 Natascha Riedinger1*†
Natascha Riedinger1*† Benjamin Brunner2†
Benjamin Brunner2† Sebastian Krastel3
Sebastian Krastel3 Gail L. Arnold2†Laura M. Wehrmann4†Michael J. Formolo5†
Gail L. Arnold2†Laura M. Wehrmann4†Michael J. Formolo5† Antje Beck6†
Antje Beck6† Steven M. Bates7
Steven M. Bates7 Susann Henkel8,9
Susann Henkel8,9 Sabine Kasten8,9Timothy W. Lyons7
Sabine Kasten8,9Timothy W. Lyons7- 1Boone Pickens School of Geology, Oklahoma State University, Stillwater, OK, USA
- 2Department of Geological Sciences, University of Texas at El Paso, El Paso, TX, USA
- 3Institute of Geosciences, University of Kiel, Kiel, Germany
- 4School of Marine and Atmospheric Sciences, Stony Brook University, Stony Brook, NY, USA
- 5ExxonMobil Upstream Research Company, Spring, TX, USA
- 6GEOMAR—Helmholtz Centre for Ocean Research Kiel, Kiel, Germany
- 7Department of Earth Sciences, University of California, Riverside, CA, USA
- 8Department of Geosciences, Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, Germany
- 9MARUM—Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany
The interplay between sediment deposition patterns, organic matter type and the quantity and quality of reactive mineral phases determines the accumulation, speciation, and isotope composition of pore water and solid phase sulfur constituents in marine sediments. Here, we present the sulfur geochemistry of siliciclastic sediments from two sites along the Argentine continental slope—a system characterized by dynamic deposition and reworking, which result in non-steady state conditions. The two investigated sites have different depositional histories but have in common that reactive iron phases are abundant and that organic matter is refractory—conditions that result in low organoclastic sulfate reduction rates (SRR). Deposition of reworked, isotopically light pyrite and sulfurized organic matter appear to be important contributors to the sulfur inventory, with only minor addition of pyrite from organoclastic sulfate reduction above the sulfate-methane transition (SMT). Pore-water sulfide is limited to a narrow zone at the SMT. The core of that zone is dominated by pyrite accumulation. Iron monosulfide and elemental sulfur accumulate above and below this zone. Iron monosulfide precipitation is driven by the reaction of low amounts of hydrogen sulfide with ferrous iron and is in competition with the oxidation of sulfide by iron (oxyhydr)oxides to form elemental sulfur. The intervals marked by precipitation of intermediate sulfur phases at the margin of the zone with free sulfide are bordered by two distinct peaks in total organic sulfur (TOS). Organic matter sulfurization appears to precede pyrite formation in the iron-dominated margins of the sulfide zone, potentially linked to the presence of polysulfides formed by reaction between dissolved sulfide and elemental sulfur. Thus, SMTs can be hotspots for organic matter sulfurization in sulfide-limited, reactive iron-rich marine sedimentary systems. Furthermore, existence of elemental sulfur and iron monosulfide phases meters below the SMT demonstrates that in sulfide-limited systems metastable sulfur constituents are not readily converted to pyrite but can be buried to deeper sediment depths. Our data show that in non-steady state systems, redox zones do not occur in sequence but can reappear or proceed in inverse sequence throughout the sediment column, causing similar mineral alteration processes to occur at the same time at different sediment depths.
Introduction
Dynamic depositional systems strongly impact sedimentary geochemical processes; sediments formed under such geochemical regimes do not always show the typical sequence of terminal electron acceptor processes predicted for steady state environments (e.g., Claypool and Kaplan, 1974; Froelich et al., 1979; Berner, 1981). Depending on the availability and reactivity of the solid-phase electron acceptors, the redox sequence can even be reversed in some cases. For example, iron and manganese reduction can reappear or persist at depth below the zone dominated by sulfate reduction (e.g., Postma and Jakobsen, 1996; Kasten et al., 1998; Hensen et al., 2003; Riedinger et al., 2014; Treude et al., 2014). In particular, non-steady state depositional conditions can have a strong impact on the inventory of sulfide minerals in anoxic marine environments (e.g., Kasten et al., 1998, 2003; Aller et al., 2010; Borowski et al., 2013; Aller, 2014; Peketi et al., 2015). Iron sulfide-containing sediments that are eroded and/or transported down-slope in oxygenated seawater can be subject to reoxidation, leading to the conversion of iron monosulfides and fine-grained pyrite to (amorphous) ferric hydroxides (Luther et al., 1982; Morse, 1991). Shielded from sulfidic conditions in the upper sediment column due to rapid burial—and in the presence of mostly reworked, unreactive organic matter—those oxidized reactive iron phases are then preserved in deeper subsurface sediments (Hensen et al., 2003; März et al., 2008; Riedinger et al., 2014). The continental margin off Uruguay and Argentina is characterized by such highly dynamic depositional conditions (e.g., Riedinger et al., 2005; Henkel et al., 2011, 2012; Krastel et al., 2011, 2013), and these locations are likely to be representative of environments that are common throughout the world along continental margins.
Sediments along the continental margin off Uruguay and Argentina, at the western rim of the Argentine Basin, are not only characterized by redistribution/reworking, including recycling of organic matter, but also by an input of iron (as iron (oxyhydr)oxides). Those inputs of reactive iron greatly outpace sedimentary sulfide production, which results in an iron-dominated system (Haese et al., 2000; Hensen et al., 2003; Riedinger et al., 2005). This situation arises because residual, less reactive organic matter, which has already been degraded at the shallower sites of initial deposition, is reworked and transported and further remineralized along with sulfide minerals, which are oxidized to yield high amounts of Fe(III). Hensen et al. (2003) concluded that the non-steady state processes and associated iron oxidation in this dynamic system favors the retention of any reduced sulfur generated following redeposition, which was corroborated by the quantification of sulfur burial based on a transport and reaction model (Riedinger et al., 2005) and subsequent direct measurements of sulfur constituents (Riedinger et al., 2014). Although these studies demonstrated that sedimentary sulfur cycling in an iron oxide-dominated, dynamic marine system strongly affects sulfur sequestration and subseafloor microbial processes, some questions remain unresolved; in particular, it is unclear why a wide variety of different reduced sulfur compounds is formed and if there is a “classical” zonation—analogous to the standard redox tower—for diagenetic sulfur transformations. To study the fate of various sulfur species, their contents and isotopic signatures, as well as the iron inventory, in more detail in these dynamic depositional systems, we collected samples via gravity cores from the lower slope in the western Argentine Basin during the RV Meteor Expedition M78/3 in May-July 2009.
Sulfur Cycling in Marine Sediments
The main driver of the marine sedimentary sulfur cycle is the microbial reduction of sulfate (e.g., Goldhaber and Kaplan, 1974; Froelich et al., 1979; Jørgensen, 1982; Bowles et al., 2014). The two major catabolic microbial sulfate reduction pathways are organoclastic sulfate reduction and sulfate reduction coupled to anaerobic oxidation of methane (AOM); both processes release hydrogen sulfide to the pore water (e.g., Goldhaber and Kaplan, 1974; Jørgensen, 1982; Hoehler et al., 1994; Boetius et al., 2000). Microbial sulfate reduction discriminates against the heavier sulfur isotopes, leading to a relative depletion in 34S in the produced hydrogen sulfide and a corresponding relative 34S enrichment in the remaining sulfate pool (Jones and Starkey, 1957; Harrison and Thode, 1958; Thode et al., 1961; Kaplan and Rittenberg, 1964; Rees, 1970; Goldhaber and Kaplan, 1974, 1980; Froelich et al., 1979; Bottrell and Raiswell, 2000; Brunner and Bernasconi, 2005; Sim et al., 2011; Wing and Halevy, 2014). Typically, with increasing sediment depth, as the sulfate pool becomes smaller, the remaining sulfate becomes isotopically heavier (e.g., Goldhaber and Kaplan, 1974; Torres et al., 1996). This trend is reflected in the production of hydrogen sulfide that also becomes isotopically heavier. As a consequence, with increasing sediment depth, any sulfur phase resulting from sulfide oxidation or directly precipitated iron sulfide will also become enriched in 34S (e.g., Goldhaber and Kaplan, 1980). Furthermore, at the sulfate-methane transition (SMT) where sulfate is almost entirely consumed, iron sulfides are precipitated with the heaviest isotope composition compared to the upper sediments (Jørgensen et al., 2004; Borowski et al., 2013; Lin et al., 2016).
In most continental margin sediments, pyrite is the most abundant iron sulfide species (e.g., Goldhaber and Kaplan, 1975; Cornwell and Morse, 1987; Morse and Cornwell, 1987) and a long-term sink for sulfur (Berner, 1982, 1989; Berner and Raiswell, 1983). Pyrite abundance is controlled mainly by the rate of microbial sulfate reduction, which depends on the amount and quality of buried organic matter or the upward flux of methane, and the availability of reactive iron to (re)oxidize and/or precipitate the produced hydrogen sulfide (e.g., Goldhaber and Kaplan, 1974; Jørgensen, 1977, 1982, 1990; Berner, 1984; Raiswell et al., 1988; Lyons, 1997). In iron-dominated sedimentary systems, hydrogen sulfide is effectively scavenged by dissolved Fe2+ and other, solid reactive iron phases, resulting in the accumulation of intermediate/metastable sulfur and iron sulfide phases, such as elemental sulfur, mackinawite, and greigite, which can be further transformed into pyrite (e.g., Goldhaber and Kaplan, 1975; Canfield, 1989; Kasten et al., 1998; Rickard and Luther, 2007; Fu et al., 2008; Peiffer et al., 2015). In systems limited by hydrogen sulfide availability, intermediate sulfur phases such as thiosulfate and elemental sulfur can be microbially disproportionated into 34S-enriched sulfate and 34S-depleted hydrogen sulfide (e.g., Thamdrup et al., 1993; Jørgensen and Nelson, 2004; Böttcher et al., 2005). This process further increases the offset between isotopically heavy sulfate and light sulfide (Canfield and Thamdrup, 1994). Thus, the concentration and sulfur isotope composition of sulfur and iron sulfide phases can be used to trace ongoing and past biogeochemical sulfur cycling in marine sediments (e.g., Goldhaber and Kaplan, 1974; Goldhaber et al., 1977; Howarth, 1979; Jørgensen, 1979; Howarth and Jørgensen, 1984; Jørgensen et al., 2004).
In addition to the interaction of sulfur with iron, a further major sink of sulfur in marine systems is the incorporation into organic compounds (e.g., Sinninghe Damsté et al., 1988; Anderson and Pratt, 1995; Brüchert and Pratt, 1996; Canfield et al., 1998; Werne et al., 2004). During early diagenesis, organic matter can be sulfurized via reaction with hydrogen sulfide and/or intermediate sulfide oxidation products, such as polysulfides (e.g., Aizenshtat et al., 1983; Vairavamurthy and Mopper, 1987; Kohnen et al., 1989; Sinninghe Damsté and deLeeuw, 1990; Vairavamurthy et al., 1992; Anderson and Pratt, 1995; Adam et al., 1998; Werne et al., 2000; Filley et al., 2002). In the uppermost surface sediments of organic-rich, sulfide-dominated marine sedimentary systems, sulfurization of labile organic compounds, such as humic acids (François, 1987; Ferdelman et al., 1991), can precede the formation of pyrite (Mossmann et al., 1991; Vairavamurthy et al., 1992, 1995; Filley et al., 2002; Werne et al., 2008). The isotopic composition of the precursor inorganic sulfur species, such as hydrogen sulfide, polysulfides, or elemental sulfur, should thus be recorded in the resulting organic sulfur compounds (Werne et al., 2008; Amrani, 2014). Unfortunately, the bulk organic sulfur fraction, with an isotopic signature representing a mixture of the distinct organic sulfur compounds, does not allow us to distinguish between different sulfurization pathways (Werne et al., 2003); only compound-specific approaches may provide some further insights (Amrani and Aizenshtat, 2004; Raven et al., 2016).
Methods
Sedimentary Setting
The sedimentary environment of the Argentine Basin, including the continental margin off Argentina and Uruguay (Figure 1), is controlled by dynamic depositional processes, such as gravity-controlled sediment transport and strong current circulation (Ewing and Leonardi, 1971; Klaus and Ledbetter, 1988; Hernández-Molina et al., 2009; Preu et al., 2013). In the upper waters, the southward flowing Brazil Current and the northward flowing Malvinas (Falkland) Current meet in front of the Rio de la Plata (Peterson and Stramma, 1991). The Brazil Malvinas Confluence (BMC) leads to an increase in primary production over a distinct area related to the mixing of these tropical and Antarctic water masses, which results in strong gradients in nutrient, salinity, and temperature (Antoine et al., 1996; Behrenfeld and Falkowski, 1997; Chiessi et al., 2007) and elevated organic carbon input into the sediment along the shelf and upper slope. Southward-flowing North Atlantic Deep Water (NADW, 2,000 and 4,000 m) and northward-flowing Antarctic Bottom Water (AABW >4,000 m) are parallel to the continental margin; below 4,000 m water depth, the AABW transports mostly fine-grained sediment.
Figure 1. Site location of studied cores (yellow filled circles) in the Argentine Basin.
Predominantly terrigenous material, delivered from the numerous fluvial tributaries along the coast of Argentina and Uruguay (Iriondo, 1984; Piccolo and Perillo, 1999), is transported downslope from the continental shelf via gravity-controlled mass flows, including turbidity currents and density flows (Biscaye and Dasch, 1971; Ewing et al., 1971; Klaus and Ledbetter, 1988; Sachs and Ellwood, 1988; Romero and Hensen, 2002; Hensen et al., 2003; Henkel et al., 2011, 2012; Krastel et al., 2011; Gruetzner et al., 2012; Voigt et al., 2013). These mass flows also transport reworked organic matter further downslope, resulting in burial of refractory organic carbon (Hedges and Keil, 1995) in the deeper parts of the basin. Thus, high mean sedimentation rates, including mass transport deposits (MTD), combined with low levels of reactive organic carbon, leads to rapid burial of (highly) reactive iron minerals present as both primary terrigenous phases and reworked and oxidized authigenic components (Hensen et al., 2003; Riedinger et al., 2005, 2014).
Sampling and Sample Processing
Gravity cores were collected east of the Rio de la Plata mouth from water depths of 3821 m and 3687 m (GeoB 13824-1 38°13.14′ S, 53°21.29′ W; GeoB 13863-1 39°18.70′ S, 53°57.16′ W; Figure 1) during R/V Meteor expedition M78/3 (Krastel and Wefer, 2011). The cores were taken at two sites from different depositional settings (Figure 2). GeoB 13824 was collected at the foot of the Mar del Plata Canyon (Krastel and Wefer, 2011), which is characterized by high accumulation of sediments that bypassed the shelf through the canyon, as well as potential erosion events. Core GeoB 13863 was recovered from a site on the lower slope south of the Rio de la Plata where recent sediment input is mainly influenced by currents and minor amounts of mass gravity flow. The site experienced profound variations in depositional conditions over glacial/interglacial timescales—most likely related to changes in sea level (Riedinger et al., 2005).
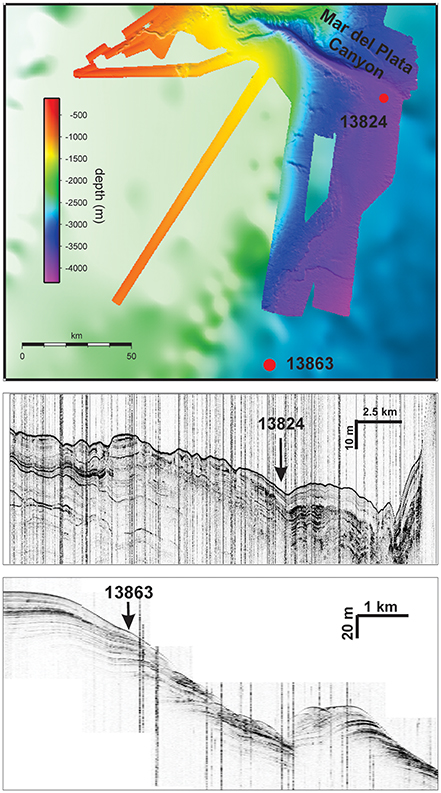
Figure 2. Multibeam bathymetric map of the study site area and seismic profiles across the investigated locations. Seismic profiles are displayed for Site GeoB 13824 from S to N, and for Site GeoB 13863 from NE to SW.
After retrieval, the cores were cut immediately into 1-m segments on deck. For methane analysis, 3 cc syringe samples were taken from every core segment and transferred into 20 mL headspace vials pre-filled with 10 mL of a 5 M NaCl solution and stored at 4°C. Additionally, samples for determinations of sulfate reduction rates (SRR) were taken from every core segment (for Hole GeoB 13824 only). Pore water samples were extracted on segment-halves in a cold room (~4°C) via the Rhizon method (Seeberg-Elverfeldt et al., 2005; Dickens et al., 2007). For sulfate and hydrogen sulfide concentrations and isotope analyses, 5 mL subsamples of pore water were added to a 2.5% zinc acetate (ZnAc) solution in order to fix all sulfide present as zinc sulfide (ZnS). The pH and Eh were measured using punch-in electrodes. Solid phase samples were taken at 20–30 cm intervals and placed and sealed under a nitrogen atmosphere in aluminum bags and stored frozen (−20°C) until processed onshore. The geochemical data reported here can be accessed via the information system PANGAEA operated by the World Data Centers for Marine Environmental Sciences (https://doi.pangaea.de/10.1594/PANGAEA.856812).
Pore Water Analyses
Total dissolved sulfide concentrations (ΣH2S = H2S + HS− + S2−) were analyzed onboard the ship spectrophotometrically using the methylene blue method (Cline, 1969). GeoB 13824-1 samples for sulfate () concentrations were also analyzed onboard at 1:50 and 1:100 dilutions using a high performance liquid chromatography (HPLC) Sykam solvent delivery system coupled to a Waters 430 conductivity detector. Sulfate concentrations for Site GeoB 13863 were measured at the Max Planck Institute for Marine Microbiology (MPI-MM) in Bremen, Germany, via suppressed ion chromatography at a 1:100 dilution with double de-ionized water on a Metrohm 761 compact IC. Standard calibrations were performed using seawater provided by the International Association for the Physical Sciences of the Oceans (IAPSO) and in-house standards. The error of replicate analyses of sulfide and sulfate was <2 and 3%, respectively. Methane (CH4) was measured with a Hewlett Packard 5890A gas chromatograph using a splitless injector, a stainless steel Porapak-Q column and a flame ionization detector at the MPI-MM. Chromatographic response on the GC instrument was calibrated against three different standards with variable concentrations of CH4. The measured concentrations were corrected for sediment porosity.
Potential polysulfide concentrations (Σ-Calc., n = 2,…,8) were calculated using the thermodynamic constants of Kamyshny et al. (2007) with the program MinteQ®, assuming an activity of 1 for solid elemental sulfur and using the measured sulfide concentration. Values were calculated for seawater at 2°C and in situ pH of 8. Compared to seawater at pH 8.2 at 25°C, where 1.15 mol of S0 can be solubilized per mol of sulfide (Kamyshny, 2009), only 0.21 mol of S0 can be solubilized per mol of sulfide at 2°C because of the lower dissolution of S0 as polysulfides at lower temperature (Kamyshny et al., 2007). For a chosen sulfide concentration, the value for Σ-Calc. is lower than the value of solubilized S0 because the latter calculates total concentration of zero-valent sulfur from polysulfide species with different sulfur chain length, while the former sums the concentration of polysulfides. For example, 0.21 mol of S0 can be solubilized in presence of 0.6 mM sulfide at 2°C, which corresponds to a total polysulfide concentration of 0.037 mM.
Sulfate Reduction Rates
Using the 35S radiotracer method (Jørgensen, 1978), sulfate reduction rate (SRR) experiments were carried out onboard the ship in a refrigerated container (4°C). Sediment slurries injected with 35 were incubated under in situ pressure (38 MPa; for further details regarding pressure incubations, see Vossmeyer et al., 2012). After the pressure was released, the incubation experiments were terminated by transferring the samples into 15 mL tubes containing a 20% ZnAc solution. These samples were kept frozen at −20°C during transport and storage. TSRR were determined via the single-step cold chromium reduction (Kallmeyer et al., 2004), and measurements were carried out by scintillation counting at the Center for Geobiology, Aarhus University, Denmark. The detection limit for the SRR was between 0.2 and 1.3 pmol cm−3 d−1.
Solid Phase Analyses
Multi-acid total digestions (hydrofluoric, HF; hydrochloric, HCl; and nitric, HNO3) were performed on ~50 mg of dry sediment sample using a microwave system (CEM Mars Xpress) at the Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research, Bremerhaven, Germany (AWI). The accuracy of the measurements was verified using NIST SRM 2702 and in-house (MAX) standards. Major elements were analyzed via inductively coupled plasma-atomic emission spectrometry (ICP-AES, Thermo Scientific IRIS Intrepid instrument). The reference standard contents were within the accepted analytical error for all elements measured. Contents of total carbon (TC) and total inorganic carbon (TIC) were determined by measuring dried and homogenized samples using an ELTRA CS 500 carbon sulfur analyzer equipped with acidification and furnace modules. The accuracy was ±3% and ±4%, respectively. The amount of total organic carbon (TOC) was calculated by subtracting the TIC fraction from TC.
Sequential iron extractions were carried out under anoxic conditions using frozen subsamples. Ascorbate, dithionite, and oxalate steps were applied on ca. 150–200 mg samples to determine, respectively, the fractions present as adsorbed ferrous iron and highly reactive/bioavailable ferric iron (Febio), crystalline iron oxides such as goethite and hematite (Feoxide), and magnetite (Femagn; Ferdelman, 1988; Poulton and Canfield, 2005; Raiswell et al., 2010; Wehrmann et al., 2014; Henkel et al., 2016). The solutions were flushed with N2 prior to extraction. All solutions were freshly prepared prior to extraction, and reagent blanks were taken. The iron concentration was analyzed by inductively coupled plasma-mass spectrometry (ICP-MS; Agilent 7500ce) after dilution in trace-metal grade 2% HNO3. Replicate sample extractions yielded reproducibility within 8%, and all iron concentrations of reagent blanks were below detection limit.
All solid sulfur phases were analyzed sequentially (for detailed method descriptions see Riedinger et al., 2014). Elemental sulfur (S0) was extracted from approximately 2–3 g of wet (freshly thawed) sediment by shaking for ~12 h in 10 mL pure methanol with a sample-to-extractant ratio of ~1/10 (Zopfi et al., 2004). The headspace was flushed with N2 to avoid oxidation of the reduced species. The concentration of S0 was analyzed at the MPI-MM using a Sykam pump (S1100), a UV–Vis Detector (Sykam S3200), a Zorbax ODS-column (125 × 4 mm, 5 μm; Knauer, Germany) and 100% methanol (HPLC grade) at a flow rate of 1 mL per minute. Elemental sulfur was eluted after 3.5 min and detected at 265 nm, with a detection limit of about 1 μM and an analytical precision of ±0.5% SD. Based on replicate sample extractions and in-house standards, the precision and accuracy of the elemental sulfur measurements is better than 0.001 wt.%. Therefore, our samples with reported contents below 0.001 wt.% may represent elemental sulfur-free samples. This consideration is important in the discussion of potential presence or absence of polysulfide. For determination of acid volatile sulfide (AVS; mainly iron monosulfides—“FeS”) and chromium reducible sulfur (CRS; which, due to the preceding removal of AVS and S0 corresponds mainly to pyrite), the samples were treated with the sequential, two-step acid/Cr(II) method (Fossing and Jørgensen, 1989). The sulfide produced in each step was trapped as ZnS in a 5% Zn-acetate solution and analyzed following dilution using the methylene blue method (Cline, 1969). Reproducibility was better than 7% based on an in-house standard. The fraction of total organic sulfur (TOS) was determined on the solid residue following the Cr(II) step (Werne et al., 2003). The samples were filtered and rinsed with double distilled water, dried and analyzed using a carbon-sulfur elemental analyzer (ELTRA CS 500). All solid-phase data are reported in dry weight units.
The degree of pyritization (DOP; Raiswell and Canfield, 1998) was calculated from the analyzed iron phases by dividing pyrite Fe (FeCRS; calculated from pyrite S) by the total highly reactive Fe present as iron oxides, iron monosulfide and pyrite Fe (FeCRS/[Febio+Feoxide+Femag+FeAVS+FeCRS]). Highly reactive iron (FeHR) was defined according to Raiswell and Canfield (1998) as the sum of Febio, Feoxide, FeAVS, and FeCRS.
Sediment porosity was determined according to standard IODP procedures (Blum, 1997) at the MARUM—the Center for Marine Environmental Sciences at the University of Bremen, Germany—using helium-displacement penta-pycnometers. The data were corrected for evaporated seawater, specifically the mass of precipitated salts, as described by Krastel et al. (2011).
Sulfur Isotopes
For isotope analyses of sulfate, filtered pore water aliquots were acidified, and sulfate was precipitated as barium sulfate (BaSO4) by addition of barium chloride solution (BaCl2, 1 M). For determination of the sulfur isotope compositions of ΣH2S, S0, AVS, and CRS, ZnS precipitates were converted to Ag2S by addition of AgNO3 and subsequent washing with NH4OH to remove colloidal silver. Zinc sulfide was obtained from elemental sulfur by using the hot acid/Cr(II) distillation method. Sulfur isotope ratios of TOS were measured on bulk sediments (Werne et al., 2003). Isotope compositions of pore water sulfate-sulfur were determined at the MPI-MM, all other isotope measurements were carried out at the University of California-Riverside (UCR). The measurements were performed by sample combustion with elemental analyzers that were connected via continuous helium flow to Thermo-Finnigan Delta® gas source isotope ratio mass spectrometers. All sulfur isotope measurements were calibrated with reference materials NBS 127 (δ34S = +21.1‰) and IAEA-SO-6 (δ34S = −34.1‰), and the standard error (1σ) of the measurements was <0.2‰ for δ34S. The sulfur isotope composition is reported with respect to Vienna Canyon Diablo Troilite (V-CDT). In the case of the sulfur isotope measurements of total dissolved sulfide, the sample sizes were very small (~1/10th typical analysis weight, 0.04 mg), which increases the uncertainty for the reported isotope composition. Based on repeated measurements of very small amounts of our laboratory standard, we estimate the standard error (1σ) for very small samples to be <2‰ for δ34S.
Results
The sulfidic zone is defined as the interval where dissolved hydrogen sulfide accumulates in the pore water. The intervals above and below the sulfidic zone are defined as the postoxic and methanic zones, respectively (after Berner, 1981). Due to the typical loss of the top few centimeters of the core during recovery, the uppermost oxic zone was not sampled.
Pore Water
The pore water concentration profiles at both sites show a similar trend, with a linear decrease in sulfate concentrations from about 27.5 mM at the top of the cores to complete depletion at the SMT located at ~5.5 mbsf at Site GeoB 13824 and ~5 mbsf at Site GeoB 13863 (Figure 3). Below this transition, methane increases with depth at the two sites to concentrations of 6.1 mM and 9.9 mM, respectively. Pore water accumulations of free sulfide are restricted to a narrow interval close to the SMT—the sulfidic zone. At Site GeoB 13824, this sulfidic zone lies between 3.8 and 6.9 mbsf with maximum ΣH2S of 668 μM; at Site GeoB 13863, the sulfidic zone is confined to 3.8–5.7 mbsf, and ΣH2S reaches 597 μM. The dissolved iron (Fe2+) concentrations at Site GeoB 13824 show a decrease from the top of the core (15.3 μM) down to about 3.5 mbsf (1.2 μM). Below this depth and throughout the sulfidic zone, dissolved iron was close to or below the detection limit (0.5 μM). Below the sulfidic zone, dissolved iron concentrations strongly increase with depth, with the exception of one small excursion at 8.5–9 mbsf (marked by a drop to 15 μM), with concentrations reaching 72.6 μM (Figure 3A). The dissolved iron profile at Site GeoB 13863 shows a similar trend to that observed at Site 13824, with slightly elevated values in the upper sediment layers and a maximum concentration of 17.3 μM at 0.8 mbsf and Fe2+ below detection in the sulfidic zone. Below this zone, dissolved iron concentrations increase again to a maximum of 50 μM before they decrease slightly with depth but remain above 25 μM (Figure 3B; Riedinger et al., 2014).
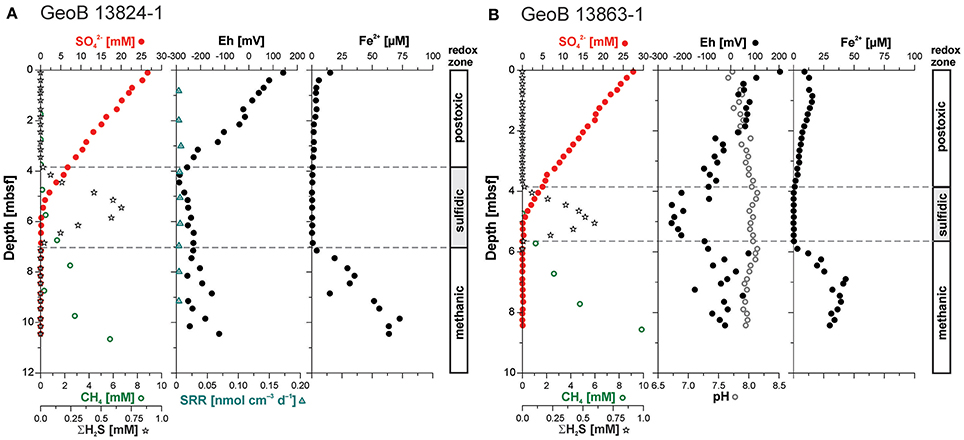
Figure 3. Sulfate (; red filled circles), methane (CH4; green open circles), hydrogen sulfide (ΣH2S; open stars), and ferrous iron (Fe2+) pore water concentration profiles as well as Eh (filled circle) and pH (open circle) profiles from (A) Site GeoB 13824 and (B) GeoB 13863. No pH measurements available for Site 13824. Profiles of , ΣH2S, CH4, and Fe2+ for Site GeoB 13863 after Riedinger et al. (2014). Sulfate reduction rates (SRR) are low throughout the sediment column at Site GeoB 13824 (no data available for Site GeoB 13863). Insufficient sample resolution precluded determinations of SRR for surface sediments and actual zone of sulfate reduction coupled anaerobic methane oxidation at the sulfate-methane transition (SMT). At both sites H2S accumulation in the pore water is limited to a narrow interval located near the SMT. The dashed lines indicate the upper and lower boundary of this sulfidic zone. Redox zones are defined after Berner (1981).
At Site GeoB 13824, the redox potential (Eh) decreases linearly with depth, from 142 mV at the sediment surface to −288 mV at 4.45 mbsf. Below this depth, the redox potential increases slightly again with depth but stays below −120 mV. In contrast to the Eh profile at Site GeoB 13824, the redox potential at Site GeoB 13863 shows a strong correlation with dissolved sulfide concentration, including an Eh decrease to −244 mV in the sulfidic zone. Above and below this zone, Eh-values increase to 204 and 74 mV, respectively. The pH at this site does not show strong variation and stays in the range of 7.66–8.14, with slightly elevated values in the sulfidic zone. No pH measurements are available for Site GeoB 13824.
Sulfate Reduction Rates
Sulfate reduction rates (SRR) are only available for sediments from Site GeoB 13824. The rates are low (<12 pmol cm−3 d−1) throughout the core with values close to or below the detection limit. As a likely consequence of the low sampling resolution, the expected higher SRR at the sediment surface, as well as at the SMT as related to sulfate reduction coupled to anaerobic methane oxidation, were not captured.
Solid Phase
The investigated sediments are dominated by detrital material with low contents of calcium carbonate (CaCO3) restricted to the upper 1.8 m at Site GeoB 13824 and the upper 0.5 m at Site GeoB 13863, along with high amounts of total iron ranging from 2.6 to 4.7 wt.% and aluminum from 6 to 9 wt.% (Figure 4). The Fe/Al ratios at both sites stay constant throughout the sediment column, with an average value of 0.5, similar to an average crustal value (Taylor and McLennan, 1985). At Site GeoB 13824, the CaCO3 contents show a peak between 5 and 6 mbsf, corresponding to the depth of the SMT. The sediments at Site GeoB 13863 do not display such a peak likely because of our sampling protocol. Specifically, prior to sampling, discrete carbonate crystals were removed from about 5 mbsf, within the SMT, for separate analysis and are thus not included in the bulk element contents. Thus, for both sites, carbonate is present at the depth of the SMT, as depicted in the sediment log (Figure 4). TOC contents at Site GeoB 13824 show only slight variation throughout the core, with a range from 1.43 to 0.76 wt.%; sediments in the uppermost meter and at ~2.5 mbsf show the highest contents (>1 wt.%). At Site GeoB 13863, the TOC contents range from 1.15 wt.% (in the uppermost 20 cm) to 0.44 wt.% and average 0.78 wt.% (Riedinger et al., 2014).
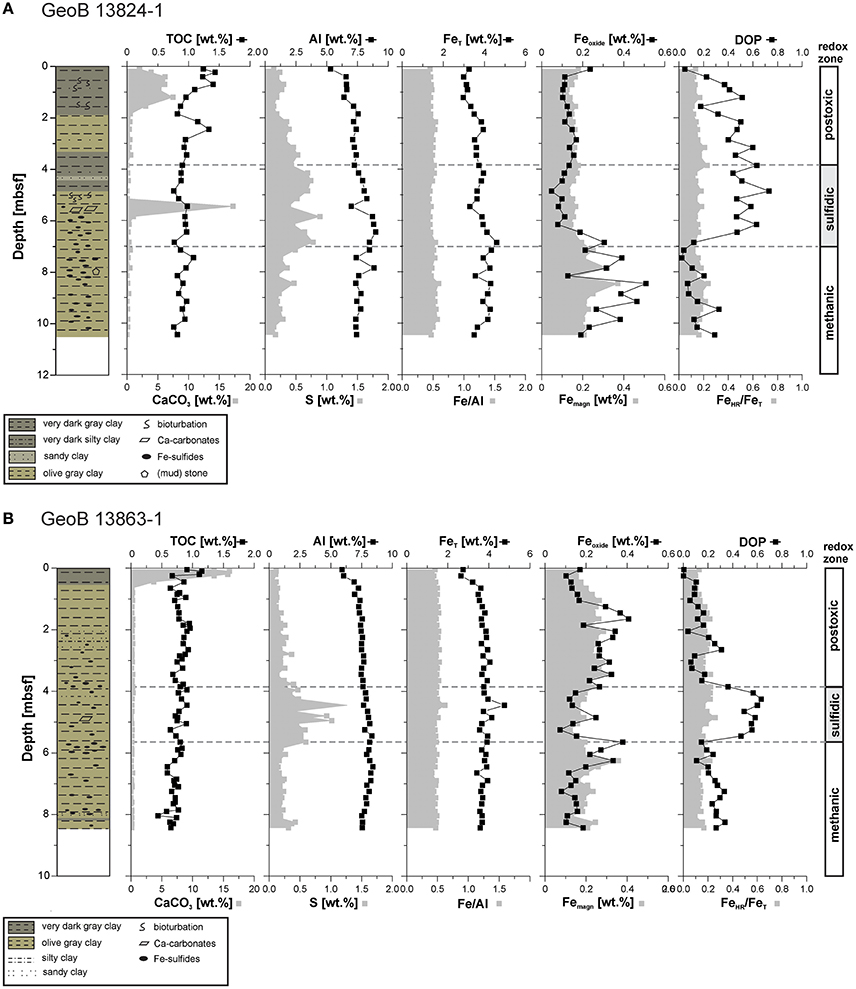
Figure 4. Solid phase concentration profiles of total organic carbon (TOC), calcium carbonate, Al, S, total Fe (FeT), Fe/Al ratio, Fe oxide phases (Feoxide, incl. labile and crystallized Fe-oxide), magnetite (Femagn), degree of pyritization (DOP), and highly recative Fe to total Fe ratio (FeHR/FeT) determined on samples from sites (A) GeoB 13824 and (B) GeoB 13863. TOC and FeT/Al ratios for Site GeoB 13863 after Riedinger et al. (2014). Lithology after Krastel and Wefer (2011).
Total sulfur contents at Site GeoB 13824 are between 0.14 and 0.53 wt.% above and below the sulfidic zone. Within the sulfidic zone, values increase to up to 0.89 wt.%. Similarly, the total sulfur contents at Site GeoB 13863 show an increase within the sulfidic zone (up to 1.26 wt.%), while above and below this zone the values are low (0.09–0.42 wt.%). The iron oxide phases (Feoxide) at Site GeoB 13824, including labile iron (oxyhydr)oxides and crystalline phases (hematite and goethite), are in the range of 0.24 to 0.05 wt.% in the upper 6 m and strongly increase (up to 0.51 wt.%) in the deeper sediments—starting at the lower boundary of the sulfidic zone, with a drop at 8.15 mbsf to a value of 0.13 wt.%. The concentration profile for iron in magnetite (Femagn) resembles that for Feoxide, with a less pronounced decrease in the sulfidic layer (0.13 wt.%) and a smaller increase below the sulfidic zone (up to 3.7 wt.%). The Feoxide and Femagn contents at Site GeoB 13863 scatter between 0.07 and 0.41 wt.%, with increased values above and below the sulfidic zone at 1.2–4 mbsf and 5.6–6.5 mbsf, respectively. While the ratio of highly reactive iron (FeHR) to total iron (FeT) is similar at both study sites, with values between 0.1 and 0.25, the DOP shows a strong difference: DOP at Site GeoB 13824 is elevated between 1 and 6.2 mbsf, with values reaching 0.73 (with one outlier of 0.18 at 1.5 mbsf). DOP at Site GeoB 13863 displays increased ratios up to only 0.63 in the sulfidic zone. Outside these intervals of increased DOP at both sites, the ratio stays low (<0.4) throughout the remaining sediment column.
Stable Sulfur Isotope Composition of Pore Water and Solid Phase Sulfur Compounds
The stable sulfur isotope composition of pore water sulfate (δ34S-SO4) displays a typical enrichment in 34S with depth—with a starting value of δ34S-SO4 (+21.1‰) at the sediment surface at both sites equal to the global seawater values (+21.1‰ e.g., Rees, 1970; Böttcher et al., 2007). The δ34S-SO4 values increase with depth to +64.7‰ at 5.2 mbsf and +54.0‰ at 4.5 mbsf at Sites GeoB 13824 and 13863, respectively (Figure 5). The few data points for the sulfur isotope composition of total free sulfide (δ34S-H2S) show no clear trend and scatter between +16.4 and +43.0‰ at both sites (Figure 5). Some of the scatter in the δ34S-H2S data may be the result of very low concentrations (because of limited sample volume) and associated instrumental uncertainty at these levels. Consequently, interpretations of δ34S-H2S data should be viewed with caution.
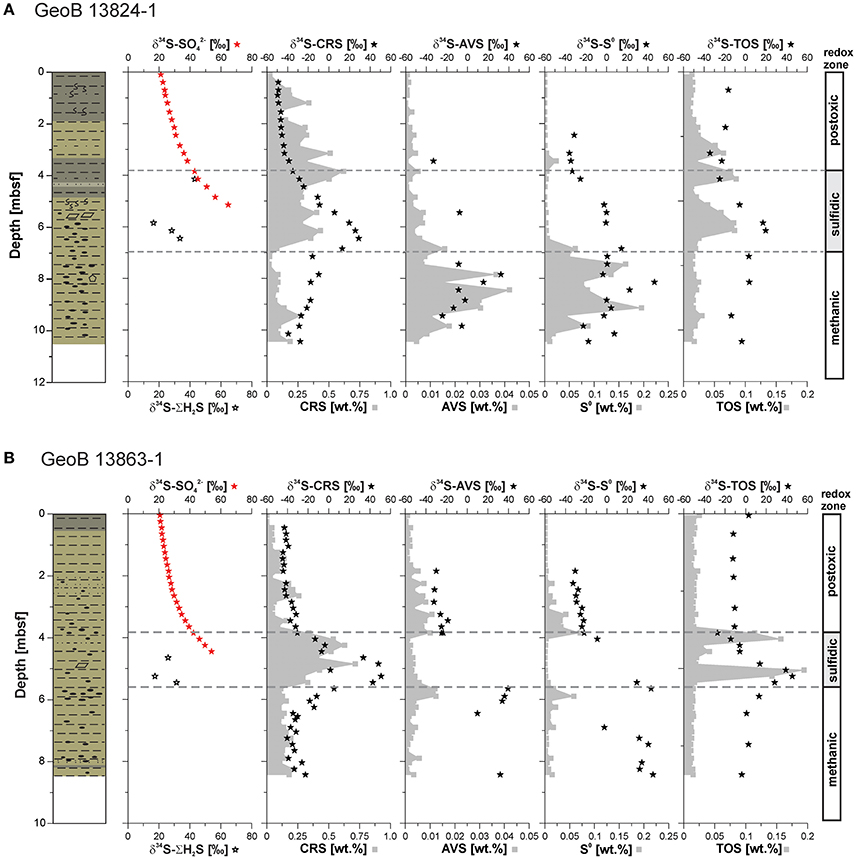
Figure 5. Concentration profiles of chromium reducible sulfur (CRS), acid volatile sulfide (AVS), elemental sulfur (S0), and total organic sulfur (TOS) as well as their sulfur stable isotope profiles in addition to sulfate (δ34S-), and hydrogen sulfide (δ34S-ΣH2S), (sulfur isotope compositions are reported in delta notation relative to Vienna-Cañon Diablo Troilite, VCDT) determined on samples from sites (A) GeoB 13824 and (B) GeoB 13863. , AVS, CRS, and S0 concentrations for Site GeoB 13863 after Riedinger et al. (2014). Lithology after Krastel and Wefer (2011); for lithology legend see Figure 4.
Pyrite contents (CRS) at Site GeoB 13824 are elevated above and within the sulfidic zone, reaching values of up to 0.5 wt.%. In contrast, in the uppermost meter of the core and below the sulfidic zone, pyrite contents remain below 0.2 wt.% with one exception (0.27 wt.%) at 9.45 mbsf (Figure 5A). The stable sulfur isotope composition of pyrite (δ34S-CRS) shows a trend similar to the δ34S-SO4 profile, starting with −49.1‰ at the surface and showing the highest enrichment in 34S (+29.0‰) at 6.5 mbsf—just below the SMT. Below the sulfidic zone, the δ34S-CRS data show a slight progressive depletion in 34S with depth, from −9.8 to −39.3‰ in the lowermost sediment layer. In contrast to pyrite, the contents of iron monosulfide phases (AVS) are low (<0.016 wt.%) in the upper 7.5 meters at Site GeoB 13824 and increase below the sulfidic zone to a maximum of 0.042 wt.% at 8.45 mbsf (Figure 5A). The stable sulfur isotope composition of AVS (δ34S-AVS) at the same site varies between −33.0 and +32.4‰, with the highest values found below the sulfidic zone. The contents of S0 show similar trends to AVS, with highest contents below the sulfidic zone reaching 0.164 wt.%. Although the stable sulfur isotope composition of elemental sulfur (δ34S-S0) scatters throughout the sediment core, the heaviest values are observed below the sulfidic zone, with values of up to +46.5‰. TOS contents are low (<0.03 wt.%) in the upper 2.45 meters at this site and below the sulfidic zone (Figure 5A). Distinct peaks in TOS are observed slightly above of the sulfidic zone, between 2.5 and 3.5 mbsf, at the top (3.5 to 4.5 mbsf) and within the sulfidic zone (5–6.5 mbsf). The stable sulfur isotope composition of TOS (δ34S-TOS) is more negative above the SMT (between −34.2 and −16.4‰) compared to values below of the SMT (−13.6 to +19.8‰).
The pyrite content at Site GeoB 13863 in the sulfidic zone ranges up to 0.72 wt.%, while below and above this zone, the pyrite contents stay below 0.34 wt.% (Figure 5B). Similar to the pyrite concentration profile, the δ34S-CRS data display a positive excursion in the sulfidic zone, reaching +50.9‰. Above and below the sulfidic zone, δ34S-CRS data stay below −11‰, with heavier values below and lighter values above. In contrast to pyrite contents, increased amounts of AVS are limited to the intervals above and below the sulfidic zone, with values of up to 0.012 wt.% (Figure 5B). Outside of these intervals, AVS stays below 0.008 wt.%. Due to the low contents, no δ34S-AVS measurements could be carried out on samples from the sulfidic zone. Below the sulfidic zone, however, δ34S-AVS values are measurable and high (up to +39.5‰), while values are between −32.2 to −18.8‰ above the sulfidic zone. Profiles very similar to AVS are observed for elemental sulfur, with low contents (<0.015 wt.%) except for narrow intervals directly above and below the sulfidic zone where contents increase to up to 0.071 wt.%. δ34S-S0 values are negative above the sulfidic zone (< −20‰), whereas elemental sulfur is enriched in 34S below this zone with values reaching +44.8‰. Two TOS peaks are observed within the sulfidic zone, one at the upper and one at the lower boundary, with contents of up to 0.194 wt.% (Figure 5B). Values above and below the sulfidic zone are constant at ~0.02 wt.%. Above the SMT, δ34S-TOS is essentially constant around −10‰ with two exceptions, one at the surface (+3.0‰) and one at 3.85 mbsf (−27.3‰). Below the SMT, δ34S-TOS values are around 0‰ (from −3.7 to +2.8‰), except for the lower TOS peak (at the lower boundary of the sulfidic zone) where the 34S is enriched (up to +45.3‰).
Discussion
The investigated sediments are dominated by terrigenous inputs of silt and clay with high amounts of total iron, including abundant reactive ferric iron minerals (Figure 4). The observed concentration profiles of the various iron sulfide phases reflect the ongoing alteration of iron oxides via diverse reaction pathways several meters below the seafloor. The high amounts of reactive iron are the cause for the lack or low concentrations of dissolved hydrogen sulfide over most of the sediment column (for a detailed discussion on iron cycling in these sediments see Riedinger et al., 2014). Produced sulfide is immediately scavenged, either via oxidation coupled to the reduction of the iron oxides or by precipitation with dissolved ferrous iron produced by the reduction of ferric iron (Berner and Westrich, 1985; Hartgers et al., 1997; Riedinger et al., 2014). Sulfide can only build up in the pore water when the rates of sulfide formation exceed those of iron-phase alteration, leading to the establishment of a sulfidic zone (Figure 3). Because of the low reactivity of the organic matter (Riedinger et al., 2014), SRR are extremely low even in the presence of abundant sulfate in the upper meters (Figure 3A), with average rates of 5.8 pmol cm−3 d−1—about one order of magnitude lower than rates usually found in sediments from similar water depths (e.g., Fossing et al., 2000; Sawicka et al., 2012). Thus, sulfide production is mainly restricted to the SMT. The presence of free hydrogen sulfide only in the center of the SMT and the sulfide-limited conditions above and below result in a distinct sequence of metastable sulfur phases and pyrite across the sulfidic zone. Ultimately, this geochemical regime leads to the accumulation of metastable sulfur phases, such as authigenic monosulfides (Figures 5, 6) and elemental sulfur, as well as sulfurization of organic matter several meters below the sea floor.
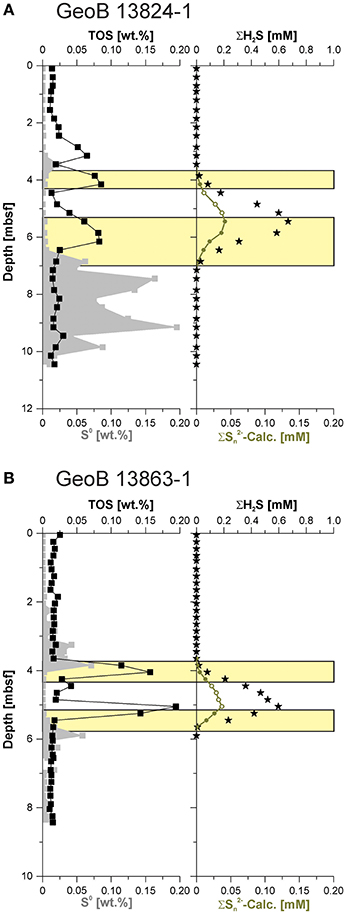
Figure 6. Elemental sulfur (S0), total organic sulfur (TOS), hydrogen sulfide (ΣH2S), and calculated polysulfide (Σ-Calc.) concentration from (A) Site GeoB 13824 and (B) GeoB 13863. The green filled circles indicate the potential availability of Σ-Calc., while open symbols suggest maximal possible Σ-Calc. if elemental sulfur was available. The yellow bars display the intervals of potential polysulfide occurrence.
Pyrite Formation at the Center of the Sulfidic Zone
Elemental sulfur and iron monosulfides form at the upper and lower boundary of the sulfidic zone. In contrast, elevated pyrite concentrations are found at the center of this zone. This relationship suggests that the pyrite is mainly formed in the presence of excess hydrogen sulfide. The relatively high (for an iron-dominated system) DOP ≥ 0.6 for the sediments within the sulfidic zone at both sites indicates almost complete alteration of the iron oxide phases, including magnetic minerals. Canfield and Berner (1987) postulated that magnetic minerals (such as magnetite) can be replaced by pyrite if sulfide concentrations remain high for relatively long periods of time (several hundreds of years), leading to an alteration (or loss) of the magnetic signal. The distribution of iron sulfide phases at Site GeoB 13863 agrees well with the results from numerical reactive-transport modeling for the depositional scenario specific to this location (see Riedinger et al., 2005). Specifically, the modeling predicts elevated pyrite contents that are limited to the sulfidic zone, coinciding with a strong decrease in magnetic susceptibility (Riedinger et al., 2005). Similar observations are also reported for other iron-dominated areas such as the Amazon Fan (Jørgensen and Kasten, 2006), Zambesi Fan (März et al., 2008) or cold-seep systems off southwestern Taiwan (Hsu et al., 2014).
Pyrite Outside of the Sulfidic Zone
Relatively high DOP values are also observed in the postoxic zone at Site GeoB 13824. This relationship could be attributed to sulfide accumulation from organoclastic sulfate reduction related to slightly higher TOC amounts and a longer duration of pore water steady-state conditions at this site compared to Site GeoB 13863. Alternatively, it is possible that at Site GeoB 13824, the SMT was previously located at a shallower location in the sediment. This, however, is not supported by the δ34S-CRS data, which remain low in the postoxic zone. A different possibility is that lower availability of reactive iron phases at Site GeoB 13824 relative to Site GeoB 13863 would also result in higher sulfide accumulation rates and thus more complete pyritization of the reactive phases.
In light of the extremely low measured SRR and the presence of high amounts of reactive iron phases, we are forced to question whether all of the pyrite in the surface sediments formed in situ. The highly 34S-depleted pyrite isotope signature (~ −30 to −50‰ in the upper 4 m; Figure 5) is very similar to other sulfur isotope signatures in shallow marine environments (e.g., Bottrell et al., 2009). We suggest that a major portion of the pyrite found in the surface sediments is derived from sediment reworking of shallow-water sediments, where organoclastic sulfate reduction and associated pyrite formation is more prevalent.
Formation of Iron Monosulfide and Elemental Sulfur
Intermediate sulfur phases are mainly observed below the sulfidic zone at Site GeoB 13824. At Site GeoB 13863, these intermediate sulfur phases also occur at the upper and the lower boundary of the sulfidic zone (Figure 5). Formation of intermediate sulfur phases at the upper and lower boundaries of the sulfidic zone—that is, simultaneous formation in sediments of different ages—reflects the presence of high amounts of available iron (oxyhydr) oxide phases that react with sulfide to form zero-valent sulfur and ferrous iron (e.g., Aplin et al., 1993), while the precipitation of iron monosulfides is driven by the reaction of hydrogen sulfide with ferrous iron. The ferrous iron can result from iron (oxyhydr) oxide reduction coupled to in situ sulfide oxidation, but it can also diffuse to the sulfidic zone—originating from the upper sediment layers and associated organoclastic iron reduction and the lowermost sediments via iron-reduction coupled to AOM as discussed by Riedinger et al. (2014) for Site GeoB 13863. In absence of free sulfide, or at low pH (Kamyshny et al., 2004, 2007, 2008; Kamyshny and Ferdelman, 2010), elemental sulfur is the stable zero-valent sulfur phase. At the investigated sites, the pH is high (7.66–8.14 in the sulfidic zone at Site GeoB 13863). This pH range makes it likely that elemental sulfur is replaced by polysulfides at the fringes of the sulfidic zone (Rickard and Luther, 2007; Kamyshny, 2009), which is corroborated by the near absence of elemental sulfur within the sulfidic zone (Figures 5, 6).
The co-occurrence of elevated elemental sulfur and AVS below the sulfidic zone indicates that in a sulfide-limited system, metastable minerals can persist and be buried into deeper sediment depths (Figure 5). This relationship indicates that intermediate sulfur phases can be more stable in situ than expected based on laboratory and/or modeling results. The availability of reactive iron oxide minerals in these deeper sediments can inhibit the transformation of iron monosulfide and elemental sulfur to pyrite, a reaction that likely only proceeds via an aqueous phase (e.g., polysulfides) (e.g., Rickard and Luther, 2007). The buried elemental sulfur might provide a deep-subsurface sulfur source for microbial communities, such as sulfur disproportionating organisms (e.g., Thamdrup et al., 1993), or for the reduction to sulfide in the methanic zone by archaea (Stetter and Gaag, 1983)—and may even lead to the accumulation of a deep sulfate pool (Riedinger et al., 2010; Treude et al., 2014).
Sulfurization of Organic Matter
The low δ34S-TOS values in the uppermost sediments at both sites (Figure 5) are comparable to those in other continental margin settings (e.g., Bottrell et al., 2009). In the presence of excess hydrogen sulfide, sulfurization of selected labile organic compounds appears to precede the formation of pyrite in marine surface sediments (François, 1987; Ferdelman et al., 1991; Mossmann et al., 1991; Vairavamurthy et al., 1992, 1995; Amrani and Aizenshtat, 2004). The limited availability of such labile organic compounds in the investigated uppermost sediments concurrent with high amounts of reactive iron suggest, however, that a the marginal sulfurized organic compounds, depleted in 34S, were mainly transported to this site, most likely from reworked shallow slope and/or shelf sediments (see also Riedinger et al., 2014).
Polysulfides are known catalysts for the sulfurization of organic compounds (e.g., François, 1987; Werne et al., 2000, 2008). Thus, enhanced sulfurization of organic matter can be expected to occur in geochemical zones where polysulfide formation is prevalent. Elevated TOS contents are not found within the entire sulfidic zone but rather occur in two distinct peaks. At site GeoB 13863, these two peaks align almost perfectly with the transition between the sulfide-free and sulfide-rich portions of the sediment, exactly at the position where one would expect to observe the accumulation of polysulfide—because of the peak elemental sulfur formation and subsequent reaction with sulfide—as shown by the calculated potential polysulfide concentrations (Figure 6B). The δ34S-TOS data indicate different sources of sulfur for the upper and lower TOS peaks, likely reflecting the δ34S-H2S at those depths. There appears to be a third TOS peak in the sulfide-free postoxic zone at site GeoB 13824; however, the same overall picture emerges—an absence of elemental sulfur (and to a lesser degree absence of AVS) coincides with a peak in TOS (Figure 5A). The TOS content of sediments located in the sulfidic zone between the two TOS enrichment peaks fall to almost background levels, likely indicating that sulfurization of particulate organic matter occurs at the sulfidic fringes of the sulfide zone but not in the center of the SMT where hydrogen sulfide displays the highest concentrations. Polysulfides have been shown to be stronger nucleophiles for the sulfurization of organic matter, and nucleophilic substitution has also been recognized to be the dominant sulfurization mechanism (Amrani, 2014, and references therein). If sulfurization of organic matter is indeed tied to polysulfides, our findings imply that the center of the SMT has lower polysulfide concentrations than the border region of the SMT. This could be explained by the absence of polysulfides due to a lack of oxidants.
Implications for the Geochemistry of Dynamic, Iron-Dominated Sedimentary Systems
The lack of dissolved hydrogen sulfide in the pore water of the surface sediments at our sites (Figure 3) indicates that the sulfide produced by organoclastic sulfate reduction reacts immediately with dissolved ferrous iron and thus little or no polysulfide is produced. In the absence of polysulfides, organic substances appear to be protected from sulfurization in a major portion of the sediment column. This observation is in good agreement with similar findings, for example, from the Cariaco Basin (Werne et al., 2003, 2008). Since sulfide accumulation in the pore water is often restricted to a distinct zone, as produced by sulfate reduction coupled to anaerobic methane oxidation, the fringes of the SMT become hotspots for polysulfide formation and consequent organic matter sulfurization (Quijada et al., 2016). With the SMT being a zone of enhanced organic matter sulfurization, there is potential for enhanced preservation of organic molecules in this zone, which implies that biomarkers from organisms that thrive in such zones have an elevated preservation potential (Hebting et al., 2006).
The formation of S0 at the SMT margins due to diffusion of sulfide into ferric-rich sediments, which then reacts with sulfide to form polysulfide, coincides with the occurrence of AVS that is the product of sulfide reacting with counter-diffusing ferrous iron. This raises the interesting question about the competition for polysulfide for AVS to pyrite transformation vs. organic sulfurization and how that plays out in the core of the SMT vs. the margins.
Competition between Sulfurization of Organic Matter and Pyrite Formation
There are two major pyrite formation mechanisms: the polysulfide and the hydrogen sulfide pathways (for a review, see Butler et al., 2004). The polysulfide pathway (Berner, 1970; Rickard, 1975; Sørensen and Jørgensen, 1984; Schoonen and Barnes, 1991; Canfield and Thamdrup, 1994; Rickard and Luther, 2007) consists of the transformation of solid iron monosulfide to an aqueous species, which can react with polysulfide to form pyrite, and a shorter-chained polysulfide species, according to
The hydrogen sulfide pathway follows as similar pattern, with an initial transformation of solid iron monosulfide to an aqueous iron monosulfide, which then reacts with hydrogen sulfide, whereby the hydrogen ions are reduced to dihydrogen gas (e.g., Rickard and Luther, 1997; Butler and Rickard, 2000; Rickard and Luther, 2007), according to
The latter reaction only proceeds with H2S, and not bisulfide (HS−). This means that for the reaction to occur, neutral or slightly acidic conditions are preferred; however, the reaction can also take place under slightly basic conditions (Rickard and Luther, 1997).
At our sites, sulfurization of reworked organic matter at the edges of the SMT appears to out-compete pyrite formation, as is evidenced by the peaks in TOS at the fringes of the sulfidic zone—specifically, organic matter is sulfurized, while AVS is preserved. In the center of the sulfidic zone pyrite predominates and no further generation of sulfurized organic matter occurs. Here, we consider two speculative explanations for this observation. The scenario presumes that polysulfide is also present in the sulfidic zone, but instead of being used for sulfurization of organic matter it is consumed in the formation of pyrite (Equation 1). In this scenario, one would have to speculate that under high sulfide concentrations, the competition between sulfurization of organic matter and pyrite formation is tilted in advantage of the latter.
Alternatively, one could consider the predominance of pyrite within the center of the sulfidic zone an indicator for absence of polysulfide, this would explain why no further generation of sulfurized organic matter occurs. One would then conclude that pyrite formation proceeds via the hydrogen sulfide pathway. It is important to note that in our sediments the pH is up to 8.14 in the center of the sulfidic zone—conditions that are not favorable for the hydrogen sulfide reaction (Equation 2) to occur, because most of the sulfide prevails as bisulfide. To solve this contradiction, one could speculate that the precipitation of authigenic calcite or dolomite from bicarbonate driven by the AOM coupled to sulfate reduction at the SMT (e.g., Kelts and McKenzie, 1982; Malone et al., 2002; Moore et al., 2004; Meister et al., 2007; Nöthen and Kasten, 2011; Wehrmann et al., 2011; Riedinger et al., 2014) liberates hydrogen ions, which pushes sulfide speciation slightly toward H2S. This sulfide then becomes available for pyrite formation (Equation 2). Thus, the precipitation of authigenic carbonate minerals, as observed at the SMT at both sites (Figure 4), could facilitate the formation of pyrite at the SMT via the hydrogen sulfide reaction (Equation 2). The combination of carbonate precipitation with pyrite formation balances hydrogen ion production and consumption, with no net change in pH, which remains high at a value around 8.
Interpretation of the Geochemical Evolution of Dynamic Sediments Based on Inventory and Isotope Composition of Sulfur Species
Sulfur and iron transformations in the sediments of the Argentine Basin occur in a dynamic sedimentary system. Rapid overall sedimentation is sometimes interrupted by phases of sediment winnowing caused by ocean currents. Episodic mass wasting processes can lead to instantaneous sediment accumulation (e.g., Hensen et al., 2003; Riedinger et al., 2005; Henkel et al., 2011, 2012). These changes cause non-steady state conditions in the subsurface sediment/pore-water system (e.g., Kasten et al., 2003). A key question is if the analysis of the inventory of iron and sulfur species and the stable sulfur isotope composition of the sulfur constituents can fingerprint similar processes in other settings. To illustrate the potential of this approach, we present a three-stage geochemical scenario for the two investigated sites (Figure 7). The first stage is dominated by high sedimentation, as has been discussed previously by Hensen et al. (2003) and Riedinger et al. (2005) in the context of numerical modeling approaches. The high sedimentation rates could be due to one or multiple depositional events. During this time, reworked sediment from upslope settings accumulate, carrying TOS and pyrite with sulfur isotope signatures that correspond to these shallower settings (Figure 7A). These phases are intermingled with refractory organic matter and ample amounts of iron (oxyhydr)oxides. A large portion of the rapidly buried organic matter reaches the methanic zone, resulting in an increase in the methane flux from below. During this phase, the sulfate concentration profile displays a “concave up” shape (Figure 7A; Hensen et al., 2003; Kasten et al., 2003).
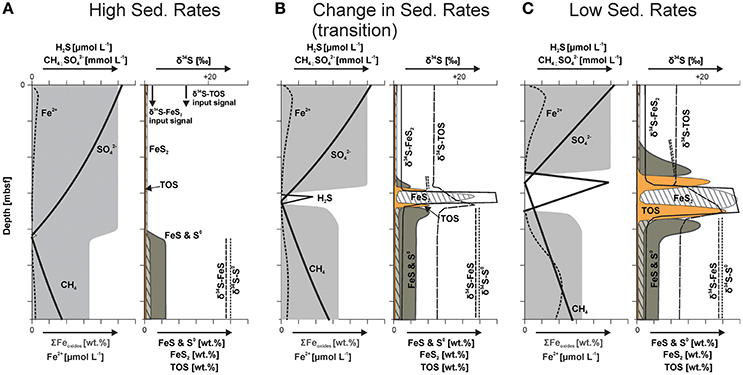
Figure 7. Schematic model displaying iron oxide alteration and sulfur phase distribution with its isotopic signature related to changes in depositional conditions (modified after Riedinger, 2005). (A) During high sedimentation (one or multiple depositional events) the SMT moves rapidly upward, a non-steady state condition that manifests itself as concave-up sulfate profile (Hensen et al., 2003). Under these conditions, sulfide (H2S) produced by sulfate () reduction coupled to anaerobic oxidation of methane (CH4) at the SMT is consumed faster than it can build up in the sediment by reacting with highly reactive iron oxide phases (ΣFeoxides; area shaded in light gray) and dissolved ferrous iron (Fe2+). This results in the formation of intermediate sulfur phases (elemental sulfur, S0, and monosulfide phases, FeS) with distinct stable sulfur isotope signatures (δ34S). Only minor amounts of pyrite (FeS2) are formed, the observed pyrite and TOS were mainly derived by sedimentation. (B) Once sedimentation rates decrease, H2S builds up at the SMT, which leads to the formation of pyrite in the sulfidic zone, as well as to the sulfurization of organic compounds (TOS) at the upper and lower rim of the sulfidic zone. Adjacent to the sulfidic zone, iron monosulfides and elemental sulfur are formed. All of these phases show stable sulfur isotope signatures that reflect the isotope composition of sulfide at the respective position in the sediment. These signatures are distinctively different from the isotope signatures of the same sulfur constituents that formed elsewhere in the sediment column, or were derived by sedimentation. (C) At low sedimentation rates, hydrogen sulfide can accumulate at the SMT and spread outwards. The result is a broadening of the sulfidic zone and consequently broader CRS and TOS peaks.
As the SMT migrates upward, the production of sulfide by sulfate reduction coupled to AOM sweeps across sediment that contains abundant reactive iron phases. The sulfide reacts immediately with these iron phases (ΣFeoxides; area shaded in light gray in Figure 7)—producing zero-valent sulfur phases (i.e., elemental sulfur) and ferrous iron, which in turn reacts with sulfide to form iron monosulfide. These newly formed sulfur phases carry the sulfur isotope signature of sulfide formed at the upward migrating SMT. Due to the refractory nature of the organic matter, organoclastic sulfate reduction is of minor importance, and only minor amounts of pyrite are formed, which may marginally alter (slight enrichment in 34S) the sulfur isotope signature of the reworked pyrite delivered by sedimentation.
The second stage in our geochemical scenario is reached when sedimentation rates (strongly) decrease and the sulfate flux is primarily controlled by diffusion again, which causes a slowing down of the upward migration of the SMT. Under these conditions, the production of sulfide at the SMT is no longer out-competed by sulfide consumption through reaction with reactive iron phases. As a consequence, sulfide starts to build up in the pore water, and a sulfidic zone is established, which broadens over time. At the center of the sulfidic zone, pyrite is formed via the reaction of iron monosulfide with H2S, probably in conjunction with calcite precipitation. At the fringes of the sulfidic zone, iron monosulfides, and elemental sulfur are formed through the interaction between dissolved Fe2+ and reactive iron oxide species and sulfide diffusing away from the center of the sulfidic zone. At these reaction fronts, found at the upper and lower boundaries of the sulfidic zone, additional hydrogen sulfide reacts with the produced elemental sulfur—resulting in the formation of dissolved polysulfides, which in turn sulfurize the organic matter. The sulfurization results in two TOS enrichment fronts, one near the upper and one at the lower boundary of the sulfidic zone, with less and more 34S-enriched signatures, respectively (Figures 7B,C). This process also explains why TOS and elemental sulfur phases do not overlap.
The third stage in the scenario is reached when the sedimentation rates become very low (returning to mainly hemipelagic sedimentation). The system then adjusts to a quasi-steady state in terms of the pore water, and the upward moving SMT is now “fixed” at a certain/relative depth (moving upward slowly at the same rate as sedimentation). Hydrogen sulfide accumulating at the SMT then spreads further out, resulting in a broadening of the sulfidic zone and broader CRS and TOS peaks (Figure 7C). This is the stage that is captured in the investigated sediments, with steady state pore water profiles as displayed by sulfate, while the solid phases are integrated and mostly capture the preceding non-steady state conditions. Numerical modeling results for sediments from nearby locations suggest a timespan of several hundred to a few thousand years for the pore water sulfate concentration profile to regain the observed steady state conditions (Hensen et al., 2003).
This three-stage scenario explains most of the observed patterns at Sites GeoB 13824 and 13863. Some refinements are required, however, to address phenomena specific to individual sites. For example, there is a third TOS peak in the postoxic zone of Site GeoB 13824, indicating that substantial polysulfide formation also took place at a shallower depth in the past. This relationship could be explained by an excursion of the SMT into shallower sediments, followed by a deepening of the SMT due to higher downward flux of sulfate into the SMT or a lower upward flux of methane. Such fluctuations can take place, for example, when sediment is removed by winnowing or if less sulfate is consumed by organocastic sulfate reduction during low sedimentation due to an increase in the refractory nature of organic matter. If the system experienced a constant low sedimentation rate over a long period of time, some of the observed isotope excursions could be overprinted. For example, the isotope signatures associated with the upper fringe of the SMT signal would be overprinted by the upward movement of the redox zones with a lower SMT fringe signature, which may add CRS and TOS with distinctly different isotope signatures to the bulk signal. In a different scenario, where high sedimentation rates follow a short period of low sedimentation, the CRS and TOS associated with the SMT would be buried, and the observed isotope excursions would be altered slowly over time if at all. The different isotope signatures, and the potential stacking of such signatures during shifts in methane or sulfate fluxes, opens the possibility of using high-resolution sulfur isotope signatures from organic matter to reconstruct past changes in the location of the SMT (Wehrmann et al., 2013).
Summary and Conclusions
Our study provides a refined view of the zonation of biogeochemical processes that occur within and in the vicinity of the sulfidic zone around the SMT in sedimentary systems dominated by reactive iron. Pyrite is formed in the center of the sulfidic zone. At the upper and lower fringe of the sulfidic zone, the interval where pyrite formation prevails is bounded by a zone in which sulfurization of organic matter dominates. The polysulfides are supplied by the reaction of free sulfide with zero-valent sulfur, which in turn is supplied by the oxidation of sulfide with iron (oxyhydr) oxides. Considering the refractory nature of the organic matter, it is interesting that sulfurization outcompetes pyrite formation. Above and below the sulfidic zone, elemental sulfur, and iron monosulfide phases build up, a process that is fueled by sulfide oxidation, which yields S0 and ferrous iron and induces the precipitation of iron monosulfides through reaction with ferrous iron. Because of the distinct locations of these processes with respect to the center of the SMT, the produced sulfur phases record sulfur isotope signatures for sulfide that are representative for the isotope trends observed in the SMT—that is, a strong enrichment in 34S from the top to the bottom of the SMT (e.g., Rudnicki et al., 2001; Brunner et al., 2016; Turchyn et al., 2016). These distinct isotope signatures allow the reconstruction of the complex history of biogeochemical sulfur cycling in dynamic sediments. The power of this approach has previously been demonstrated for pyrite enrichment-fronts that indicate the location of past (paleo or fossil) SMTs (Borowski et al., 2013; Lin et al., 2016) but can now be expanded and refined by the inclusion of the inventory and stable sulfur isotope composition of other sulfur constituents, such as the sulfur isotope composition of the organic phase (δ34S-TOS). We believe these analyses can be greatly augmented by employing high-resolution SIMS techniques to identify the former presence of upper and lower fringes of SMTs, particularly in cases where such signatures were stacked on top of each other during fluctuations in the depth of the SMT. Such zones may have two isotopically distinct populations of pyrite and TOS, corresponding to the upper and lower SMT fringe. Techniques such as sulfur isotope analyses using SIMS have the potential to reveal such variations and clustering of isotope signatures that remain undetected in a bulk sample (Xiao et al., 2010; Bontognali et al., 2012; Farquhar et al., 2013; Fischer et al., 2014).
Our findings not only facilitate the reconstruction of the geochemical history of dynamic sediments, they also shed new light on several interlinked themes in the study of iron-sulfur cycling in marine sediments in iron dominated systems: (i) the occurrence of elemental sulfur and iron monosulfides at the upper and lower boundary of the sulfidic zone indicates that in a sulfide-limited system these metastable minerals are not (immediately) converted into pyrite and thus can be buried into deeper sediment depths where they can fuel deep biosphere processes (e.g., sulfate reducers via S0 disproportionation); (ii) the sulfurization of organic compounds predominantly occurs when polysulfides become available in the margins of the sulfidic zone, leading to a preferential fixation and long-term preservation of biomarkers that occur in this zone. Our data provide evidence that organic matter sulfurization can occur at any sediment depth as long as hydrogen sulfide and oxidants are co-mingled to form dissolved polysulfides; (iii) pyrite is formed in the core of the sulfidic zone, likely via the reaction of iron monosulfide with H2S, a process that produces H2 gas and that may require or induce precipitation of authigenic carbonate minerals and (iv) considerable amounts of reactive iron persist in sediment over long time due to rapid relocations of the SMT (thus short sulfide exposure time), which shifts hot spots of sulfide generation to specific intervals in the sediment column, where the reactive iron is more rapidly depleted due to the presence of free sulfide compared to the intervals below and above of this zone.
A further conclusion of our study is that the sulfur isotope signature of iron sulfide and organic sulfur phases in marine sedimentary systems is influenced by depositional as well as in situ geochemical processes—that is, there is a link between deposition in the shallow subsurface sediments and long-term signals being buried and preserved in the sedimentary record. Thus, dynamic depositional systems are characterized by non-traditional redox zonation, where inverse redox zonation sequences or overlap of specific zones can occur, resulting in multiple intervals of specific redox processes in different sediment strata. In other words, comparable geochemical reactions proceed in sediments of different sediment depth and ages.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the captain, the crew, and the scientists of the R/V Meteor M78/3 expedition carried out within the framework of the Research Centre/Cluster of Excellence “The Ocean in the Earth System” (MARUM—Center for Marine Environmental Sciences at the University of Bremen) funded by the Deutsche Forschungsgemeinschaft (DFG). For sampling and analysis assistance we thank M. Strasser, J. Sawicka, and J. Tomasini; and for laboratory assistance B. Patel. For insightful comments and inspiration we are grateful to R. Raiswell, B. Dickenson, S. Harris, and B. K. Frieda. We greatly appreciate the constructive comments from our two reviewers. Funding was provided by the U.S. National Science Foundation (NSF OCE-1234778) to NR and TWL, and an appointment to the National Aeronautics and Space Administration (NASA) Postdoctoral Program to NR. This is an Oklahoma State University—Boone Pickens School of Geology contribution #2016-44.
References
Adam, P., Phillippe, E., and Albrecht, P. (1998). Photochemical sulfurization of sedimentary organic matter: a widespread process occurring at early diagenesis in natural environments? Geochim. Cosmochim. Acta 62, 265–271. doi: 10.1016/s0016-7037(97)00332-3
Aizenshtat, Z., Stoler, A., Cohen, Y., and Nielsen, H. (1983). “The geochemical sulphur enrichment of recent organic matter by polysulfides in the Solar-Lake,” in Advances in Organic Geochemistry 1981, eds M. Bjorøy, P. Albrecht, C. Cornford, K. de Groot, G. Eglington, E. Galimov, D. Leythaeuser, R. Pelet, J. Rullkotter, and G. Speers (Chichester: Wiley), 279–288.
Aller, R. C. (2014). “Sedimentary diagenesis, depositional environments, and benthic fluxes,” in Treatise on Geochemistry, 2nd Edn., eds H. D. Holland and K. K. Turekian (Oxford: Elsevier), 293–334.
Aller, R. C., Madrid, V., Chistoserdov, A., Aller, J. Y., and Heilbrun, C. (2010). Unsteady diagenetic processes and sulfur biogeochemistry in tropical deltaic muds: implications for oceanic isotope cycles and the sedimentary record. Geochimica et Cosmochimica Acta 74, 4671–4692. doi: 10.1016/j.gca.2010.05.008
Amrani, A. (2014). Organosulfur compounds: molecular and isotopic evolution from biota to oil and gas. Annu. Rev. Earth Planet. Sci. 42, 733–768. doi: 10.1146/annurev-earth-050212-124126
Amrani, A., and Aizenshtat, Z. (2004). Mechanisms of sulfur introduction chemically controlled: δ34S imprint. Organ. Geochem. 35, 1319–1336. doi: 10.1016/j.orggeochem.2004.06.019
Anderson, T. F., and Pratt, L. M. (1995). “Isotope evidence for the origin of organic sulfur and elemental sulfur in marine sediments,” in Geochemical Transformations of Sedimentary Sulfur, eds. M. A. Vairavamurthy and M. A. A. Schoonen. ACS Symposium Series 612 (Washington, DC), 378–396.
Antoine, D., Andre, J.-M., and Morel, A. (1996). Oceanic primary production: 2. Estimation at global scale from satellite (coastal zone color scanner) chlorophyll. Glob. Biogeochem. Cycles 10, 57–69. doi: 10.1029/95GB02832
Aplin, A. C., Macquaker, J. H., Lovley, D. R., Lyons, S. T., and Aargaard, P. (1993). C-S-Fe geochemistry of some modern and ancient anoxic marine muds and mudstones. Philos. Trans. R. Soc. Lond. Ser. A 344, 89–100. doi: 10.1098/rsta.1993.0077
Behrenfeld, M. J., and Falkowski, P. G. (1997). Photosynthetic rates derived from satellite-based chlorophyll concentration. Limnol. Oceanogr. 42, 1–20. doi: 10.4319/lo.1997.42.1.0001
Berner, R. (1989). Biogeochemical cycles of carbon and sulfur and their effect on atmospheric oxygen over Phanerozoic time. Palaeogeogr. Palaeoclimatol. Palaeoecol. 75, 97–122. doi: 10.1016/0031-0182(89)90186-7
Berner, R. A. (1981). A new geochemical classification of sedimentary environments. J. Sed. Petrol. 51, 359–365.
Berner, R. A. (1982). Burial of organic carbon and pyrite sulfur in the modern ocean: its geochemical and environmental significance. Am. J. Sci. 282, 451–473. doi: 10.2475/ajs.282.4.451
Berner, R. A. (1984). Sedimentary pyrite formation: an update. Geochim. Cosmochim. Acta 48, 605–615. doi: 10.1016/0016-7037(84)90089-9
Berner, R. A., and Raiswell, R. (1983). Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time - a new theory. Geochim. Cosmochim. Acta 47, 855–862. doi: 10.1016/0016-7037(83)90151-5
Berner, R. A., and Westrich, J. T. (1985). Bioturbation and the early diagenesis of carbon and sulfur. Am. J. Sci. 285, 193–206. doi: 10.2475/ajs.285.3.193
Biscaye, P. E., and Dasch, E. J. (1971). The rubidium, strontium, strontium-isotope system in deep-sea sediments: Argentine Basin. J. Geophys. Res. 76, 5087–5096. doi: 10.1029/jc076i021p05087
Blum, P. (1997). Physical Properties Handbook: A Guide to the Shipboard Measurement of Physical Properties of Deep-Sea Cores. College Station, TX: Ocean Drilling Program; Ocean Drilling Project Technical Note.
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi: 10.1038/35036572
Bontognali, T. R. R., Sessions, A. L., Allwood, A. C., Fischer, W. W., Grotzinger, J. P., Summons, R. E., et al. (2012). Sulfur isotopes of organic matter preserved in 3.45-billion-year-old stromatolites reveal microbial metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 15146–15151. doi: 10.1073/pnas.1207491109
Borowski, W. S., Rodriguez, M. N., Paull, C. K., and Ussler, I. I. I. W. (2013). Are 34S-enriched authigenic sulfide minerals a proxy for elevated methane flux and gas hydrates in the geologic record? Mar. Petrol. Geol. 43, 381–395. doi: 10.1016/j.marpetgeo.2012.12.009
Böttcher, M. E., Brumsack, H.-J., and Dürselen, C.-D. (2007). The isotopic composition of modern seawater sulfate: I. Coastal waters with special regard to the North Sea. J. Mar. Syst. 67, 73–82. doi: 10.1016/j.jmarsys.2006.09.006
Böttcher, M. E., Thamdrup, B., Gehre, M., and Theune, A. (2005). 34S/32S and 18O/16O fractionation during sulfur disproportionation by Desulfobulbus propionicus. Geomicrobiol. J. 22, 219–226. doi: 10.1080/01490450590947751
Bottrell, S. H., and Raiswell, R. (2000). “Sulphur isotopes and microbial sulphur cycling in sediments,” in Microbial Sediments, eds. R. E. Riding and S. M. Awramik (Berlin, Springer-Verlag), 96–104.
Bottrell, S. H., Mortimer, R. J. G., Davies, I. N., Harvey, S. M., and Krom, M. D. (2009). Sulphur cycling in organic-rich marine sediments from a Scottish fjord. Sedimentology 56, 1159–1173. doi: 10.1111/j.1365-3091.2008.01024.x
Bowles, M. W., Mogollón, J. M., Kasten, S., Zabel, M., and Hinrichs, K. U. (2014). Global rates of marine sulfate reduction and implications for sub-sea-floor metabolic activities. Science 344, 889–891, doi: 10.1126/science.1249213
Brüchert, V., and Pratt, L. M. (1996). Contemporaneous early diagenetic formation of organic and inorganic sulfur in estuarine sediments from St. Andrew Bay, Florida, USA. Geochim. Cosmochim. Acta 60, 2325–2332. doi: 10.1016/0016-7037(96)00087-7
Brunner, B., and Bernasconi, S. M. (2005). A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria. Geochim. Cosmochim. Acta 69, 4759–4771. doi: 10.1016/j.gca.2005.04.015
Brunner, B., Arnold, G. L., Røy, H., Müller, I. A., and Jørgensen, B. B. (2016). Off limits: sulfate below the sulfate-methane transition. Front. Earth Sci. 4:75. doi: 10.3389/feart.2016.00075
Butler, I. B., and Rickard, D. (2000). Framboidal pyrite formation via the oxidation of iron (II) monosulfides by hydrogen sulfide. Geochim. Cosmochim. Acta 64, 2665–2672. doi: 10.1016/S0016-7037(00)00387-2
Butler, I. B., Böttcher, M. E., Rickard, D., and Oldroyd, A. (2004). Sulfur isotope partitioning during experimental formation of pyrite via the polysulfide and hydrogen sulfide pathways: implications for the interpretation of sedimentary and hydrothermal pyrite isotope records. Earth Planet. Sci. Lett. 228, 495–509. doi: 10.1016/j.epsl.2004.10.005
Canfield, D. E. (1989). Reactive iron in marine sediments. Geochim. Cosmochim. Acta 53, 619–632. doi: 10.1016/0016-7037(89)90005-7
Canfield, D. E., and Berner, R. A. (1987). Dissolution and pyritization of magnetite in anoxic marine-sediments. Geochim. Cosmochim. Acta 51, 645–659. doi: 10.1016/0016-7037(87)90076-7
Canfield, D. E., and Thamdrup, B. (1994). The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulphur. Science 266, 1973–1975. doi: 10.1126/science.11540246
Canfield, D. E., Boudreau, B. P., Mucci, A., and Gundersen, J. K. (1998). The early diagenetic formation of organic sulfur in the sediments of Mangrove Lake, Bermuda. Geochim. Cosmochim. Acta 62, 767–781. doi: 10.1016/S0016-7037(98)00032-5
Chiessi, C. M., Ulrich, S., Mulitza, S., Pätzold, J., and Wefer, G. (2007). Signature of the Brazil-Malvinas Confluence (Argentine Basin) in the isotopic composition of planktonic foraminifera from surface sediments. Mar. Micropaleontol. 64, 52–66. doi: 10.1016/j.marmicro.2007.02.002
Claypool, G. E., and Kaplan, I. R. (1974). “The origin and distribution of methane in marine sediments,” in Natural Gases in Marine Sediments, ed I. R. Kaplan (New York, NY: Plenum), 99–139.
Cline, J. D. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Cornwell, J. C., and Morse, J. W. (1987). The characterization of iron sulfide minerals in anoxic marine sediments. Mar. Chem. 22, 193–206. doi: 10.1016/0304-4203(87)90008-9
Dickens, G. R., Kölling, M., Smith, D. C., and Schnieders, L. (2007). Rhizon sampling of pore waters on scientific drilling expeditions: an example from the IODP expedition 302, Arctic Coring Expedition (ACEX). Sci. Drill. 4, 22–25. doi: 10.5194/sd-4-22-2007
Ewing, M., and Leonardi, A. G. (1971). Sediment transport and distribution in the Argentine Basin. 5. Sedimentary structure of the Argentine margin, basin, and related provinces. Phys. Chem. Earth 8, 125–251. doi: 10.1016/0079-1946(71)90017-6
Ewing, M., Eittreim, S. L., Ewing, J. I., and Le Pichon, X. (1971). Sediment transport and distribution in the Argentine Basin. 3. Nepheloid layer and processes of sedimentation. Phys. Chem. Earth 8, 51–77. doi: 10.1016/0079-1946(71)90015-2
Farquhar, J., Cliff, J., Zerkle, A. L., Kamyshny, A., Poulton, S. W., Claire, M., et al. (2013). Pathways for Neoarchean pyrite formation constrained by mass-independent sulfur isotopes. Proc. Natl. Acad. Sci. U.S.A. 110, 17638–17643. doi: 10.1073/pnas.1218851110
Ferdelman, T. G. (1988). The Distribution of Sulphur, Iron, Manganese, Copper and Uranium in a Salt Marsh Sediment Core as Determined by a Sequential Extraction Method. MS thesis, University of Delaware.
Ferdelman, T. G., Church, T. M., and Luther, G. W. (1991). Sulfur enrichment of humic substances in a Delaware salt marsh sediment core. Geochim. Cosmochim. Acta 55, 979–988. doi: 10.1016/0016-7037(91)90156-Y
Filley, T., Freeman, K., Wilkin, R., and Hatcher, P. (2002). Biogeochemical controls on reaction of sedimentary organic matter and aqueous sulfides in Holocene sediments of Mud Lake, Florida. Geochim. Cosmochim. Acta 66, 937–954. doi: 10.1016/S0016-7037(01)00829-8
Fischer, W. W., Fike, D. A., Johnson, J. E., Raub, T. D., Guan, Y., Kirschvink, J. L., et al. (2014). SQUID–SIMS is a useful approach to uncover primary signals in the Archean sulfur cycle. Proc. Natl. Acad. Sci. U.S.A. 111, 5468–5473. doi: 10.1073/pnas.1322577111
Fossing, H., and Jørgensen, B. B. (1989). Measurement of bacterial sulfate reduction in sediments. Evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222. doi: 10.1007/BF00002889
Fossing, H., Ferdelman, T. G., and Berg, P. (2000). Sulfate reduction and methane oxidation in continental margin sediments influenced by irrigation (South–East Atlantic off Namibia). Geochim. Cosmochim. Acta 64, 897–910. doi: 10.1016/S0016-7037(99)00349-X
François, R. (1987). A study of sulphur enrichment in the humic fraction of marine sediments during early diagenesis. Geochim. Cosmochim. Acta 51, 17–27. doi: 10.1016/0016-7037(87)90003-2
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090. doi: 10.1016/0016-7037(79)90095-4
Fu, Y., Von Dobeneck, T., Franke, C., Heslop, D., and Kasten, S. (2008). Rock magnetic identification and geochemical process models of greigite formation in Quaternary marine sediments from the Gulf of Mexico (IODP Hole U1319A). Earth Planet. Sci. Lett. 275, 233–245. doi: 10.1016/j.epsl.2008.07.034
Goldhaber, M. B., Aller, R. C., Cochran, J. K., Rosenfeld, J. K., Martens, C. S., and Berner, R. A. (1977). Sulfate reduction, diffusion, and bioturbation in Long Island Sound sediments: report of the FOAM group. Am. J. Sci. 277, 193–237. doi: 10.2475/ajs.277.3.193
Goldhaber, M. B., and Kaplan, I. R. (1974). “The sulfur cycle,” in The Sea, Vol. 5, Marine Chemistry, ed E. D. Goldberg (New York, NY; London; Sydney, NSW; Toronto, ON: John Wiley & Sons, Inc.), 569–655.
Goldhaber, M. B., and Kaplan, I. R. (1975). Controls and consequences of sulfate reduction rates in recent marine sediments. Soil Sci. 119, 42–55. doi: 10.1097/00010694-197501000-00008
Goldhaber, M. B., and Kaplan, I. R. (1980). Mechanisms of sulfur incorporation and isotope fractionation during early diagenesis in sediments of the Gulf of California. Mar. Chem. 9, 95–143. doi: 10.1016/0304-4203(80)90063-8
Gruetzner, J., Uenzelmann-Neben, G., and Franke, D. (2012). Variations in sediment transport at the central Argentine continental margin during the Cenozoic. Geochem. Geophys. Geosyst. 13:Q10003. doi: 10.1029/2012gc004266
Haese, R. R., Schramm, J., Rutgers van der Loeff, M. M., and Schulz, H. D. (2000). A comparative study of iron and manganese diagenesis in continental slope and deep sea basin sediments off Uruguay (SW Atlantic). Int. J. Earth Sci. 88, 619–629. doi: 10.1007/s005310050292
Harrison, A. G., and Thode, H. G. (1958). Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Trans. Faraday Soc. 54, 84–92.
Hartgers, W. A., Lopez, J. F., Sinninghe Damste, J. S., Reiss, C., Maxwell, J. R., and Grimalt, J. O. (1997). Sulfur-binding in recent environments. II. Speciation of sulfur and iron and implications for the occurrence of organo-sulfur compounds. Geochim. Cosmochim. Acta 61, 4769–4788. doi: 10.1016/s0016-7037(97)00279-2
Hebting, Y., Schaeffer, P., Behrens, A., Adam, P., Schmitt, G., Schneckenburger, P., et al. (2006). Biomarker evidence for a major preservation pathway of sedimentary organic carbon. Science 312, 1627–1631. doi: 10.1126/science.1126372
Hedges, J., and Keil, R. G. (1995). Sedimentary organic matter preservation: an assessment and speculative synthesis. Mar. Chem. 49, 81–115.
Henkel, S., Kasten, S., Poulton, S. W., and Staubwasser, M. (2016). Determination of the stable iron isotopic composition of sequentially leached iron phases in marine sediments. Chem. Geol. 421, 93–102. doi: 10.1016/j.chemgeo.2015.12.003
Henkel, S., Schwenk, T., Hanebuth, T. J. J., Strasser, M., Riedinger, N., Formolo, M., et al. (2012). “Pore water geochemistry as a tool for identifying and dating recent mass-transport deposits,” in Submarine Mass Movements and Their Consequences: 5th International Symposium (Advances in Natural and Technological Hazards Research, 31), eds Y. Yamada, K. Kawamura, K. Ikehara, Y. Ogawa, R. Urgeles, D. Mosher, J. Chaytor, and M. Strasser (Springer Netherlands), 87–97.
Henkel, S., Strasser, M., Schwenk, T., Hanebuth, T. J. J., Hüsener, J., Arnold, G. L., et al. (2011). An interdisciplinary investigation of a recent submarine mass transport deposit at the continental margin off Uruguay. Geochem. Geophys. Geosyst. 12:Q08009. doi: 10.1029/2011GC003669
Hensen, C., Zabel, M., Pfeifer, K., Schwenk, T., Kasten, S., Riedinger, N., et al. (2003). Control of sulfate pore-water profiles by sedimentary events and the significance of anaerobic oxidation of methane for burial of sulfur in marine sediments. Geochim. Cosmochim. Acta 67, 2631–2647. doi: 10.1016/s0016-7037(03)00199-6
Hernández-Molina, F. J., Paterlini, M., Violante, R., Marshall, P., de Isasi, M., Somoza, L., et al. (2009). Contourite depositional system on the Argentine Slope: an exceptional record of the influence of Antarctic water masses. Geology 37, 507–510. doi: 10.1130/G25578A.1
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S. (1994). Field and laboratory studies of methane oxidation in an anoxic marine sediment: evidence for a methanogen-sulfate reducer consortium. Global Biogeochem. Cycles 8, 451–463. doi: 10.1029/94GB01800
Howarth, R. W. (1979). Pyrite: its rapid formation in a Saltmarsh and its importance to ecosystem metabolism. Science 203, 49–51. doi: 10.1126/science.203.4375.49
Howarth, R. W., and Jørgensen, B. B. (1984). Formation of 35S-labellede lemental sulfur and pyrite in coastal marine sediments (Limfjorden and Kysing Fjord, Denmark) during short term 35SO42–reduction measurements. Geochim. Cosmochim. Acta 48, 1807–1818. doi: 10.1016/0016-7037(84)90034-6
Hsu, T., Jiang, W., and Wang, Y. (2014). Authigenesis of vivianite as influenced by methane-induced sulfidization in cold-seep sediments off southwestern Taiwan. J. Asian Earth Sci. 89, 88–97. doi: 10.1016/j.jseaes.2014.03.027
Iriondo, M. H. (1984). “The quaternary of Northeastern Argentina,” in Quaternary of South America and Antarctic Peninsula, Vol 2, ed J. Rabassa (A. A. Balkema) (Berlin; Heidelberg; New York, NY: Springer), 51–78.
Jones, G. G., and Starkey, R. L. (1957). Fractionation of stable isotopes of sulfur by microorganisms and their role in deposition of native sulfur. Appl. Microbiol. 5, 111–118.
Jørgensen, B. B. (1977). The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark). Limnol. Oceanogr. 22, 814–832. doi: 10.4319/lo.1977.22.5.0814
Jørgensen, B. B. (1978). A comparison of methods for the quantification of bacterial sulfate reduction in coastal marine sediments. I. Measurement with radiotracer technique. Geomicrobiol. J. 1, 11–27. doi: 10.1080/01490457809377721
Jørgensen, B. B. (1979). A theoretical model of the sulfur isotope distribution in marine sediments. Geochim. Cosmochim. Acta 43, 363–374. doi: 10.1016/0016-7037(79)90201-1
Jørgensen, B. B. (1982). Mineralization of organic matter in the sea bed–the role of sulphate reduction. Nature 296, 643–645. doi: 10.1038/296643a0
Jørgensen, B. B. (1990). A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249, 152–154. doi: 10.1126/science.249.4965.152
Jørgensen, B. B., and Kasten, S. (2006). “Sulfur cycling and methane oxidation,” in Marine Geochemistry, eds H. D. Schulz and M. Zabel (Heidelberg: Springer-Verlag), 574.
Jørgensen, B. B., and Nelson, D. C. (2004). Sulfide oxidation in marine sediments: geochemistry meets microbiology. GSA Special Pap. 379, 63–81. doi: 10.1130/0-8137-2379-5.63
Jørgensen, B. B., Böttcher, M. E., Lüschen, H., Neretin, L. N., and Volkov, I. I. (2004). Anaerobic methane oxidation and a deep H2S sink generate isotopically heavy sulfides in Black Sea sediments. Geochim. Cosmochim. Acta 68, 2095–2118. doi: 10.1016/j.gca.2003.07.017
Kallmeyer, J., Ferdelman, T. G., Weber, A., Fossing, H., and Jørgensen, B. B. (2004). A cold chromium distillation procedure for radiolabeled sulfide applied to sulfate reduction measurements. Limnol. Oceanogr. Methods 2, 171–180. doi: 10.4319/lom.2004.2.171
Kamyshny, A. Jr., Goifman, A., Gun, J., Rizkov, D., and Lev, O. (2004). Equilibrium distribution of polysulfide ions in aqueous solutions at 25°C: a new approach for the study of polysulfides' equilibria. Environ. Sci. Technol. 38, 6633–6644. doi: 10.1021/es049514e
Kamyshny, A. Jr., Zilberbrand, M., Ekeltchik, I., Voitsekovski, T., Gun, J., and Lev, O. (2008). Speciation of polysulfides and zerovalent sulfur in sulfide-rich water wells in southern and central Israel. Aquat. Geochem. 14, 171–192. doi: 10.1007/s10498-008-9031-6
Kamyshny, A. Jr. (2009). Solubility of cyclooctasulfur in pure water and sea water at different temperatures. Geochim. Cosmochim. Acta 73, 6022–6028. doi: 10.1016/j.gca.2009.07.003
Kamyshny, A. Jr., Ferdelman, T. G. (2010). Dynamics of zero-valent sulfur species including polysulfides at seep sites on intertidal sand flats (Wadden Sea, North Sea). Mar. Chem. 121, 17–26. doi: 10.1016/j.marchem.2010.03.001
Kamyshny, A. Jr., Gun, J., Rizkov, D., and Voitsekovski, T., Lev, O. (2007). Equilibrium distribution of polysulfide ions in aqueous solutions at different temperatures by rapid single phase derivatization. Environ. Sci. Technol. 41, 6633–6644. doi: 10.1021/es062637+
Kaplan, I. E., and Rittenberg, S. C. (1964). Microbiological fractionation of sulfur isotopes. J. Gen. Microbiol. 34, 195–212. doi: 10.1099/00221287-34-2-195
Kasten, S., Freudenthal, T., Gingele, F. X., and Schulz, H. D. (1998). Simultaneous formation of iron-rich layers at different redox boundaries in sediments of the Amazon deep-sea fan. Geochim. Cosmochim. Acta 62, 2253–2264. doi: 10.1016/S0016-7037(98)00093-3
Kasten, S., Zabel, M., Heuer, V., and Hensen, C. (2003). “Processes and signals of nonsteady-state diagenesis in deep-sea sediments and their pore waters,” in The South Atlantic in the Late Quaternary: Reconstruction of Material Budget and Current Systems, eds G. Wefer, S. Mulitza, and V. Ratmeyer (Berlin; Heidelberg: Springer), 431–459.
Kelts, K., and McKenzie, J. A. (1982). Diagenetic Dolomite Formation in Quaternary Anoxic Diatomaceous Muds of Deep-Sea Drilling Project Leg-64, Gulf of California. Initial Rep. Deep Sea Drill. Proj. 64, 553–569.
Klaus, A., and Ledbetter, M. T. (1988). Deep-sea sedimentary processes in the Argentine Basin revealed by high-resolution seismic records (3.5 kHz echograms). Deep Sea Res. 35, 899–917. doi: 10.1016/0198-0149(88)90067-2
Kohnen, M. E. L., Sinninghe Damste, J. S., ten Haven, H. L., and de Leeuw, J. W. (1989). Early incorporation of polysulphides in sedimentary organic matter. Nature 341, 640–641. doi: 10.1038/341640a0
Krastel, S., and Wefer, G. (2011). Sediment Transport off Uruguay and Argentina: From the Shelf to the Deep Sea - Cruise No. M78/3 - May 19 - July 06, 2009 - Montevideo (Uruguay) - Montevideo (Uruguay). DFG-Senatskommission für Ozeanographie. METEOR-Berichte, M78/3:58. doi: 10.2312/cr_m78_3
Krastel, S., Lehr, J., Winkelmann, D., Schwenk, T., Preu, B., Strasser, M., et al. (2013). “Mass wasting along Atlantic continental margins: a comparison between NW-Africa and the de la Plata River region (northern Argentina and Uruguay),” in Submarine Mass Movements and Their Consequences, Vol. 37, Advances in Natural and Technological Hazards Research, eds S. Krastel, J.-H. Behrmann, D. Völker, M. Stipp, C. Berndt, and R. Urgeles (Springer International Publishing), 459–470.
Krastel, S., Wefer, G., Hanebuth, T. J. J., Antobreh, A. A., Freudenthal, T., Preu, B., et al. (2011). Sediment dynamics and geohazards off Uruguay and the de la Plata River region (Northern-Argentina). Geo Mar. Lett. 31, 271–283. doi: 10.1007/s00367-011-0232-4
Lin, Z., Suna, X., Peckmann, J., Lub, Y., Xu, L., Strauss, H., et al. (2016). How sulfate-driven anaerobic oxidation of methane affects the sulfur isotopic composition of pyrite: a SIMS study from the South China Sea. Chem. Geol. 440, 26–41. doi: 10.1016/j.chemgeo.2016.07.007
Luther, G. W., Giblin, A., Howarth, R. W., and Ryans, R. A. (1982). Pyrite and oxidized iron mineral phases formed from pyrite oxidation in salt marsh and estuarine sediments. Geochim. Cosmochim. Acta 46, 2665–2669. doi: 10.1016/0016-7037(82)90385-4
Lyons, T. W. (1997). Sulfur isotope trends and pathways of iron sulfide formation in upper Holocene sediments of the anoxic Black Sea. Geochim. Cosmochim. Acta 61, 3367–3382. doi: 10.1016/S0016-7037(97)00174-9
Malone, M. J., Claypool, G., Martin, J. B., and Dickens, G. R. (2002). Variable methane fluxes in shallow marine systems over geologic time: the composition and origin of pore waters and authigenic carbonates on the New Jersey shelf. Mar. Geol. 189, 175–196. doi: 10.1016/s0025-3227(02)00474-7
März, C., Hoffmann, J., Bleil, U., de Lange, G. J., and Kasten, S. (2008). Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean). Mar. Geol. 255, 118–130. doi: 10.1016/j.margeo.2008.05.013
Meister, P., McKenzie, J. A., Vasconcelos, C., Bernasconi, S., Frank, M., Gutjahr, M., et al. (2007). Dolomite formation in the dynamic deep biosphere: results from the Peru Margin. Sedimentology 54, 1007–1032. doi: 10.1111/j.1365-3091.2007.00870.x
Moore, T. S., Murray, R. W., Kurtz, A. C., and Schrag, D. P. (2004). Anaerobic methane oxidation and the formation of dolomite. Earth Planet. Sci. Lett. 229, 141–154. doi: 10.1016/j.epsl.2004.10.015
Morse, J. W. (1991). Oxidation kinetics of sedimentary pyrite in seawater. Geochim. Cosmochim. Acta 55, 3665–3667. doi: 10.1016/0016-7037(91)90064-C
Morse, J. W., and Cornwell, J. C. (1987). Analysis and distribution of iron sulfide minerals in recent anoxic marine sediments. Mar. Chem. 22, 55–69. doi: 10.1016/0304-4203(87)90048-X
Mossmann, J.-R., Aplin, A. C., Curtis, C. D., and Coleman, M. L. (1991). Geochemistry of inorganic and organic sulphur in organic-rich sediments from the Peru Margin. Geochim. Cosmochim. Acta 55, 3581–3595. doi: 10.1016/0016-7037(91)90057-C
Nöthen, K., and Kasten, S. (2011). Reconstructing changes in seep activity by means of pore water and solid phase Sr/Ca and Mg/Ca ratios in pockmark sediments of the Northern Congo Fan. Mar. Geol. 287, 1–13. doi: 10.1016/j.margeo.2011.06.008
Peiffer, S., Behrends, T., Hellige, K., Larese-Casanova, P., Wan, M., and Pollok, K. (2015). Pyrite formation and mineral transformation pathways upon sulfidation of ferric hydroxides depend on mineral type and sulfide concentration. Chem. Geol. 400, 44–55. doi: 10.1016/j.chemgeo.2015.01.023
Peketi, A., Mazumdar, A., Joao, H. M., Patil, D. J., Usapkar, A., and Dewangan, P. (2015). Coupled C–S–Fe geochemistry in a rapidly accumulating marine sedimentary system: diagenetic and depositional implications. Geochem. Geophys. Geosyst. 16, 2865–2883. doi: 10.1002/2015GC005754
Peterson, R. G., and Stramma, L. (1991). Upper-level circulation in the South Atlantic Ocean. Progr. Oceanogr. 26, 1–73. doi: 10.1016/0079-6611(91)90006-8
Piccolo, M. C., and Perillo, G. M. E. (1999). “The Argentina estuaries: a review,” in Estuaries of South America, eds G. M. E. Perillo, M. C. Piccolo, and M. Pino-Quivira (Springer), 101–132. doi: 10.1007/978-3-642-60131-6_6
Postma, D., and Jakobsen, R. (1996). Redox zonation: equilibrium constraints on the Fe (III)/SO4-reduction interface. Geochimica et Cosmochimica Acta 60, 3169–3175. doi: 10.1016/0016-7037(96)00156-1
Poulton, S. W., and Canfield, D. E. (2005). Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221. doi: 10.1016/j.chemgeo.2004.09.003
Preu, B., Hernández-Molina, F. J., Violante, R., Piola, A. R., Paterlini, C. M., Schwenk, T., et al. (2013). Morphosedimentary and hydrographic features of the northern Argentine margin: the interplay between erosive, depositional and gravitational processes and its conceptual implications. Deep Sea Res. Part I 75, 157–174. doi: 10.1016/j.dsr.2012.12.013
Quijada, M., Riboulleau, A., Faure, A., Michels, R., and Tribovillard, N. (2016). Organic matter sulfurization on protracted diagenetic timescales: The possible role of anaerobic oxidation of methane. Mar. Geol. 381, 54–66. doi: 10.1016/j.margeo.2016.08.010
Raiswell, R., Buckley, F., Berner, R. A., and Anderson, T. F. (1988). Degree of pyritization of iron as a paleoenvironmental indicator of bottom-water oxygenation. J. Sediment. Res. 58, 812–819.
Raiswell, R., and Canfield, D. E. (1998). Sources of iron for pyrite formation. Am. J. Sci. 298, 219–245. doi: 10.2475/ajs.298.3.219
Raiswell, R., Vu, H. P., Brinza, L., and Benning, L. G. (2010). The determination of labile Fe in ferrihydrite by ascorbic acid extraction: methodology, dissolution kinetics and loss of solubility with age and de-watering. Chem. Geol. 278, 70–79. doi: 10.1016/j.chemgeo.2010.09.002
Raven, M. R., Session, A. L., Fischer, W. W., and Adkins, J. F. (2016). Sedimentary pyrite d34S differs from porewater sulfide in Santa Barbara Basin: proposed role of organic sulfur. Geochim. Cosmochim. Acta 186, 120–134. doi: 10.1016/j.gca.2016.04.037
Rees, C. E. (1970). The sulphur isotope balance of the ocean: an improved model. Earth Planet. Sci. Lett. 17:366. doi: 10.1016/0012-821X(69)90051-X
Rickard, D. T. (1975). Kinetics and mechanism of pyrite formation at low temperatures. Am. J. Sci. 275, 636– 652. doi: 10.2475/ajs.275.6.636
Rickard, D., and Luther, G. W. III. (1997). Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulfide in aqueous solutions between 25 and 125°C: the mechanism. Geochim. Cosmochim. Acta 61:135. doi: 10.1016/s0016-7037(96)00322-5
Rickard, D., and Luther, G. W. III. (2007). Chemistry of iron sulfides. Chem. Rev. 107, 514–562. doi: 10.1021/cr0503658
Riedinger, N. (2005). Preservation and Diagenetic Overprint of Geochemical and Geophysical Signals in Ocean Margin Sediments Related to Depositional Dynamics. Berichte, 242, Fachbereich Geowissenschaften, Universität Bremen, 91.
Riedinger, N., Brunner, B., Formolo, M. J., Solomon, E., Kasten, S., Strasser, M., et al. (2010). Oxidative sulfur cycling in the deep biosphere of the Nankai Trough, Japan. Geology 38, 851–854. doi: 10.1130/G31085.1
Riedinger, N., Formolo, M. J., Lyons, T. W., Henkel, S., Beck, A., and Kasten, S. (2014). An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments. Geobiology 12, 172–181. doi: 10.1111/gbi.12077
Riedinger, N., Pfeifer, K., Kasten, S., Garming, J. F. L., Vogt, C., and Hensen, C. (2005). Diagenetic alteration of magnetic signals by anaerobic oxidation of methane related to a change in sedimentation rate. Geochim. Cosmochim. Acta 69, 4117–4126. doi: 10.1016/j.gca.2005.02.004
Romero, O., and Hensen, C. (2002). Oceanographic control of biogenic opal and diatoms in surface sediments of the South Western Atlantic. Mar. Geol. 186, 263–280. doi: 10.1016/S0025-3227(02)00210-4
Rudnicki, M. D., Elderfield, H., and Spiro, B. (2001). Fractionation of sulfur isotopes during bacterial sulfate reduction at elevated temperatures. Geochim. Cosmochim. Acta 65, 777–789. doi: 10.1016/S0016-7037(00)00579-2
Sachs, S. D., and Ellwood, B. B. (1988). Controls on magnetic grain size variations and concentration in the Argentine Basin, South Atlantic Ocean. Deep-Sea Res. 35, 929–942. doi: 10.1016/0198-0149(88)90069-6
Sawicka, J. E., Jørgensen, B. B., and Brüchert, V. (2012). Temperature characteristics of bacterial sulfate reduction in continental shelf and slope sediments. Biogeosciences 9, 3425–3435. doi: 10.5194/bg-9-3425-2012
Schoonen, M. A. A., and Barnes, H. L. (1991). Reactions forming pyrite and marcasite from solution: II. Via FeS precursors below 100°C. Geochim. Cosmochim. Acta 55, 1505–1514. doi: 10.1016/0016-7037(91)90123-m
Seeberg-Elverfeldt, J., Schlüter, M., Feseker, T., and Kölling, M. (2005). Rhizon sampling of porewaters near the sediment-water interface of aquatic systems. Limnol. Oceanogr. Methods 3, 361–371. doi: 10.4319/lom.2005.3.361
Sim, M. S., Bosak, T., and Ono, S. (2011). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Sinninghe Damsté, J. S., Rijpstra, W. I. C., de Leeuw, J. W., and Schenck, P. A. (1988). Origin of organic sulphur compounds and sulphur containing high molecular weight substances in sediments and immature crude oils. In Advances in Organic Geochemistry 1987 (eds. L. Mattavelli and L. Novelli). Organ. Geochem. 13, 593–606. doi: 10.1016/0146-6380(88)90079-4
Sinninghe Damsté, J. S., and deLeeuw, J. W. (1990). Analysis, structure and geochemical significance of organically-bound sulphur in the geosphere: state of the art and future research. Organ. Geochem. 16, 1077–1101. doi: 10.1016/0146-6380(90)90145-P
Sørensen, J., and Jørgensen, B. B. (1984). Early diagenesis in sediments from Danish coastal waters: microbial activity and Mn-Fe-S geochemistry. Geochim. Cosmochim. Acta 51, 1583–1590. doi: 10.1016/0016-7037(87)90339-5
Stetter, K. O., and Gaag, G. (1983). Reduction of molecular sulphur by methanogenic bacteria. Nature 305, 310–311. doi: 10.1038/305309a0
Taylor, S. R., and McLennan, S. M. (1985). The Continental Crust: Its Composition and Evolution. Oxford: Blackwell.
Thamdrup, B., Finster, K., Hansen, J. W., and Bak, F. (1993). Bacterial disproportionation of Elemental Sulfur coupled to chemical reduction of iron or manganese. Appl. Environ. Microbiol. 59, 101–108.
Thode, H. G., Monster, J., and Dunford, H. B. (1961). Sulfur isotope geochemistry. Geochim. Cosmochim. Acta 25, 159–174. doi: 10.1016/0016-7037(61)90074-6
Torres, M. E., Brumsack, H. J., Bohrmann, G., and Emeis, K. C. (1996). Barite fronts in continental margin sediments: a new look at barium remobilization in the zone of sulfate reduction and formation of heavy barites in diagenetic fronts. Chem. Geol. 127, 125–139. doi: 10.1016/0009-2541(95)00090-9
Treude, T., Krause, S., Maltby, J., Dale, A. W., Coffin, R., and Hamdan, L. J. (2014). Sulfate reduction and methane oxidation activity below the sulfate-methane transition zone in Alaskan Beaufort Sea continental margin sediments: implications for deep sulfur cycling. Geochim. Cosmochim. Acta 144, 217–237. doi: 10.1016/j.gca.2014.08.018
Turchyn, A. V., Antler, G., Byrne, D., Miller, M., and Hodell, D. A. (2016). Microbial sulfur metabolism evidenced from pore fluid isotope geochemistry at Site U1385. Global Planet. Change 141, 82–90. doi: 10.1016/j.gloplacha.2016.03.004
Vairavamurthy, A., and Mopper, K. (1987). Geochemical formation of organic sulphur compounds (thiols) by addition of H2S to sedimentary organic matter. Nature 329, 623–625. doi: 10.1038/329623a0
Vairavamurthy, A., Mopper, K., and Taylor, B. (1992). Occurrence of particle-bound polysulfides and significance of their reaction with organic matters in marine sediments. Geophys. Res. Lett. 19, 2043–2046. doi: 10.1029/92GL01995
Vairavamurthy, M. A., Orr, W. L., and Manowitz, B. (1995). Geochemical transformations of sedimentary sulfur: an introduction. Am. Chem. Soc. 612, 1–14. doi: 10.1021/bk-1995-0612.ch001
Voigt, I., Henrich, R., Preu, B. M., Piola, A. R., Hanebuth, T. J. J., Schwenk, T., et al. (2013). A submarine canyon as a climate archive—interaction of the Antarctic Intermediate Water with the Mar del Plata Canyon (Southwest Atlantic). Mar. Geol. 341:46–57. doi: 10.1016/j.margeo.2013.05.002
Vossmeyer, A., Deusner, C., Kato, C., Inagaki, F., and Ferdelman, T. G. (2012). Substrate-specific pressure-dependence of microbial sulfate reduction in deep-sea cold seep sediments of the Japan Trench. Front. Microbiol. 3:253. doi: 10.3389/fmicb.2012.00253
Wehrmann, L. M., Arndt, S., März, C., Ferdelman, T. G., and Brunner, B. (2013). The evolution of early diagenetic signals in Bering Sea subseafloor sediments in response to varying organic carbon deposition over the last 4.3 Ma. Geochim. Cosmochim. Acta 109, 175–196. doi: 10.1016/j.gca.2013.01.025
Wehrmann, L. M., Formolo, M. J., Owens, J. D., Raiswell, R., Ferdelman, T. G., Riedinger, N., et al. (2014). Iron and manganese speciation and cycling in glacially influenced high-latitude fjord sediments (West Spitsbergen, Svalbard): evidence for a benthic recycling-transport mechanism. Geochim. Cosmochim. Acta 141, 628–655. doi: 10.1016/j.gca.2014.06.007
Wehrmann, L. M., Risgaard-Petersen, N., Schrum, H. N., Walsh, E. A., Huh, Y., Ikehara, M., et al. (2011). Coupled organic and inorganic carbon cycling in the deep subseafloor sediment of the northeastern Bering Sea Slope (IODP Exp. 323). Chem. Geol. 284, 251–261. doi: 10.1016/j.chemgeo.2011.03.002
Werne, J. P., Hollander, D. J., Behrens, A., Schaeffer, P., Albrecht, P., and Sinninghe Damsté, J. S. S. (2000). Timing of early diagenetic sulfurization of organic matter: a precursor–product relationship in Holocene sediments of the anoxic Cariaco Basin, Venezuela. Geochim. Cosmochim. Acta 64, 1741–1751. doi: 10.1016/s0016-7037(99)00366-x
Werne, J. P., Hollander, D. J., Lyons, T. W., and Sinninghe Damsté, J. S. (2004). Organic sulfur biogeochemistry: recent advances and future research directions. GSA Special Pap. 379, 135–150. doi: 10.1130/0-8137-2379-5.135
Werne, J. P., Lyons, T. W., Hollander, D. J., Formolo, M., and Sinninghe Damsté, J. S. (2003). Reduced sulfur in euxinic sediments of the Cariaco Basin: sulfur isotope constraints on organic sulfur formation. Chem. Geol. 195, 159–179. doi: 10.1016/s0009-2541(02)00393-5
Werne, J. P., Lyons, T. W., Hollander, D. J., Schouten, S., Hopmans, E. C., and Sinninghe Damsté, J. S. (2008). Investigating pathways of diagenetic organic matter sulfurization using compound-specific sulfur isotope analysis. Geochim. Cosmochim. Acta 72, 3489–3502, doi: 10.1016/j.gca.2008.04.033
Wing, B. A., and Halevy, I. (2014). Intracellular metabolite levels shape sulfur isotope fractionation during microbial sulfate respiration. Proc. Natl. Acad. Sci. U.S.A. 111, 18116–18125. doi: 10.1073/pnas.1407502111
Xiao, S., Schiffbauer, J. D., McFadden, K. A., and Hunter, J. (2010). Petrographic and SIMS pyrite sulfur isotope analyses of Ediacaran chert nodules: implications for microbial processes in pyrite rim formation, silicification, and exceptional fossil preservation. Earth Planet. Sci. Lett. 297, 481–495. doi: 10.1016/j.epsl.2010.07.001
Zopfi, J., Ferdelman, T. G., and Fossing, H. (2004). “Distribution and fate of sulfur intermediates – sulfite, tetrathionate, thiosulfate, and elemental sulfur – in marine sediments,” in Sulfur Biochemistry – Past and Present, Vol. 379, Geological Society of America Special Paper, eds J. P. Amend, K. J. Edwards, and T. W. Lyons (Boulder, CO: The Geological Society of America), 97–116.
Keywords: subsurface sulfur cycle, biogeochemistry, non-steady state, sulfur isotopes, sulfate-methane transition
Citation: Riedinger N, Brunner B, Krastel S, Arnold GL, Wehrmann LM, Formolo MJ, Beck A, Bates SM, Henkel S, Kasten S and Lyons TW (2017) Sulfur Cycling in an Iron Oxide-Dominated, Dynamic Marine Depositional System: The Argentine Continental Margin. Front. Earth Sci. 5:33. doi: 10.3389/feart.2017.00033
Received: 19 December 2016; Accepted: 13 April 2017;
Published: 09 May 2017.
Edited by:
Tanja Bosak, Massachusetts Institute of Technology, USAReviewed by:
Gilad Antler, University of Cambridge, UKMustafa Yucel, Middle East Technical University, Turkey
Copyright © 2017 Riedinger, Brunner, Krastel, Arnold, Wehrmann, Formolo, Beck, Bates, Henkel, Kasten and Lyons. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natascha Riedinger, bmF0YXNjaGEucmllZGluZ2VyQG9rc3RhdGUuZWR1
†Formerly at the Max Planck Institute of Marine Microbiology, Bremen, Germany