightly slow under aerobic conditions in comparison to its wild-type counterpart (Figure 8A). However, when testing the response of MsΔfhbII under oxidative stress, it exhibited enhanced growth sensitivity towards oxidants, most prominently in the presence of cumene hydroperoxide (CHP), which inhibited the growth of MsΔfhbII completely at 20 mM, whereas wild-type M. smegmatis was able to resume its growth after a period of initial lag under similar conditions (Figure 8B).
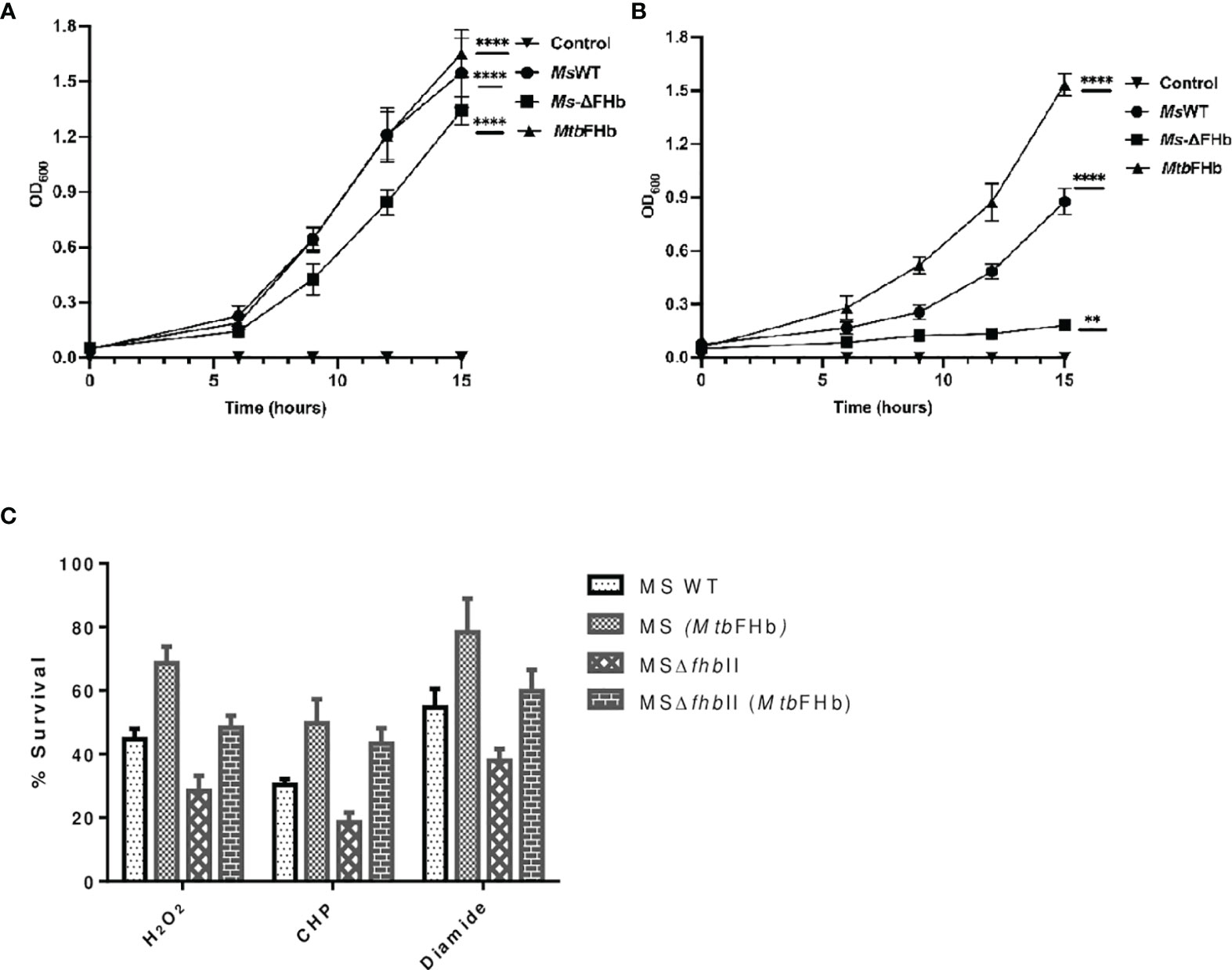
Figure 8 Implications of Mtb fhb gene expression on growth properties of MsΔfhbII and wild-type strains of M. smegmatis. Growth profile of wild type and MsΔfhbII strains of M. smegmatis overexpressing Mtb fhb gene (A) under aerobic and (B) under oxidative stress conditions in the presence of 20 mM CHP. (C) Survival of MsΔfhbII mutant of M. smegmatis after complementation of Mtb fhb gene during exposure to different oxidants, H2O2 (20 mM), CHP (10 mM), and diamide (10 mM). For CFU count, Mtb cells were grown to mid-log phase and washed twice in 7H9 medium and single-cell suspension was then prepared by centrifuging the culture at 8,000 rpm for 10 min in the presence of sterilized glass beads. The O.D. of cells was adjusted to 0.03. Cells were then exposed to oxidants for 30 min and CFU was checked after plating and counting colonies after 2 days. Data represent mean values with standard deviations (error bars) from three independent experiments. Data are shown as mean ± standard deviation of three independent experiments. Two-way ANOVA was employed to analyze statistical significance level with p< 0.05. MsWT: Wild-type M. smegmatis; Ms+MtbFHb: M. smegmatis carrying Mtb fhb gene; MSΔfhbII: M. smegmatis carrying deletion of fhbII gene; MSΔfhbII+MtbFHb: Mtb fhb gene complemented in MSΔfhbII. **p < 0.01, ****p < 0.0001.
Expression of the Mtb fhb gene Promotes Tolerance Towards Different Oxidants and Reduces Lipid Peroxidation in MsΔfhbII and Wild-Type M. smegmatis
In an attempt to understand the physiological role of MtbFHb, we complemented the fhb gene of Mtb in MsΔFHbII and also overexpressed it in wild-type M. smegmatis. Expression of the Mtb fhb gene in wild type and MsΔfhbII strains of M. smegmatis was checked through qRT-PCR, which indicated nearly 2.5- to 3-fold increase in the transcript level of the Mtb fhb gene with respect to the control (Figure S7). Also, no signal for MsfhbII gene was observed in the MsΔfhbII strain of M. smegmatis. As shown in Figure 8C, complementation of the Mtb fhb gene significantly increased the resistance of the MsΔfhbII strain towards different oxidants in comparison to its isogenic control cells. Moreover, overexpression of the Mtb fhb gene also enhanced the resistance of wild-type M. smegmatis towards various oxidants. The lipid content of mycobacterial cell wall is unusually high and its unsaturated fatty acids may get decomposed under oxidative stress; thus, it can be hypothesized that being a membrane-associated protein, MtbFHb may protect the damage of lipids and the integrity of cell wall due to its antioxidant activity. Since D-lactate is produced as a by-product of lipid peroxidation and MtbFHb is able to metabolize D-lactate, we assessed the connection of MtbFHb with lipid peroxidation to understand its role under oxidative stress. Therefore, we checked the level of lipid peroxidation in the Mtb fhb gene expressing MsΔfhbII and wild-type M. smegmatis under oxidative stress along with their isogenic control cells. The level of lipid hydroperoxide appeared significantly higher in the MsΔFHbII strain as compared to wild-type M. smegmatis under oxidative stress (Table 1). When the Mtb fhb gene was over-expressed in these strains, the level of lipid hydroperoxide reduced to 25% to 30%.

Table 1 Cellular level of lipid hydroperoxide in fhb gene expressing cells of wild type and Δ MsFHbII strains of M. smegmatis.
Discussion
Mtb is armed with a number of antioxidant defense mechanisms and compensatory metabolic pathways that allow the pathogen to cope with hazardous intracellular environment where copious amounts of reactive oxygen and nitrogen species are produced via the host defense system. Thiol-disulfide oxidoreductases like thioredoxin reductase (trxB) and mycothiol disulfide reductase are vital components of the antioxidant defense system of Mtb that regulate the activity of many cellular proteins through reversible reduction of their disulfide bond and play a central role in redox homeostasis (Kumar et al., 2011; Trivedi et al., 2012). MtbFHb sets the first example of a heme-containing oxidoreductase having two distinct disulfide reductase activities that may play a pivotal role in redox homeostasis and defense from toxic oxidants during the intracellular regime of Mtb.
Phylogenetically, MtbFHb and its homologs form a separate cluster from conventional type I FHbs (Gupta et al., 2011). Our study demonstrated that MtbFHb acts as a unique electron-transferring protein that utilizes NADPH and D-lactate as electron donors to carry out two different disulfide reductase activities. Conservation of thioredoxin reductase-like FAD-binding site in homologs of MtbFHb suggested that these FHbs may be acting as novel heme containing disulfide-reducing enzymes. This is supported by the observation that MtbFHb and its homolog in M. smegmatis (MsFHbII) are able to carry out insulin and NADPH-dependent DTNB reduction very similar to trxB of Mtb. Both of these FHbs are functional as disulfide reductase in an oxygen-bound state, suggesting that their globin domain is acting as an oxygen sensor to regulate its functional activity. Generally, thioredoxin and thioredoxin reductase together with NADPH constitute a redox complex in which thioredoxin reductase catalyzes the electron transfer from NADPH to thioredoxin via FAD. The reduced thioredoxin with a dithiol/disulfide active site (CXXC) then reduces disulfide linkage and also serves as an electron donor in different metabolic processes. The globin domain in MtbFHb appears to accept electrons from the reductase and may donate electrons to different cellular processes, very similar to the Trx/TrxR hybrid protein of M. leprae (Wang et al., 1998). Although the reductase domain of MtbFHb carries three cysteine residues, it lacks a conventional CXXC active site. Mutation in Cys188 did not alter disulfide reductase activity of MtbFHb, and this cysteine is not conserved in MsFHbII as well as in other homologs of MtbFHb in mycobacteria, which indicated that it may not be involved in disulfide reductase activities of these FHbs. Two other cysteine residues, Cys289 and Cys360, of MtbFHb are fully conserved among type II FHbs, and mutation in any one of these residues appeared detrimental for the protein expression. It is quite likely that these two cysteines are crucial for the conformational stability and functional activity of these FHbs as a disulfide reductase by remaining close enough to maintain functional conformation and create an active site dithiol of the protein. This assumption was substantiated when Cys289 and Cys360 residues in the MtbFHbcys188ala mutant were alkylated by IMA, which inhibited its NADPH-dependent DTNB reduction. These results demonstrated that Cys289 and Cys360 residues are important for the function of MtbFHb as a disulfide reductase. It has been observed earlier that the mere presence of CXXC motif may not ensure a thioredoxin reductase-like property and distantly placed cysteines may also participate in forming an active site (Alam et al., 2007). Since MtbFHb carries two distinct overlapping FAD-binding sites and FAD binds with the reductase domain of MtbFHb in 1:1 stoichiometry (Gupta et al., 2012), it can be envisaged that these co-factor binding sites are utilized one at a time. It is quite intriguing how these sites and co-factors are selected for a specific reductase activity. MtbFHb catalyzes the oxidation of D-lactate into pyruvate in a FAD-dependent manner having specific D-lactate oxidation activity as 28.3 µM/min/mg (Gupta et al., 2012). Our experimental data suggest that D-lactate creates a conformational change and attenuates NADPH-dependent disulfide reductase activity of MtbFHb, changing its specificity towards reduction of di-mycothiol. Raman spectroscopic studies on MtbFHb have shown that a structural change in the heme domain of the protein induces significant conformational changes within the reductase domain and inter-domain interactions are required for the stabilization of the sixth ligand within the heme active site (Gupta et al., 2012). Thus, it is likely that the D-lactate-mediated conformational change in MtbFHb abrogates its interactions with NADPH and changes its substrate specificity from general dithiol to di-mycothiol. This is supported by the observation that the synthetic analog of mycothione (des-myoinositol) gets reduced by MtbFHb only in the presence of D-lactate but not in the presence of NADPH. Mycobacteria carry millimolar concentration of mycothiol as the low-molecular-weight thiol, which is maintained in the cell in a reduced state by a mycothiol disulfide reductase (Mtr) that uses NADPH as an electron donor; however, its catalytic activity for mycothiol disulfide is one to two orders slower than other disulfide reductases (Patel and Blanchard, 1998), which may not be sufficient to maintain the level of reducing environment in the cell during rapid oxidation of mycothiol under severe oxidative stress that Mtb faces within the macrophagic environment (Shastri et al., 2018). Thus, the NADPH-dependent disulfide reductase activity of MtbFHb can participate in conjunction with thioredoxin reductase and Mtr to create a robust antioxidant system that can play a vital role in protecting the viability and maintenance of redox balance by reduction of oxidized mycothiol and its recycling during intracellular infection when Mtb gets exposed to copious amounts of reactive oxygen and nitrogen species.
The dual function of MtbFHb as NADPH-dependent disulfide reductase and D-lactate-dependent mycothione reductase appears unique and may be highly advantageous when Mtb gets exposed to a strong oxidative burst during macrophage infection. MtbFHb carries high-affinity lipid binding sites and remains associated with membrane lipids (Gupta et al., 2012). Under high oxidative stress, membrane and cell wall lipids are among the most significant targets of oxidative damage. Since the lipid content of the cell envelope of Mtb is unusually high, decomposition of unsaturated fatty acids of Mtb cell wall into toxic methylglyoxal may generate D-lactate as a by-product of lipid peroxidation (Rachman et al., 2006), which may be highly toxic due to increased OH+ generation (Ali et al., 2000). It has been shown that the level of methylglyoxal increases significantly during mycobacterial infection of macrophages (Rachman et al., 2006). It may result in the accumulation of D-lactate in the cell as Mtb lacks a gene for the D-lactate dehydrogenase (Pinchuk et al., 2009). No functional D-lactate dehydrogenase activity has been demonstrated so far in Mtb that can metabolize accumulated D-lactate. Therefore, the proximity of MtbFHb with the cell membrane and its D-lactate-dependent mycothiol disulfide reductase activity may be highly advantageous for the protection of cell wall integrity and maintenance of reducing environment under high oxidative stress. This is supported by the observation that MtbFHb expressing cells of M. smegmatis are less prone to lipid peroxidation and more resistant to oxidative stress as compared to control cells. Additionally, expression of MtbFHb in MtbFHbII deleted mutant of M. smegmatis reduces lipid peroxidation and provides enhanced protection against different oxidants. During oxidation of D-lactate by MtbFHb, the oxygen bound heme iron gets reduced and converted to Fe2+ state, suggesting that the heme domain is acting as an electron acceptor. Since MtbFHb remains associated with the cell membrane, it is likely that it donates electrons to the menaquinone to feed the electron transport and use it as an electron sink for the removal of toxic D-lactate, generated during membrane lipid peroxidation. The H2O2 scavenging ability of pyruvate (Biagini et al., 2001; Guarino et al., 2019), produced during oxidation of D-lactate, may also contribute to the antioxidant activity of MtbFHb.
It is interesting to note that besides Mtb, only two virulent mycobacteria, M. avium and M. africanum, have D-LDH-type FAD-binding motifs similar to MtbFHb, whereas it is absent or mutated in other mycobacteria (Figure S1). It is likely that fast removal of D-lactate, accumulated due to membrane lipid peroxidation, is required for highly virulent mycobacteria to protect their membrane integrity and survival within the intracellular environment where large amounts of reactive oxygen and nitrogen species are produced. This is supported by the observation that the transcriptional activity of Rv0385 is induced several folds in Mtb during oxidative stress (Gupta et al., 2012) when accumulation of D-lactate may occur in the cell. Although MsFHbII also carries overlapping D-LDH-type FAD-binding sites similar to MtbFHb, the reduction of heme is extremely slow in the presence of D-lactate. Sequence composition of one of the intergenic regions of the LDH-type FAD-binding motif of MsFHbII is different from MtbFHb and carries non-polar residues that may affect solvent accessibility and the interactions of protein co-factor with the electron donor (Dym et al., 2000), thereby reducing its efficiency of electron transfer. It is likely that NADPH-dependent disulfide reductase and reduced efficiency of D-lactate oxidation by non-pathogenic mycobacteria like M. smegmatis may be sufficient to minimize their stress level.
Taken together, the present study provides the first report on dual function of MtbFHb as a NADPH-dependent disulfide reductase and D-lactate-dependent mycothione reductase, which is modulated by two distinct overlapping FAD-binding sites. The D-lactate-dependent di-mycothiol reductase activity of MtbFHb, observed during this study, appears unique and may be highly advantageous for Mtb. Under highly oxidative environment, when D-lactate accumulates due to excessive lipid peroxidation and rapid oxidation of mycothiol occurs, the substrate specificity of MtbFHb gets diverted towards di-mycothiol, possibly due to conformational change in MtbFHb by D-lactate that creates an active site that is highly specific for the reduction of di-mycothiol. It may provide a dual advantage to Mtb during exposure of highly oxidative environment by eliminating the toxicity of D-lactate accumulation in the cell, simultaneously maintaining the reducing environment of cell by facilitating reduction of mycothiol disulfide. Thus, MtbFHb along with Mtr can constitute a robust system for the protection of Mtb within the oxidative environment, simultaneously balancing the redox environment of the cell during pathogenesis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
KD conceived and designed the project. KD, RJ, and AK provided the resources and analyzed the data. NT, MH, and AC carried out experimental work. AS synthesized the des-myo-inositol disulfide mycothione and performed LC-MS analysis. MS was written by KD. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank CSIR-IMTECH and the Panjab University, Chandigarh for providing research facilities for carrying out this study. We also acknowledge the help provided by Dr. Amit Singh from Indian Institute of Sciences, Bangluru, India, by providing strains and plasmids for this study. Financial assistance provided by the Department of Science and Technology, ICMR, and the National Academy of Sciences India (NASI) is thankfully acknowledged. This is a CSIR-IMTECH communication No. 38/2021.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.796727/full#supplementary-material
References
Alam, S., Garg, S. K., Agrawal, P. (2007). Molecular Function of WhiB4/Rv3681c of Mycobacteriium Tuberculosis H37Rv: A [4Fe-4S] Cluster Co-Ordinating Protein Disulphide Reductase. Mol. Microbiol. 63, 1414–1431. doi: 10.1111/j.1365-2958.2007.05589.x
Ali, M. A., Yasui, F., Matsugo, S., Konishi, T. (2000). The Lactate-Dependent Enhancement of Hydroxyl Radical Generation by the Fenton Reaction. Free Radic. Res. 32, 429–438. doi: 10.1080/10715760000300431
Arner, E. S. J., Holmgren, A. (2000). Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur. J. Biochem. 267, 6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x
Biagini, G. A., Park, J. H., Lloyd, D., Edward, M. R. (2001). The Antioxidant Potential of Pyruvate in the Amitochondriate Diplomonads Giardia Intestinalis and Hexamita Inflate. Microbiology 147, 3359–3365. doi: 10.1099/00221287-147-12-3359
Bloom, B. R., Murray, C. J. (1992). Tuberculosis: Commentary on a Re-Emergent Killer. Science 257, 1055–1064. doi: 10.1126/science.257.5073.1055
Bonamore, A., Boffi, A. (2008). Flavohemoglobin: Structure and Reactivity. IUBMB Life 60, 19–28. doi: 10.1002/iub.9
Bryk, R., Lima, C. D., Erdjument-Bromage, H., Tempst, P., Nathan, C. (2002). Metabolic Enzymes of Mycobacteria Linked to Antioxidant Defence by a Thioredoxin- Like Protein. Science 295, 1073–1077. doi: 10.1126/science.1067798
Buchmeier, N. A., Newton, G. L., Koledin, T., Fahey, R. C. (2003). Association of Mycothiol With Protection of Mycobacterium Tuberculosis From Toxic Oxidants and Antibiotics. Mol. Microbiol. 47, 1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x
Cha, M. K., Kim, W. C., Lim, C. J., Kim, K., Kim, I. H. (2004). Escherichia Coli Periplasmic Thiol Peroxidase Acts as Lipid Hydroperoxide Peroxidase and the Principal Antioxidative Function During Anaerobic Growth. J. Biol. Chem. 279, 8769–8778. doi: 10.1074/jbc.M312388200
Chan, J., Tanaka, K., Carroll, D., Flynn, J., Bloom, B. R. (1995). Killing of Virulent Mycobacterium Tuberculosis by Reactive Nitrogen Intermediated Produced by Activated Macrophages. Infect. Immun. 63, 736–740. doi: 10.1128/iai.63.2.736-740.1995
Cook, G. M., Berney, M., Gebhard, S., Heinemann, M., Cox, R. A., Danilchanka, O., et al. (2009). Physiology of Mycobacteria. Adv. Microb. Physiol. 52, 81– 319. doi: 10.1016/S0065-2911(09)05502-7
Dye, C. (2006). Global Epidemiology of Tuberculosis. Lancet 367, 938–940. doi: 10.1016/S0140-6736(06)68384-0
Dym, O., Eisenberg, D. (2001). Sequence-Structure Analysis of FAD-Containing Proteins. Protein Sci. 10, 1712–1728. doi: 10.1110/ps.12801
Dym, O., Pratt, E. A., Ho, C., Eisenberg, D. (2000). The Crystal Structure of D- Lactate Dehydrogenase, a Peripheral Membrane Respiratory Enzymes. Proc.Natl. Acad. Sci. U. S. A. 97, 9413–9418. doi: 10.1073/pnas.97.17.9413
Garbe, T. R., Barati, J., Barmini, S., Zhang, Y., Zied, C. A., Tang, D., et al. (1994). Transformation of Mycobacterial Species Using Hygromycin Resistance as Selectable Marker. Microbiology 140, 133–138. doi: 10.1099/13500872-140-1-133
Guarino, V. A., Oldham, W. M., Loscalzo, J., Zhang, Y. Y. (2019). Reaction Rate of Pyruvate and Hydrogen Peroxide: Assessing Antioxidant Capacity of Pyruvate Under Biological Conditions. Sci. Rep. 9, 55951–55959. doi: 10.1038/s41598-019-55951-9
Gupta, S., Pawaria, S., Lu, C., Yeh, S. R., Dikshit, K. L. (2011). Novel Flavohemoglobins of Mycobacteria. IUMB Life 63, 337–345. doi: 10.1002/iub.460
Gupta, S., Pawaria, S., Lu, C., Yeh, S. R., Dikshit, K. L. (2012). An Unconventional Hexacoordinated Flavohemoglobin of Mycobacterium Tuberculosis. J. Biol. Chem. 287, 16435–16446. doi: 10.1074/jbc.M111.329920
Kumar, A., Farhana, A., Guidry, L., Saini, V., Hondalus, M., Steyn, A. J. (2011). Redox Homeostasis in Mycobacteria: The Key to Tuberculosis Control? Expert Rev. Mol. Med. 13, e39.
Lama, A., Pawaria, S., Dikshit, K. L. (2006). Oxygen Binding and NO Scavenging Properties of Truncated Hemoglobin, HbN, of Mycobacterium Smegmatis. FEBS Letts. 580, 4031–4041. doi: 10.1016/j.febslet.2006.06.037
Livak, K. J., Schmittgen, T. D. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Manca, C., Paul, S., BarryIII, C. F., Freedman, V. H., Kapalan, G. (1999). Mycobacterium Tuberculosis Catalase and Peroxidase Activities and Resistance to Oxidative Killing in Human Monocytes In Vitro. Inf. Immun. 67, 74–79. doi: 10.1128/IAI.67.1.74-79.1999
Newton, G. L., Arnold, K., Price, M. S., Sherrill, C., Delcardayre, S. B., Aharonowitz, Y., et al. (1996). Distribution of Thiols in Microorganisms: Mycothiol Is a Major Thiol in Most Actinomycetes. J.Bacteriol. 178, 1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996
Ouellet, H., Ouellet, Y., Richard, C., Labarre, M., Wittenberg, B., Wittenberg, J., et al. (2002). Truncated Hemoglobin HbN Protects Mycobacterium Bovis From Nitric Oxide. Proc. Natl. Acad. Sci. U. S. A. 99, 5902–5907. doi: 10.1073/pnas.092017799
Patel, M. P., Blanchard, J. S. (1998). Synthesis of Des-Myo-Inositol Mycothiol and Demonstration of a Mycobacterial Specific Reductase Activity. J. Am. Chem. Soc 120, 11538–11539. doi: 10.1021/ja983111v
Patel, M. P., Blanchard, J. S. (1999). Expression, Purification, and Characterization of Mycobacterium Tuberculosis Mycothione Reductase. Biochemistry. 38, 11827– 11833. doi: 10.1021/bi991025h
Pathania, R., Navani, N. K., Gardner, A. M., Gardner, P. R., Dikshit, K. L. (2002). Nitric Oxide Scavenging and Detoxification by the Mycobacterium Tuberculosis Haemoglobin, HbN in Escherichia Coli. Mol. Microbiol. 45, 1303–1314. doi: 10.1046/j.1365-2958.2002.03095.x
Pinchuk, G. E., Rodionov, D. A., Yang, C., Li, X., Osterman, A. L., Dervyn, E., et al. (2009). Genomic Reconstruction of Shewanella Oneidensis MR-1 Metabolism Reveals a Previously Uncharacterized Machinery for Lactate Utilization. Proc. Natl. Acad. Sci. U. S. A. 106, 2874–2879. doi: 10.1073/pnas.0806798106
Rachman, H., Kim, N., Ulrichs, T., Baumann, S., Pradl, L., Nasser Eddine, A., et al. (2006). Critical Role of Methylglyoxal and AGE in Mycobacteria- Induced Macrophage Apoptosis and Activation. PloS One 1, e29. doi: 10.1371/journal.pone.0000029
Reyes, A. M., Pedre, B., De Armas, M. I., Tossounian, M. A., Radi, R., Messens, J., et al. (2018). Chemistry and Redox Biology of Mycothiol. Antioxid. Redox Signal. 28, 487–504. doi: 10.1089/ars.2017.7074
Shastri, M. D., Shukla, S. D., Chong, W. C., Dua, K., Peterson, G. M., Patel, R. P., et al. (2018). Role of Oxidative Stress in Pathology and Management of Human Tuberculosis. Oxid. Med. Cell. Longev. 2018, 1–10. doi: 10.1155/2018/7695364
Singh, S., Thakur, N., Oliveira, A., Petruk, A. A., Hade, M. D., Sethi, D., et al. (2014). Mechnanistic Insight Into the Enzymatic Reduction of Truncated Haemoglobin N of Mycobacterium Tuberculosis: Role of the CD Loop and Pre-A Motif in Electron Cycling. J. Biol. Chem. 289, 21573–21583. doi: 10.1074/jbc.M114.578187
Song, H., Wolschendorf, F., Niederweis, M. (2009). Construction of Unmarked Deletion Mutants in Mycobacteria. Methods Mol. Biol. 465, 279–295. doi: 10.1007/978-1-59745-207-6_19
Stover, C. K., de la Cruz, V. F., Fuerst, T. R., Burlein, J. E., Benson, L. A., Bennett, L. T., et al. (1991). New Use of BCG for Recombinant Vaccines. Nature 351, 456–460. doi: 10.1038/351456a0
Thakur, N., Gupta, S., Hade, M. D., Dikshit, K. L. (2014). Type I Flavohemoglobin of Mycobacterium Smegmatis Is a Functional Nitric Oxide Dioxygenase. IUBMB Life 66, 396–404. doi: 10.1002/iub.1275
Thakur, N., Kumar, A., Dikshit, K. L. (2018). Type II Flavohemoglobin of Mycobacterium Smegmatis Oxidizes D-Lactate and Mediate Electron Transfer. Int. J. Biol. Macromol. 112, 868–875. doi: 10.1016/j.ijbiomac.2018.02.010
Trivedi, A., Singh, N., Bhat, S. A., Gupta, P., Kumar, A. (2012). Redox Biology of Tuberculosis Pathogenesis. Adv. Microb. Physiol. 60, 263–324. doi: 10.1016/B978-0-12-398264-3.00004-8
Ung, K. S. E., Av-Gay, Y. (2006). Mycothiol-Dependent Mycobacterial Response To Oxidative Stress. FEBS Letts. 580, 2712–2716. doi: 10.1016/j.febslet.2006.04.026
Voskuil, M., Bartek, I., Visconti, K., Schoolink, G. (2011). The Response of Mycobacterium Tuberculosis to Reactive Oxygen and Nitrogen Species. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00105
Wang, P. F., Marcinkeviciene, J., Williams, C. H., Jr, Blanchard, J. S. (1998). Thioredoxin Reductase-Thioredoxin Fusion Enzyme From Mycobacterium Leprae: Comparison With the Separately Expressed Thioredoxin Reductase. Biochemistry 37, 16378–16389. doi: 10.1021/bi980754e
Keywords: flavohemoglobin, Mycobacterium tuberculosis, FAD, D-lactate, thioredoxin reductase, oxidative stress
Citation: Thakur N, Sharma AN, Hade MD, Chhaya A, Kumar A, Jolly RS and Dikshit KL (2022) New Insights Into the Function of Flavohemoglobin in Mycobacterium tuberculosis: Role as a NADPH-Dependent Disulfide Reductase and D-Lactate-Dependent Mycothione Reductase. Front. Cell. Infect. Microbiol. 11:796727. doi: 10.3389/fcimb.2021.796727
Received: 17 October 2021; Accepted: 31 December 2021;
Published: 10 February 2022.
Edited by:
Subramanian Dhandayuthapani, Texas Tech University Health Sciences Center El Paso, United StatesReviewed by:
Sarika Mehra, Indian Institute of Technology Bombay, IndiaHugues Ouellet, The University of Texas at El Paso, United States
Copyright © 2022 Thakur, Sharma, Hade, Chhaya, Kumar, Jolly and Dikshit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kanak L. Dikshit, a2FuYWtkaWtzaGl0QHB1LmFjLmlu