 Hannah S. Weber
Hannah S. Weber Bo Thamdrup
Bo Thamdrup Kirsten S. Habicht†
Kirsten S. Habicht†- Nordic Center for Earth Evolution and Department of Biology, University of Southern Denmark, Odense, Denmark
Sulfur isotope signatures provide key information for the study of microbial activity in modern systems and the evolution of the Earth surface redox system. Microbial sulfate reducers shift sulfur isotope distributions by discriminating against heavier isotopes. This discrimination is strain-specific and often suppressed at sulfate concentrations in the lower micromolar range that are typical to freshwater systems and inferred for ancient oceans. Anaerobic oxidation of methane (AOM) is a sulfate-reducing microbial process with a strong impact on global sulfur cycling in modern habitats and potentially in the geological past, but its impact on sulfur isotope signatures is poorly understood, especially in low-sulfate environments. We investigated sulfur cycling and 34S fractionation in a low-sulfate freshwater sediment with biogeochemical conditions analogous to early Earth environments. The zone of highest AOM activity was associated in situ with a zone of strong 34S depletions in the pool of reduced sulfur species, indicating a coupling of sulfate reduction (SR) and AOM at sulfate concentrations < 50 μmol L−1. In slurry incubations of AOM-active sediment, the addition of methane stimulated SR and induced a bulk sulfur isotope effect of ~29‰. Our results imply that sulfur isotope signatures may be strongly impacted by AOM even at sulfate concentrations two orders of magnitude lower than at present oceanic levels. Therefore, we suggest that sulfur isotope fractionation during AOM must be considered when interpreting 34S signatures in modern and ancient environments.
Introduction
Microbial sulfur transformations play an important role in the biogeochemistry of the modern Earth surface and have done so during most of Earth's history, while the interactions of the sulfur, oxygen, and carbon cycles have undergone dramatic changes (Canfield and Teske, 1996; Canfield and Raiswell, 1999; Canfield et al., 2006). Stable isotope signatures archived in the geological record have been pivotal for reconstructing milestones of Earth's biogeochemical history and, consequently, the evolution of life, and they are useful for studies of modern sulfur cycling (Canfield and Raiswell, 1999; Canfield et al., 2000; Canfield, 2001).
Accurate interpretation of isotope signatures depends on a detailed understanding of the isotopic fractionation associated with different sulfur transformations. Fractionation of sulfur isotopes at low temperature is primarily controlled by microbes mediating dissimilatory SR and sulfur disproportionation. Microbial sulfate reducers are observed to discriminate kinetically against 34 with enrichment factors, εSRR, in the range of –3 to –70‰ (εSRR = [34S/32Ssulfide – 34S/32Ssulfate] × [34S/32Ssulfate]−1) in both natural settings and pure cultures (Canfield, 2001; Wortmann et al., 2001; Canfield et al., 2010; Sim et al., 2011). The magnitude of fractionation is impacted by environmental parameters such as, e.g., availability of sulfate and electron donors, and is highly dependent on the physiology and growth of the sulfate-reducing microorganisms (Bradley et al., 2016). Bradley et al. (2016) showed that sulfate reducers suppress the discrimination against heavier sulfate isotopes when sulfate concentrations in their environment fall below strain-specific concentration thresholds. A sulfate threshold of ~200 μmol L−1 was suggested for natural communities of bacterial sulfate reducers (Habicht et al., 2002), but has recently been challenged by the report of substantial isotope fractionation between sulfate and sulfide at sulfate concentrations in the range of 5–10 μmol L−1 in the chemocline of ferruginous Lake Matano (Crowe et al., 2014). Isotope fractionation between sulfate and sulfide in natural environments may, however, also result from disproportionation processes. Disproportionation of intermediate sulfur species such as elemental sulfur, S0, producing sulfide and sulfate, is associated with fractionations of up to ~40‰ between the products (isotopically heavier sulfate and lighter sulfide; Canfield et al., 1998, 2005). Thus, in iron-rich environments where S0 forms from abiotic sulfide reoxidation by ferric iron, repeated cycles of disproportionation and sulfide oxidation may increase the isotopic fractionation between sulfate and sulfide substantially above that caused by SR alone (Canfield and Thamdrup, 1994; Pellerin et al., 2015).
The global sulfur cycle is further impacted by anaerobic oxidation of methane (AOM), a microbial process controlling recent atmospheric levels of the powerful greenhouse gas methane (Reeburgh, 2007). For the interpretation of both modern and ancient stable sulfur isotope signatures, information is scarce about sulfur isotope fractionation during the process both on the biochemical and the biogeochemical level. In marine sediments, AOM is associated with a consortium of anaerobic methane-oxidizing archaea (ANME) and sulfate-reducing bacteria (SRB; Boetius et al., 2000). It was hypothesized that sulfate-AOM functions by syntrophic coupling, where ANME oxidize methane and produce a soluble interspecies electron carrier that is used by the sulfate-reducing partner (e.g., Meulepas et al., 2010). As an alternative, recent studies of ANME-SRB consortia suggest direct electron transfer between the microbial partners by nanowires and/or outer membrane cytochromes providing cell-to-cell-contact (McGlynn et al., 2015; Wegener et al., 2015). Another model suggested two novel unidentified enzymatic pathways where the ANME carries out SR and produces zero-valent sulfur as key intermediate that is further disproportionated by the associated bacterial partner (Milucka et al., 2012).
Sulfur isotope fractionation associated with SR coupled to AOM, and its impact on sulfur isotope signatures in the environment, has gained limited scientific attention so far. A few studies demonstrated that sulfur-cycling AOM influences δ34S signatures in the environment (Jørgensen et al., 2004; Borowski et al., 2013) and concurrently alters the oxygen isotopic value in sulfate (Antler et al., 2014, 2015; Avrahamov et al., 2014; Deusner et al., 2014; Sivan et al., 2014). Two studies investigated sulfate-dependent AOM in slurry incubations fed with Black Sea microbial mats and seep sediments and suggested that AOM-driven sulfur isotope fractionation factors fall in the range of ~20 to 40‰ in gas seeps (Deusner et al., 2014; Sivan et al., 2014) and may exceed 60‰ in marine sulfate-methane-transition zones (SMTZs; Deusner et al., 2014).
So far, however, studies of sulfur isotope fractionation associated with AOM have focused mainly on sulfate concentrations in the millimolar range, whereas, from a geological perspective, sulfur cycling, and isotope fractionation at low sulfate concentrations is also of great relevance. Seawater sulfate levels in the Archaean Eon (before 2.5 billion years ago) are estimated to < 0.01% of modern seawater, probably restricting biological productivity (Crowe et al., 2014), while estimated atmospheric methane levels were two to three orders of magnitudes higher than at present (Catling et al., 2001). As iron dominated oceanic chemistry during this time and through much of Earth's history, a coupling of AOM to iron instead of sulfate has been proposed as a globally important process on early Earth (Konhauser et al., 2005; Beal et al., 2009). Such a process was suggested to occur in modern sediment (Konhauser et al., 2005; Beal et al., 2009; Sivan et al., 2011), but has not been conclusively proven at the cellular level. In contrast, it has been demonstrated that cryptic sulfur cycling involving the generation and consumption of sulfur intermediates can catalyse large-scale iron turnover even at micromolar levels of sulfate (Hansel et al., 2015). Consequently, an apparent coupling of AOM and Fe reduction could possibly result from sulfate-dependent AOM fueled by rapid reoxidation of sulfide (Beal et al., 2011; Norði et al., 2013).
Freshwater systems low in sulfate are frequently used as early Earth analogs to study components of early ocean biogeochemistry (Canfield et al., 2010; Crowe et al., 2011; Knossow et al., 2015). One such site is the anoxic sediment of Lake Ørn, with sulfate concentrations < 200 μmol L−1, maximum methane concentrations of 1.4 mmol L−1 and reactive iron oxide concentrations of up to 65 μmol cm−3 (Norði et al., 2013). Here, 90% of the methane was found to be consumed by AOM before it reached the sediments surface, and AOM was active even at sulfate concentrations as low as 3 μmol L−1 (Norði et al., 2013). However, it was not clear from this study whether AOM was coupled to iron reduction or to SR supported by a cryptic sulfur cycle.
In the present study, we hypothesized that AOM in Lake Ørn sediment can be directly coupled to sulfur turnover at very low sulfate concentrations and even has the potential to induce fractionation of stable sulfur isotope species under these conditions. We demonstrate that sulfur cycling in the AOM zone in Lake Ørn sediment is associated with substantial isotope fractionation and, based on experimental incubations, we infer a direct coupling of sulfur cycling and AOM at sulfate concentrations below 50 μmol L−1.
Materials and Methods
Sediment Sampling
Sediment was sampled in April and November 2012 at the center of Lake Ørn (water depth: 4 m), using a hand-operated Kajak sampler. In both April and November, three sediment cores were collected, each, for triplicate depth profiles of biogeochemical parameters. In April, samples were taken for depth profiles of stable sulfur isotopes (δ34S of reduced sulfur) and in November samples were taken to obtain concentrations of HCl-extractable iron (Fe), sulfate, sulfide, methane, zero-valent sulfur, stable sulfur isotopes (δ34S of reduced sulfur), porosity, rates of sulfate reduction (SR), and anaerobic oxidation of methane (AOM). Moreover, in November, we sampled (1) four additional cores and pooled the porewater from the corresponding horizons of all seven cores for measurement of δ34S of sulfate and (2) 24 additional cores for a sediment slurry incubation experiment. At sampling days, the bottom water temperature was 7 and 8°C, in November and April, respectively and the overlying water column was oxygenated. The biogeochemistry of the sediment was described previously (Norði et al., 2013).
Slurry Incubation Experiment
Twenty-four cores (diameter = 5.2 cm) were dissected under a constant nitrogen flow into an upper (0–7 cm; “sulfide reoxidation zone”) and a lower (7–21 cm, “AOM zone”) depth interval, transferred into two and three Schott glass bottles, respectively, and mixed 1:1 with medium (anoxic water, 3 mmol L−1 NaHCO3; pH = 6.6; 200 μmol L−1 and 50 μmol L−1 Na2SO4, respectively). For the lower depth interval, one slurry bottle was amended with S0 to a final concentration of ~100 μmol cm−3. Slurries were then split and transferred into 0.5 L (sulfide reoxidation zone) and 1 L (AOM zone) Schott bottles, sealed with butyl rubber stoppers (anoxic as flushed and stored under helium atmosphere before usage) and the headspace was flushed with N2:CO2 (90:10; 0.5 bar) for 15 min. Afterwards, half of the bottles of the sulfide reoxidation zone slurries and two-thirds (including the elemental sulfur amended ones) of the AOM zone slurries were flushed with methane for 15 min. The bottles were individually packed into anaerobic plastic bags, which were flushed with helium. The bottles were stored at 5°C in darkness and carefully inverted for mixing every few days until further processing.
One bottle from each treatment was processed after 1, 6, 19, 43, 70 days, respectively, and for sulfide reoxidation zone, additionally after 103 days, as follows: the bottles were inverted, transferred into an anaerobic glove bag (filled with nitrogen) and samples were taken through the rubber stopper via a syringe and needle (flushed with helium prior to usage) for concentration determination of porewater parameters (sulfide, iron, sulfate), and filtered through a nylon syringe filter (Q-Max; pore size = 0.2 μm) directly into the corresponding vials. The bottles were then removed from the anaerobic glove bag and opened under constant nitrogen flow for concentration determination of solid and gaseous parameters (methane, iron, porosity, zero-valent sulfur, δ34S signatures) and SR measurements in triplicates.
Solid, Porewater, and Gaseous Parameters
The cores obtained for depth profile characterization were all dissected in 2 cm intervals (April) or 2 cm (0–6 cm) and 3 cm (6–21 cm) intervals (November) within 2 days after sampling under constant nitrogen flow at 5°C. From each interval, one sample was taken per parameter, which resulted in triplicate depth profiles for each sampling point (April and November).
For porewater analysis, centrifugation vials were filled completely, centrifuged at 4000 g for 10 min (precooled, Centrifuge 5810; Eppendorf) and the supernatant was filtered anoxically (glove bag filled with nitrogen) through a syringe filter (pore size = 0.2 μm; syringe, needle, and filter flushed and stored in helium prior to usage). For dissolved Fe, 1 mL porewater was mixed with 20 μL 6 N hydrochloric acid (HCl), stored at −20°C, and Fe2+ concentrations were determined using the Ferrozine reagent (Stookey, 1970; Thamdrup et al., 1994). For analysis of sulfate and sulfide concentrations, 1 ml porewater was mixed with 20 μL of 20% zinc acetate and stored at −20°C. Sulfate concentrations were quantified via ion chromatography (Dionex ICS-1500) with a detection limit of 1 μmol L−1 and sulfide concentrations were quantified by the methylene blue method (detection limit = 0.5 μmol L−1; Cline, 1969).
For analysing methane concentrations, 2 mL of sediment was subsampled in triplicates via a cut-off syringe directly into a 20 mL serum vial, prefilled with 5 mL 2.5% sodium hydroxide (NaOH), closed, and shaken immediately and stored upside down for at least 24 h to equilibrate the methane into the headspace. Methane was quantified by injecting 100 μL of the headspace into a gas chromatograph with an FID detector (Perkin Elmer).
The density was analyzed by subsampling 1 mL of sediment with a cut-off syringe into a pre-weight centrifugation vial and the water content/porosity as the weight loss from after drying at 90°C for 48 h. Solid-phase Fe was extracted by transferring 0.5 mL sediment into 5 ml 1 N HCl and shaking the samples on a rotary shaker for 24 h in darkness (Raiswell et al., 1994). The method quantitatively dissolves ferrihydrite (Fe2O3⋅0.5(H2O)), and ferrous phases such as siderite (FeCO3), vivianite (Fe3(PO4)2⋅8H2O) and iron monosulfides (FeS), but not crystalline iron oxides or pyrite (Raiswell et al., 1994). After extraction, we centrifuged the samples at 4000 g for 10 min (Centrifuge 5810; Eppendorf), filtered the supernatant through a nylon syringe filter (pore size = 0.2 μm) and quantified the concentration using the Ferrozine method (Stookey, 1970; Thamdrup et al., 1994). A previous study reported a total iron content of ~3000 μmol g−1 in Lake Ørn sediment (O'Connell et al., 2015), suggesting that the 1 N HCl-extractable iron corresponds to < 20% of the total iron pool. For quantification of zero-valent sulfur, sediment was mixed with 20% zinc acetate and stored at −20°C until further processing (Ferdelmann et al., 1997). For analysis, 0.2–0.5 g sediment of each sample was mixed with 10 mL methanol and shaken on a rotary shaker for 24 h. Thereafter, the methanol-extract was centrifuged at 4000 g for 10 min (Centrifuge 5810; Eppendorf) and 1 mL of the supernatant was used for concentration measurements by HPLC-analysis (Agilent 1100 series, Hewlett Packard). For the incubations with S0 addition, methanol treatment showed insufficient extraction of S0 and data are therefore unreliable and not shown. For δ34S and concentration analysis of reduced sulfur species (acid volatile sulfur, AVS, and chromium reducible sulfur, CRS) and δ34S analysis of zero-valent sulfur and sulfate, sediment was mixed with 20% zinc acetate and stored at −20°C. After thawing, sediment was centrifuged immediately at 4000 g for 10 min (Centrifuge 5810; Eppendorf) to separate the supernatant from the sediment. For the δ34S sulfate depth profiles, supernatant was pooled from seven cores. Thereafter, the supernatant was filtered through a polycarbonate filter (pore size = 0.45 μm), acidified with 6 N HCl to a pH = 1 and barium chloride (BaCl2, 1 M) was added in surplus to form barium sulfate (BaSO4). The sample was then heated up to 80°C to slowly evaporate water and finally the remaining BaSO4 was boiled with 25 mL of a reducing mixture of hydriodic acid (HI), concentrated hydrochloric acid (HCl), and 50% hyphosphorous acid (H3PO4; Thode et al., 1961) for 6 h in a distillation flask connected to a reflux condenser. Liberated hydrogen sulfides were driven out by a constant N2 flow through a water trap into 0.5 mol L−1 cadmium acetate dihydrate (Cd(CH3COO)2⋅2H2O) to produce cadmium sulfide (CdS). CdS was converted with 1 M silver nitrate (AgNO3) into silver sulfide (Ag2S), warmed up to 50°C for 24 h (facilitates coagulation), filtered onto a cellulose ester filter (pore size = 0.45 μm) and dried at room temperature. The reduced sulfur compounds AVS and CRS were obtained using the hot two-steps chromium distillation (Canfield et al., 1986; Fossing and Jørgensen, 1989), where AVS and CRS were reduced via HCl and hot chromium (Cr(II)) solution and liberated hydrogen sulfides were trapped as Ag2S. To obtain δ34S values from zero-valent sulfur, a Soxhlet extraction was carried out from samples at incubation start, using 5–10 g sediment and acetone as extractant. In brief, over an extraction period of 24 h, dissolved zero-valent sulfur was reduced and precipitated on copper wires (Cu) as copper sulfide (CuS; Allen and Parkes, 1995). Thereafter, CuS wires were distilled using the hot chromium distillation (Canfield et al., 1986; Fossing and Jørgensen, 1989) and liberated hydrogen sulfides were trapped as Ag2S. A Soxhlet extraction was also carried out with zero-valent sulfur that was added to the incubations of the AOM zone. The sulfur isotope compositions of AVS, CRS, zero-valent sulfur, and sulfate were quantified with a gas-source Isotope Ratio Mass spectrometer (Delta V ADVANTAGE), where SO2 gas was produced from combustion of the samples in a FlashEA 1112 Element Analyser and data were reported in Isodat Workspace 3.0 as per mill difference relative to the VCDT and three in-house (Ag2S) standards (S.D. 0.5‰). The δ34S value of zero-valent sulfur “off the shelf” was 16.7‰.
Subsampling for SR and AOM Rates
For SR and AOM rates, 5 mL sediment/slurry was filled into glass tubes in triplicates, closed with plunger and septum (stored under helium prior to sampling) and pre-incubated for 1 h before injection of radiotracers (Jørgensen, 1978).
For SR rates, per sample 20 μl carrier-free 35 tracer (100 kBq) was injected through the septum and the samples incubated at 5°C in darkness for 6 h. The incubations were stopped by transfer to zinc acetate (20%; 10 mL) and freezing at −20°C. Killed controls were taken in parallel by transferring sediment/slurry to zinc acetate after incubation and then adding tracer. Radiolabeled sulfur species (sulfate and AVS + CRS) were determined via the cold chromium distillation (Kallmeyer et al., 2004; Røy et al., 2014).
For AOM rates, 14CH4 (dissolved in water; 20 μl; 0.5 kBq; supplied from American Radiolabeled Chemicals, cleaned with hopcalite and sodium hydroxide) was injected through the septum into the sediment/slurry and samples incubated at 5°C in darkness for 20 h. To stop incubations the samples were transferred into glass vials prefilled with 15 ml sodium hydroxide (2.5% w/v) and immediately closed with butyl rubber stoppers, shaken, and stored upside down. Methane concentrations were determined as described above and activities of 14CH4 and 14CO2 were quantified (Treude et al., 2005), using phenylethylamine and sodium hydroxide (1:1) as CO2 trap. Killed controls were taken with each time point, where tracer was injected after determining the microbial activity.
Radioactivity was measured on a QuantaSmart-4.00 Scintillation counter (count time: 10 min; coincidence time: 18 ns; delay before burst: 75 ns). For determining the detection limits for the SR and AOM rate measurements, the mean plus three times the standard deviation of the killed controls were subtracted from the product pools, 35S sulfides, and 14CO2, respectively.
Model
A curve-fitting model to obtain εAOM was built for the AOM zone incubations based on the assumption that the total sulfate consumption rate of AOM was the sum of the net sulfate production from the unamended incubations and the net sulfate consumption of the methane-amended incubations. We used T0 values of the sulfate concentration and δ34S sulfate from the AOM zone incubation amended with methane and calculated theoretical enrichments of δ34S sulfate over a course of 80 one-day time steps for different values of εAOM between 20 and 40‰ at 1‰ increments. The model assumes no isotope fractionation associated with sulfate production from sulfide oxidation and approximates the rate of 32 production and consumption by the measured bulk rates. In more detail, for time step T0, we first calculated the ratio 34∕32R between 34 and 32 by 34∕32R = ((δ34ST0/1000) + 1) × 34∕32Rstandard with δ34ST0 = 22.2‰ and 34∕32Rstandard = 0.0045005 (VCDT). Thereafter, we obtained the 32 and 34 concentrations by [32]T0 = []T0/(34∕32R + 1) with []T0 = 39 μmol L−1 and [34]T0 = []T0 – [32]T0. For each following time step Tn, [32]Tn was calculated by [32], where ProdSO42− = 3.5 nmol cm−3 day−1 is the net sulfate production rate in the slurry incubation without methane amendment and ConSO42− = 4 nmol cm−3 day−1 is the net sulfate consumption in the slurry incubation with methane addition. Furthermore, we calculated [34] for the same time steps using [34]Tn = [34]Tn−1 + ((Tn – Tn−1) × ProdSO42− × 34∕32Rsulfides) – ((Tn – Tn−1) × 34ProdTn−1), where 34∕32Rsulfides = 0.0044856 (δ34S = 3.0‰), which corresponds to the value of the sulfide pool of AOM zone slurry incubations. 34ProdTn−1 was calculated for each time step by 34ProdTn−1 = (ConSO42−/α) × ([34]/[32]). For α, we used values in the range of 1.020 and 1.040, which corresponds to εAOM values between 20 and 40‰. After calculating α-dependent [34], and [32] for all time steps, we calculated δ34 using δ34 = ((([34]/[32])/34∕32Rstandard) – 1) × 1000 and matched the resulting δ34S values with the δ34S sulfate enrichment measured in the methane-amended incubations to determine the value of α yielding the best fit (least sum of squares). The εAOM value was determined from the best fit α.
Results and Discussion
Large AOM Zone in Lake Ørn Sediment
The biogeochemical zonation of Lake Ørn sediment showed a well-developed AOM zone similar to a previous report (Norði et al., 2013). Thus, methane diffusing upwards from the methanogenic zone below 20 cm depth was largely consumed well below the oxic realm (upper 4 mm; Norði et al., 2013), as indicated by the concave trend of methane concentrations over depth and low μmolar levels above 7 cm (Figure 1A, Table 1). Characteristic of this sediment is a very high iron content and a deep penetration of poorly crystalline, HCl-extractable ferric iron, which reached down to 20 cm, while concentrations of HCl-extractable ferrous iron increased with depth (Figure 1B, Table 1). Sulfate depth profiles resembled each other in five of seven cores with surface concentrations of ~200 μmol L−1 and a detection depth of 17 cm (Figure 1A, Table 1). In two of the cores, the sulfate profile deviated and one of them had a sulfate concentration of ~70 μmol L−1 at maximal sampling depth (18–21 cm). Sulfate and methane coexisted in a broad SMTZ at 7–17 cm. Rates of dissimilatory SR were highest in the depth intervals 2–4 cm and 12–15 cm, and AOM rates increased steeply until 12–15 cm, locating the AOM zone to 8–21 cm (Figure 1C, Table 1), in good agreement with the SMTZ (Figure 1A). The rates ratio of SR to AOM in the AOM zone was ~2:1. Thus, the measured SR rate exceeded the potential sulfate demand of AOM suggesting that at least some SR was coupled to organotrophy.
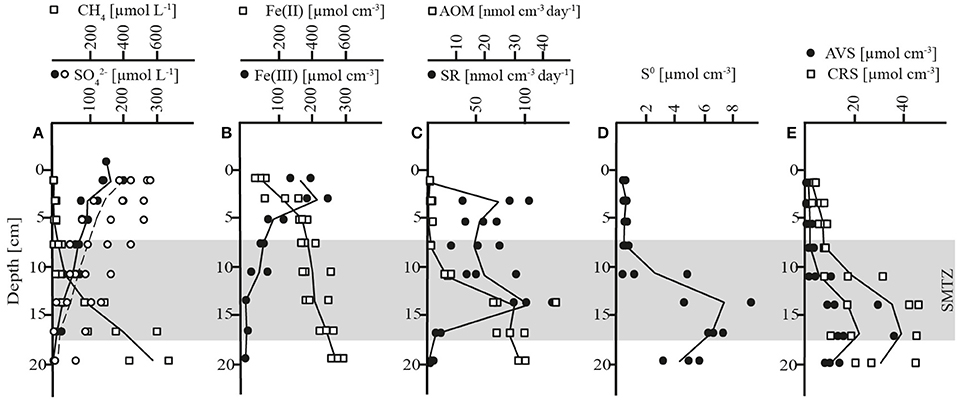
Figure 1. Concentration depth profiles and microbial rates from November. (A) methane (CH4) and sulfate (), whereas closed circles represent sulfate data from triplicate cores and open circles represent sulfate data from four additional cores (B) HCl-extractable iron (Fe(II) and Fe(III)) (C) rates of sulfate reduction (SR) and anaerobic oxidation of methane (AOM) (D) zero-valent sulfur (S0) (E) acid volatile sulfur (AVS); chromium reducible sulfur (CRS); shadowed area: sulfate-methane-transition-zone (SMTZ).
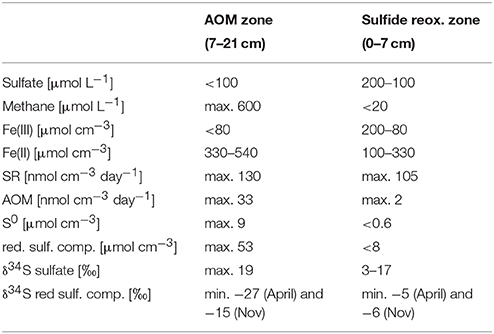
Table 1. Porewater and solid-phase parameters of triplicate depth profiles in November 2012, summarized for the AOM zone (7–21 cm) and the sulfide reoxidation zone (0–7 cm).
Microbially produced hydrogen sulfide did not accumulate to detectable levels in the porewater, which implies a rapid turnover time (10 min or less relative to the measured SR) in combination with ferric iron reduction and iron sulfide precipitation. The distributions of reduced sulfur species S0, acid volatile sulfides (AVS, here: FeS) and chromium reducible sulfides (CRS), differed markedly between the two zones of highest SR rates and divided the sediment column into two distinct horizons: an upper layer (0–7 cm) with low concentrations and shallow gradients of these compounds, and a deeper layer at 7–21 cm, coincident with the AOM zone, where all three phases accumulated, reaching highest concentrations at the base of the SMTZ at 15–18 cm depth (Figures 1D,E, Table 1). The lack of accumulation of reduced sulfur species in the upper layer, despite active SR, indicated an extremely close coupling of SR, and sulfide reoxidation processes with ferric iron as the most likely electron acceptor (Schippers and Jørgensen, 2002; Holmkvist et al., 2011). Potential reactions include the abiotic oxidation of sulfide to zero-valent sulfur with iron (hydr)oxides (Pyzik and Sommer, 1981; Yao and Millero, 1996), followed by disproportionation to sulfide and sulfate (Thamdrup et al., 1993). Alternatively, sulfate may have been generated through direct oxidation of reduced sulfur compounds coupled to iron reduction without involvement of S0 disproportionation, as discussed below. In the AOM zone, the availability of reactive ferric iron was lower and the pool of ferrous iron was larger than in the upper layer, and reduced sulfur compounds accumulated (Figures 1B,E, Table 1). Zero-valent sulfur concentrations increased steeply below 7 cm, resulting in a peak of 7 μmol cm−3, corresponding to almost half of the AVS concentration (Figures 1D,E, Table 1), in the zone of highest AOM activity. A transient accumulation of S0 is frequently observed during early diagenesis, where it is assumed to result from the reoxidation of H2S with ferric iron (Thamdrup et al., 1994; Hansel et al., 2015). The large fraction of S0 in Lake Ørn sediment is unusual, however, as S0 is typically an order of magnitude less abundant than FeS in sulfate-reducing sediments (e.g., Thamdrup et al., 1994).
In summary, the geochemical profiles and rate measurements demonstrate active sulfur cycling throughout the sulfate-containing zone of the sediment, with highly efficient recycling of sulfide in the upper layer, where the ferric iron content is highest, and precipitation of reduced sulfur compounds in the AOM zone, although the accumulation of S0 indicates some recycling there as well. The enhanced rate of SR in the AOM zone could suggest a coupling of the two processes, but the profiles alone do not allow us to discern between such a coupling and, e.g., a direct coupling of AOM to iron reduction.
Depletion of 34S in Sulfides in AOM Zone
The AOM zone coincided with a distinct zone of strong 34S depletion in the reduced sulfur compounds with lowest δ34S values of −27‰ in April and −15‰ in November (Figure 2, Table 1). In April, δ34S values were up to 21 and 29‰ higher above and below the AOM zone, respectively, than within it. In November, the SMTZ was shifted a few cm downwards in comparison to April and coincided with a distinct, but less pronounced pool in 34S-depleted sulfides. Here, δ34S values above and below the AOM zone were up to 9 and 16‰ higher than within the zone, respectively. The shift of the 34S depletion zone was likely dynamically related to microbial activity and therefore rather a result of seasonal dynamics than of historical origin. The δ34S profile for sulfate, determined in November (Figure 2A) showed increasing enrichment of 34S in the sulfate pool from ~3 to ~17‰ from the bottom water to 7 cm depth and a small further enrichment to ~19‰ in the AOM zone. Accordingly, the isotopic shift between sulfate and reduced sulfur compounds, in the following abbreviated as Δ34S ( − HS−), increased with depth: In the upper zone of sulfide reoxidation, where sulfate concentrations were >100 μmol L−1, Δ34S ( − HS−) was between ~15 and 29‰. In the AOM zone where sulfate concentrations decreased from 100 μmol L−1 to the detection limit, isotope differences between the two pools were between 21 and 34‰ While the Δ34S ( − HS−) values thus indicate substantial fractionation associated with the accumulation of reduced sulfur species in the AOM zone, this was not reflected in a concomitant increase in δ34S of sulfate with depth in this zone, as should be expected (Figure 2A). We attribute this discrepancy to the procedure used to obtain sufficient material for the sulfate isotope measurements, which involved the combination of pore water from seven parallel cores for a single measurement, whereas δ34S of reduced sulfur, as well as all other geochemical parameters, were measured in triplicates in the same three of the seven cores. Unfortunately, two of the additional cores used for δ34S of sulfate had strongly deviating sulfate distribution with no evidence of sulfate consumption below 10 cm depth. As sulfate from these cores completely dominated the combined sulfate sample in the two deepest depth intervals, we speculate that it also caused the leveling off of the δ34S sulfate profile. Thus, the Δ34S ( − HS−) values provide more robust evidence of isotope fractionation than the sulfate δ34S gradient.
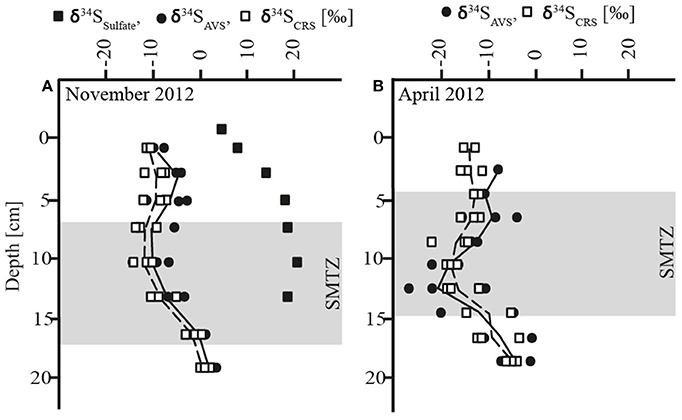
Figure 2. (A) Depth profiles of δ34S of sulfate, acid volatile sulfur (AVS) and chromium reducible sulfur (CRS) in November (B) Depth profiles of acid volatile sulfur (AVS) and chromium reducible sulfur (CRS) in April; shadowed area: sulfate-methane-transition-zone (SMTZ).
The δ34S signatures and the distinct offset between the oxidized and reduced sulfur pools must be understood as a “bulk” signal and as a result of a concert of different metabolic pathways of the sulfur cycling community. The primary control of sulfur isotope fractionation is generally organotrophic SR and the process was likely to be active in both the sulfide reoxidation and in the AOM zone. An earlier study showed that microbial sulfate reducers, including Danish lake sediment communities, limit 34S isotope fractionation at sulfate concentrations < 200 μmol L−1, reaching ≤ 5‰ below 100 μmol L−1 (Habicht et al., 2002). Based on this finding and due to decreasing sulfate concentrations, the impact of bacterial SR on sulfur isotope fractionation in Lake Ørn sediment was expected to be low in general, greatest in the sulfide reoxidation zone, and decreasing with increasing depth, which contrasts with our observations. However, the distinct 34S depletion in the reduced sulfur compounds in Lake Ørn was not only associated with the zone of very low-sulfate concentrations, but also with the highest AOM activities (Figures 1C, 2). This finding suggests that the observed sulfur isotope fractionation may arise specifically through AOM-dependent sulfur cycling. A recent study showed that the sulfate concentration dependence of sulfur isotope fractionation varies markedly between bacterial species (Bradley et al., 2016). Thus, AOM-dependent sulfate reducers could potentially be capable of 34S fractionation at sulfate levels where fractionation is suppressed in organotrophic sulfate reducers. The potential for a distinct fractionation pattern associated with SR during AOM is further emphasized by the recent hypothesis that AOM-dependent SR occurs through a unique S0-producing pathway, as hypothesized (Milucka et al., 2012). Antler et al. (2015) recently suggested that AOM-dependent SR imparts a unique δ18O/δ34S ratio to the residual sulfate in estuarine and marine sediments, but this was based on sulfate at millimolar concentrations, and a fractionation factor relative to the reduced products was not determined.
In addition to microbial organiclastic and methanoclastic SR, the oxidative cycling of intermediate sulfur species may have affected the stable sulfur isotope signatures in Lake Ørn. In particular, S0 was unusually abundant in the AOM zone (Figure 1D). The source S0 may have been sulfate-dependent AOM either by a direct intracellular archaeal production (Milucka et al., 2012) or indirectly via a sulfide reoxidation process with ferric iron (Figure 5). Therefore, microbial disproportionation fueled by methanoclastic and organiclastic SR was potentially an active process contributing to the 34S enrichment in the sulfate pool and 34S depletion in the reduced sulfur compounds (Canfield and Thamdrup, 1994), both in the sulfide reoxidation zone and in the AOM zone.
To further investigate which sulfur cycling microbial processes were active and altering the δ34S signatures in Lake Ørn sediment, we set up a series of anoxic slurry incubations from the zones of sulfide reoxidation and AOM. We studied sulfur turnover and δ34S signatures in incubations of both zones where methane was removed at incubation start in comparison with incubations where methane was added to enable sulfate-dependent AOM. The potential for S0 disproportionation was investigated in the AOM zone by addition of zero-valent sulfur to methane-amended incubations.
An Iron-Fueled Cryptic Sulfur Cycle
In slurries where methane was removed at incubation start, a net accumulation of sulfate was measured in both the sulfide reoxidation zone (0–7 cm) and the AOM zone (7–21 cm) with rates of 3.6 ± 0.9 and 3.3 ± 0.3 nmol cm−3 day−1, respectively (Figures 3A, 4A, Table S1). In both zones, there were no significant changes in S0 concentrations over time (Figures 3B, 4B, Table S1) and soluble hydrogen sulfide remained around the detection limit. At the same time, extractable ferrous iron concentrations increased and ferric iron decreased. Methane did not build up over the course of the experiment (Figures S1A, S2A). Despite net sulfate accumulation, SR as measured with the 35S assay was active at rates averaging 1 nmol cm−3 day−1 in the sulfide reoxidation zone and 10 nmol cm−3 day−1 in the AOM zone, and was hence substantially lower in the slurries than in the depth profiles (Figures 1C, 3C, 4C, Table 1, Table S1). The δ34S signatures showed depletion of 34S in the growing sulfate pool from 3 to −2‰ in the sulfide reoxidation zone and from 21 to 8‰ in the AOM zone (Figures 3D, 4D). At the same time, δ34S values of reduced sulfur compounds remained around −9‰ (AVS) and −12‰ (CRS) in the top zone and around −4‰ (AVS) and −6‰ (CRS) in the AOM zone with no significant change over time (Figures 3E,F, 4E,F).
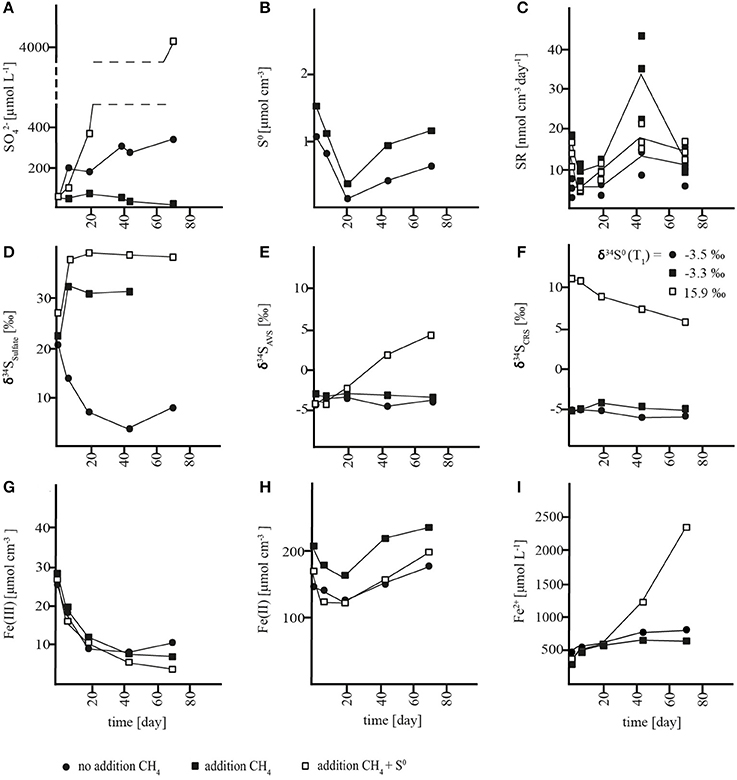
Figure 3. Concentration changes over time in AOM zone slurry incubations. (A) sulfate (), (B) zero-valent sulfur (S0), (C) sulfate reduction rates (SR), (D) δ34S values of sulfate, (E) δ34S values of acid volatile sulfur (AVS) (F) δ34S values of chromium reducible sulfur (CRS), (G) solid-phase HCl-extractable iron (Fe(III)), (H) solid-phase HCl-extractable iron (Fe(II)), and (I) porewater iron (Fe2+).
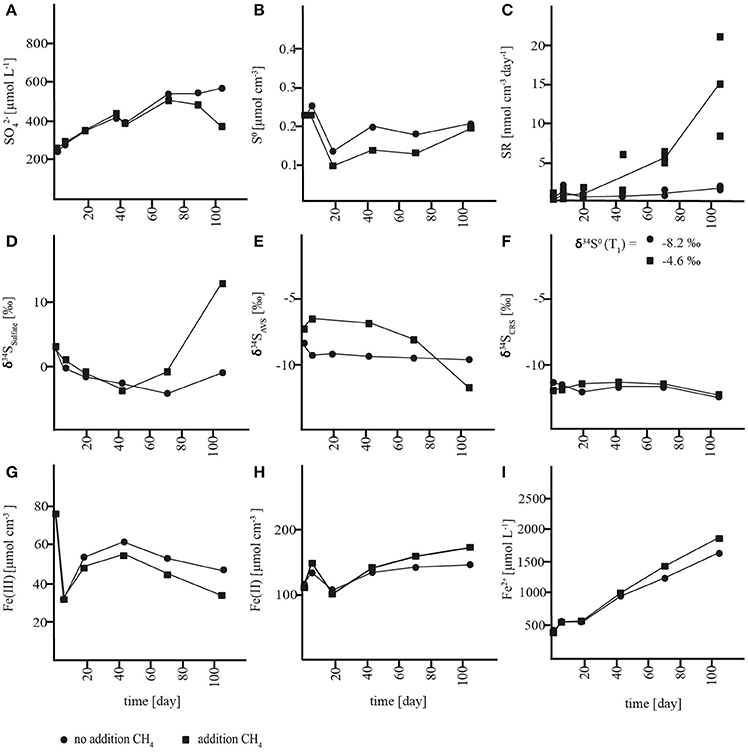
Figure 4. Concentration changes over time in sulfide reoxidation zone slurry incubations. (A) sulfate (), (B) zero-valent sulfur (S0), (C): sulfate reduction rates (SR), (D) δ34S values of sulfate, (E) δ34S values of acid volatile sulfur (AVS) (F) δ34S values of chromium reducible sulfur (CRS), (G) solid-phase HCl-extractable iron (Fe(III)), (H) solid-phase HCl-extractable iron (Fe(II)), and (I) porewater iron (Fe2+).
The net production of sulfate with parallel reduction of ferric iron and increase of ferrous iron concentrations in the slurries indicated an iron-driven sulfate production mechanism. Sulfate production caused by oxidation of reduced sulfur compounds due to oxygen contamination can be excluded due to anoxic conditions. Therefore, by analogy to our interpretation of the lacking accumulation of reduced sulfur compounds at 0–7 cm (see above), we propose that anaerobic sulfide oxidation was the source of sulfate through either (1) coupled partial oxidation and disproportionation or (2) a direct oxidation to sulfate. In the first scenario, abiotic oxidation of sulfide to S0 would be coupled to iron reduction followed by microbial disproportionation of S0. Subsequently, S0 disproportionation would lead to the production of sulfate and sulfide (Thamdrup et al., 1993). As mentioned above, S0 disproportionation leads to enrichment of sulfate and depletion of reduced sulfur compounds in 34S (Canfield and Thamdrup, 1994), which is contrary to the observed 34S depletion in sulfate and rather constant δ34S values of the reduced sulfur compounds (Figures 3E,F, 4E,F). Furthermore, a change in the δ34S of the reduced sulfur compounds should have been evident especially in the sulfide oxidation zone incubations, where initial concentrations of reduced sulfur species were very low. By contrast, direct abiotic or microbial sulfide oxidation to sulfate is generally associated with negligible isotope fractionation (Habicht et al., 1998 and references therein). Thus, a direct oxidation of reduced sulfur compounds to sulfate is a likely explanation for the decreasing δ34S of sulfate with no change in δ34S of the reduced species. Indeed, a mixing plot of δ34S versus sulfate concentration from the sulfide reoxidation zone showed that the sulfate produced corresponded to an end-member source with a δ34S value of −11.2 ± 1.4‰ (Figure S3A), which equates to the δ34S value of reduced sulfur compounds (−11‰). This result indicated that sulfate is supplied by direct sulfide oxidation to the sulfide oxidation zone, and that this oxidation included sulfide produced during SR, the activity of which was documented with the 35S assay (Figure 4C), such that SR did not impact the δ34S fingerprint. The AOM zone, on the other hand, showed an end-member value of +3.9 ± 3.0‰ (Figure S3B), which was well-above the δ34S value of reduced sulfur compounds of −5‰. This offset points to the involvement of additional processes in sulfur cycling, and the trend may be explained by S0 disproportionation acting as an additional source of sulfate concomitantly with sulfur oxidation, or by incomplete reoxidation of sulfide formed during SR.
To further investigate the potential for S0 disproportionation and to elucidate its effect on stable sulfur isotope signatures in Lake Ørn sediment, we added zero-valent sulfur (~100 μmol cm−3) to parallel methane-containing slurries of the AOM zone. As expected for S0 disproportionation, sulfate concentrations increased (Thamdrup et al., 1993) dramatically at a rate of 61 ± 7 nmol cm−3 day−1. Ferrous iron also accumulated while ferric iron concentrations decreased (Figures 3G–I). After depletion of ferric iron, soluble hydrogen sulfide started to build up (Figure S1D) as expected during disproportionation (Thamdrup et al., 1993) and in further support of our conclusion that porewater sulfide levels in Lake Ørn are controlled by iron with a large buffer capacity. SR rates, measured with the 35S assay, averaged 13 nmol cm−3 day−1, which was comparable to the rate in the previously described incubations of the AOM zone (Figure 3C, Table S1). With increasing sulfate concentrations in the S0-amended slurry incubations, sulfate was enriched in 34S from 27 to 38‰, corresponding to an isotopic shift of ~22‰ between added S0 and sulfate, while the CRS pool, including S0, was depleted in 34S (Figures 3A,D,F). The isotopic shift between S0 and sulfate is within the range previously observed for sulfur disproportionation in freshwater cultures (Canfield et al., 1998). The results indicate that S0 disproportionation was the dominant microbial pathway of sulfur transformation in the incubation with additional zero-valent sulfur and therefore had a general potential to occur in situ.
In conclusion, sulfide in Lake Ørn sediment was efficiently recycled to sulfate by direct, iron-driven anoxic sulfide oxidation, as well as by S0 disproportionation. This indicates that the deep sulfate-containing zone and the thick SMTZ in the sediment were maintained by this internal cryptic sulfur cycle, thus setting the scene for sulfur-cycling AOM (Figure 5).
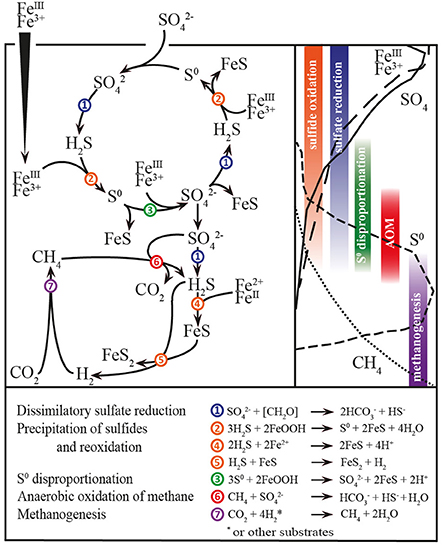
Figure 5. Scheme of abiotical and microbial processes in Lake Ørn sediment. Left up: Sulfur, carbon, and iron cycling; numbers indicate processes; right up: depth distribution of porewater and solid parameters and depth distributions of processes; down: stoichiometric balanced microbial and abiotical process reactions.
Impact of Methane on Sulfur Turnover and δ34S Signatures
In order to investigate the impact of methane on sulfur cycling and δ34S signatures, methane was added to slurries from the sulfide reoxidation and the AOM zone (1.4 mmol L−1). In the AOM zone, this resulted in substantial changes in sulfur turnover and δ34S biogeochemistry relative to the incubations without methane addition. Instead of accumulating, sulfate was consumed at ~0.5 ± 0.3 nmol cm−3 day−1 until depletion at day 73, which corresponds to a gross consumption of ~4 nmol cm−3 day−1 (Figure 3A, Table S1), assuming that the sulfate production observed in the absence of methane was not affected by methane addition. In the sulfide reoxidation zone, sulfate was produced at the same rate as without methane addition during the first 43 days, but this was followed by less intense net sulfate production and eventually a net sulfate consumption of ~4 nmol cm−3 day−1 after day 73 (Figure 4A; Table S1). Changes of methane concentrations could not be determined in the methane-amended slurries due to the large methane pool of the headspace. SR rates measured by the 35S assay increased over time and were substantially higher than SR rates from the unamended incubations, with mean rates of 11 nmol cm−3 day−1 in the top zone and 23 nmol cm−3 day−1 in the AOM zone (Figures 3C, 4C, Table S1). In the latter, SR decreased abruptly at the last time point, as sulfate was depleted. The surplus sulfate consumption in comparison to the unamended incubations was similar for the two zones, 10–13 nmol cm−3 day−1. Neither in the AOM zone nor the sulfide reoxidation zone significant changes in S0 concentrations or accumulation of soluble H2S were detected (Figures 3B, 4B). Iron consumption and production rates were comparable to incubations without addition of methane in the AOM zone, but were enhanced in the sulfide reoxidation zone (Figures 3G–I, 4G–I).
The addition of methane to the Lake Ørn slurries could have influenced the sulfur and iron cycles in three possible scenarios. In the first scenario, methane addition may have stimulated iron reduction that resulted in depletion of bioreducible Fe(III). Thereafter, organotrophic iron reducers may have been limited and their competitive suppression of organotrophic SR was overturned. Thus, in this scenario, methane addition would have resulted in enhanced organotrophic SR, which caused a net consumption of sulfate and a concurrent 34S enrichment of the sulfate pool. We consider this explanation as rather unlikely, as methane addition did not affect Fe(III) concentrations in the AOM zone, and although an effect was observed in the sulfide reoxidation zone, Fe(III) was not depleted to an extent that would typically limit the process in sediments (Figures 3G–I, 4G–I; Thamdrup, 2000).
In the second scenario, methanogens may have been active and competing with sulfate reducers for organic carbon, potentially in both investigated zones, but to a higher extent in the AOM zone. The addition of methane may have suppressed the methanogens, and therefore the activity of organiclastic SR increased, explaining the higher SR rates in the methane-amended slurries in comparison with the non-amended ones. In this scenario, the impact of methane on the sulfate concentrations and 34S signatures would be explained solely by the activity of organotrophic SR and does not require any involvement of sulfur-cycling AOM. We argue against this scenario by highlighting (1) that methanogenesis was not detected in the incubations without methane amendment and (2) that organotrophic iron reduction was a dominant process in Lake Ørn sediment as shown by Norði et al. (2013) and by the apparently methane-independent rapid decrease in ferric iron and increase of ferrous iron in our incubations (Figures 3G–I, 4G–I). Therefore, it is more likely that methanogens were inhibited by competition with iron reducers for common substrates (Lovley and Phillips, 1986; Roden and Wetzel, 1996) than that they were outcompeting organotrophic sulfate reducers for the same reason. Even if methanogenesis was occurring, an inhibition of the process at the methane levels used in our incubations has to our knowledge never been reported.
We therefore propose a third scenario in which methane addition stimulated the activity of sulfate-dependent AOM that caused net sulfate consumption and a significant alteration in the δ34S fingerprint. We propose that the enhanced 35SR rates in the methane-amended slurries reflect sulfate-dependent AOM activity. In the sulfide reoxidation zone, we interpret the first 43 incubation days as a lag phase and the rest of the incubation as an AOM phase although an earlier stimulation of SR may have been masked by complete reoxidation. We consider scenario three, the stimulation of sulfate-dependent AOM by methane addition, as the most likely and therefore propose that sulfate-dependent AOM results in substantial stable sulfur isotope fractionation at low micromolar sulfate levels. In comparison to the δ34S depletion of sulfate in methane-unamended sediment, δ34S values of sulfate showed 34S enrichment of up to 17‰ in the top zone and 9‰ in the AOM zone relative to initial values in the methane-amended incubations. At the same time, reduced sulfur compounds became depleted in 34S in the top zone, but did not change substantially in the AOM zone, where changes would have been masked by the high initial concentrations (Figures 3E,F, 4E,F, S1B,C, S2B,C).
In conclusion, organotrophic SR probably took place in all slurry incubations and did apparently limit 34S fractionation under incubation conditions. Microbial S0 disproportionation showed potential to occur and caused an isotopic effect in S0 amended incubations. A substantial difference in sulfur cycling and δ34S signatures resulted due to the addition of methane, which we interpret as a direct effect of sulfate-dependent AOM. We applied a curve-fitting model for the AOM zone incubation with methane, in order to estimate a bulk fractionation factor for the sulfur fractionating microbial processes (Figure S4). The model was based on the sum of net sulfate production from the unamended incubations and the net sulfate consumption of the methane-amended incubations and yielded an isotope effect of 29‰ (Figure S4).
Conclusions and Implications
AOM strongly regulates sulfur and methane dynamics in modern marine systems and probably did so in the geological past (Reeburgh, 2007; Knittel and Boetius, 2009; Norði et al., 2013). In the low-sulfate freshwater system Lake Ørn, we showed that the iron and sulfur cycles are tightly interconnected and sulfide reoxidation processes generate cryptic sulfur cycling that supplies sulfate internally to the microbial community. Slurry incubations complementing the biogeochemical depth profiles showed that organotrophic SR and S0 disproportionation apparently co-occur with AOM in the broad SMTZ. In the slurry incubations, the availability of methane caused a dramatic change in sulfate concentration trends and resulted in an offset between sulfate and reduced sulfur compounds (29‰) comparable to the offset between these two pools in the AOM zone of the depth profiles (21–34‰).
Studies addressing 34S isotope fractionation during AOM are rare and the physiology of sulfur cycling during AOM and the involved enzymes are not well-understood. Previous studies reported that sulfur-cycling AOM can induce sulfur isotope fractionation in marine environments and at sulfate concentrations in the millimolar range (Jørgensen et al., 2004; Antler et al., 2014, 2015; Avrahamov et al., 2014; Deusner et al., 2014; Sivan et al., 2014). A few of these studies assigned εAOM values of 20–30‰ for AOM in gas seeps (Deusner et al., 2014; Sivan et al., 2014) and suggested that AOM may cause fractionations of up to 60‰ in marine SMTZs (Jørgensen et al., 2004; Deusner et al., 2014).
However, the present study is, to our knowledge, the first report of substantial isotope fractionation during SR coupled to AOM at low micromolar sulfate levels in aquatic sediments (≤ 50 μmol L−1 in the AOM zone incubation and in situ). A previous study with natural communities of organotrophic sulfate reducers stated a suppression of 34S fractionation at sulfate concentrations < 200 μmol L−1 (Habicht et al., 2002), which contradicts our findings at first glance. In that study, however, the Danish lake sediment microbial communities were fueled directly by the degradation of organic matter detritus and were not associated to anaerobic methane oxidation as in Lake Ørn, which suggests a specific association of fractionation to AOM at low sulfate concentrations. In line with this, a sulfur isotope difference of up to 23‰ between sulfate and sulfide was also demonstrated recently in the water column of ferruginous Lake Matano in Indonesia, where sulfate concentrations were in the range of 5–10 μmol L−1, and the authors assigned the isotope effect to microbial SR (Crowe et al., 2014). Organic carbon mineralization rates are expectedly low in this extremely oligotrophic lake, and an earlier study suggested AOM to be active at the water depth of 34S fractionation (Crowe et al., 2011). Hence, the sulfur isotope fractionation in Lake Matano might potentially be also a result of sulfur cycling associated with AOM. These results, and the recent finding of high variability of fractionation between different sulfate reducers (Bradley et al., 2016), highlight the importance of interpreting 34S isotope fractionation during SR on a cellular and enzymatic level.
Sulfate-dependent AOM is mainly known from marine environments and associated with a consortium of archaea and bacteria (e.g., Hinrichs et al., 1999; Boetius et al., 2000). So far, it has not been possible to obtain a pure culture of the consortium or its partners and therefore, involved sulfur-cycling enzymatic pathways and their behavior towards 34S fractionation could not be identified. Until recently, it was hypothesized that sulfur cycling and 34S fractionation is restricted to the bacterial partner and follows enzymatic pathways known from bacterial sulfate reducers. However, Milucka et al. (2012) found that the archaeal ANME-2 partner of a marine AOM consortium was able to reduce sulfate and produce and accumulate zero-valent sulfur, which was subsequently disproportionated as disulfide by the bacterial Desulfosarcina/Desulfococcus partner. In Lake Ørn, a large accumulation of zero-valent sulfur, unusual to sediments with organotrophic SR, occurred in the zone of highest AOM activity. If zero-valent sulfur in Lake Ørn was provided by sulfur-cycling AOM following the model of Milucka et al. (2012), the methane-induced isotopic effect could have been shaped both by archaeal SR and by bacterial S0 disproportionation.
The iron-rich and low-sulfate SMTZ of Lake Ørn sediment can serve as a model system for the study of ancient sulfur and methane turnover. In the Archaean Eon, atmospheric oxygen concentrations were low (Bekker et al., 2004), sulfate was probably only a trace constituent (Crowe et al., 2014) and there is geological evidence for microbial production of methane (Canfield et al., 2005). Large-scale deposition of iron-rich sediments (banded iron formations, BIFs) during the Archaean Eon (>2.5 billion years ago) and parts of the Proterozoic (2.5–0.54 billion years ago) are evidence for the global importance of iron in Earth history (Canfield et al., 2005). Due to suggested high iron contents, AOM coupled to iron reduction has been suggested previously as an important methane sink during low-sulfate conditions in the Archaean Eon (Beal et al., 2009) but evidence for direct coupling of AOM and iron reduction is still pending. However, our findings suggest that sulfate-dependent AOM coupled indirectly to iron via a cryptic sulfur cycle could have been a possible metabolic pathway in the geological past and that this process is likely to have impacted δ34S signatures of sedimentary sulfides.
To create a clearer picture of δ34S signatures in modern and ancient environments, further investigations concerning 34S fractionation during AOM, especially in low-sulfate environments are crucial. Previous studies showed that sulfur-cycling AOM impacts not only sulfur (δ34SSO4), but also oxygen isotopes (δ18OSO4) in the dissolved sulfate pool and apparently creates an unique pattern among them that can help to distinguish between organotrophic and methanotrophic SR to be the main drivers of the footprints (e.g., Antler et al., 2014, 2015; Avrahamov et al., 2014; Deusner et al., 2014). Therefore, future studies may probably be best approached by combined investigations of oxygen and sulfur isotopes.
Author Contribution
HW, BT and KH designed research, analyzed and interpreted the data, and wrote the paper. HW performed field and laboratory based research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Michael Forth and Laura Bristow for their help sampling the sediment and Heidi Grøn Jensen, Susanne Møller, and Dina Holmgaard Skov for technical assistance in the laboratory. This study was supported by the National Danish Research Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/feart.2016.00061
References
Allen, R. E., and Parkes, R. J. (1995). Digestion procedures for determining reduced sulfur species in bacterial cultures and in ancienct and recent sediments. Geochem. Transform. Sediment. Sulfur 612, 243–257. doi: 10.1021/bk-1995-0612.ch013
Antler, G., Turchyn, A. V., Herut, B., Davies, A., Rennie, V. C. F., and Sivan, O. (2014). Sulfur and oxygen isotope tracing of sulfate driven anaerobic methane oxidation in estuarine sediments. Estuar. Coast. Shelf Sci. 142, 4–11. doi: 10.1016/j.ecss.2014.03.001
Antler, G., Turchyn, A. V., Herut, B., and Sivan, O. (2015). A unique isotopic fingerprint of sulfate-driven anaerobic oxidation of methane. Geology 43, 619–622. doi: 10.1130/G36688.1
Avrahamov, N., Antler, G., Yechieli, Y., Gavrieli, I., Joye, S. B., Saxton, M., et al. (2014). Anaerobic oxidation of methane by sulfate in hypersaline groundwater of the Dead Sea aquifer. Geobiology 12, 511–528. doi: 10.1111/gbi.12095
Beal, E. J., Claire, M. W., and House, C. H. (2011). High rates of anaerobic methanotrophy at low sulfate concentrations with implications for past and present methane levels. Geobiology 9, 131–139. doi: 10.1111/j.1472-4669.2010.00267.x
Beal, E. J., House, C. H., and Orphan, V. J. (2009). Manganese- and iron-dependent marine methane oxidation. Science 325, 184–187. doi: 10.1126/science.1169984
Bekker, A., Holland, H. D., Wang, P.-L., Rumble, I. I. I. D., Stein, H. J., Hannah, J. L., et al. (2004). Dating the rise of atmospheric oxygen. Nature 427, 117–120. doi: 10.1038/nature02260
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi: 10.1038/35036572
Borowski, W. S., Rodriguez, N. M., Paull, C. K., and Ussler, W. I. (2013). Are 34S-enriched authigenic sulfide minerals a proxy for elevated methane flux and gas hydrates in the geologic record? Mar. Pet. Geol. 43, 381–395. doi: 10.1016/j.marpetgeo.2012.12.009
Bradley, A. S., Leavitt, W. D., Schmidt, M., Knoll, A. H., Girguis, P. R., and Johnston, D. T. (2016). Patterns of sulfur isotope fractionation during microbial sulfate reduction. Geobiology 14, 91–101. doi: 10.1111/gbi.12149
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M., and Berner, R. A. (1986). The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155. doi: 10.1016/0009-2541(86)90078-1
Canfield, D. E., and Thamdrup, B. (1994). The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulfur. Science 266, 1973–1975.
Canfield, D. E., and Teske, A. (1996). Late Proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies. Nature 382, 127–132. doi: 10.1038/382127a0
Canfield, D. E., Thamdrup, B., and Fleischer, S. (1998). Isotope fractionation and sulfur metabolism by pure and enrichment cultures of elemental sulfur-disproportionating bacteria. Limnol. Oceanogr. 43, 253–264. doi: 10.4319/lo.1998.43.2.0253
Canfield, D. E., and Raiswell, R. (1999). The evolution of the sulfur cycle. Am. J. Sci. 299, 697–723. doi: 10.2475/ajs.299.7-9.697
Canfield, D. E., Habicht, K. S., and Thamdrup, B. (2000). The archean sulfur cycle and the early history of atmospheric oxygen. Science 288, 658–661. doi: 10.1126/science.288.5466.658
Canfield, D. E. (2001). Biogeochemistry of sulfur isotopes. Rev. Mineral. Geochem. 43, 607–636. doi: 10.2138/gsrmg.43.1.607
Canfield, D. E., Thamdrup, B., and Kristensen, E. (2005). “Advances,” in Marine Biology Aquatic Geomicrobiology, Vol. 48, eds A. J. Southward, P. A Tyler, C. M, Young, and L. A. Fuiman (San Diego, CA: Elsevier), 314–374.
Canfield, D. E., Rosing, M. T., and Bjerrum, C. (2006). Early anaerobic metabolisms. Philos. Trans. R. Soc. Lond. Ser. B 361, 1819–1836. doi: 10.1098/rstb.2006.1906
Canfield, D. E., Farquhar, J., and Zerkle, A. L. (2010). High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology 38, 415–418. doi: 10.1130/G30723.1
Catling, D. C., Zahnle, K. J., and McKay, C. P. (2001). Biogenic methane, hydrogen escape, and the irreversible oxidation of Early Earth. Science 293, 839–842. doi: 10.1126/science.1061976
Cline, J. D. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. Methods 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Crowe, S. A., Paris, G., Katsev, S., Jones, C., Kim, S.-T., Zerkle, A. L., et al. (2014). Sulfate was a trace constituent of Archean seawater. Science 346, 735–739. doi: 10.1126/science.1258966
Crowe, S. A., Katsev, S., Leslie, K., Sturm, A., Magen, C., Nomosatryo, S., et al. (2011). The methane cycle in ferruginous Lake Matano. Geobiology 9, 61–78. doi: 10.1111/j.1472-4669.2010.00257.x
Deusner, C., Holler, T., Arnold, G. L., Bernasconi, S. M., Formolo, M. J., and Brunner, B. (2014). Sulfur and oxygen isotope fractionation during sulfate reduction coupled to anaerobic oxidation of methane is dependent on methane concentration. Earth Planet. Sci. Lett. 399, 61–73. doi: 10.1016/j.epsl.2014.04.047
Ferdelmann, T. G., Lee, C., Pantoja, S., HArder, J., Bebout, B. M., and Fossing, H. (1997). Sulfate reduction and methanogenesis in a Thioploca-dominated sediment off the coast of Chile. Gechim. Cosmochim. Acta 61, 3065–3079. doi: 10.1016/S0016-7037(97)00158-0
Fossing, H., and Jørgensen, B. B. (1989). Measurement of bacterial sulfate reduction in sediments: evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222. doi: 10.1007/BF00002889
Habicht, K. S., Canfield, D. E., and Rethmeier, J. (1998). Sulfur isotope fractionation during bacterial reduction and disproportionation of thiosulfate and sulfite. Geochim. Cosmochim. Acta 62, 2585–2595. doi: 10.1016/S0016-7037(98)00167-7
Habicht, K. S., Gade, M., Thamdrup, B., Berg, P., and Canfield, D. E. (2002). Calibration of sulfate levels in the Archean Ocean. Science 298, 2372–2374. doi: 10.1126/science.1078265
Hansel, C. M., Lentini, C. J., Tang, Y., Johnston, D. T., Wankel, S. D., and Jardine, P. M. (2015). Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments. ISME J. 9, 2400–2412. doi: 10.1038/ismej.2015.50
Hinrichs, K. U., Hayes, J. M., Sylva, S. P., Brewer, P. G., and DeLong, E. F. (1999). Methane-consuming archaebacteria in marine sediments. Nature 398, 802–805. doi: 10.1038/19751
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B. (2011). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim. Cosmochim. Acta 75, 3581–3599. doi: 10.1016/j.gca.2011.03.033
Jørgensen, B. B. (1978). Comparison of methods for quantification of bacterial sulfate reduction in coastal marine sediments. 1. Measurement with radiotracer techniques. Geomicrobiol. J. 1, 11–27. doi: 10.1080/01490457809377721
Jørgensen, B. B., Böttcher, M. E., Lüschen, H., Neretin, L. N., and Volkov, I. I. (2004). Anaerobic methane oxidation and a deep H2S sink generate isotopically heavy sulfides in Black Sea sediments. Geochim. Cosmochim. Acta 68, 2095–2118. doi: 10.1016/j.gca.2003.07.017
Kallmeyer, J., Ferdelman, T. G., Weber, A., Fossing, H., and Jørgensen, B. B. (2004). A cold chromium distillation procedure for radiolabeled sulfide applied to sulfate reduction measurements. Limnol. Oceanogr. Methods 2, 171–180. doi: 10.4319/lom.2004.2.171
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi: 10.1146/annurev.micro.61.080706.093130
Knossow, N., Blonder, B., Eckert, W., Turchyn, A. V., Antler, G., and Kamyshny, A. Jr. (2015). Annual sulfur cycle in a warm monomictic lake with sub-millimolar sulfate concentrations. Geochem. Trans. 16, 1–24. doi: 10.1186/s12932-015-0021-5
Konhauser, K. O., Newman, D. K., and Kappler, A. (2005). The potential significance of microbial Fe(III) reduction during deposition of Precambrian banded iron formations. Geobiology 3, 167–177. doi: 10.1111/j.1472-4669.2005.00055.x
Lovley, D. R., and Phillips, E. J. P. (1986). Organic matter mineralization with reduction of ferric iron in anaerobic sediments. Appl. Environ. Microbiol. 51, 683–689.
McGlynn, S. E., Chadwick, G. L., Kempes, C. P., and Orphan, V. J. (2015). Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature 526, 531–535. doi: 10.1038/nature15512
Meulepas, R. J. W., Jagersma, C. G., Khadem, A. F., Stams, A. J. M., and Lens, P. N. L. (2010). Effect of methanogenic substrates on anaerobic oxidation of methane and sulfate reduction by an anaerobic methanotrophic enrichment. Appl. Microbiol. Biotechnol. 87, 1499–1506. doi: 10.1007/s00253-010-2597-0
Milucka, J., Ferdelman, T. G., Polerecky, L., Franzke, D., Wegener, G., Schmid, M., et al. (2012). Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 491, 541–546. doi: 10.1038/nature11656
Norði, K. Á., Thamdrup, B., and Schubert, C. J. (2013). Anaerobic oxidation of methane in an iron-rich Danish freshwater lake sediment. Limnol. Oceanogr. 58, 546–554. doi: 10.4319/lo.2013.58.2.0546
O'Connell, D. W., Mark Jensen, M., Jakobsen, R., Thamdrup, B., Joest Andersen, T., Kovacs, A., et al. (2015). Vivianite formation and its role in phosphorus retention in Lake Ørn, Denmark. Chem. Geol. 409, 42–53. doi: 10.1016/j.chemgeo.2015.05.002
Pellerin, A., Hao Bui, T., Rough, M., Mucci, A., Canfield, D. E., and Wing, B. A. (2015). Mass-dependent sulfur isotope fractionation during reoxidative sulfur cycling: a case study from Mangrove Lake, Bermuda. Geochim. Cosmochim. Acta 149, 152–164. doi: 10.1016/j.gca.2014.11.007
Pyzik, A., and Sommer, S. E. (1981). Sedimentary iron monosulfides: kinetics and mechanism of formation. Geochim. Cosmochim. Acta 45, 687–698. doi: 10.1016/0016-7037(81)90042-9
Raiswell, R., Canfield, D. E., and Berner, R. A. (1994). A comparison of iron extraction methods for the determination of degree of pyritisation and the recognition of iron-limited pyrite formation. Chem. Geol. 111, 101–110. doi: 10.1016/0009-2541(94)90084-1
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi: 10.1021/cr050362v
Roden, E. E., and Wetzel, R. G. (1996). Organic carbon oxidation and suppression of methane production by microbial Fe(III) oxide reduction in vegetated and unvegetated freshwater wetland sediments. Limnol. Oceanogr. 41, 1733–1748. doi: 10.4319/lo.1996.41.8.1733
Røy, H., Weber, H. S., Tarpgaard, I. H., Ferdelman, T. G., and Jørgensen, B. B. (2014). Determination of dissimilatory sulfate reduction rates in marine sediment via radioactive 35S tracer. Limnol. Oceanogr. Methods 12, 196–211. doi: 10.4319/lom.2014.12.196
Schippers, A., and Jørgensen, B. B. (2002). Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments. Geochim. Cosmochim. Acta 66, 85–92. doi: 10.1016/S0016-7037(01)00745-1
Sim, M. S., Bosak, T., and Ono, S. (2011). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Sivan, O., Adler, M., Pearson, A., Gelman, F., Bar-Or, I., John, S. G., et al. (2011). Geochemical evidence for iron-mediated anaerobic oxidation of methane. Limnol. Oceanogr. 56, 1536–1544. doi: 10.4319/lo.2011.56.4.1536
Sivan, O., Antler, G., Turchyn, A. V., Marlow, J. J., and Orphan, V. J. (2014). Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps. Proc. Natl. Acad. Sci. U.S.A. 111, E4139–E4147. doi: 10.1073/pnas.1412269111
Stookey, L. L. (1970). Ferrozine-a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781. doi: 10.1021/ac60289a016
Thamdrup, B. (2000). “Bacterial manganese and iron reduction in aquatic sediments,” in Advances in Microbial Ecology, ed B. Schink (New York, NY: Kluwer Academic/Plenum Publishers), 41–84.
Thamdrup, B., Finster, K., Hansen, J. W., and Bak, F. (1993). Bacterial disproportionation of elemental sulfur coupled to chemical reduction of iron or manganese. Appl. Environ. Microbiol. 59, 101–108.
Thamdrup, B., Fossing, H., and Jørgensen, B. B. (1994). Manganese, iron and sulfur cycling in a coastal marine sediment, Aarhus Bay, Denmark. Geochim. Cosmochim. Acta 58, 5115–5129. doi: 10.1016/0016-7037(94)90298-4
Thode, H. G., Monster, J., and Dunford, H. B. (1961). Sulphur isotope geochemistry. Geochim. Cosmochim. Acta 25, 150–174. doi: 10.1016/0016-7037(61)90074-6
Treude, T., Krüger, M., Boetius, A., and Jørgensen, B. B. (2005). Environmental control on anaerobic oxidation of methane in the gassy sediments of Eckernförde Bay (German Baltic). Limnol. Oceanogr. 50, 1771–1786. doi: 10.4319/lo.2005.50.6.1771
Wegener, G., Krukenberg, V., Riedel, D., Tegetmeyer, H. E., and Boetius, A. (2015). Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature 526, 587–589. doi: 10.1038/nature15733
Wortmann, U. G., Bernasconi, S., and Böttcher, M. E. (2001). Hypersulfidic deep biosphere indicates extreme sulfur isotope fractionation during single-step microbial sulfate reduction. Geology 29, 647–650. doi: 10.1130/0091-7613(2001)029<0647:HDBIES>2.0.CO
Keywords: anaerobic oxidation of methane, sulfur isotope fractionation, freshwater sediment, iron, early Earth analog
Citation: Weber HS, Thamdrup B and Habicht KS (2016) High Sulfur Isotope Fractionation Associated with Anaerobic Oxidation of Methane in a Low-Sulfate, Iron-Rich Environment. Front. Earth Sci. 4:61. doi: 10.3389/feart.2016.00061
Received: 06 January 2016; Accepted: 09 May 2016;
Published: 01 June 2016.
Edited by:
Orit Sivan, Ben-Gurion University of the Negev, IsraelReviewed by:
Aubrey L. Zerkle, University of St. Andrews, UKGilad R. Antler, Aarhus University, Denmark
Copyright © 2016 Weber, Thamdrup and Habicht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah S. Weber, aGFubmFoQGJpb2xvZ3kuc2R1LmRr
†Present Address: Kirsten S. Habicht, Unisense A/S, Aarhus, Denmark