 Andrew L. Rose
Andrew L. Rose- School of Environment, Science and Engineering, Southern Cross University, Lismore, NSW, Australia
Redox conditions in natural waters are a fundamental control on biogeochemical processes and ultimately many ecosystem functions. While the dioxygen/water redox couple controls redox thermodynamics in oxygenated aquatic environments on geological timescales, it is kinetically inert in the extracellular environment on the much shorter timescales on which many biogeochemical processes occur. Instead, electron transfer processes on these timescales are primarily mediated by a relatively small group of trace metals and stable radicals, including the reactive oxygen species superoxide. Such processes are of critical biogeochemical importance because many of these chemical species are scarce nutrients, but may also be toxic at high concentrations. Furthermore, their bioavailability and potentially toxicity is typically strongly influenced by their redox state. In this paper, I examine to what extent redox conditions in oxygenated natural waters are expected to be reflected in the redox states of labile redox-active compounds that readily exchange electrons with the dioxygen/superoxide redox couple, and potentially with each other. Additionally, I present the hypothesis that the relative importance of the dioxygen/superoxide and superoxide/hydrogen peroxide redox couples exerts a governing control on local redox conditions in oxygenated natural waters on biogeochemically important timescales. Given the recent discovery of widespread extracellular superoxide production by a diverse range of organisms, this suggests the existence of a fundamental mechanism for organisms to tightly regulate local redox conditions in their extracellular environment in oxygenated natural waters.
Introduction
Electron transfer and transport in natural waters is a fundamental control on chemical conditions, biological processes, and ecosystem structure. Oxidation and reduction (redox) reactions involve the transfer of electrons, and associated chemical energy, from one chemical species to another. In most natural surface waters, chemical energy is originally captured from solar energy by photosynthesis, and then redistributed by chemical and physical transport processes. This chemical energy is the substrate for life on Earth. Chemical redox reactions combined with physical transport of redox active species therefore dictate, at a fundamental level, the functioning of biogeochemical systems on Earth from cellular to ecosystem scales.
In most natural surface waters, oxygen controls redox conditions on geological timescales. The Great Oxidation Event approximately 2.4 billion years ago saw a massive shift in redox conditions on Earth resulting from photosynthetic release of O2 into the atmosphere, following the evolution of cyanobacteria (Sverjensky and Lee, 2010). However, the O2/H2O redox couple is extremely kinetically inert because the dioxygen molecule possesses an unusual electronic structure, with its lowest energetic state (ground state) possessing two unpaired electrons with parallel spins, i.e., its ground state is a triplet state biradical (Sawyer, 1991). Quantum mechanical restrictions (the Pauli exclusion principle) dictate that, under typical environmental conditions, triplet state dioxygen can react only extremely slowly with molecules possessing singlet state electronic configurations (e.g., most organic compounds), but much more readily with molecules possessing unpaired electrons (e.g., free radicals and transition metal ions; Fridovich, 1998).
Usually, there are relatively few free radical species present in oxygenated natural waters at sufficiently high concentration to facilitate the thermal, abiotic reduction of O2 at substantial rates. In natural waters, free radicals typically include only a select few trace metals, certain organic radicals, and a few other trace species such as nitric oxide (Goldstein and Czapski, 1995). Enzymatic reactions (respiration) inside living cells catalyze reduction of oxygen to water, and in aquatic environments supporting sufficient biomass, this can be the major pathway for oxygen consumption, even resulting in complete hypoxia when respiration rates are high. Conversely, photosynthetic organisms can enzymatically catalyse the sunlight-driven oxidation of water by CO2 (or other compounds). In the absence of biological catalysis, sunlight can also mediate abiotic photochemical oxygen consumption (Amon and Benner, 1996; Andrews et al., 2000). Overall, however, rates of oxygen consumption and production are balanced over long timescales, and O2 and H2O coexist at high concentrations in the majority of natural surface waters, defying thermodynamic expectations.
The electronic structure of O2 also dictates that the transfer of four electrons required to reduce O2 to H2O or oxidize H2O to O2 must occur via single electron transfer steps (Sawyer, 1991). The sequential reduction of O2 to H2O under near neutral pH conditions results first in the formation of superoxide () then hydrogen peroxide (H2O2), and finally hydroxyl (HO•), which collectively belong to a group known as reactive oxygen species (ROS). Despite the overall thermodynamic impetus, reduction of O2 to does not guarantee the complete reduction of O2 to H2O as the electronic structure of again favors reaction only with molecules possessing unpaired electrons (Sawyer, 1991). In addition, under conditions typical of natural waters is relatively reducing; i.e., is relatively easily oxidized back to O2, while its reduction to H2O2 is unfavorable due to the thermodynamic instability of the immediate product of the reaction (Sawyer, 1991). In contrast, H2O2 and HO• are powerful oxidants such that once formed, they will ultimately be almost exclusively further reduced to H2O. Thus, reduction of to H2O2 represents a major barrier to the overall reduction of O2 to H2O, and whether is being primarily oxidized to O2 or primarily reduced to H2O2 in a particular environment has the potential to exert a major influence on local redox chemistry (Rose et al., 2010).
In this paper, I explore the influence of ROS on local redox conditions in oxygenated natural waters on environmentally relevant timescales and discuss their significance to understanding the redox biogeochemistry of natural waters on a fundamental level, with a particular emphasis on the role of . Furthermore, while acknowledging the importance of biological processes, the focus of the paper is on non-enzymatic reactions in the extracellular environment, which is the dominant pathway for redox cycling of a substantial number of redox active elements (Borch et al., 2009). On the basis of the theory presented, I also present the hypothesis that that the relative importance of the dioxygen/superoxide and superoxide/hydrogen peroxide redox couples exerts a governing control on local redox conditions in oxygenated natural waters on biogeochemically important timescales. While the approach taken can be applied to any natural waters, the scope of this paper is restricted primarily to carbonate-buffered, oxygen saturated waters with a nominal pH of 8.1 and dissolved O2 concentration of 250 μM.
Redox Thermodynamics of Oxygenated Natural Waters
The redox thermodynamics of oxygenated natural waters are dominated by the chemistry of oxygen, which is present in both its most common oxidized state O2 and its most common reduced state H2O; the latter, of course, also provides the solvent in which aqueous reactions occur. For a chemical species to be thermodynamically able to exist in aqueous solution in a particular redox state, the redox potential of the half reaction that defines its oxidation or reduction to another redox state must lie between the redox potentials for oxidation of H2O to O2, or reduction of H+ to H2 (Fraústo Da Silva and Williams, 2001). Under standard conditions (unit activity of all reactants and products at 298 K), the standard redox potentials (E0) for oxidation of H2O to O2 and reduction of H+ to H2 are +1270 and 0 mV, respectively, when defined with respect to the standard hydrogen electrode. However, the redox potential E varies as defined by the Nernst equation:
where n is the number of electrons transferred in the half-reaction, {Ox} refers to the activity of the oxidized species, {Red} refers to the activity of the reduced species, E and E0 are in mV, and the coefficient of 59.16 mV applies at 25°C.
Thus at pH 8.1, which is typical for carbonate buffered natural surface waters and where {H+} = 10−8.1 M, the redox potentials for oxidation of H2O to O2 and reduction of H+ to H2 are instead +790 and −480 mV, respectively, due to the presence of protons in the respective reduction half reactions. While these values represent the ultimate thermodynamic constraints on stability in aqueous solution, in practice the former is conditional on a mechanism existing to catalyze the four electron oxidation of H2O to O2. As previously discussed, in the absence of catalysis the oxidation of H2O to O2 must proceed initially via the one-electron oxidation of H2O to HO•. This step is much less thermodynamically favorable than the overall oxidation, with a redox potential of +2060 mV under standard conditions at pH 8.1. In most oxygenated natural waters, photosynthesis is the only significant process that can facilitate multi-electron transfer steps during oxidation of H2O to O2. Thus, in the extracellular environment, couples with redox potentials for one-electron transfers lying between −480 and +2060 mV are practically stable with respect to their aqueous environment at pH 8.1.
Figure 1 illustrates the thermodynamic stability at pH 8.1 of various redox states of the major redox active elements C, N and S, along with several biogeochemically important trace metals, with regard to the oxidation or reduction of water. Stability in aqueous solution at pH 8.1 is shown with respect to both the theoretical limit imposed by H2O/O2 couple (hereafter referred to as absolute stability) and the practical limit imposed by the H2O/HO• couple (hereafter referred as conditional stability). It is clear that a limited number of redox states for both major elements and trace metals are permissible under these conditions.
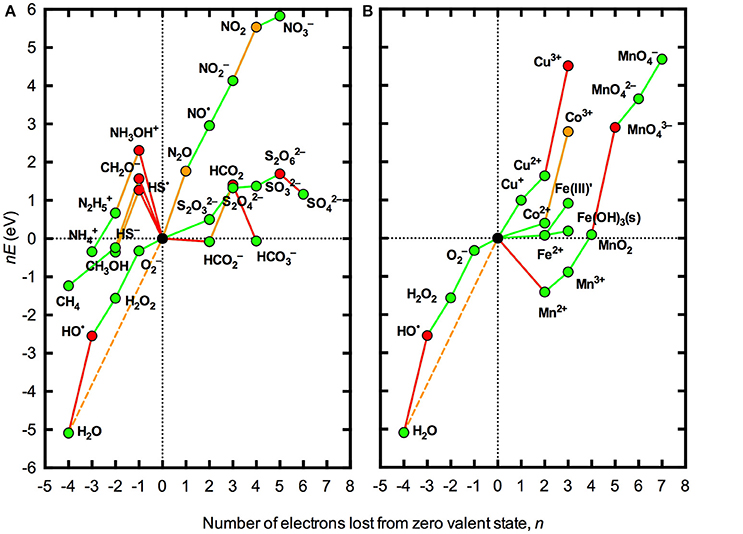
Figure 1. Frost diagrams for some common redox active elements in natural waters under standard conditions at pH 8.1. (A) Representative species for the major elements C, N, and S. (B) Trace metals Co, Cu, Fe, and Mn. All redox potentials (E) are defined with reference to the standard hydrogen electrode at pH 8.1 (Pt,H2|aH+ = 10−8.1). The compounds shown represent the dominant acid-base species at pH 8.1, allowing for hydrolysis of trace metals. The zero valent states for the elements shown are a representative carbohydrate “CH2O” equivalent to 1/6 of a glucose molecule (Morel and Hering, 1993), N2 (g), S8 (s), Co0 (s), Cu0 (s), Fe0 (s), Mn0 (s), and O2 (aq). Where the necessary thermodynamic data were available, one-electron transfer steps are shown. Red lines indicate redox couples that are unstable with respect to one-electron transfer processes involving the H2O/HO• couple (lines with positive slope) or the H2/H+ couple (lines with negative slope); orange lines indicate redox couples that are unstable with respect to multi-electron transfer processes involving the H2O/O2 couple; and green lines indicate redox couples that are thermodynamically permissible in aqueous solution at pH 8.1. The dashed orange line represents the redox potential associated with the four electron transfer for complete reduction of O2 to H2O. Red symbols represent species that are thermodynamically unable to exist due to instability with respect to one-electron transfer processes involving the H2O/HO• couple (lines with positive slope) or the H2/H+ couple; orange symbols represent species that are thermodynamically able to exist in the absence of processes to catalyze multi-electron transfer processes involving the H2O/O2 couple; and green symbols represent species that are thermodynamically able to coexist with both the H2/H+ and H2O/O2 couples. Thermodynamic relationships used to construct the diagrams are listed in the Appendix in Supplementary Material. The format of the Frost diagrams was adapted from Figures 1.7 and 1.8 in Fraústo Da Silva and Williams (2001).
Stability in aqueous solution with respect to the H2/H+, H2O/O2, or H2O/HO• couples does not mean that a particular species is stable against any redox transformation, however. Most notably, the presence of O2 should drive other redox active elements toward the most oxidized thermodynamically stable species. However, for the major redox active elements C, N, and S (Figure 1A), transformations between many of the various stable redox states are not possible via one-electron steps, as the intermediate species are thermodynamically unstable in aqueous solution. Thus, highly reduced species such as CH4, , and HS− are unable to be oxidized by O2 directly without catalysis. In contrast, transformation among most of the permissible redox states for the trace metals shown in Figure 1B is able to occur via one-electron transfers, enabling direct oxidation or reduction by O2, H2O, and/or ROS.
Of course, this reasoning applies only to direct reactions between these various redox couples and O2, H2O, or ROS: reactions between two redox species can occur independently of O2, H2O, or ROS, and in many such reactions multi-electron transfers are possible (and indeed favored), as detailed by Luther (2010). Furthermore, one-electron transfers between C, N or S species and O2, H2O, or ROS can be facilitated via trace metals, as has been shown to facilitate sulfide oxidation by O2 in natural waters (Luther et al., 2011). Nonetheless, the role of such reactions in oxygenated waters is limited by the fact that where such alternate pathways to oxidation exist, accumulation of reduced compounds to relatively high concentrations is unlikely to occur.
While this approach is useful to constrain the redox thermodynamics of oxygenated natural waters on a broad level, a more realistic analysis must also account for the complex speciation of many elements. In particular, trace metals potentially exist as a range of different solid phases or in complexes with a vast array inorganic and organic ligands. The redox potentials of solid phases or complex species can be very different to those for the simple aquated species; for example, the redox potential for reduction of Fe(III) to Fe(II) at pH 7 ranges from –750 mV when complexed with the strong Fe(III) binding ligand enterobactin to 1150 mV when complexed with the strong Fe(II) binding ligand phenanthroline (Pierre et al., 2002). The difference in thermodynamic behavior of the “average” speciation of dissolved inorganic Fe(III) in seawater, denoted as Fe(III)′, and amorphous Fe(OH)3 solid is illustrated in Figure 1.
A second limitation with the analysis presented in Figure 1 is that standard conditions are defined by unit activities of all species (except, in this case, H+). As seen in the Nernst equation (Equation 1), the redox potential varies with the ratio of the oxidized and reduced species in the couple such that the slope of the lines in Figure 1 will change by 59 meV for each order of magnitude difference between the activities of the oxidized and reduced species in the couple. Thus, while a particular process may be thermodynamically favorable (or unfavorable) under standard conditions, this may not necessarily be the case under the conditions actually encountered in a particular natural water body where the concentrations of the oxidized and reduced species may be orders of magnitude apart. The effect of using typical concentrations of O2 and ROS found in many natural surface waters rather than standard conditions for thermodynamic analysis is illustrated in Figure 2. It is evident that accounting for actual concentrations of these species, rather than using unit activities, brings the redox potentials of all one-electron steps in the reduction of O2 to H2O much closer to the overall redox potential for the four-electron process. As a result, some species that were seen to be conditionally unstable (with respect to the H2O/O2 couple) but practically stable (with respect to H2O/HO•) under standard conditions (Figure 1) are practically unstable under the conditions shown in Figure 2, for example N2O and Co3+. However, the latter case applies to the inorganic redox couple, whereas in marine waters Co is known to exist predominantly in organically complexed form (Vraspir and Butler, 2009), again illustrating the importance of accounting for complex speciation.
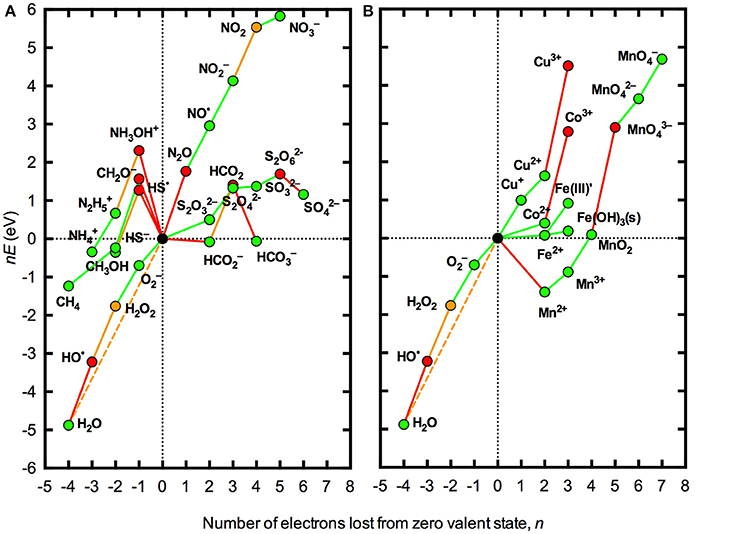
Figure 2. Frost diagrams for some common redox active elements in natural waters at pH 8.1 accounting for typical activities of oxygen redox species in natural waters. (A) Representative species for the major elements C, N, and S. (B) Trace metals Co, Cu, Fe, and Mn. Typical activities were taken as [O2] = 250 μM, [] = 100 pM (Rose et al., 2008; Hansard et al., 2010), [H2O2] = 100 nM (Yuan and Shiller, 2005; Mostofa and Sakugawa, 2009; Vermilyea et al., 2010; Rusak et al., 2011) and [HO•] = 1 fM (Zhou and Mopper, 1990). These values are based primarily on measurements from seawater, since this is where most measurements of ROS concentrations in natural surface waters have been made. All other conditions and the lines and symbols used are the same as in Figure 1.
Timescales of Redox Processes in Oxygenated Natural Waters
Thermodynamic analysis is based on the requirement that sufficient time has passed for equilibrium to have been reached. While redox equilibrium is likely to be reached on geological timescales, it is unlikely to occur on timescales on the order of seconds to days, because environmental conditions in natural waters typically vary significantly over these timescales due to processes such as diel variations in solar radiation, hydrologic fluctuations (e.g., inputs of rainwater or variations in streamflow), and biological activity. Furthermore, chemistry imposes substantial constraints on the kinetics of some redox reactions, which allows thermodynamically incompatible redox species to coexist at potentially high concentrations, as exemplified by the coexistence of O2 and H2O in oxygenated natural waters.
Redox active compounds in natural waters vary considerably in terms of their reactivity, and hence longevity. At one end of this scale are relatively kinetically inert compounds that typically undergo redox reactions on timescales of greater than a day, and can therefore potentially persist at relatively high concentrations in oxygenated natural waters even in thermodynamically unfavorable redox states. Many of the commonly occurring compounds of C, N, and S in aquatic environments behave in this way. The ability of such compounds to persist in thermodynamically unstable redox states at relatively high concentrations provides a critical store of chemical free energy in the environment that can be harnessed by organisms for biological use if the kinetic inertness of such compounds can be overcome. “Activation” of thermodynamically unstable but kinetically inert redox active compounds typically occurs through photochemical catalysis or reaction with free radicals (e.g., via enzymes containing trace metal or organic radical centers), however such reactions are often spatially and/or temporally compartmentalized, for example during daylight hours or inside cells. In the extracellular environment, “activation” tends to be a relatively slow process due to the relatively low abundance of potential catalysts such as free (non-organically complexed) trace metals in many natural waters. Therefore, these kinetically inert compounds usually have a limited ability to mediate electron transfer and transport processes in the extracellular environment in oxygenated natural waters on timescales of less than a day.
At the other end of the scale are compounds that undergo redox reactions on timescales of less than a second, and which thus typically exist only at sub-picomolar concentrations in most natural waters. The short lifetimes and hence low concentrations of these transient species mean that they are unable to diffuse far from the site of their production. Thus, while potentially important in specific microenvironments, their low concentrations and extreme localisation means that such highly reactive species are typically unable to influence redox conditions in natural waters on a large spatial scale. Between these two extremes exist redox active compounds that have half-lives (or turnover times) of between a second and a day. This range has particular significance because it corresponds to the timescale of many critical biological and environmental processes. Furthermore, compounds with lifetimes in this range are typically sufficiently long-lived to persist at biologically significant concentrations. This longevity also enables such redox active compounds to facilitate physical transport of electrons on biologically significant spatial scales of at least a few millimeters.
A second critical property of certain redox active compounds is their ability to undergo reversible redox cycling. In the extracellular environment in oxygenated natural waters, this is only possible with the existence of two or more thermodynamically stable redox states (with respect to the H2/H+ and H2O/HO• redox couples) that can be accessed by one-electron transfers. As can be seen for the selection of elements shown in Figure 2, such compounds are relatively uncommon (although the selection shown in Figure 2 is not exhaustive). Compounds which possess this ability to undergo reversible redox cycling and for which this cycling typically occurs with a turnover time between a second and a day are hereafter referred to as labile redox active compounds (LRACs). LRACs possess a unique combination of properties: sufficient reactivity to accept and donate electrons on biologically important timescales but sufficient longevity to transport these labile electrons over biologically important spatial scales, and the ability to do so repeatedly. This confers an ability for LRACs to potentially act as “electron shuttles” in oxygenated natural waters, and thereby influence overall local redox conditions in a biologically significant way.
In the extracellular environment in oxygenated natural waters, relatively few compounds satisfy the definition of LRACs. Under conditions typical of natural waters, such compounds are primarily redox active trace metals, organic moieties and potentially a few trace non-metal compounds that can exist in two or more redox states that can be accessed via one electron transfers. Table 1 lists a range of trace metals that may meet the definition of LRACs in natural waters on the basis of their ability to undergo reversible redox cycling. However, the ability to undergo redox cycling in this manner does not necessarily mean that these compounds do in fact function as LRACs, since the kinetics of redox cycling in many cases remains relatively poorly understood. Furthermore, the list in Table 1 is not exhaustive. In particular, several other elements (mostly metals and metalloids) are well known to exist in natural waters in multiple redox states separated by two or more electron transfers (e.g., As and Se), however there is a lack of information on the kinetics and mechanisms of redox transformations for many of these species that is needed to determine if such transformations may be able to occur via one-electron transfers.
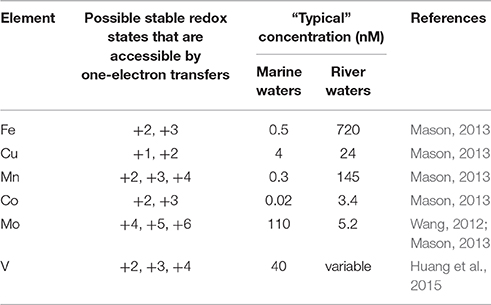
Table 1. Trace metals that can potentially be interconverted between multiple redox states through one electron transfers in oxygenated natural waters.
Other LRACs in natural waters may include certain reduced sulfur species such as thiols and metal sulfide clusters, which may be found at measurable concentrations in oxic waters (Bowles et al., 2003; Luther and Rickard, 2005; Rickard and Luther, 2006); organic moieties such as semiquinones, which appear to be ubiquitous components of humic type natural organic compounds (Aeschbacher et al., 2010); and the ROS . In the case of the anion, resonance stabilization of the unpaired electron results in a so-called “three electron bond” (Neuman, 1934; Pauling, 1979) that renders the molecule to be very selective in its reactions in aqueous solution (Sawyer, 1991). Thus, the O2/ couple undergoes reversible electron transfer on a timescale characteristic of LRACs, but only with the relatively small set of compounds that satisfy the strict requirement to possess unpaired electrons.
The ROS H2O2 also has a typical lifetime in oxygenated waters on the order of a few days (Cooper and Zepp, 1990; Yuan and Shiller, 2005; Vermilyea et al., 2010), which is close to the timescale characteristic of LRACs. However, one electron transfers involving H2O2 are not readily reversible. The reduction of H2O2 to HO• is essentially irreversible owing to the strongly oxidizing nature of HO•, while oxidation of H2O2 to appears very unusual in natural waters. Consequently, H2O2 behaves predominantly as a relatively long-lived trace oxidant rather than a LRAC, with limited ability to facilitate “electron shuttling,” unlike .
Redox Cycling of Labile Redox Active Species in Oxygenated Natural Waters
LRACs collectively act as carriers for a pool of labile electrons that can effectively maintain locally reducing conditions relative to the overall redox potential set by the O2/H2O system in oxygenated natural waters. The size of this pool of electrons, and hence the intensity of the reducing conditions, will be determined by the steady-state balance of electrons entering and leaving the pool. As these electrons can potentially be exchanged on timescales of seconds to days, the redox speciation of various LRACs would be expected to reflect the prevailing redox conditions within this pool on these timescales.
Sources of Electrons to the Pool of Labile Redox Active Compounds
The input of electrons into the LRAC pool in oxygenated natural waters is primarily driven through the slow oxidation of kinetically inert reduced compounds and/or physical transport of reduced LRACs from more reducing environments such as sediments or rainwater (Figure 3). As kinetically inert reduced compounds are resistant to oxidation otherwise, this release of electrons into the LRAC pool is achieved mostly through biological or photochemical catalysis.
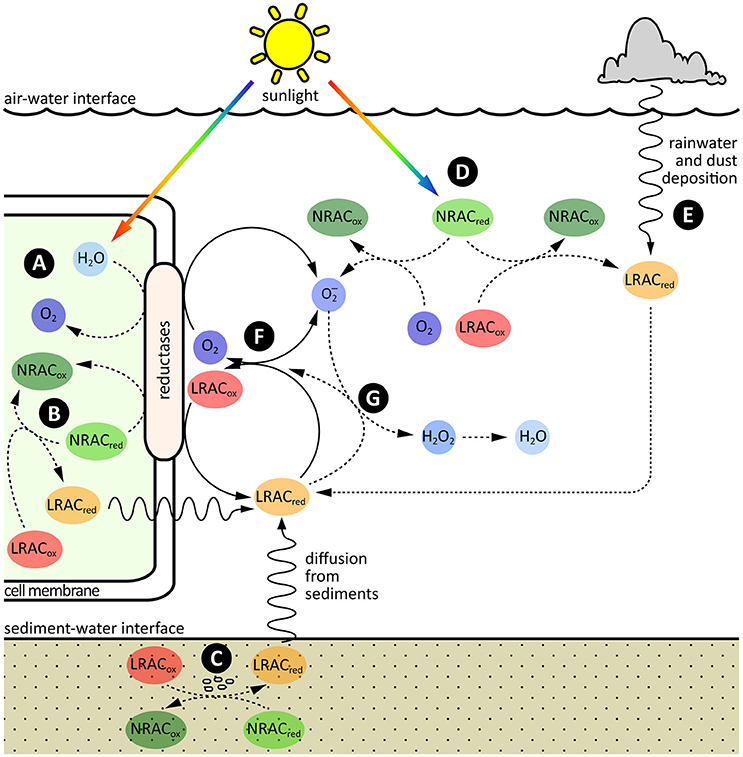
Figure 3. Sources, sinks and cycling of electrons in the LRAC pool. The major sources of electrons are: (A) Photosynthetic oxidation of water inside cells, with subsequent donation of electrons to LRACs in the extracellular environment through membrane associated oxidoreductase enzymes, or excretion of reduced LRACs through the cell membrane; (B) Respiratory oxidation of organic carbon or other non-labile redox active compounds (NRACs) inside cells, with subsequent donation of electrons to LRACs in the extracellular environment through membrane associated oxidoreductase enzymes, or excretion of reduced LRACs through the cell membrane; (C) Respiratory oxidation of other reduced NRACs in reducing environments such as sediments, with subsequent physical transport of reduced LRACs into overlying oxygenated surface waters; (D) Abiotic, photochemical oxidation of reduced NRACs in sunlit surface waters; and (E) Abiotic, photochemical oxidation of water or reduced NRACs in the atmosphere, with subsequent deposition of reduced LRACs into oxygenated surface waters through dust or rain. (F) Once in the LRAC pool, electrons can potentially be readily exchanged between a range of different LRAC redox couples, including the O2/ couple. (G) The primary sink of electrons in the pool is through oxidation of by reduced LRACs, resulting in the essentially irreversible formation of H2O2 and, ultimately, water.
The precise mechanisms by which each of these processes occur are, in general, relatively poorly understood. There is now strong evidence for biological input of electrons into the extracellular LRAC pool in a wide range of marine and freshwater aquatic environments, as demonstrated by extensive measurements of biologically mediated extracellular production (Diaz et al., 2013; Zhang et al., 2016). However, it remains unclear whether O2 is reduced to directly by cell surface oxidoreductase enzymes, or whether reductases first reduce other LRACs that subsequently react with dioxygen to yield . It may also be possible that some reductase enzymes are able to reduce multiple LRACs, as has been shown for the ferric reductase system of the yeast Saccharomyces cerevisiae (Lesuisse et al., 1996; Kosman, 2003), or that some organisms release soluble reduced LRACs such as small quinone molecules directly, as has been shown for the bacterium Escherichia coli (Korshunov and Imlay, 2006). In the case of non-photosynthetic organisms, the electrons must presumably originate from respiratory processes, however there is evidence that in at least some photosynthetic organisms, the electrons are primarily derived from photosynthesis (Marshall et al., 2002).
Mechanisms of abiotic photochemical processes in sunlit surface waters are also not fully resolved. Natural organic matter is thought to undergo photochemical oxidation via photoexcitation and subsequent electron transfer processes, potentially involving quinone, phenol and or other similar moieties within the heterogeneous molecular structure (Zhang et al., 2012; Sharpless and Blough, 2014). Simpler organic molecules, such as siderophores containing carboxylic acid moieties, may undergo direct photodecarboxylation reactions, also potentially liberating electrons (Barbeau et al., 2003). In addition, complex formation between organic compounds and metals such as iron and copper can lead to formation of charge transfer complexes that increase the efficiency of photochemical processes, and potentially result in direct formation of reduced LRACs through photochemical ligand-to-metal charge transfer (Jones et al., 1985; Barbeau, 2006).
Cycling of Electrons among the Pool of Labile Redox Active Compounds
The defining characteristic of LRACs is their ability to undergo reversible redox cycling on relatively short timescales, while persisting at biologically significant concentrations. While the kinetics of electron transfer between many specific redox couples in natural waters have been well studied, knowledge of electron transfer between multiple LRAC species simultaneously is poor. Because O2 is present at high concentrations in all oxygenated natural waters while most other LRACs are present at trace concentrations, transfer of electrons to O2 by reduced LRACs likely represents a major pathway for electron transfer among the LRAC pool. The oxygenation of reduced metal LRACs including Fe(II), Mn(II), V(II), Cu(I), and Co(II) is thought to occur via the Haber-Weiss mechanism (Haber and Weiss, 1934):
where Mn+ represents the reduced form of the metal and M(n+1)+ its oxidized form.
The kinetics of this reaction vary by many orders of magnitude between different metals, and also between different complex species of the same metal. For example, the second order rate constant is around 4 M−1.s−1 for oxygenation of inorganic Fe(II) in 0.7 M NaCl and 2 mM bicarbonate at pH 8.1 (Santana-Casiano et al., 2005) and around 2 M−1.s−1 for inorganic Cu(I) under similar conditions (González-Dávila et al., 2009), but <10−4 M−1.s−1 for Mn(II) in seawater at pH 8.1 (Morgan, 2005). Similarly, rate constants for the oxygenation kinetics of Fe(II) in seawater at pH 8.1 when complexed by natural organic ligands extracted from coastal soils ranged from 2 to 1000 M−1.s−1 (Rose and Waite, 2003). Thus, the rate at which electrons are cycled between LRACs and O2 can vary substantially depending on the LRAC involved.
The oxygenation of reduced trace metals is expected to produce , however the initial product of the reaction may involve coordination of in the inner sphere of the metal. Oxygenation of V2+, Co2+, and Fe2+ appears to proceed via an outer-sphere mechanism, resulting in production of non-coordinated , while Mn2+ and hydrolysed Fe(II) species appear to react via an inner-sphere process, such that would initially be coordinated to the metal center (Rosso and Morgan, 2002). While it is likely that is ultimately released from inner sphere complexes, the details of this process are not well known.
As previously stated, can function as a mild one-electron reductant under conditions typical of natural waters (Sawyer, 1991). Thus, many of the oxygenation reactions discussed above are reversible, with the net effect that electrons are passed between O2, , and various other LRACs. Kinetics of reduction by under conditions relevant to natural surface waters have been documented for a range of Fe(III) complexes and colloids (Rush and Bielski, 1985; Rose and Waite, 2005; Fujii et al., 2006), Cu(II) (Zafiriou et al., 1998; Voelker et al., 2000), and Mn(III) (Hansard et al., 2011). One electron reduction of various complexes of Co(III), Ni(III), and Mo(V) by has also been observed in aqueous solution (Afanas'ev, 1989), although in some cases under conditions that are not necessarily relevant to oxygenated natural waters around neutral pH.
Due to the ubiquitousness of the O2/ couple in oxygenated natural waters, the mechanisms discussed above are thus expected to be ubiquitous drivers of redox cycling of LRACs in these systems. Recently, González et al. (2016) demonstrated that the Fe(II)/Fe(III) and Cu(I)/Cu(II) redox couples can exchange electrons directly, but that a substantial proportion of electron transfer between the couples was mediated by the O2/ couple under oxygenated conditions. Direct reactions between other LRAC couples are also theoretically possible, and could potentially include multiple electron transfers that could facilitate redox cycling of couples for which stable redox states are not accessible through one electron transfers. However, to date there are few reports of such reactions at the trace concentrations of LRACs found in most in natural waters.
Sinks of Electrons from the Pool of Labile Redox Active Compounds
Because oxidation of kinetically inert reduced compounds is largely irreversible in the extracellular environment, electrons must leave the LRAC pool via other pathways. The major potential sinks for electrons in the LRAC pool are therefore expected to be physical transport of reduced LRACs out of the local environment, scavenging of reduced LRACs through non-redox processes, or redox processes yielding kinetically non-labile products for which reduced LRACs are precursors.
Mechanisms for loss due to physical transport potentially include biological uptake of reduced LRACs such as Fe(II), or transport into more oxidizing environments. In the latter case electrons will be lost from the local LRAC pool, but will still remain associated with the overall LRAC pool until removed by one of the other processes discussed. Scavenging of reduced LRACs through non-redox processes can potentially occur by complexation (and hence stabilization of the reduced LRAC against reoxidation), radical addition reactions, or precipitation of reduced LRACs into non-reactive particulate forms. However, these processes are likely to be relatively minor in many oxygenated natural waters, as the reduced forms of many LRACs (especially trace metals) tend to form weaker complexes and remain more soluble than their corresponding oxidized forms, while scavenging through radical addition reactions is not known to occur at significant rates.
The final pathway in which reduced LRACs react to produce kinetically non-labile species is only possible where the LRAC species can exist in multiple, thermodynamically accessible redox states. While Table 1 does not provide an exhaustive list of possible LRACs, it is clear that the number of potential LRACs satisfying this requirement is small. As discussed earlier, abiotic oxidation Mn2+ is known to be extremely slow (Morgan, 2005), such that reduction of Mn(III) to Mn(II) represents one such possible sink for electrons in the LRAC pool. However, as Mn concentrations are typically relatively low in natural waters (Table 1), the capacity of Mn to act as an ongoing sink for electrons in the LRAC pool is extremely limited—under steady-state conditions with a continuous input of electrons to the LRAC pool through NRAC oxidation, the rate of Mn(III) reduction to Mn(II) would ultimately need to be balanced by the rate of Mn(II) reoxidation. In the event that the rate of Mn(II) reoxidation was negligibly slow, then all Mn would be reduced to Mn(II) such that the rate of Mn(III) reduction would approach zero, and thus Mn would cease to act as a sink of electrons from the LRAC pool. The same reasoning applies whenever the total concentration of all allowable redox states of an LRAC is small, such that none of the compounds shown in Table 1 can act as a major sink of electrons from the LRAC pool under typical conditions in oxygenated natural waters.
ROS are a unique exception to this constraint, as the exceedingly high total concentration of oxygen ensures that ROS have a practically limitless capacity to remove electrons from the LRAC pool. While the O2/ couple can readily exchange electrons with other LRACs, further reduction of yields H2O2 which, as previously discussed, does not readily undergo reoxidation to in most natural waters, and therefore acts as a sink for electrons from the LRAC pool. Furthermore, H2O2 can undergo further irreversible reduction through reaction with reduced LRACs to generate HO•, which can undergo one further irreversible reduction step to water.
Importance of ROS for Local Redox Chemistry in Oxygenated Natural Waters
At least in part because of their ability to exchange electrons on short timescales while maintaining selectivity in reactions, LRACs are integral components of many redox-active enzymes in biological systems (Fraústo Da Silva and Williams, 2001). Thus, the bioavailability of LRACs is a critical influence on ecosystem scale structure and function in natural waters, given that acquisition of LRACs from the extracellular environment is critical for cells to synthesize and maintain these enzymes. For many LRACs, especially trace metals, there is a fine balance between scarcity (sometimes to the point of growth limitation) under conditions of low bioavailability and toxicity under conditions of high bioavailability. This is directly related to redox conditions, which typically strongly influence the bioavailability of trace metals and potentially other LRACs (Fraústo Da Silva and Williams, 2001; Borch et al., 2009).
The ability of to influence the bioavailability of Fe in oxygenated waters by mediating its redox cycling has already been demonstrated under a range of scenarios (Kustka et al., 2005; Rose et al., 2005; Garg et al., 2007; Rose, 2012). Biologically produced extracellular has also been shown to drive redox cycling of Mn in fungi (Hansel et al., 2012) and bacteria (Learman et al., 2011) under oxygenated conditions. However, from the discussion above it is evident that may be expected to play a more general role in controlling the loss of electrons from the LRAC pool, and therefore in controlling local redox conditions. While functioning as a reductant within the O2/ couple, plays a major role in facilitating redox cycling among the LRAC pool, however once reduced to H2O2, it facilitates irreversible loss of electrons from the LRAC pool. Strictly speaking, itself is unable to directly accept electrons in aqueous solution, however it exists in equilibrium with its conjugate acid HOO•, which is a powerful oxidant. Although is dominant at pH 8.1, given that the pKa for the acid-base pair is 4.8 (Bielski et al., 1985), the existence of even small concentrations of HOO• in equilibrium with implies that loss of electrons from the LRAC pool via this pathway is unavoidable. However, provided that the rate of loss is relatively low, then ongoing inputs of electrons through the processes described earlier can potentially maintain substantial steady-state concentrations of reduced LRACs.
Assuming that reduction of to H2O2 is the primary pathway for loss of electrons from the LRAC pool, the rate of loss, Ploss, will be given by:
where represents the fraction of that is present in the protonated HOO• form, kox represents the rate constant for oxidation of a particular reduced LRAC by HOO•, αred represents the fraction of a particular LRAC that is in the reduced state, and [LRAC]T represents the total concentration of that LRAC.
The three major assumptions underlying the development of Equation (3) are that: (i) reduction of to H2O2 represents the major pathway for loss of electrons from the LRAC pool; (ii) the LRAC pool rapidly reaches redox (pseudo)equilibrium when subjected to a perturbation; and (iii) the species comprising the LRAC pool can be accurately defined. The first assumption is essentially based on the principle that O2 is the primary oxidant of reduced LRACs; under conditions where other oxidants may be present at significant concentrations, Equation (3) will not be valid and the hypothesis presented is unlikely to be correct. As discussed in Section Sinks of Electrons from the Pool of Labile Redox Active Compounds, other processes that might remove electrons from the LRAC pool are unlikely to be significant in many oxygenated natural waters. The second assumption is based primarily on the notion that the O2/ couple can mediate relatively rapid electron transfer between various LRAC couples. While this is relatively well known for some LRAC couples, as discussed in Section Cycling of Electrons among the Pool of Labile Redox Active Compounds the kinetics of this process can potentially vary by several orders of magnitude for different LRAC couples. The third assumption is based on the principle that kinetically inert redox active compounds and highly reactive transient species can be neatly and precisely distinguished from LRACs, and that their contribution to electron transfer within the LRAC pool can be ignored. This is conceptually difficult, since in reality the timescales of redox processes involving different couples can vary almost continuously, and the distinction between LRAC and non-LRAC couples is somewhat arbitrary. Furthermore, even if such a distinction can be accurately made, the kinetics of redox processes involving different couples is strongly influenced by complex speciation and pH, which can vary enormously between different types of natural waters, as discussed in Section Cycling of Electrons among the Pool of Labile Redox Active Compounds.
While it would seem reasonable to suggest that these assumptions might be valid at least in certain aquatic environments, it is clear that a substantial amount of additional information is required to establish under what conditions, if any, this will be the case. In particular, site-specific information on the kinetics of redox transformations between the full range of redox couples present, including accounting for complex speciation, is required to test the underlying assumptions of the theory presented and the validity of the resulting Equation (3) for any particular natural water body. Explicitly testing the hypothesis that the relative importance of the O2/ and /H2O2 redox couples exerts a governing control on local redox conditions will require such information to be gathered from a wide variety of natural waters to determine the range of aquatic environments in which this hypothesis might hold.
Should this theory prove to hold in a particular aquatic system, the consequences for redox biogeochemistry are profound. From Equation (3), it follows that under steady-state conditions (where the rate of input of electrons into the LRAC pool must equal the rate of loss):
1. Maintaining a higher steady-state concentration requires a higher rate of input of electrons into the LRAC pool for a given set of kox and [LRAC]T. However this dependence is not linear, as increasing the steady-state concentration will also increase values of αred, assuming that the redox state of the O2/ couple reflects the redox state of the entire LRAC pool. In other words, a relatively low rate of electron input is needed to maintain slightly reducing local redox conditions within the LRAC pool (relative to the overall redox potential set by the O2/H2O system), but greatly increasing rates are needed to maintain more strongly reducing conditions.
2. Systems containing higher total LRAC concentrations will require higher rates of input of electrons into the LRAC pool to maintain a given steady-state concentration. In other words, a relatively low rate of electron input is needed to maintain slightly reducing local redox conditions in systems containing low total LRAC concentrations compared with systems containing high total LRAC concentrations.
By adjusting rates of extracellular electron transport, through direct production of or via other reduced LRACs, organisms could therefore have the ability to tightly regulate local redox conditions, at least within a limited window of redox potentials, in the extracellular environment on the critical biogeochemical timescales of seconds to days. Given the increasing evidence for the ubiquity of extracellular production by aquatic microorganisms (Diaz et al., 2013; Zhang et al., 2016), this suggests that, at least in some oxygenated natural waters, ROS (and in particular ) may play a critical role in mediating overall local redox conditions, with major implications for the biogeochemistry of these systems.
Author Contributions
All work for this manuscript was conducted by AR.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/feart.2016.00096/full#supplementary-material
References
Aeschbacher, M., Sander, M., and Schwarzenbach, R. P. (2010). Novel electrochemical approach to assess the redox properties of humic substances. Environ. Sci. Technol. 44, 87–93. doi: 10.1021/es902627p
Afanas'ev, I. B. (1989). Superoxide Ion Chemistry and Biological Implications. Boca Raton, FL: CRC Press.
Amon, R. M. W., and Benner, R. (1996). Photochemical and microbial consumption of dissolved organic carbon and dissolved oxygen in the Amazon River system. Geochim. Cosmochim. Acta 60, 1783–1792. doi: 10.1016/0016-7037(96)00055-5
Andrews, S. S., Caron, S., and Zafiriou, O. C. (2000). Photochemical oxygen consumption in marine waters: A major sink for colored dissolved organic matter? Limnol. Oceanogr. 45, 267–277. doi: 10.4319/lo.2000.45.2.0267
Barbeau, K. A. (2006). Photochemistry of organic iron(III) complexing ligands in oceanic systems. Photochem. Photobiol. 82, 1505–1516. doi: 10.1111/j.1751-1097.2006.tb09806.x
Barbeau, K., Rue, E. L., Trick, C. G., Bruland, K. W., and Butler, A. (2003). Photochemical reactivity of siderophores produced by marine heterotrophic bacteria and cyanobacteria based on characteristic Fe(III) binding groups. Limnol. Oceanogr. 48, 1069–1078. doi: 10.4319/lo.2003.48.3.1069
Bielski, B. H. J., Cabelli, D. E., Arudi, R. L., and Ross, A. B. (1985). Reactivity of HO2/ radicals in aqueous solution. J. Phys. Chem. Ref. Data 14, 1041–1100. doi: 10.1063/1.555739
Borch, T., Kretzschmar, R., Kappler, A., Cappellen, P. V., Ginder-Vogel, M., Voegelin, A., et al. (2009). Biogeochemical redox processes and their impact on contaminant dynamics. Environ. Sci. Technol. 44, 15–23. doi: 10.1021/es9026248
Bowles, K. C., Ernste, M. J., and Kramer, J. R. (2003). Trace sulfide determination in oxic freshwaters. Anal. Chim. Acta 477, 113–124. doi: 10.1016/S0003-2670(02)01370-3
Bratsch, S. G. (1989). Standard electrode potentials and temperature coefficients in water at 298.15 K. J. Phys. Chem. Ref. Data 18, 1–21. doi: 10.1063/1.555839
Cooper, W. J., and Zepp, R. G. (1990). Hydrogen peroxide decay in waters with suspended soils: evidence for biologically mediated processes. Can. J. Fish. Aquat. Sci. 47, 888–893. doi: 10.1139/f90-102
Diaz, J. M., Hansel, C. M., Voelker, B. M., Mendes, C. M., Andeer, P. F., and Zhang, T. (2013). Widespread production of extracellular superoxide by heterotrophic bacteria. Science 340, 1223–1226. doi: 10.1126/science.1237331
Fraústo Da Silva, J. R. R., and Williams, R. J. P. (2001). The Biological Chemistry of the Elements: The Inorganic Chemistry of Life. Oxford: Oxford University Press.
Fujii, M., Rose, A. L., Waite, T. D., and Omura, T. (2006). Superoxide-mediated dissolution of amorphous ferric oxyhydroxide in seawater. Environ. Sci. Technol. 40, 880–887. doi: 10.1021/es051622t
Garg, S., Rose, A. L., Godrant, A., and Waite, T. D. (2007). Iron uptake by the ichthyotoxic Chattonella marina (Raphidophyceae): impact of superoxide generation. J. Phycol. 43, 978–991. doi: 10.1111/j.1529-8817.2007.00394.x
Goldstein, S., and Czapski, G. (1995). Kinetics of nitric oxide autoxidation in aqueous solution in the absence and presence of various reductants. The nature of the oxidizing intermediates. J. Am. Chem. Soc. 117, 12078–12084.
González, A. G., Pérez-Almeida, N., Magdalena Santana-Casiano, J., Millero, F. J., and González-Dávila, M. (2016). Redox interactions of Fe and Cu in seawater. Mar. Chem. 179, 12–22. doi: 10.1016/j.marchem.2016.01.004
González-Dávila, M., Santana-Casiano, J. M., González, A. G., Pèrez, N., and Millero, F. J. (2009). Oxidation of copper(I) in seawater at nanomolar levels. Mar. Chem. 115, 118–124. doi: 10.1016/j.marchem.2009.07.004
Haber, F., and Weiss, J. (1934). The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. A Mathe. Phys. Sci. 147, 332–351. doi: 10.1098/rspa.1934.0221
Hansard, S. P., Easter, H. D., and Voelker, B. M. (2011). Rapid reaction of nanomolar Mn(II) with superoxide radical in seawater and simulated freshwater. Environ. Sci. Technol. 45, 2811–2817. doi: 10.1021/es104014s
Hansard, S. P., Vermilyea, A. W., and Voelker, B. M. (2010). Measurements of superoxide radical concentration and decay kinetics in the Gulf of Alaska. Deep Sea Res. I Oceanogr. Res. Papers 57, 1111–1119. doi: 10.1016/j.dsr.2010.05.007
Hansel, C. M., Zeiner, C. A., Santelli, C. M., and Webb, S. M. (2012). Mn(II) oxidation by an ascomycete fungus is linked to superoxide production during asexual reproduction. Proc. Natl. Acad. Sci. U.S.A. 109, 12621–12625. doi: 10.1073/pnas.1203885109
Huang, J.-H., Huang, F., Evans, L., and Glasauer, S. (2015). Vanadium: Global (bio)geochemistry. Chem. Geol. 417, 68–89. doi: 10.1016/j.chemgeo.2015.09.019
Jones, G. J., Waite, T. D., and Smith, J. D. (1985). Light-dependent reduction of copper(II) and its effect on cell-mediated thiol-dependent superoxide production. Biochem. Biophys. Res. Commun. 128, 1031–1036. doi: 10.1016/0006-291X(85)90151-2
Korshunov, S., and Imlay, J. A. (2006). Detection and quantification of superoxide formed within the periplasm of Escherichia coli. J. Bacteriol. 188, 6326–6334. doi: 10.1128/JB.00554-06
Kosman, D. J. (2003). Molecular mechanisms of iron uptake in fungi. Mol. Microbiol. 47, 1185–1197. doi: 10.1046/j.1365-2958.2003.03368.x
Kustka, A., Shaked, Y., Milligan, A., King, D. W., and Morel, F. M. M. (2005). Extracellular production of superoxide by marine diatoms: contrasting effects on iron redox chemistry and bioavailability. Limnol. Oceanogr. 50, 1172–1180. doi: 10.4319/lo.2005.50.4.1172
Learman, D. R., Voelker, B. M., Vazquez-Rodriguez, A. I., and Hansel, C. M. (2011). Formation of manganese oxides by bacterially generated superoxide. Nat. Geosci. 4, 95–98. doi: 10.1038/ngeo1055
Lesuisse, E., Casteras-Simon, M., and Labbe, P. (1996). Evidence for the Saccharomyces cerevisiae ferrireductase system being a multicomponent electron transport chain. J. Biol. Chem. 271, 13578–13583. doi: 10.1074/jbc.271.23.13578
Luther, G. W. III. (2010). The role of one- and two-electron transfer reactions in forming thermodynamically unstable intermediates as barriers in multi-electron redox reactions. Aquat. Geochem. 16, 395–420. doi: 10.1007/s10498-009-9082-3
Luther, G. W. III. Findlay, A. J., Macdonald, D. J., Owings, S. M., Hanson, T. E., Beinart, R. A., et al. (2011). Thermodynamics and kinetics of sulfide oxidation by oxygen: a look at inorganically controlled reactions and biologically mediated processes in the environment. Front. Microbiol. 2:62. doi: 10.3389/fmicb.2011.00062
Luther, G. W. III., and Rickard, D. T. (2005). Metal sulfide cluster complexes and their biogeochemical importance in the environment. J. Nanopart. Res. 7, 389–407. doi: 10.1007/s11051-005-4272-4
Marshall, J.-A., Hovenden, M., Oda, T., and Hallegraeff, G. M. (2002). Photosynthesis does influence superoxide production in the ichthyotoxic alga Chattonella marina (Raphidophyceae). J. Plankton Res. 24, 1231–1236. doi: 10.1093/plankt/24.11.1231
Morel, F. M. M., and Hering, J. G. (1993). Principles and Applications of Aquatic Chemistry. New York, NY: Wiley.
Morgan, J. J. (2005). Kinetics of reaction between O2 and Mn(II) species in aqueous solutions. Geochim. Cosmochim. Acta 69, 35–48. doi: 10.1016/j.gca.2004.06.013
Mostofa, K. M. G., and Sakugawa, H. (2009). Spatial and temporal variations and factors controlling the concentrations of hydrogen peroxide and organic peroxides in rivers. Environ. Chem. 6, 524–534. doi: 10.1071/EN09070
Neuman, E. W. (1934). Potassium superoxide and the three-electron bond. J. Chem. Phys. 2, 31–33. doi: 10.1063/1.1749353
Pauling, L. (1979). The discovery of the superoxide radical. Trends Biochem. Sci. 4, N270–N271. doi: 10.1016/0968-0004(79)90203-2
Pierre, J. L., Fontecave, M., and Crichton, R. R. (2002). Chemistry for an essential biological process: the reduction of ferric iron. BioMetals 15, 341–346. doi: 10.1023/A:1020259021641
Rickard, D., and Luther, G. W. III. (2006). Metal sulfide complexes and clusters. Rev. Mineral. Geochem. 61, 421–504. doi: 10.2138/rmg.2006.61.8
Rose, A. L. (2012). The influence of extracellular superoxide on iron redox chemistry and bioavailability to aquatic microorganisms. Front. Microbiol. 3:124. doi: 10.3389/fmicb.2012.00124
Rose, A. L., Godrant, A., Furnas, M., and Waite, T. D. (2010). Dynamics of nonphotochemical superoxide production in the Great Barrier Reef lagoon. Limnol. Oceanogr. 55, 1521–1536. doi: 10.4319/lo.2010.55.4.1521
Rose, A. L., Salmon, T. P., Lukondeh, T., Neilan, B. A., and Waite, T. D. (2005). Use of superoxide as an electron shuttle for iron acquisition by the marine cyanobacterium Lyngbya majuscula. Environ. Sci. Technol. 39, 3708–3715. doi: 10.1021/es048766c
Rose, A. L., and Waite, T. D. (2003). Effect of dissolved natural organic matter on the kinetics of ferrous iron oxygenation in seawater. Environ. Sci. Technol. 37, 4877–4886. doi: 10.1021/es034152g
Rose, A. L., and Waite, T. D. (2005). Reduction of organically complexed ferric iron by superoxide in a simulated natural water. Environ. Sci. Technol. 39, 2645–2650. doi: 10.1021/es048765k
Rose, A. L., Webb, E. A., Waite, T. D., and Moffett, J. W. (2008). Measurement and implications of nonphotochemically generated superoxide in the equatorial Pacific Ocean. Environ. Sci. Technol. 42, 2387–2393. doi: 10.1021/es7024609
Rosso, K. M., and Morgan, J. J. (2002). Outer-sphere electron transfer kinetics of metal ion oxidation by molecular oxygen. Geochim. Cosmochim. Acta 66, 4223–4233. doi: 10.1016/S0016-7037(02)01040-2
Rusak, S. A., Peake, B. M., Richard, L. E., Nodder, S. D., and Cooper, W. J. (2011). Distributions of hydrogen peroxide and superoxide in seawater east of New Zealand. Mar. Chem. 127, 155–169. doi: 10.1016/j.marchem.2011.08.005
Rush, J. D., and Bielski, B. H. J. (1985). Pulse radiolytic studies of the reactions of HO2/ with Fe(II)/Fe(III) ions. The reactivity of HO2/ with ferric ions and its implication on the occurrence of the Haber-Weiss reaction. J. Phys. Chem. 89, 5062–5066. doi: 10.1021/j100269a035
Santana-Casiano, J. M., González-Dávila, M., and Millero, F. J. (2005). Oxidation of nanomolar levels of Fe(II) with oxygen in natural waters. Environ. Sci. Technol. 39, 2073–2079. doi: 10.1021/es049748y
Sharpless, C. M., and Blough, N. V. (2014). The importance of charge-transfer interactions in determining chromophoric dissolved organic matter (CDOM) optical and photochemical properties. Environ. Sci. Proc. Impacts 16, 654–671. doi: 10.1039/c3em00573a
Sverjensky, D. A., and Lee, N. (2010). The Great Oxidation Event and mineral diversification. Elements 6, 31–36. doi: 10.2113/gselements.6.1.31
Vermilyea, A. W., Hansard, S. P., and Voelker, B. M. (2010). Dark production of hydrogen peroxide in the Gulf of Alaska. Limnol. Oceanogr. 55, 580–588. doi: 10.4319/lo.2009.55.2.0580
Voelker, B. M., Sedlak, D. L., and Zafiriou, O. C. (2000). Chemistry of superoxide radical in seawater: reactions with organic Cu complexes. Environ. Sci. Technol. 34, 1036–1042. doi: 10.1021/es990545x
Vraspir, J. M., and Butler, A. (2009). Chemistry of marine ligands and siderophores. Ann. Rev. Mar. Sci. 1, 43–63. doi: 10.1146/annurev.marine.010908.163712
Wang, D. (2012). Redox chemistry of molybdenum in natural waters and its involvement in biological evolution. Front. Microbiol. 3:427. doi: 10.3389/fmicb.2012.00427
Wardman, P. (1989). Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 18, 1637–1755. doi: 10.1063/1.555843
Yuan, J., and Shiller, A. M. (2005). Distribution of hydrogen peroxide in the northwest Pacific Ocean. Geochem. Geophys. Geosys. 6:Q09M02. doi: 10.1029/2004GC000908
Zafiriou, O. C., Voelker, B. M., and Sedlak, D. L. (1998). Chemistry of the superoxide radical () in seawater: reactions with inorganic copper complexes. J. Phys. Chem. A 102, 5693–5700. doi: 10.1021/jp980709g
Zhang, T., Hansel, C. M., Voelker, B. M., and Lamborg, C. H. (2016). Extensive dark biological production of reactive oxygen species in brackish and freshwater ponds. Environ. Sci. Technol. 50, 2983–2993. doi: 10.1021/acs.est.5b03906
Zhang, Y., Del Vecchio, R., and Blough, N. V. (2012). Investigating the mechanism of hydrogen peroxide photoproduction by humic substances. Environ. Sci. Technol. 46, 11836–11843. doi: 10.1021/es3029582
Keywords: superoxide, oxygen, reactive oxygen species, labile redox-active compounds, redox conditions, natural waters
Citation: Rose AL (2016) The Influence of Reactive Oxygen Species on Local Redox Conditions in Oxygenated Natural Waters. Front. Earth Sci. 4:96. doi: 10.3389/feart.2016.00096
Received: 16 June 2016; Accepted: 25 October 2016;
Published: 11 November 2016.
Edited by:
Leanne C. Powers, Skidaway Institute of Oceanography, USAReviewed by:
Julian Blasco, Spanish National Research Council, SpainPaul R. Erickson, ETH Zurich, Switzerland
Copyright © 2016 Rose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew L. Rose, YW5kcmV3LnJvc2VAc2N1LmVkdS5hdQ==