 Adi Torfstein
Adi Torfstein Alexandra V. Turchyn
Alexandra V. Turchyn- 1Institute of Earth Sciences, Hebrew University of Jerusalem, Jerusalem, Israel
- 2Interuniversity Institute for Marine Sciences, Eilat, Israel
- 3Department of Earth Sciences, University of Cambridge, Cambridge, United Kingdom
We report the δ34S and δ18O(SO4) measured in gypsum, pyrite, and elemental sulfur through a 456-m thick sediment core from the center of the Dead Sea, representing the last ~200 kyrs, as well as from the exposed glacial outcrops of the Masada M1 section located on the margins of the modern Dead Sea. The results are used to explore and quantify the evolution of sulfur microbial metabolism in the Dead Sea and to reconstruct the lake's water column configuration during the late Quaternary. Layers and laminae of primary gypsum, the main sulfur-bearing mineral in the sedimentary column, display the highest δ34S and δ18O(SO4) in the range of 13–28 and 13–30‰, respectively. Within this group, gypsum layers deposited during interglacials display lower δ34S and δ18O(SO4) relative to those associated with glacial or deglacial stages. The reduced sulfur phases, including chromium reducible sulfur, and secondary gypsum crystals are characterized by extremely low δ34S in the range of −27 to +7‰. The δ18O(SO4) of the secondary gypsum in the M1 outcrop ranges from 8 to 14‰. The relationship between δ34S and δ18O(SO4) of primary gypsum suggests that the rate of microbial sulfate reduction was lower during glacial relative to interglacial times. This suggests that the freshening of the lake during glacial wet intervals, and the subsequent rise in sulfate concentrations, slowed the rate of microbial metabolism. Alternatively, this could imply that sulfate-driven anaerobic methane oxidation, the dominant sulfur microbial metabolism today, is a feature of the hypersalinity in the modern Dead Sea. Sedimentary sulfides are quantitatively oxidized during epigenetic exposure, retaining the lower δ34S signature; the δ18O(SO4) of this secondary gypsum is controlled by oxygen atoms derived equally from atmospheric oxygen and from water, which is likely a unique feature in this hyperarid environment.
Introduction
Overview
The biological use of sulfur in its multiple valence states is a primary tracer of the redox dynamics in lacustrine environments both in the modern environment and over geological time (Claypool et al., 1980; Strauss, 1997; Canfield, 2004; Bottrell and Newton, 2006; and references therein). The microbially-mediated reduction of sulfate to sulfide, termed “microbial sulfate reduction,” involves a large sulfur isotopic fractionation where the lighter 32S isotope is preferentially reduced relative to the heavier 34S. The magnitude of this sulfur isotope fractionation can range between 2 and 70‰ and is largely a function of the rate of microbial sulfate reduction, which itself is a function of temperature, sulfate concentration, and organic carbon supply (Kaplan and Rittenberg, 1964; Chambers and Trudinger, 1979; Canfield, 1998; Habicht et al., 1998; Wortmann et al., 2007; Sim et al., 2011). Changes in the redox conditions of bottom waters or within sediments can impart measureable sulfur isotopic differences in sulfate and sulfide minerals preserved in the sedimentary record; these isotopic changes have been used to reconstruct past redox conditions and the behavior of the biogeochemical sulfur cycle. In particular, the sulfur isotope composition of sulfate minerals has been used to study the evolution of closed or semi-closed water bodies such as the Black Sea (e.g., Lyons, 1997; Neretin et al., 2003), Lake Baikal (Watanabe et al., 2004), the Messinian Mediterranean (e.g., Lu et al., 2001), Lake Eyre (e.g., Baas-Becking and Kaplan, 1956), Death Valley (Yang et al., 1997), the Estancia Basin in New Mexico (Szynkiewicz et al., 2009), and the lacustrine deposits of the Green River Formation (e.g., Tuttle and Goldhaber, 1993), among others. The isotopic signature of the sedimentary archive reflects past patterns of sulfur isotope composition of the lake, allowing for the reconstruction of water column configuration, water sources, and in turn, regional climatic-hydrologic trends (e.g., Claypool et al., 1980; Strauss, 1997).
Typically, arid continental basins are characterized by low precipitation-to-evaporation ratios which result in the development of hypersaline water bodies and high deposition rates of evaporate minerals such as carbonate and gypsum (e.g., Great Salt Lake of Utah, Lake Chad of Central Africa, the Caspian Sea, and the Dead Sea; Eugster, 1980). Under such conditions, climate perturbations can induce larger freshwater influx triggering density stratification of the water column that disconnects the lower body from the atmosphere, leading to the development of anoxic conditions. Because sulfur is a redox-sensitive element the study of the sulfur cycle in terminal lacustrine bodies can impart important information on regional and global hydrological patterns.
Of the above mentioned lacustrine bodies, the Dead Sea is the most saline deep lake in the world today [~300 m water depth, water level 430 m below mean sea level (mbmsl), salinity ~340 g/l], and its water levels and limnological history have been shown to reflect regional climate patterns, themselves controlled by northern hemisphere climate change during the Quaternary (e.g., Stein, 2001; Bartov et al., 2003; Enzel et al., 2003; Bookman Ken-Tor et al., 2004; Torfstein et al., 2013b). Due to its high salinity, any input of freshwater induces density stratification that can persist for several years. Long term lake stratification also developed in the geological past, when freshwater input was significantly higher than present, and was sustained for several thousands of years. Such a stratified water column can develop intense anoxic conditions in the lower layer (e.g., Froelich et al., 1979; Arndt et al., 2013). This deepwater anoxia drives changes in microbial communities and biogeochemical cycles of redox sensitive elements like iron, manganese, and sulfur.
In spite of the high salinity in the modern Dead Sea, evidence for microbial sulfate reduction, likely coupled to anaerobic methane oxidation, has been found in ground water and sedimentary deposits (Oldenburg et al., 2000a,b; Gavrieli et al., 2001; Avrahamov et al., 2014; Thomas et al., 2016). Microbial sulfate reduction has been inferred from the coexistence of aqueous sulfate and sulfide, as well as measurements of sulfur isotopes that suggest a sulfur isotope fractionation of about 30‰ in the modern Dead Sea (Gavrieli et al., 2001). As the lake was hypersaline over the course of the Quaternary (Torfstein et al., 2009), with a similar configuration to the modern lake, it is likely that microbial sulfate reduction dominated the oxidation of organic carbon in the past as well. There is further evidence for microbial sulfate reduction in late Quaternary deposits exposed along the lake margins, in the form of a sulfur isotope offset between gypsum and sulfur nodules found within gypsum. This offset has been interpreted to reflect microbial sulfate reduction under carbon-limited conditions (Torfstein et al., 2005, 2008; Bishop et al., 2013). Thus far, evidence of past microbial sulfate reduction has been limited to the marginal and exposed sites along the Dead Sea, precluding the direct reconstruction of the composition and history of the deep anoxic layer in the deep center of the lake.
Here, we focus on the isotopic composition of sulfur (δ34S) and sulfate-bound oxygen [δ18O(SO4)] in primary and secondary gypsum, reduced sulfide minerals, and native sulfur. Samples are studied in the deep center of the Dead Sea and compared with an exposed marginal lacustrine sedimentary record (Figure 1). In particular, we report for the first time results from the recent Dead Sea Deep Drilling Project (DSDDP), which recovered a continuous 456 m thick core from the deep center of the modern lake spanning the last ~200,000 years (Neugebauer et al., 2014; Torfstein et al., 2015). We use the results to reconstruct the evolution of redox conditions in the deep-water column, understand the rate of microbial sulfate reduction and how it links to the source of carbon, and explore the oxidation pathways of sulfide minerals in the Dead Sea Basin (DSB).
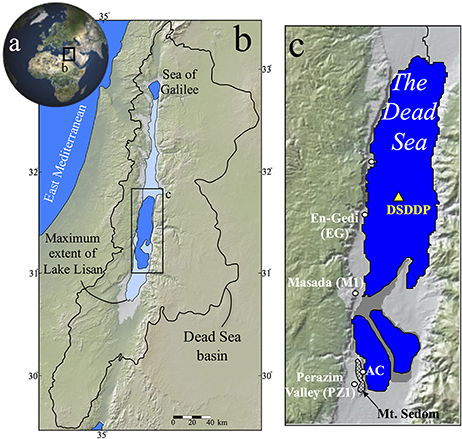
Figure 1. Location map.
The Dead Sea
Limnology
The Quaternary Dead Sea lacustrine bodies evolved from the Pliocene Sedom Lagoon, which derived its chemical constituents from ingressing seawater and interaction with the carbonate wall-rock, and resulted in the development of the unique calcium-chloride composition of the DSB brines (Zak, 1967; Starinsky, 1974; Stein et al., 2000, 2002). After the disconnection of the Sedom lagoon from the open sea around 3 Ma (Torfstein et al., 2009; Belmaker et al., 2013; Matmon et al., 2014), the limnological and geochemical history of the closed and terminal water bodies that subsequently occupied the DSB (the early- to mid-Pleistocene Lake Amora, last glacial Lake Lisan, and the Holocene Dead Sea) was controlled by the interaction between the brine and freshwater input (Katz et al., 1977; Stein, 2001; Gavrieli and Stein, 2006; Katz and Starinsky, 2008).
The Dead Sea brines are characterized by a distinct enrichment in calcium and chloride (16,700 and 219,700 mg/l, respectively; Stein et al., 1997), while incoming freshwater runoff, which largely drains carbonate terrains, is enriched in bicarbonate (~200 mg/l; Stein et al., 1997). The high calcium concentrations in the lake impose very low concentrations of bicarbonate and sulfate through the saturation of calcium carbonate and gypsum (CaSO4·2H2O), respectively. Incoming freshwater, delivering bicarbonate, swiftly precipitates as calcium carbonate (dominantly aragonite), while dissolved sulfate (also introduced into the mixolimnion from freshwater sources) accumulates until gypsum saturation is eventually reached. At that point, gypsum crystals settle through the water column to the anoxic monimolimnion, where gypsum is under saturated due to the removal of sulfate through microbial sulfate reduction and dissolves (Torfstein et al., 2005).
Sedimentology
The Dead Sea lacustrine deposits include sequences of alternating aragonite and detrital laminae (aad facies, Figure 2), which represent annual deposition cycles of primary inorganic aragonite precipitated from the lake during summer evaporation and silt-size detritus washed into the lake during winters (Begin et al., 1974; Katz et al., 1977; Stein et al., 1997). The aad facies reflects a wet hydrological regime with high freshwater and thus bicarbonate input, and is associated with relatively higher lake levels. Increasing aridity is reflected by decreased amounts of aragonite relative to detritus, then precipitation of gypsum, followed by halite. Brown silt layers, a few cm thick or more, mark enhanced activity of freshwater floods during relatively low-level intervals.
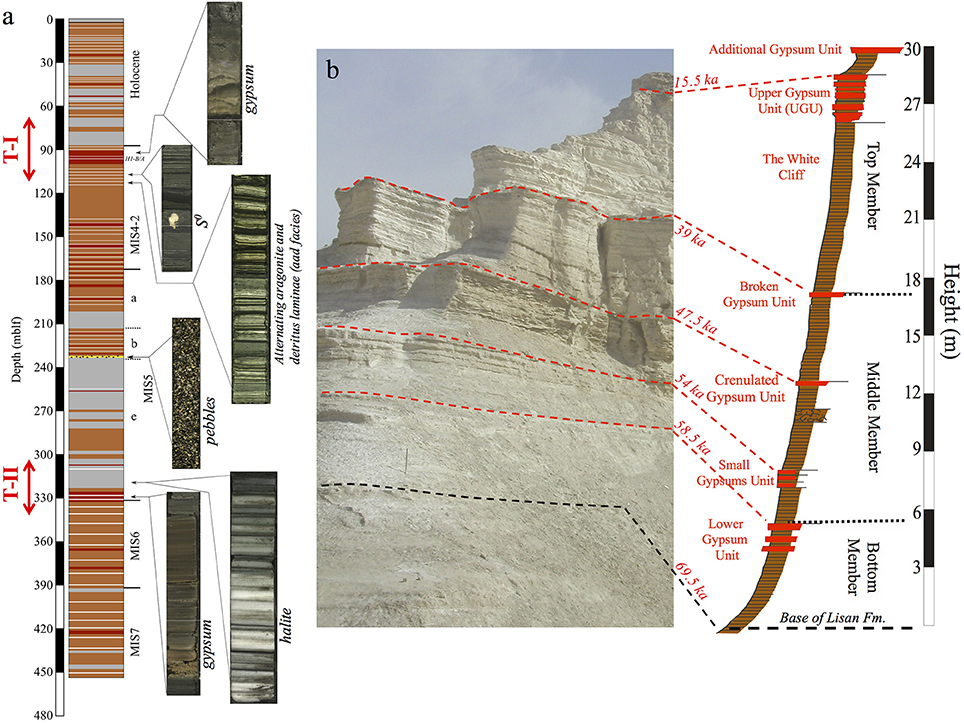
Figure 2. (a) Generalized columnar section of the Dead Sea Deep Drilling Project (DSDDP) core, (b) Masada M1 section. Core photos of several sediment facies are presented in (a). Legend for columnar section: gray, halite; red, gypsum; white stripes, aad facies; brown, marl; yellow layer (at 234 mblf), pebble layer. The timing of Termination –II and –I is marked by red arrow left of the columnar section. Ages of marker layers in (b) are from Torfstein et al. (2013a). Note that the Masada M1 section of the Lisan Formation, corresponds to depths of ~177–90 mblf in the DSDDP core.
These lithological types appear throughout the DSDDP core (Figures 2–4) as well as in sediment outcrops along the margins of the modern Dead Sea, such as the M1 sedimentary section located on the foothills of the Masada archeological site. The main interval that is exposed in the M1 sedimentary section is the last glacial Lisan Formation (70–14 ka; Kaufman, 1971; Haase-Schramm et al., 2004). Massive halite units that reflect major lake level drawdowns occur uniquely in the deep DSDDP core, and are synchronous with interglacial cycles (Torfstein et al., 2015).

Figure 3. (a) Native sulfur nodule in aragonite-detritus sequence ~20 cm above the Lower Gypsum Unit (M1), (b) Nodules in aragonite-detritus outcrop. The black coating of the extracted nodules and the yellow native sulfur nodule on the right, (c) Native sulfur nodules in massive gypsum layer, (d) Native sulfur nodule in its crust, comprised of secondary gypsum and Fe-oxides, (e) Aragonite-detritus sequence of the Lisan Formation at M1. The abundant secondary gypsum concretions and bulges throughout the sequence, (f,g) Native sulfur nodules at the base of a field sample extracted from between the gypsum units comprising the Lower Gypsum Unit at M1, (h) Thin section under polarized light exhibiting micro secondary gypsum concretions offsetting surrounding aragonite laminae (from Haliva-Cohen et al., 2012). The concretion at the lower part of (h) is ~1 mm thick from bottom to top, (i) SEM magnification of primary gypsum crystals, (j) SEM magnification of aragonite needles, (k) SEM magnification of a native sulfur particle.

Figure 4. DSDDP core photos of (A) Termination-I, and (B) Termination-II. Depth in meters below lake floor (mblf), and ages (ka) are marked in black and blue, respectively (ages after Torfstein et al., 2015). Red and gray rectangles highlight the location of gypsum and halite intervals, respectively.
Sulfur and Sulfate Bound Oxygen
The sulfur isotopic composition (measured as 34S/32S reported relative to the Vienna Canon Diablo Troilite standard in parts-per-thousand, or permil units) of sulfate or other sulfur species in sedimentary systems is often controlled by microbial reduction of sulfate that imposes a large sulfur isotope fractionation among the various sulfur species (Rees, 1973; Cypionka et al., 1998; Habicht et al., 1998). Microbial sulfate reduction coupled to organic carbon oxidation is described through the simplified equation:
Specifically, sulfur isotopes in sulfate increase monotonically as the 32S atom is slowly distilled into the product sulfide during microbial sulfate reduction; the rate at which the 34S/32S ratio in the residual sulfate increases is a function largely of the sulfate reduction rate as well as temperature, organic carbon source, microbial community, pressure, and other environmental factors. Microbial sulfate reduction also fractionates the sulfate-bound oxygen isotopes [δ18O(SO4) is the ratio of 18O/16O reported vs. Vienna-Standard Mean Ocean Water in permil units], whereby the residual sulfate is preferentially enriched in the heavy isotope (18O).
In the microbial cell however, oxygen isotopes in intermediate valence state sulfur species such as sulfite, can equilibrate with water and these intermediate valence state species may return to the extracellular sulfate pool (Brunner et al., 2005; Brunner and Bernasconi, 2005; Antler et al., 2013). Thus, the relative change in the sulfur and oxygen isotope composition of sulfate over the onset of microbial sulfate reduction has been shown to reflect the degree of recycling of sulfur within cells (over the amount which is unidirectionally reduced). Because of this, the oxygen isotope ratio of sulfate has become a powerful tool for exploring sedimentary and aqueous sulfur redox dynamics (Brunner et al., 2005; Wortmann et al., 2007; Antler et al., 2013; Knossow et al., 2015). The oxygen isotope composition of sulfate is “fixed” the last time that any reduced valence state sulfur species is oxidized to sulfate (the oxygen atoms in sulfate do not subsequently readily equilibrate or exchange with water). Most sulfide oxidation pathways involve only small sulfur isotope fractionations, while the oxygen isotope composition of the resulting sulfate depends on the variable contribution of oxygen atoms from water and from atmospheric oxygen (Balci et al., 2007; Calmels et al., 2007). Thus, the δ18O(SO4) often reflects the pathway by which the reduced sulfur was last oxidized plus any equilibration with water along the oxidation or reduction pathway. Together, the measurement of sulfur and oxygen isotopes in sulfate provide a powerful tool for reconstructing the microbially-mediated redox cycling of sulfur in a range of marine, marginal marine, and lacustrine environments (e.g., Claypool et al., 1980; Aharon and Fu, 2000; Bottrell and Newton, 2006; Leavitt et al., 2013; Antler et al., 2015).
Previous studies have explored the sulfur cycle in the Dead Sea and its precursors over the late Quaternary but have been limited to the marginal outcrops of the Dead Sea. The density-driven stratification during increased freshwater influx to the Dead Sea partitioned aqueous sulfur into two distinct and separated reservoirs; 34S depleted sedimentary sulfide minerals (resulting from water column microbial sulfate reduction) and 34S-enriched sulfate that remains in the water column and is found in gypsum beds that precipitate during lake overturn. The longer and stronger the persistence of anoxic conditions in the stratified lake during wet periods, the thicker and more 34S-enriched the gypsum that precipitates when conditions turn arid and the lake level declines resulting in water column mixing (Torfstein et al., 2008, 2005). This interpretation of the evolution of the biogeochemical sulfur cycle over hydrological changes in the Dead Sea derives entirely from measurements of 34S-enriched gypsum (ranging between ~ −25 and +30‰) from the lake margin deposits; thus far no sedimentary sulfide minerals have been found in the marginal outcrops. By contrast, secondary gypsum, found in the Lisan Formation (Fm.) in the form of fine laminae or micro-concretions, displays 34S-depleted compositions unlike the primary gypsum beds (Torfstein et al., 2005, 2008). These secondary gypsum crystals have been interpreted to derive from the oxidation of the (missing) reduced sulfur minerals initially formed by water column microbial sulfate reduction in the Dead Sea.
Methods
Sample Collection
Most of the samples studied here were retrieved from the Dead Sea Deep Drilling Project (DSDDP) core, which was acquired by the International Continental Drilling Program (ICDP) campaign during 2010–2011 (Neugebauer et al., 2014; Torfstein et al., 2015). Additional samples are from the Masada (M1) section, Arubotaim Cave (AC) section, and En Gedi (EG) section exposed on the western margins of the Dead Sea (Figure 1), representing the last glacial Lisan Fm., mid-Pleistocene Amora Fm., and Holocene Ze'elim Fm., respectively. The samples consist of primary gypsum layers, secondary gypsum laminae and micro-concretions, soluble salts, and marly silt (mud) samples, from which sulfide phases were extracted (see below).
Sample Processing and Analysis
The primary and secondary gypsum minerals were dissolved in a sodium chloride saturated solution (~50 g/L). A super-saturated barium chloride solution (~30 g/L) was added to the effluent to precipitate the aqueous sulfate from gypsum dissolution as barite (BaSO4). The barite was weighed into tin and silver capsules and analyzed for sulfur and oxygen isotopes, respectively, at the University of Cambridge in the Godwin Laboratory. For sulfur isotopes vanadium pentoxide was added as a combustion agent. Sulfur isotopes were analyzed through combustion to SO2 in a Flash Element Analyzer coupled by continuous helium flow to a Delta Advantage mass spectrometer (CF-GS-IRMS – Thermo Finnegan). Samples for sulfur isotopes were bracketed by international standard NBS-127 (samples were corrected to NBS-127 δ34S = 20.3‰, stdev = 0.2‰ based on repeat analysis of the standards and blind replicates). Oxygen isotopes in sulfate were analyzed in triplicate through pyrolysis in a TC/EA coupled by continuous helium flow to a Delta Advantage mass spectrometer (GS-CF-IRMS – Thermo Finnegan). Samples were bracketed by NBS-127 [δ18O(SO4) = 8.6‰], with reported uncertainties reflecting the average and standard deviation of triplicate analyses of each sample.
Acid Volatile Sulfide (AVS) and Chromium Reducible Sulfur (CRS) were extracted from silt mud samples throughout the DSDDP core. The acid volatile sulfides are largely understood to be iron monosulfides, while the chromium reduction targets the iron disulfide minerals, primarily pyrite. The samples had been pre-washed by submerging in deionised water and agitating for 24 h then dried prior to extraction. Approximately 2–3 g of sample material then was weighed and transferred to a reaction vessel which was placed upon a hotplate and flushed with nitrogen. The AVS fraction was extracted with the addition of 35 ml of 0.1 M stannous chloride in 6 M hydrochloric acid. The mixture was heated and kept at sub-boiling temperature for 35 min. The CRS fraction was extracted through 35 ml of 1 M chromous chloride in 0.5 M hydrochloric acid. Pyrite and elemental sulfur are both reduced to hydrogen sulfide by chromous chloride, which is oxidized to chromic chloride (CrCl3) in the process. The mixture was heated for 2–3 h. The two chemical digestions done on the sample produced both hydrogen sulfide gas (from the sulfide minerals) as well as chlorine gas (from the presence of hydrochloric acid in the reagents). To remove the chlorine, an in-line nitric acid trap containing 0.1 M nitric acid in deionised water was used such that the gas stream was bubbled through the trap, dissolving the chlorine. The hydrogen sulfide gas was passed into a further trap containing a solution of 0.2 M silver nitrate in 10% ammonium hydroxide, forming silver sulfide. The silver sulfide from both the CRS and AVS extractions was then washed and vacuum filtered with 0.45 μm filter paper and left to dry.
The silver sulfide was prepared for sulfur isotope analysis in the same way as the barite. An additional standard (IAEA-S3 − δ34S = −32.3‰) was also run. Following established protocol, the raw data from the mass spectrometer were adjusted relative to the difference between the measured and expected values of the standards, using a linear interpolation between the average of each set of standards.
Several bulk mud samples from the DSDDP core were exposed to long dissolution (several weeks while agitated) in NaCl to ascertain whether the overnight leaching and likely non-quantitative dissolution of gypsum imparted a sulfur or oxygen isotope fractionation on the gypsum. The isotope composition of both long and short dissolution was the same.
Results
The isotopic composition of sulfur and oxygen in primary gypsum layers in the DSDDP core (Supplementary Table 1) and the M1 outcrop (Supplementary Table 2) reveal strong glacial—interglacial variability (Figures 5–7). Both the penultimate and last glacial terminations (T-II and T-I, respectively) are characterized by the precipitation of massive primary gypsum units (~137–134 and 17–14 ka; Figure 4) and both of these deglacial intervals display temporal δ34S maxima of ~23‰ (MIS6/5) and 28‰ (last deglacial). During the subsequent interglacial stages, i.e., the last interglacial and the Holocene, the δ34S of primary gypsum units dropped to ~12.9–15.7‰. Higher δ34S is observed across the two glacial stages (MIS6 and 4-2), with a gradual rise with time toward the deglacial maxima. The 34S-enrichment of primary gypsum during glacial maxima is most evident in the last glacial cycle, in agreement with previous observations from exposed-marginal outcrops (Torfstein et al., 2005, 2008). Primary gypsum units were not identified across the MIS6 interval in the DSDDP core. Overall, coeval samples from the DSDDP core and marginal outcrops display overlapping sulfur isotopic compositions (Figure 6).
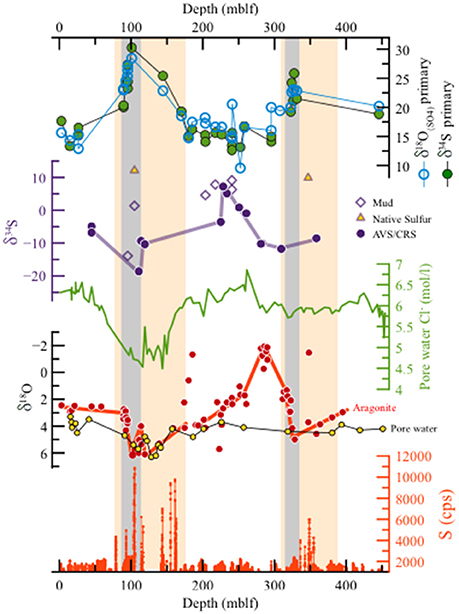
Figure 5. The DSDDP core. (a) Abundance of sulfur from XRF scanning, (b) δ18O of aragonite laminae (red, from Torfstein et al., 2015) and pore waters (yellow, from Lazar et al., 2014) (note the reverse y axis). (c) Chloride concentrations in pore waters (from Lazar et al., 2014), (d) δ34S (‰) of reduced sulfur minerals. Full circles represent the AVS and CRS fractions, and empty diamonds mark sulfate extracted from in mud intervals. Yellow triangles mark native sulfur nodules. (e) δ34S and δ18O(SO4) of primary gypsum. The similar trends of both isotopic systems, with a gradual rise toward maximum values in the two deglacial stages (marked by gray rectangles). Interglacial stages are characterized by consistently low values ca. 14–20‰. The orange rectangles mark the depth interval corresponding to two glacial cycles (MIS6 and last glacial). δ34S of primary gypsum and AVS/CRS fractions displays decoupling, whereby the highest δ34S of primary gypsum correspond to the interval of low δ34S of AVS/CRM minerals, while during interglacial stages, characterized by relatively low primary gypsum δ34S, the AVS/CRS δ34S is relatively high. This pattern supports the isotopic fractionation decoupling of sulfur phases in the high and stratified lake (during wet glacials) compared to a very weak decoupling during arid interglacials, when the lake is relatively low, mixed and the water column probably oxidized more frequently. High sulfur concentrations are observed during deglacials, representing the deposition of massive gypsum layers.
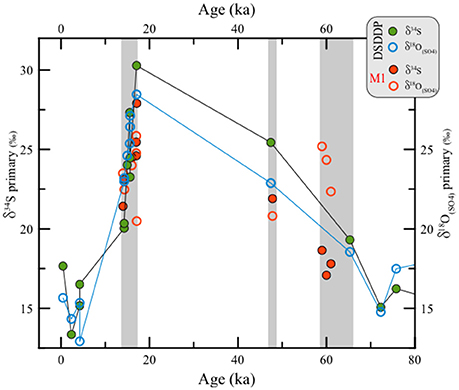
Figure 6. Comparison between δ34S vs. δ18O(SO4) in primary gypsum from the last glacial interval at the DSDDP core and the Masada M1 site. For reference, the displayed ages of different gypsum units pertain to the Lower Gypsum Unit (61–58.5 ka), Curled Gypsum Unit (49–47.5 ka), Upper Gypsum Unit (17.1–15.5 ka) and the Additional Gypsum Unit (14.5 ka) (see stratigraphic definitions of Torfstein et al., 2008).
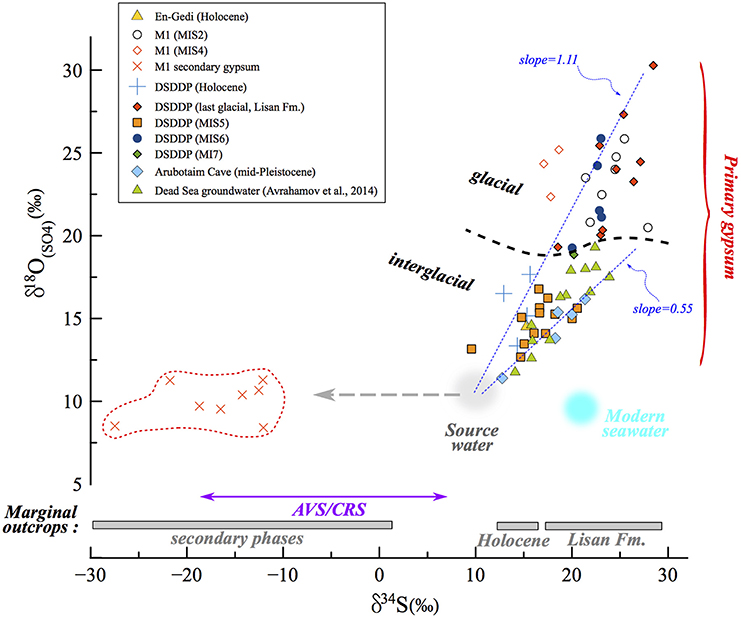
Figure 7. δ34S vs. δ18O(SO4) in the Dead Sea lacustrine environment. The compositions of primary and secondary minerals branch out from the same source sulfate composition, that of influxing freshwater (gray circle). The correlation reveals a clear difference (marked by dashed black curve) between interglacial deposits and those associated with full glacial and deglacial stages. In the land-locked Dead Sea basin, the distribution and evolution patterns of δ34S and δ18O(SO4) are completely independent from the seawater composition (blue circle). In some samples only δ34S was measured (gypsum from marginal outcrops from Torfstein et al., 2008 and AVS/CRS from the DSDDP core), and the range of these data are given in the bottom part of the diagram. The slope of the δ34S vs. δ18O(SO4) has been shown to reflect the rate of microbial sulfate reduction, whereby the steeper the slope, the slower the rate. Accordingly, slopes calculated for interglacial periods (e.g., Amora Fm., Dead Sea groundwater) are systematically lower than those calculated for glacial period; two examples of linear regressions are presented (dashed blue curves): Amora Fm. with a slope of 0.55 and the glacial MIS2 samples yielding a higher slope of 1.11. This implies that the rate of microbial sulfate reduction processes was significantly slower during glacials relative to interglacials.
The δ18O(SO4) of primary gypsum co-varies with δ34S (i.e., low, rising and maximum values during interglacial stages, glacial stages and deglacial terminations, respectively) ranging between 12.7 and 30.3‰ in the DSDDP core and between 20.5 and 25.8‰ in the M1 outcrop (Figures 6, 7). Although, the M1 site is exposed at 370–340 mbsl, approximately 400 m higher than the DSDDP site location, the δ34S and δ18O(SO4) from gypsum measured at both locations are similar in magnitude and in pattern.
No secondary gypsum crystals were found in the DSDDP core, suggesting that this phase forms only after aerial exposure of the sedimentary section on the lake margins. The AVS and CRS sedimentary fraction from the DSDDP core (full circles in Figure 5d) are 34S-depleted ranging between −18.6 and 7.2‰ (Supplementary Table 3). These fractions also vary temporally in opposite sign to the gypsum δ34S and δ18O(SO4), with relatively higher δ34SAVS or δ34SCRS during interglacial stages [when gypsum δ34S and δ18O(SO4) is relatively low] and lower δ34SAVS or δ34SCRS during glacial and deglacial stages [when gypsum δ34S and δ18O(SO4) is relatively high]. Although, no sedimentary sulfide minerals are found in the marginal outcrop (M1), secondary gypsum from this site yields δ34S between −27.5 and 1.0‰ and δ18O(SO4) between 8.4 and 13.6‰, which is distinctly lower than the primary gypsum beds.
The discrete primary gypsum layers are continuous across the lake area, and increase in thickness toward the geographic center of the lake (Torfstein et al., 2008). The main deglacial pulse of gypsum deposition, the “Upper Gypsum Unit” (UGU, 17.1–15.5 ka; Torfstein et al., 2013a), is observed at the Masada M1 outcrop (Figure 2) where it displays a thickness of 1.07 m and δ34S in the range of 24.5–28‰ and δ18O(SO4) in the range of 20.5–26‰. By comparison, the same unit in the DSDDP core is 9.42 m thick (100.94–94.77 mblf), of which 1.35 m is comprised of primary gypsum (the rest are bracketing intervals of marl and aad, as well as a gap of 1.44 m between 100.66 and 99.22 mblf) and has a δ34S and δ18O(SO4) in the range of 25.4–28.5‰ and 23.3–30.3‰, respectively (Supplementary Table 1). Thus, the Upper Gypsum Unit shows remarkable spatial and isotopic consistency, although slightly thicker and slightly 34S- and 18O(SO4)- enriched in the deep center of the lake. Similarly, the Holocene DSDDP interval yields δ34S and δ18O(SO4) in the range of 12.9–15.7 and 13.3–17.7‰ in the primary gypsum, compared to 15.3 and 14.5‰, respectively, in the marginal Holocene Ze'elim Formation.
This correlation in isotopic composition between the DSDDP core and the marginal outcrop can also be observed in additional gypsum beds that have been previously correlated (Torfstein et al., 2015). For example, the two thin gypsum units between 89.72 and 89.47 mblf (equivalent to 14.40–14.26 ±0.5 ka) display δ34S ≈23‰ and δ18O(SO4) ≈20.2‰, compared with the “Additional Gypsum Unit” at Masada [22–24‰; this study and Torfstein et al., 2008; and δ18O(SO4) ≈23‰].
The “Lower Gypsum Unit,” marking the MIS4-3 transition (61–58.5 ka) is present in almost all marginal sites along the Dead Sea, yet its correlation to the DSDDP core is more challenging because the corresponding interval is characterized by the frequent occurrence of thinner gypsum laminae. An analysis of one of these gypsum laminae close to the base of the Lisan Fm. in the DSDDP core (170.17 mblf) yielded δ34S and δ18O(SO4) of 18.5 and 19.3‰. In this case, the δ34S is comparable with those of the Lower Gypsum Unit at M1 (17.1–18.7‰) but the δ18O(SO4) is much lower than those observed at M1 (22.4–25.2‰).
Discussion
Source Water δ34S and δ18O(SO4)
The sulfur isotopic composition of aqueous sulfate in the Dead Sea lacustrine system, and therefore the gypsum that precipitates within this system, reflects the balance between the source composition (local runoff δ34S ranges between 6 and 14‰; Gavrieli et al., 2001) and the amount of water column microbial sulfate reduction, which is controlled by the stability and length of water column stratification leading to the deep-lake anoxia. The oxygen isotopic composition of sulfate is controlled by the same processes, but is particularly sensitive to the incorporation of oxygen atoms during sulfide oxidation, and the degree of intracellular equilibration between intermediate valence state sulfur species and water. Avrahamov et al. (2014) reported one sample of marginal Dead Sea groundwaters with δ18O(SO4) = 13.6‰ (with δ34S = 15.9‰), with saline spring waters along the modern Dead Sea displaying a range of typically higher δ18O(SO4) up to 19.3‰. Khayat et al. (2006) also studied groundwater north of the Dead Sea, around Jericho, and although these are possibly contaminated by anthropogenic sources, the reported δ18O(SO4) ranged between 1.4 and 17.3‰, similar to the range measured in the DSDDP core and M1 section. Until more detailed characterization of the Dead Sea drainage is done, it is reasonable to assume an average freshwater δ18O(SO4) in the Dead Sea Basin of ~10‰ (gray circle in Figure 7) given the range of previously measured ground water, springs and other fluids in the area.
Coupling and Decoupling of δ34S and δ18O(SO4)
Our data is characterized by two primary features (Figure 5): 1. Temporal variations in the δ34S and δ18O(SO4) of the gypsum which correlate with glacial–interglacial cycles, including a strong transient positive δ34S and δ18O(SO4) perturbation during deglaciations, 2. Decoupling of the sign of δ34S between sedimentary sulfate and sulfide minerals, with anticorrelated isotopic trends during different climatic stages. We will consider the former observation first.
Although, there is a clear temporal change in the δ34S and δ18O(SO4) over glacial-interglacial cycles, before we can interpret changes in redox dynamics we need to determine whether there are changes in the oxygen isotope composition of the lake water over these intervals which may be reflected in the δ18O(SO4) of the sulfate, and thus recorded in the δ18O(SO4) of the primary gypsum. To a first order, changes in δ18O measured in aragonite follow closely the patterns of δ18O changes measured in the Soreq Cave and East Mediterranean surface water with higher δ18O corresponding to glacial stages and lower δ18O in interglacial, reflecting the control of temporal changes in the East Mediterranean source water composition on δ18O compositions both in the cave and in the lake (see further discussion in Kolodny et al., 2005).
Figure 5 shows that the aragonite δ18O is decoupled from both the δ34S and δ18O(SO4) measured in the primary gypsum. The aragonite-derived δ18O is also consistently lower than the δ18O (of the water) measured in the sedimentary pore fluids (Figure 5b). Considering an observed coeval drop in chloride concentrations in interstitial waters (Figure 5c), Lazar et al. (2014) concluded that the lake bottom waters underwent significant freshening during the last glacial cycle with chloride concentrations in the monimolimnion decreasing to about 70% of their modern value, and the δ18O of the lake water reached ~7‰, about 3‰ higher than present. Such a freshening of the lake would lower salinity and allow for higher sulfate concentrations to be maintained in the lake water (residence time of ~100–1,500 years) because they are controlled largely by gypsum saturation (Torfstein et al., 2005). Thus, the larger the lake volume during the glacial cycle, the bigger the pool of 34S- and 18O- enriched residual sulfate in the water column. When the lake destabilizes and declines during the glacial termination, the precipitating gypsum records the isotopic fingerprint and is characterized by higher δ34S and δ18O(SO4). Accordingly, the thickness of the gypsum beds that precipitate during each glacial termination, and their sulfur and oxygen isotopic composition, are positively correlated and are a qualitative metric for the intensity of the stratification and the sulfate concentration in the lake during the glacial cycle.
The second observation pertains to an anticorrelation in the δ34S between the gypsum phases and the sedimentary sulfide AVS and CRS fraction; in general, when the δ34S of the gypsum is high, the sedimentary sulfide is low, and vice versa. One interpretation of this is that when the lake was less stratified (during interglacial lowstands) there is less water column microbial sulfate reduction and thus less opportunity to separate various phases chemically or isotopically. Another possibility is that during interglacials when there is less separation in the sulfur isotope composition of sulfate and sulfide, microbial sulfate reduction is limited to the surface sediment and behaves more like a “closed system” where the available sulfate is quantitatively converted to pyrite and thus retains a similar δ34S. The observation that sedimentary sulfides from glacial and deglacial periods display significantly lower δ34S, mirrors the 34S-enrichment of coeval gypsum layers and likely reflects the strong water-column stratification and the separation of the two phases through more open-system microbial sulfate reduction.
Following lake level decline and exposure of the sedimentary column, percolation of meteoric freshwater (δ18OH2O = −6 to −3‰) caused oxidation of reduced sulfur phases in the now-exposed sediments. This explains why sedimentary sulfide minerals are found in the DSDDP core but not in the exposed outcrop. The meteoric fluids percolated along the carbonate sediments, particularly along detrital laminae, which are more permeable than aragonite laminae, and became calcium-enriched. This in turn, favored secondary gypsum crystallization after oxidation of sedimentary sulfides. The δ34S fingerprint of this gypsum is similar to that of the original sedimentary sulfide minerals (34S-depleted). By contrast, the oxygen isotope composition of the sulfate would depend on where the sulfate derived its oxygen atoms from and any isotope fractionation associated with their incorporation into the sulfate molecule. Typically, during sulfide oxidation the canonical assumption is that one oxygen atom derives from the atmosphere (+23‰) and the other three from water; this has been shown in pure culture microbial studies and applied in a range of riverine environments to understand the pathway of pyrite oxidation (e.g., Balci et al., 2007; Calmels et al., 2007). However, it appears that the secondary gypsum in the M1 section derives oxygen atoms nearly perfectly as a 50/50 mix between water and atmospheric oxygen (Figure 8), hinting that during this oxidation, two of the oxygen atoms derive from percolating meteoric water and two from atmospheric oxygen. There is a possibility that this represents a mixture of sources of oxygen atoms and isotope fractionation on their incorporation, or isotope equilibration with water in an intermediate valence state. We suggest however, similar to the interpretation of the oxygen isotope composition of sulfate in river systems and in pure culture studies, that there is negligible oxygen isotope fractionation on sulfide oxidation and that intermediate valence state sulfur species, if created, are too short lived to isotopically equilibrate with water. If our suggestion is correct, it is the first observation of an equal contribution of oxygen atoms from the two sources. We postulate that in the hyperarid conditions under which the epigenetic oxidation occurred, the incorporation mechanism of oxygen into sulfate is different than previously reported.
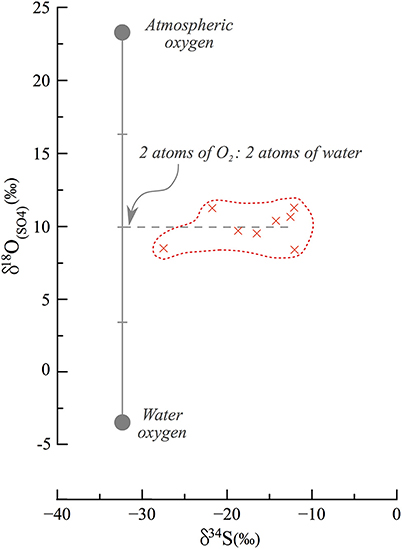
Figure 8. End member mixing and oxidation pathways in secondary gypsum. Assuming that atmospheric oxygen (23‰) and local meteoric water (−6 to −3‰) are the source of oxygen to the oxidation process of sulfides, they each contribute an equal fraction of oxygen atoms. Previous studies have typically discussed the contribution of one O2 atom from atmospheric oxygen and 3 atoms from water, and this is the first such observation of an implied equal contribution.
The Microbial Sulfur Cycle in Lake Lisan
The relative change in the sulfur and oxygen isotope composition of sulfate during microbial sulfate reduction reflects the degree of intracellular recycling of sulfur compounds and is related to the sulfate reduction rate (Brunner et al., 2005; Brunner and Bernasconi, 2005; Antler et al., 2014, 2013). Specifically, the slope between the sulfur and oxygen isotopes in sulfate over the course of sulfate reduction has been shown to correlate with the cell specific rate of sulfate reduction (Antler et al., 2013). The faster the rate of sulfate reduction, the faster the sulfur isotope composition of the residual sulfate increases relative to the oxygen isotope composition of the residual sulfate and accordingly, the slope of the cross plot of δ34S vs. δ18O(SO4) regression becomes less steep. The slope between sulfur and oxygen isotopes in sulfate has been shown to have a unique value when microbial sulfate reduction is coupled to anaerobic methane oxidation, ca. 0.39 (Antler et al., 2015). Anaerobic methane oxidation has been suggested to occur in the modern Dead Sea, but understanding the extent of methane production and oxidation during different climate intervals is a key question for understanding how the carbon budget in lakes may change with regional hydrological changes.
We take our measured δ18O(SO4) and δ34S of gypsum as representing the sulfate in the lake during stratification, assuming minimal sulfur or oxygen isotope fractionation on gypsum precipitation. The slope for the full set of sulfur and oxygen isotope data measured in primary gypsum is 0.85, but when calculating the slope for specific time intervals at each site, a difference between glacial and interglacial periods is revealed. The calculated slope ranges between 0.55 for the Amora Fm. interglacial sequence at the Arubotaim Cave site, which is only slightly lower than the slope of modern Dead Sea groundwater (0.68) (Figures 7, 9). By contrast, a much higher slope between 1.11 (MIS2) and 2.0 (MIS4) is identified at M1 during glacial intervals (for clarity, Figure 7 displays only the Amora Fm. and MIS2 slopes).
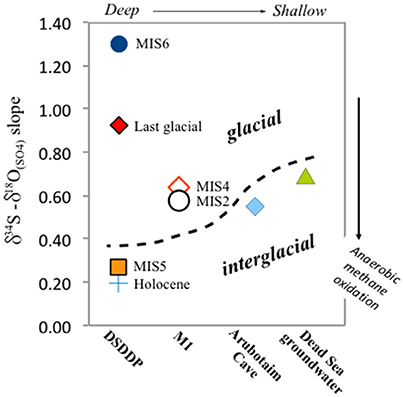
Figure 9. Slope values between δ34S and δ18O(SO4) from different time intervals at different sites.
This significant change in the slope between sulfur and oxygen isotopes in sulfate recorded in gypsum at different times implies that microbial sulfate reduction operates fundamentally differently between glacial and interglacial conditions, when water levels were high (fresher lake) and low (more saline lake), respectively. During interglacials, sulfate reduction is confined to the sediments (as it is today), and the rate derived from the δ18O(SO4) vs. δ34S crossplot suggests the rate of microbial sulfate reduction is faster relative to glacial stages. This could be because of the presence of methane, and that sulfate may be dominantly reduced through anaerobic methane oxidation, associated with a low, unique slope (~0.39; Antler et al., 2015). This fast rate of microbial sulfate reduction will also contribute to the higher overall sulfur isotope composition of the sedimentary sulfide minerals discussed above, in addition to the possible “closed system” behavior. In contrast, in glacial times when the lake was much larger, deeper, and stably stratified, microbial sulfate reduction occurred in the open water column. Although, this yields overall higher δ18O(SO4) and δ34S, the relative change in them is different relative to interglacial periods and corresponds to slower overall rates of microbial sulfate reduction. Our results suggest that the cell specific sulfate reduction rate is as much as two orders of magnitude faster in the modern basin (and more generally during interglacials) than it was during glacial periods when the lake was stratified and displayed significantly higher water level and higher lake sulfate concentrations.
These results have implications for interpreting the behavior of the microbial sulfur cycle during changes in lake salinity and regional hydrology. A conceptual summary of the evolution of these different lake parameters across Glacial—Interglacial cycles is given in Figure 10. Measuring the sulfur isotopes or oxygen isotopes in sulfate in isolation, they behave as we would have predicted; during lake stratification there is a stronger distillation of the isotopes between isotopically enriched oxidized phases in the lake and isotopically depleted reduced phases in the sediment. This is moderated during interglacials when sulfate reduction is limited to the sediments. However, the relative change in δ18O(SO4) vs. δ34S suggests that the overall cell specific rate slows down when microbial sulfate reduction occurs in the stratified water column (primarily during glacial times), in spite of overall higher sulfate concentrations. We infer that this may be due to a change in the carbon source, where during the glacial fresher lake periods, an increase in primary production in the surface water (as suggested by δ13C values in aragonite; Kolodny et al., 2005) supplies the primary carbon source for microbial sulfate reduction in the water column. In contrast, during interglacial periods, when the lake is low, mixed and more saline, primary production may be virtually absent, and the carbon source for microbial sulfate reduction is methane produced in the sediment from the reduction of previously deposited organic carbon. Microbial sulfate reduction also migrates from the water column to the sediment, and the overall rate increases. These results suggest that rates of microbial sulfate reduction may be independent of salinity even in hypersaline conditions.
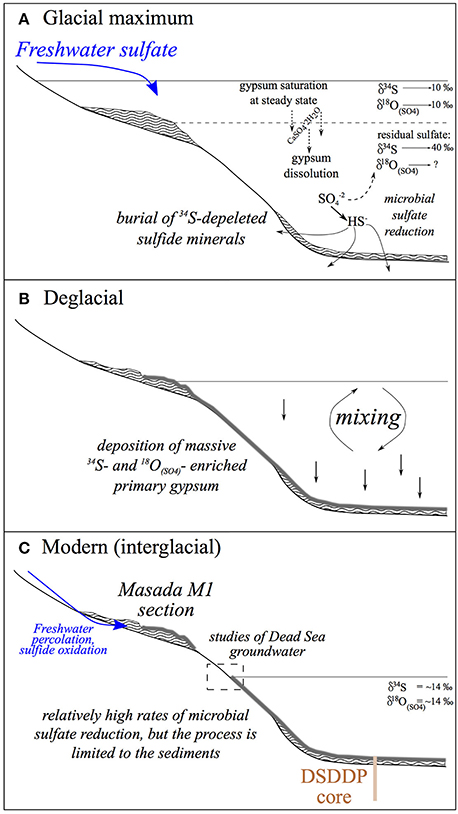
Figure 10. Conceptual evolution patterns of the Dead Sea lacustrine system across Glacial—Interglacial cycles. (A) Glacial—large freshwater influx supports a high stand and density stratification. The overall reservoir of 34S-enriched sulfate in the lake is highest during the glacial, (B) Deglacial—a regional shift from wet to dry conditions drives a lake level drop that causes water column mixing and massive precipitation of primary gypsum, (C) Modern (interglacial)—dry climatic conditions impose low water stands and a mixed water column. Microbial sulfate reduction is limited to the interstitial zone in the sediments but occurs at faster rates relative to glacial times. Marginal glacial deposits are exposed and occasional freshwater percolation through the sections results in oxidation of sedimentary sulfide to sulfate. These continue to crystalize and form secondary gypsum. This process is unique to the margins and is not recorded in the deep submerged sediments of the DSDDP.
Summary
The temporal dynamics and rates of microbial sulfate reduction were studied in the terminal, hypersaline Dead Sea lacustrine system over the last 200 kyrs, based on δ34S and δ18O(SO4) in the sedimentary record. The sulfur and oxygen isotopic composition of sulfate in the Dead Sea has evolved through multiple cycles of reduction and oxidation that are controlled by the regional hydrological regime. During wet glacial stages, influxing freshwater imposed stratified conditions, which resulted in the consumption of oxygen in the monimolimnion (lower brine) and development of anoxic conditions. These drove microbial sulfate reduction that impart the water column sulfate into reduced sulfide phases that are 34S-depleted, and residual water 34S- and 18O- enriched sulfate. Our results suggest that despite the more favorable conditions, the rate of microbial sulfate reduction during glacials is actually slower than the rate during interglacials, when the lake level is lower and the lake is mixed and oxidized, confining microbial sulfate reduction to the sediments.
Analyses of secondary gypsum samples that evolved from the oxidation of sulfidic phases, reveals extremely 34S-depleted compositions, but higher δ18O(SO4) suggesting that oxidation of sedimentary sulfide phases was facilitated by contact with meteoric surface water after the lake retreated. Thus, following their deposition, sedimentary sulfides was maintained in an oxygen-depleted environment until their recent exposure to the atmosphere. Moreover, the oxygen atoms in sulfate associated with secondary gypsum minerals are derived at a 1:1 ratio from the atmosphere and water. This ratio is globally unique to the Dead Sea basin, probably due to the hyperarid conditions that prevail in this area.
The coupling of δ34S and δ18O(SO4) in the Dead Sea system has been shown to be highly sensitive to the regional hydrological budget, and can be used to reconstruct the limno-hydrology in older sequences of the Dead Sea record, as well as in other comparable environments elsewhere.
Author Contributions
AT performed the field work and sampling. AVT processed the samples in the lab and analyzed them. Both AT and AVT performed data analyses and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DTW and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
Funding was provided by ERC Starting Investigator Grant 307582 to AVT and the Comer Science & Education Foundation grant CP69 to AT. The authors are grateful to Sam Johnson who extracted the sedimentary sulfides during a summer project. Constructive reviews by Susan Childers and David T. Wang helped to greatly improve this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/feart.2017.00062/full#supplementary-material
References
Aharon, P., and Fu, B. (2000). Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deepwater Gulf of Mexico. Geochim. Cosmochim. Acta 64, 233–246. doi: 10.1016/S0016-7037(99)00292-6
Antler, G., Turchyn, A. V., Herut, B., Davies, A., Rennie, V. C. F., and Sivan, O. (2014). Sulfur and oxygen isotope tracing of sulfate driven anaerobic methane oxidation in estuarine sediments. Estuar. Coast. Shelf Sci. 142, 4–11. doi: 10.1016/j.ecss.2014.03.001
Antler, G., Turchyn, A. V., Herut, B., and Sivan, O. (2015). A unique isotopic fingerprint of sulfate-driven anaerobic oxidation of methane. Geology 43, 619–622. doi: 10.1130/G36688.1
Antler, G., Turchyn, A. V., Rennie, V., Herut, B., and Sivan, O. (2013). Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. Cosmochim. Acta 118, 98–117. doi: 10.1016/j.gca.2013.05.005
Arndt, S., Jørgensen, B. B., LaRowe, D. E., Middelburg, J. J., Pancost, R. D., and Regnier, P. (2013). Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth Sci. Rev. 123, 53–86. doi: 10.1016/j.earscirev.2013.02.008
Avrahamov, N., Antler, G., Yechieli, Y., Gavrieli, I., Joye, S. B., Saxton, M., et al. (2014). Anaerobic oxidation of methane by sulfate in hypersaline groundwater of the dead sea aquifer. Geobiology 12, 511–528. doi: 10.1111/gbi.12095
Baas-Becking, L., and Kaplan, I. (1956). The microbiological origin of the sulphur nodules of lake eyre. Trans. R. Soc. South Aust. 79, 52–65.
Balci, N., Shanks, W. C., Mayer, B., and Mandernack, K. W. (2007). Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite. Geochim. Cosmochim. Acta 71, 3796–3811. doi: 10.1016/j.gca.2007.04.017
Bartov, Y., Goldstein, S. L., Stein, M., and Enzel, Y. (2003). Catastrophic arid episodes in the eastern mediterranean linked with the North Atlantic Heinrich events. Geology 31, 439–442. doi: 10.1130/0091-7613(2003)031<0439:CAEITE>2.0.CO;2
Begin, Z., Ehrlich, A., and Nathan, Y. (1974). Lake Lisan, the Pleistocene precursor of the dead sea. Geol. Surv. Isr. Bull. 63, 1–30.
Belmaker, R., Lazar, B., Beer, J., Christl, M., Tepelyakov, N., and Stein, M. (2013). 10Be dating of Neogene halite. Geochim. Cosmochim. Acta 122, 418–429. doi: 10.1016/j.gca.2013.08.033
Bishop, T., Turchyn, A., and Sivan, O. (2013). Fire and brimestone: the microbially mediated formation of elemental sulfur nodules from an isotope and major element study in the paleo-dead sea. PLoS ONE 8:e75883. doi: 10.1371/journal.pone.0075883
Bookman Ken-Tor, R., Enzel, Y., Agnon, A., and Stein, M. (2004). Late Holocene lake levels of the dead sea. Geol. Soc. Am. Bull. 116, 555–571. doi: 10.1130/B25286.1
Bottrell, S. H., and Newton, R. J. (2006). Reconstruction of changes in global sulfur cycling from marine sulfate isotopes. Earth Sci. Rev. 75, 59–83. doi: 10.1016/j.earscirev.2005.10.004
Brunner, B., Bernasconi, S., Kleikemper, J., and Schroth, M. (2005). A model for oxygen and sulfur isotope fractionation in sulfate during bacterial sulfate reduction processes. Geochim. Cosmochim. Acta 69, 4773–4785. doi: 10.1016/j.gca.2005.04.017
Brunner, B., and Bernasconi, S. M. (2005). A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria. Geochim. Cosmochim. Acta 69, 4759–4771. doi: 10.1016/j.gca.2005.04.015
Calmels, D., Gaillardet, J., Brenot, A., and France-Lanord, C. (2007). Sustained sulfide oxidation by physical erosion processes in the Mackenzie river basin: climatic perspectives. Geology 35, 1003–1006. doi: 10.1130/G24132A.1
Canfield, D. E. (1998). A new model for Proterozoic ocean chemistry. Nature 396, 450–453. doi: 10.1038/24839
Canfield, D. E. (2004). The evolution of the earth surface sulfur reservoir. Am. J. Sci. 304, 839–861. doi: 10.2475/ajs.304.10.839
Chambers, L. A., and Trudinger, P. A. (1979). Microbiological fractionation of stable sulfur isotopes: a review and critique. Geomicrobiol. J. 1, 249–293. doi: 10.1080/01490457909377735
Claypool, G., Holser, W., Kaplan, I., Sakai, H., and Zak, I. (1980). The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretation. Chem. Geol. 28, 199–260. doi: 10.1016/0009-2541(80)90047-9
Cypionka, H., Smock, A. M., and Boettcher, M. E. (1998). A combined pathway of sulfur compound disproportionation in desulfovibrio desulfuricans. FEMS Microbiol. Lett. 166, 181–186. doi: 10.1111/j.1574-6968.1998.tb13888.x
Enzel, Y., Bookman Ken Tor, R., Sharon, D., Gvirtzman, H., Dayan, U., Ziv, B., et al. (2003). Late holocene climates of the near east deduced from dead sea level variations and modern regional winter rainfall. Quat. Res. 60, 263–273. doi: 10.1016/j.yqres.2003.07.011
Eugster, H. (1980). Geochemistry of evaporitic lacustrine deposits. Annu. Rev. Anthropol. 8, 35–63. doi: 10.1146/annurev.ea.08.050180.000343
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090. doi: 10.1016/0016-7037(79)90095-4
Gavrieli, I., and Stein, M. (2006). On the origin and fate of the brines in the dead sea basin. Geol. Soc. Am. Spec. Pap. 401, 183–194. doi: 10.1130/2006.2401(12)
Gavrieli, I., Yechieli, Y., Halicz, L., Spiro, B., Bein, A., and Efron, D. (2001). The sulfur system in anoxic subsurface brines and its implication in brine evolutionary pathways: the Ca-chloride brines in the Dead Sea area. Earth Planet. Sci. Lett. 186, 199–213. doi: 10.1016/S0012-821X(01)00247-3
Haase-Schramm, A., Goldstein, S. L., and Stein, M. (2004). U-Th dating of lake lisan (late Pleistocene dead sea) aragonite and implications for glacial east mediterranean climate change. Geochim. Cosmochim. Acta 68, 985–1005. doi: 10.1016/j.gca.2003.07.016
Habicht, K., Canfield, D., and Rethmeier, J. (1998). Sulfur isotope fractionation during bacterial reduction and disproportionation of thiosulfate and sulfite. Geochim. Cosmochim. Acta 62, 2585–2595. doi: 10.1016/S0016-7037(98)00167-7
Haliva-Cohen, A., Stein, M., Goldstein, S. L., Sandler, A., and Starinsky, A. (2012). Sources and transport routes of fine detritus material to the late quaternary dead sea basin. Quat. Sci. Rev. 50, 55–70. doi: 10.1016/j.quascirev.2012.06.014
Kaplan, I., and Rittenberg, S. (1964). Microbiological fractionation of sulphur isotopes. J. Gen. Microbiol. 34, 195–212. doi: 10.1099/00221287-34-2-195
Katz, A., Kolodny, Y., and Nissenbaum, A. (1977). The geochemical evolution of the pleistocene lake lisan-dead sea system. Geochim. Cosmochim. Acta 41, 1609–1626. doi: 10.1016/0016-7037(77)90172-7
Katz, A., and Starinsky, A. (2008). Geochemical history of the dead sea. Aquat. Geochem. 15, 159–194. doi: 10.1007/s10498-008-9045-0
Kaufman, A. (1971). U-Series dating of dead sea basin carbonates. Geochim. Cosmochim. Acta 35, 1269–1281. doi: 10.1016/0016-7037(71)90115-3
Khayat, S., Hötzl, H., Geyer, S., Ali, W., Knöller, K., and Strauch, G. (2006). Sulphur and oxygen isotopic characters of dissolved sulphate in groundwater from the pleistocene aquifer in the southern Jordan valley (Jericho area, Palestine). Isotopes Environ. Health Stud. 42, 289–302. doi: 10.1080/10256010600839780
Knossow, N., Blonder, B., Eckert, W., Turchyn, A. V., Antler, G., and Kamyshny, A. (2015). Annual sulfur cycle in a warm monomictic lake with sub-millimolar sulfate concentrations. Geochem. Trans. 16:7. doi: 10.1186/s12932-015-0021-5
Kolodny, Y., Stein, M., and Machlus, M. (2005). Sea-rain-lake relation in the last glacial east mediterranean revealed by δ18O-δ13C in lake lisan aragonites. Geochim. Cosmochim. Acta 69, 4045–4060. doi: 10.1016/j.gca.2004.11.022
Lazar, B., Sivan, O., Yechieli, Y., Levy, E. J., Antler, G., Gavrieli, I., et al. (2014). Long-term freshening of the dead sea brine revealed by porewater Cl- and d18O in ICDP dead Sea deep-drill. Earth Planet. Sci. Lett. 400, 94–101. doi: 10.1016/j.epsl.2014.03.019
Leavitt, W. D., Halevy, I., Bradley, A. S., and Johnston, D. T. (2013). Influence of sulfate reduction rates on the phanerozoic sulfur isotope record. Proc. Natl. Acad. Sci. U.S.A. 110, 11244–11249. doi: 10.1073/pnas.1218874110
Lu, F., Meyers, W., and Schoonen, M. (2001)., S and O (SO4) isotopes, simultaneous modeling, and environmental significance of the Nijar messinian gypsum, Spain. Geochim. Cosmochim. Acta 65, 3081–3092. doi: 10.1016/S0016-7037(01)00553-1
Lyons, T. (1997). Sulfur isotopic trends and pathways of iron sulfide formation in upper holocene sediments of the anoxic black sea. Geochim. Cosmochim. Acta 61, 3367–3382. doi: 10.1016/S0016-7037(97)00174-9
Matmon, A., Fink, D., Davis, M., Niedermann, S., Rood, D., and Frumkin, A. (2014). Unraveling rift margin evolution and escarpment development ages along the dead sea fault using cosmogenic burial ages. Quat. Res. 82, 281–295. doi: 10.1016/j.yqres.2014.04.008
Neretin, L. N., Böttcher, M. E., and Grinenko, V. A. (2003). Sulfur isotope geochemistry of the black sea water column. Chem. Geol. 200, 59–69. doi: 10.1016/S0009-2541(03)00129-3
Neugebauer, I., Brauer, A., Schwab, M. J., Waldmann, N., Enzel, Y., Kitagawa, H., et al. (2014). Lithology of the long sediment record recovered by the ICDP dead sea deep drilling project (DSDDP). Quat. Sci. Rev. 102, 149–165. doi: 10.1016/j.quascirev.2014.08.013
Oldenburg, T. B. P., Rullkotter, J., Bottcher, M. E., and Nissenbaum, A. (2000a). Addendum to molecular and isotopic characterization of organic matter in recent and sub-recent sediments from the dead sea, [Organic, Geochemistry, 31 (2000). 251–265]. Org. Geochem. 31, 773–774. doi: 10.1016/S0146-6380(00)00079-6
Oldenburg, T. B. P., Rullkötter, J., Böttcher, M. E., and Nissenbaum, A. (2000b). Molecular and isotopic characterization of organic matter in recent and sub-recent sediments from the dead sea. Org. Geochem. 31, 251–265. doi: 10.1016/S0146-6380(00)00015-2
Rees, C. E. (1973). A steady-state model for sulphur isotope fractionation in bacterial reduction processes. Geochim. Cosmochim. Acta 37, 1141–1162. doi: 10.1016/0016-7037(73)90052-5
Sim, M. S., Bosak, T., and Ono, S. (2011). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Starinsky, A. (1974). Relationship between Ca-chloride Brines and Sedimentary Rocks in Israel. Ph.D. dissertation, The Hebrew University of Jerusalem.
Stein, M. (2001). The sedimentary and geochemical record of neogene-quaternary water bodies in the dead sea basin – inferences for the regional paleoclimatic history. J. Paleolimnol. 26, 271–282. doi: 10.1023/A:1017529228186
Stein, M., Agnon, A., Katz, A., and Starinsky, A. (2002). Strontium isotopes in discordant dolomite bodies of the Judea group, dead sea basin. Isr. J. Earth Sci. 51, 219–224. doi: 10.1560/61UM-UMQF-YU86-JNU0
Stein, M., Starinsky, A., Agnon, A., Katz, A., Raab, M., Spiro, B., et al. (2000). The impact of brine-rock interaction during marine evaporite formation on the isotopic Sr record in the oceans: evidence from Mt. Sedom, Israel. Geochim. Cosmochim. Acta 64, 2039–2053. doi: 10.1016/S0016-7037(00)00370-7
Stein, M., Starinsky, A., Katz, A., Goldstein, S. L., Machlus, M., and Schramm, A. (1997). Strontium isotopic, chemical, and sedimentological evidence for the evolution of lake lisan and the dead sea. Geochim. Cosmochim. Acta 61, 3875–3992. doi: 10.1016/S0016-7037(97)00191-9
Strauss, H. (1997). The isotopic composition of sedimentary sulfur through time. Palaeogeogr. Palaeoclimatol. Palaeoecol. 132, 97–118. doi: 10.1016/S0031-0182(97)00067-9
Szynkiewicz, A., Moore, C. H., Glamoclija, M., and Pratt, L. M. (2009) Sulfur isotope signatures in gypsiferous sediments of the Estancia Tularosa basins as indicators of sulfate sources, hydrological processes, microbial activity. Geochim. Cosmochim. Acta 73, 6162–6186. doi: 10.1016/j.gca.2009.07.009
Thomas, C., Ebert, Y., Kiro, Y., Stein, M., and Ariztegui, D. (2016). Microbial sedimentary imprint on the deep dead sea sediment. Depos. Rec. 118–138. doi: 10.1002/dep2.16
Torfstein, A., Gavrieli, I., Katz, A., Kolodny, Y., and Stein, M. (2008). Gypsum as a monitor of the paleo-limnological–hydrological conditions in lake lisan and the dead sea. Geochim. Cosmochim. Acta 72, 2491–2509. doi: 10.1016/j.gca.2008.02.015
Torfstein, A., Gavrieli, I., and Stein, M. (2005). The sources and evolution of sulfur in the hypersaline lake lisan (paleo-dead sea). earth planet. Sci. Lett. 236, 61–77. doi: 10.1016/j.epsl.2005.04.026
Torfstein, A., Goldstein, S., Kagan, E. J., and Stein, M. (2013a). Integrated multi-site U–Th chronology of the last glacial Lake Lisan. Geochim. Cosmochim. Acta 104, 210–231. doi: 10.1016/j.gca.2012.11.003
Torfstein, A., Goldstein, S. L., Kushnir, Y., Enzel, Y., Haug, G. H., and Stein, M. (2015). Dead sea drawdown and monsoonal impacts in the levant during the last interglacial. Earth Planet. Sci. Lett. 412, 235–244. doi: 10.1016/j.epsl.2014.12.013
Torfstein, A., Goldstein, S., Stein, M., and Enzel, Y. (2013b). Impacts of abrupt climate changes in the levant from last glacial dead sea levels. Quat. Sci. Rev. 69, 1–7. doi: 10.1016/j.quascirev.2013.02.015
Torfstein, A., Haase-Schramm, A., Waldmann, N., Kolodny, Y., and Stein, M. (2009). U-series and oxygen isotope chronology of the mid-pleistocene lake Amora (dead sea basin). Geochim. Cosmochim. Acta 73, 2603–2630. doi: 10.1016/j.gca.2009.02.010
Tuttle, M. L., and Goldhaber, M. B. (1993). Sedimentary sulfur geochemistry of the paleogene green river formation, western USA: implications for interpreting depositional and diagenetic processes in saline alkaline lakes. Geochim. Cosmochim. Acta 57, 3023–3039. doi: 10.1016/0016-7037(93)90291-4
Watanabe, T., Naraoka, H., Nishimura, M., and Kawai, T. (2004). Biological and environmental changes in lake Baikal during the late quaternary inferred from carbon, nitrogen and sulfur isotopes. Earth Planet. Sci. Lett. 222, 285–299. doi: 10.1016/j.epsl.2004.02.009
Wortmann, U., Chernyavsky, B., Bernasconi, S., Brunner, B., Böttcher, M., and Swart, P. (2007). Oxygen isotope biogeochemistry of pore water sulfate in the deep biosphere: dominance of isotope exchange reactions with ambient water during microbial sulfate reduction (ODP Site 1130). Geochim. Cosmochim. Acta 71, 4221–4232. doi: 10.1016/j.gca.2007.06.033
Keywords: Dead Sea, microbial sulfate reduction, isotope fractionation, sulfates, sulfide oxidation, paleolimnology
Citation: Torfstein A and Turchyn AV (2017) Rates and Cycles of Microbial Sulfate Reduction in the Hyper-Saline Dead Sea over the Last 200 kyrs from Sedimentary δ34S and δ18O(SO4). Front. Earth Sci. 5:62. doi: 10.3389/feart.2017.00062
Received: 26 March 2017; Accepted: 11 July 2017;
Published: 02 August 2017.
Edited by:
Tanja Bosak, Massachusetts Institute of Technology, United StatesReviewed by:
Susan Childers, Colby College, United StatesDavid T. Wang, Massachusetts Institute of Technology, United States
Copyright © 2017 Torfstein and Turchyn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adi Torfstein, YWRpLnRvcmZAbWFpbC5odWppLmFjLmls