 Ruth S. Hindshaw
Ruth S. Hindshaw Sarah M. Aciego2
Sarah M. Aciego2- 1Department of Earth Sciences, University of Cambridge, Cambridge, United Kingdom
- 2Department of Earth and Environmental Sciences, University of Michigan, Ann Arbor, MI, United States
Permafrost in the Arctic is decreasing in extent and the depth of the seasonally thawed layer, the active layer, is increasing. Increased exposure to water is increasing fluxes of organic and inorganic solutes with potential impacts for the global carbon cycle and downstream ecosystems. Understanding the relationship between solute release and active layer depth will be critical for modeling environmental impact, especially in inaccessible regions where there is a lack of data. In this study, we focus on the potential for the isotopes of lithium (Li) and uranium (U) to track active layer extent in two permafrost-dominated catchments in Svalbard: one glaciated and one unglaciated. These isotope systems can be measured to a much higher precision than concentration measurements and act as sensitive tracers of environmental change. The extent of Li isotope fractionation provides information on the balance between dissolution of primary phases and formation of secondary phases, such as clay minerals and oxides. The U activity ratio provides information on water-rock interaction times and physical properties. We observe contrasting behavior between the two catchments. The highest U activity ratios and Li isotope values (those most distinct from bedrock) are observed in summer in the unglaciated catchment, when the active layer depth is expected to be at its maximum extent, whereas negligible seasonal variation and the lowest values are observed in the glaciated catchment. We therefore propose that the extent of solute acquisition is directly linked to the active layer depth, which is restricted in the glaciated catchment due to a layer of “dead ice” underneath the glacial outwash plain, and could therefore provide a valuable tool to assess changes in active layer depth at catchment scales.
1. Introduction
The Arctic is currently experiencing a marked warming trend in comparison to other regions of the world, with average temperatures now more than 1.5°C warmer than the 1971–2000 average (Overland et al., 2013). Permafrost (soil that has remained frozen for at least 2 consecutive years) is a key feature of northern ecosystems and one consequence of increased temperatures has been a decrease in the extent of continuous permafrost (Chadburn et al., 2017) and an increase in the depth of the active-layer (the seasonally thawed upper-layer of permafrost) (Frey and McClelland, 2009; Schaefer et al., 2011). Increases in the active layer depth and degradation of permafrost will increase water-rock/soil interaction (Bring et al., 2016) and could lead to increased fluxes of organic and inorganic elements. Long-term measurements indicate that annual chemical fluxes of major elements are increasing in several large Arctic rivers (Frey et al., 2007; Frey and McClelland, 2009; Tank et al., 2016; Toohey et al., 2016). In the Mackenzie and Yukon rivers, an increase in sulfate flux has been linked to the exposure of previously frozen and unweathered material to water and the atmosphere, leading to an increase in the chemical weathering of sulfide minerals (Tank et al., 2016; Toohey et al., 2016). Similarly, a decrease in the radiogenic strontium isotopic composition of a stream in Alaska was attributed to an increase in the weathering of carbonate minerals exposed as a result of active-layer deepening (Keller et al., 2010). Other studies have pointed to the role of flushing during spring melt, which removes previously frozen solutes that became concentrated due to solute exclusion during freezing, contributing to seasonal variations (Kokelj and Burn, 2005; Lamhonwah et al., 2017; Lehn et al., 2017). Trace metal concentrations (Barker et al., 2014) and several metal isotope systems (Sr, U, Ca, Mg, Si, Keller et al., 2010; Bagard et al., 2011, 2013; Pokrovsky et al., 2013; Mavromatis et al., 2016; Lehn et al., 2017) have also been used as tracers of chemical weathering during the seasonal cycle of active-layer thaw and re-freeze. However, not all studies have found evidence for increased chemical weathering with an increase in active layer depth (Dean et al., 2016). The use of major elements to gain information on the changing conditions of the active-layer is complicated by their varied sources (e.g., different minerals) and processes (e.g., plant uptake and secondary mineral formation) in catchments, which contributes to uncertainty in their application. Therefore, there is a pressing need to develop new tracers of how biogeochemical processes are affected by permafrost thaw. Isotope tracers may provide a more robust tool as they can be measured to high precision, permitting source effects to be separated from processes. In this study we explore the utility of two different isotope systems (lithium and uranium), which are widely used in chemical weathering studies (e.g., Bourdon et al., 2003; Tomascak et al., 2016), to track the extent of chemical weathering in permafrost catchments as a proxy for the depth of the active layer.
Lithium has two isotopes (6Li and 7Li) and their ratio relative to a standard is expressed as δ7Li. The primary mechanism causing fractionation of lithium isotopes in natural systems is thought to be secondary mineral formation and/or adsorption onto particles (e.g., Huh et al., 2001; Pistiner and Henderson, 2003; Vigier et al., 2008). Other processes such as biological uptake appear to have a minimal effect on δ7Li values (Lemarchand et al., 2010; Clergue et al., 2015; Pogge von Strandmann et al., 2016). It is therefore hypothesized that the Li isotopic composition of rivers provides information on the balance between dissolution of primary minerals and secondary mineral formation occurring in a catchment (e.g., Kisakürek et al., 2005; Vigier et al., 2009; Millot et al., 2010; Dellinger et al., 2015; Pogge von Strandmann et al., 2017b). The balance between dissolution and precipitation processes varies depending on the weathering regime (Millot et al., 2010), which has been characterized by the weathering intensity: the ratio of chemical silicate weathering (W) to total denudation rate (D) (Bouchez et al., 2013). A study on the Amazon catchment, including data from other rivers, found that the relationship between the apparent fractionation factor ΔLisolution−rock and weathering intensity has a parabolic shape whereby ΔLisolution−rock increases with increasing weathering intensity to around 25‰ and then decreases as weathering intensity continues to increase (Dellinger et al., 2015). This is related to the relative supply of primary minerals by erosion that are available for dissolution relative to the scavenging of Li by secondary minerals, and eventual dissolution of these secondary phases when supply of material via erosion is low. One difficulty highlighted by Dellinger et al. (2015) is that spatial heterogeneity of δ7Li in rocks can hinder the interpretation of processes. One solution is to keep lithology constant by making multiple measurements throughout the year of the same river. There are currently no published δ7Li time-series measurements for rivers draining permafrost catchments, but three studies have spot-sampled several rivers draining areas containing permafrost in Canada, Greenland, and Tibet (Millot et al., 2010; Wimpenny et al., 2010b; Weynell et al., 2017).
Uranium isotopes are radioactive: 238U (half-life = 4.47 x 109 y) decays to 234U via 234Th and 234Pa, and 234U (half-life = 2.45 x 105 y) decays, via intermediate nuclides, to 206Pb (Bourdon et al., 2003). The 234U/238U ratio is commonly expressed as an activity ratio (234U/238U) where an activity ratio of one indicates that the decay chain is in secular equilibrium i.e., the rate of production and decay of a given nuclide is equal. Activity ratios higher than one are commonly observed in water samples and have been attributed to two process caused by alpha recoil: the displacement of a nuclide caused by the energetic release of an alpha particle. The first process is direct alpha recoil which, if it occurs within about 30 nm of a grain boundary, ejects 234Th from the mineral into solution which subsequently decays to 234U (Fleischer, 1980; Vigier and Bourdon, 2011). This process is enhanced if sediment grains are small (larger surface area). Elevated 234U in solution is also observed where sediments have been in contact with water for a long time, enhancing accumulation of 234U (Chabaux et al., 2003). The second process is preferential leaching of 234U from mineral lattice sites damaged by alpha recoil (Chabaux et al., 2003; Andersen et al., 2013). Uranium-series nuclides have been used to model a range of processes including physical erosion timescales (e.g., Vigier et al., 2006; DePaolo et al., 2012), soil formation rates (e.g., Dosseto et al., 2008; Ma et al., 2013), and water residence times (e.g., Maher et al., 2006; Schaffhauser et al., 2014). In permafrost environments U isotopes have been applied to understanding the seasonal evolution of water flow paths (Bagard et al., 2011; Koch et al., 2013) and the residence time of ice in permafrost (Ewing et al., 2015, 2016). The lithium and uranium isotope systems have previously been combined with the aim of providing coupled information on physical and chemical erosion (Pogge von Strandmann et al., 2006, 2010; Pogge von Strandmann and Henderson, 2015).
In this study we measured a time-series of water samples for δ7Li and (234U/238U) values from two adjacent catchments located in Svalbard and underlain by permafrost: one glaciated and one unglaciated. Both catchments have been well-characterized by previous studies (Hindshaw et al., 2016a, 2018) and therefore provide an ideal location to interpret the Li and U isotope measurements as the weathering sources and overall chemical weathering processes in each catchment are known. Small catchment studies such as this, where sources and processes can be more readily constrained, are a pre-requisite to understanding biogeochemical processes occurring in large catchments. We find that Li and U isotopes define a positive trend which we interpret to reflect mean water-rock residence time and may therefore be a suitable proxy for a catchment-wide measurement of active-layer depth at this fieldsite. Further research would be required to assess whether this promising relationship holds for larger permafrost-dominated catchments.
2. Field Site
Svalbard is located in the Arctic Ocean and has an Arctic climate. In 2012 (the year samples were collected) the mean temperature was −2.0 °C and precipitation was 268 mm, as recorded at Longyearbyen Airport (Nordli et al., 2014). The two studied catchments are situated next to each other (Figure 1) in the Paleogene sedimentary Central Basin of Svalbard. The sedimentary formations exposed in the catchments are from the Van Mijenfjorden group, which is Paleocene to Eocene in age (66–33.9 Ma) and contain sandstones, siltstones, and shale (Major et al., 2000). There is negligible soil formation in either catchment and the surface is dominated by moraine material and glacial sediment in the glaciated catchment and by frost-shattered shale in the unglaciated catchment (Figure 1).
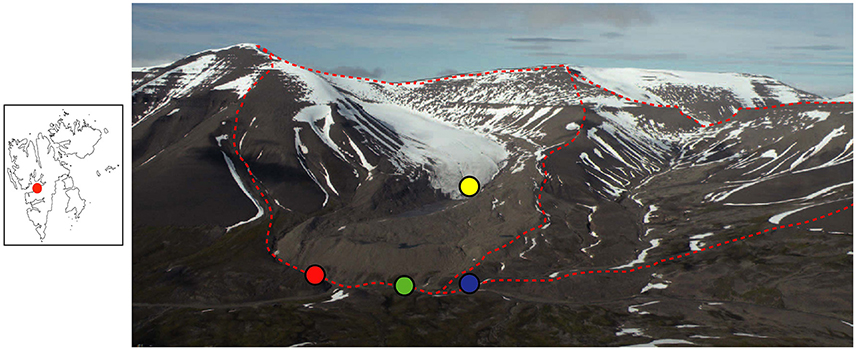
Figure 1. Panoramic view of the two study catchments. Dryadbreen is on the left and Fardalen on the right. The red dashed line demarcates the catchment boundaries. Water sampling locations are indicated as follows: red circle, Dryadbreen; blue circle, Fardalen; green circle, valley stream; yellow circle, supraglacial sample. Note the large end moraine and sandur in the glaciated catchment. The red dot in the map on the left shows the location of the study area (Latitude, 78°08′ N; Longitude, 15°30′ E) in relation to the rest of Svalbard. Reprinted from Hindshaw et al. (2016a), Copyright (2015), with permission from Elsevier.
Permafrost is continuous throughout the islands (Humlum et al., 2003) and in situ measurements document its ongoing degradation (Isaksen et al., 2007a,b). Active layer measurements are not available for the two catchments studied. However, temperature data is available for the Gruvefjellet borehole situated approximately 5 km north of the catchments at an altitude of 464 m.a.s.l. (Christiansen et al., 2010). In 2012, the temperature at the surface of this borehole first went above 0°C on 15 June (NORPERM database, Juliussen et al., 2010), implying that during the spring sampling campaign, which concluded on 18 June, the presence of an active layer in either catchment is unlikely. At Gruvefjellet the active layer depth reaches a maximum depth of about 1 m (Etzelmüller et al., 2011) and a similar temporal evolution of active layer depth is expected in the unglaciated catchment; from 0 m depth during the spring sampling to ~1 m when the summer samples were collected.
One set of samples was collected from a permafrost-affected valley Fardalen (Figure 1), which is likely to have been unglaciated for at least the last 10 kyr (Svendsen and Mangerud, 1997). The whole catchment has a south-easterly aspect that contributes to the absence of present-day glaciation. The catchment area is 3.4 km2 and ranges in elevation from 250 to 1025 m.a.s.l. The second set of samples was collected from a catchment containing the glacier Dryadbreen. The thermal regime of the glacier is expected to be cold-based with temperate patches, based on similar sized glaciers in the same area (Etzelmüller et al., 2000; Etzelmüller and Hagen, 2005). Between 1936 and 2006 the area of the glacier decreased from 2.59 to 0.91 km2 leaving large terminal and lateral ice-cored moraines and a sandur in front of the glacier (Ziaja and Pipała, 2007). The sandur surface lowered 14 m between 2001 and 2006 due to the melting of dead ice (Ziaja and Pipała, 2007). The uppermost part of the catchment faces north-north-east and the valley then curves around such that at lower elevations (<500 m) the catchment faces south-east. The catchment area is 4.8 km2 and ranges in elevation from 250 to 1031 m.a.s.l.
3. Methods
3.1. Water Sampling
Water samples were collected from the streams draining the two catchments just before their confluence with a larger river in the Fardalen valley (Figure 1). For Dryadbreen this was approximately 1 km from the front of the glacier. The main valley river was sampled after the confluence with the stream from Fardalen, but before the confluence with the stream from Dryadbreen. A supra-glacial water sample was collected from the glacier surface near the toe of the glacier. The Dryadbreen and Fardalen streams were sampled twice a day from 14 to 18th June 2012 and from 25th July to 3rd August 2012. Temperature and pH were measured in situ (Hanna HI 98160 pH meter).
The water sampling procedures for major ion chemistry are described in Hindshaw et al. (2016a). For lithium isotopes, a 1 L, 0.2 μm filtered water sample was collected from each catchment every other day. This water sample was acidified to pH 2 with single-distilled concentrated HNO3. Water samples for uranium isotope analysis were collected following the procedure outlined in Arendt et al. (2015). A pre-cleaned 15 L plastic water carrier was filled with 0.2 μm filtered water (filtered using a polycarbonate vacuum filtration unit connected to a hand pump), then 7 mL 9M HCl was added to the water followed by 3 mL of a pre-cleaned iron solution (FeCl3.6H2O in 3M HCl), resulting in an Fe concentration of 15 mg/L. The mixture was shaken and after 10 min, 10 mL 25% NH4OH was added, raising the pH to approximately 8–9 and inducing iron precipitation. After 48 h of settling, the bulk of the supernatant was gently poured off and the remaining solution was transferred to a 1 L bottle for transport from the field. The remaining liquid from the bottle containing the uranium co-precipitate was removed by centrifugation in the laboratory and the solid was digested in 1 mL 9M HCl, dried down and taken up in 10 mL 12 M HCl.
3.2. Solid Sampling
Stream suspended sediment (>0.2 μm) was retrieved from nylon filter papers during water sample collection by washing the filter paper with deionized water and then freeze drying the sample. Part of each stream suspended sediment sample was treated with 5% HCl to remove carbonates. A range of solid samples, including sedimentary rocks, bedload sediments, and glacial sediment, were also collected from the catchments. The sedimentary rock samples were first crushed (jaw crusher) and were subsequently ground to fine powders (rotary disc mill and planetary ball mill). For the sediment samples, only the latter step was required. A <2 μm size-fraction was separated from the fine powders by repeatedly rinsing and re-suspending the sample in de-ionized water with sodium phosphate as a dispersal agent, followed by sonication and centrifugation (Moore and Reynolds, 1997). For all solid samples, approximately 100 mg of material was ashed at 950°C for 120 min. The sample was then digested in a mixture of concentrated hydrofluoric and nitric acids and repeatedly dried down and re-dissolved in 6M HCl. In the final step the dried down sample was re-dissolved in 2% HNO3 for storage.
A leaching procedure designed to target authigenic phases was applied to five samples. Approximately 8 g of sediment was added to a 50 mL centrifuge tube and about 25 mL of 0.47 M acetic acid buffered with 1 M sodium acetate (AA) was was added. Samples were shaken overnight, centrifuged, and the supernatant removed. The residual sediment was washed with MQe water (18.2 Ω) and then about 25 mL of a 0.005 M hydroxylamine hydrochloride −2.6 M acetic acid mixture (HH) was added. This reagent is more dilute than commonly used (e.g., Piotrowski et al., 2004) in order to avoid leaching clay minerals (Haley et al., 2008; Chen et al., 2012). Samples were shaken for 1 h, centrifuged and the supernatant removed. The supernatant samples were centrifuged again and the top portion transferred to teflon beakers to dry down. The dried leachate was then digested in concentrated HNO3 and then taken up in 2% HNO3 in preparation for analysis.
3.3. Ionic Strength
Ionic strength (I) is a measure of concentration of a solution which takes into account the molar concentration (c) and charge (z) of all the ions in solution. This parameter was calculated in PhreeqC and is a more accurate measure of chemical behavior in non-ideal solutions as compared to the total charge of major cations (Stumm and Morgan, 1996).
3.4. Lithium and Uranium Concentrations and Isotopes
Trace element concentrations in solid and leachate samples were measured at the University of Cambridge by quadrupole inductively-coupled plasma mass spectrometry (Q-ICP-MS, Perkin Elmer Nexion 350D). The Li and U concentrations of several USGS rock standards (BCR-2, BHVO-2, AGV-2, and BIR-1) reproduced within 4% of certified values.
Lithium and uranium concentrations in water samples were determined on an Element ICP-MS (University of Cambridge) and a Thermo X series ICP-MS (University of St. Andrews). The average of these two measurements is reported. The water standard TM-25.4 (Environment Canada) was measured giving 27.9 ± 2.3 ppb (2SD, n = 8) for U and 26.4 ± 0.4 ppb (2SD, n = 8) for Li in St. Andrews, and 32.7 ± 0.6 ppb (2SD, n = 4) for U and 25.0 ± 0.2 ppb (2SD, n = 4) for Li in Cambridge. The certified values are 27.3 ± 2.1 ppb (U) and 23.7 ± 2.3 ppb (Li).
In order to separate uranium from a stream water sample matrix the 2-column procedure described in Arendt et al. (2015) was used. Uranium isotopes (234U/238U) were measured at the University of Wyoming using a Neptune PLUS (Arendt et al., 2015). Samples were run at 40 ppb in a nitric acid matrix and were bracketed with reference solution U010. Measurements were obtained in ten cycles of 5 s integrations after 30 s of baseline measurements. Repeat measurements of NIST SRM 960 (equivalent to NBL CRM-112A) and seawater gave values of 0.9637 ± 10 (2SD, n = 58) and 1.1447 ± 12 (2SD, n = 8) respectively, in agreement with the accepted values of 0.9631 ± 5 and 1.1450 ± 15 (Cheng et al., 2000; Chutcharavan et al., 2018).
Lithium was separated from other elements using a column procedure based on James and Palmer (2000). Savillex columns with an internal diameter of 6.4 mm were filled to give to resin height of 9.3 cm (3 mL resin). The resin was Bio-Rad AG 50W-X12 200-400 mesh, the elution was performed with 0.2 M HCl and the sample load was 15–20 ng. Lithium isotope ratios were measured on a Neptune multi-collector ICP-MS using an APEX IR sample introduction system and H cones. 7Li/6Li ratios were normalized to L-SVEC (NIST RM 8545) standard solution using standard-sample bracketing. Analyses were performed on 5 ppb solutions using a 1013 Ω resistor on the pre-amplifier for 6Li. The typical beam-size for samples was around 1 V on 7Li and the background was typically between 5 and 20 mV. Accuracy and precision were monitored using 6Li-N and 7Li-N solutions which gave values of −8.1 ± 0.9‰ (2SD, n = 59) and 30.2 ± 0.9‰ (2SD, n = 50), respectively. These values are in agreement with previously published values for these standards (7Li-N = 30.2‰ and 6Li-N = −8.0 to −8.3‰, Carignan et al., 2007; Millot et al., 2010). Long-term reproducibility of L-SVEC was 0.1± 0.5‰ (2SD, n = 72). Two measurements of IRMM-016 gave a δ7Li value of 0.3± 0.3‰ (2SD), within error of L-SVEC. To ensure there was no fractionation induced during chemical separation, either seawater or a rock standard was processed with every batch of 5 samples. Our value for seawater (OSIL IAPSO batch P157) is 30.8± 1.1‰ (2SD, 45 measurements of 34 discrete samples processed through column chemistry) in agreement with the compiled values of 31.1‰(Carignan et al., 2004) and 30.8‰(Rosner et al., 2007). We will apply the long-term external reproducibility of seawater to the samples measured in this study (1.1‰), as this value is greater than the 2SD of individual sample measurements.
As the solid samples in this study are shales we measured two USGS shale reference materials: SGR-1b and SCo-1 (Table 1). We measured 4.9 ± 1.9‰ (2SD, n = 10) for SGR-1b, in agreement with previously published values of 4.7 ± 0.7‰ (2SD, n = 3, Phan et al., 2016), 3.6 ± 0.4‰ (2SD, n = 3, Pogge von Strandmann et al., 2017b) and 5.0 ± 0.6‰ (2SD, n = 6, Bohlin et al., 2018). For SCo-1, we measured 0.4 ± 1.1‰ (2SD, n = 13), which is lower than the only other published value of 5.2 ± 1.5‰ (2SD, n = 3, James and Palmer, 2000). In order to investigate whether this offset indicated a problem with our method we sent an aliquot of SCo-1 powder to the University of Southampton. They measured a value 0.7 ± 0.3‰ (2SD, n = 3), providing us with confidence that the difference between our value and the value published by James and Palmer (2000) was not method related and was perhaps due to sample heterogeneity. The method was also validated using a standard addition technique (Tipper et al., 2008). Mixtures containing variable proportions of Li derived from seawater and SGR-1b were processed through column chemistry and measured (Table 1). The measured isotopic compositions of the mixtures can then be used to calculate the δ7Li values of the end-members (Tipper et al., 2008). The calculated values of 31.0 ± 0.4‰ for seawater and 5.0 ± 0.3‰ for SGR-1b are in excellent agreement with our long-term measurements of these standards (30.8 ± 1.1 and 4.9 ± 1.9‰, respectively).
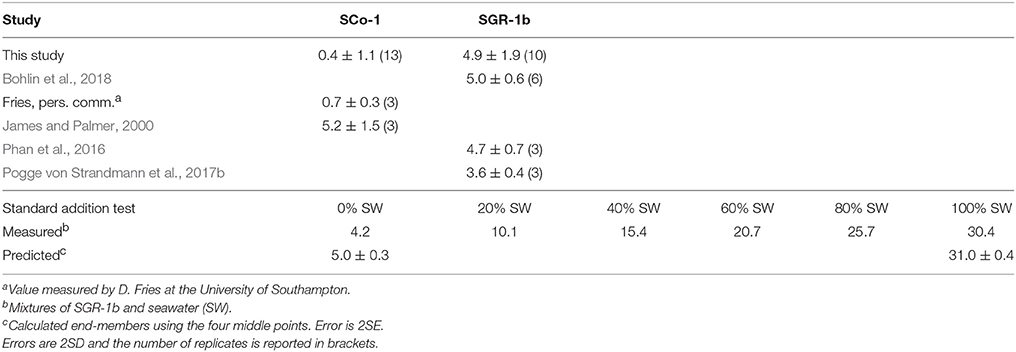
Table 1. Comparison of Li isotope values of USGS sedimentary rock standards measured in this study with literature values and measurements of seawater-SGR-1b mixtures from a standard addition test.
4. Results
4.1. Summary of Stream Water Chemistry
The chemistry of the streams was discussed in Hindshaw et al. (2016a) and a brief summary is provided here for context. The total dissolved load of the unglaciated catchment (53 ± 7 mgL−1, 1SD) was approximately double that of glaciated catchment (25 ± 4 mgL−1, 1SD) and the pH of both streams was circum-neutral (pH 6–7.5). The abundances of the major cations Ca and Mg were very similar in both catchments but in the unglaciated catchment Mg was consistently more abundant compared to the glaciated catchment where Ca was most abundant. In terms of major anions, sulfate comprised 72–86 and 35–45% of the summer anion budget (in meq) in the unglaciated and glaciated catchments respectively which, together with δ34S isotope measurements, revealed that sulfuric acid generated from pyrite weathering was a significant weathering agent in both catchments. Based on the relative proportions of cations, sulfate and bicarbonate it was proposed that the stream water chemistry of the unglaciated catchment was consistent with a sulfide oxidation coupled to silicate dissolution weathering process whereas in the glaciated catchment both carbonates and silicates weathered via both sulfuric and carbonic acids.
4.2. Uranium
Uranium concentrations ranged from 9 to 84 pmol/L in the water samples (Table 2) and there was no systematic difference in concentrations either between the catchments or between the spring and summer samples. 5 pmol/L U was measured in the supraglacial water sample. All water samples had activity ratios greater than 1, which is typical for rivers (Chabaux et al., 2003). The (234U/238U) values vary from 1.55 to 1.87, with the lowest value from the main stream (MS) and the highest value from the Fardalen stream in summer (Figure 2B, Table 2). Activity ratios were higher in Fardalen in summer compared to spring but the reverse was true in Dryadbreen. The activity ratios are similar to those observed in other high-latitude catchments (~1.6, Andersen et al., 2007) and thaw waters from permafrost (1.2–1.9, Ewing et al., 2016).
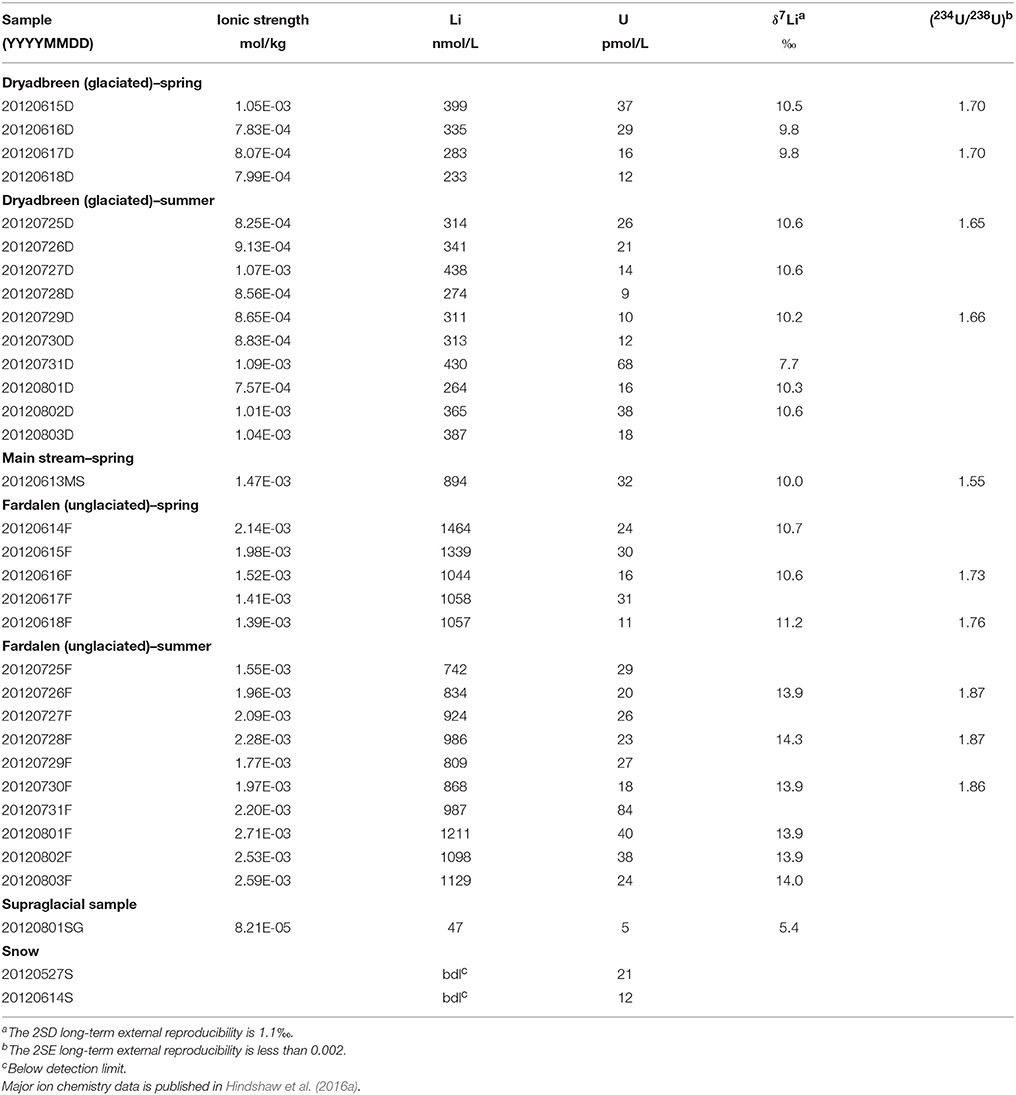
Table 2. Ionic strength and Li and U concentration and isotopic data.
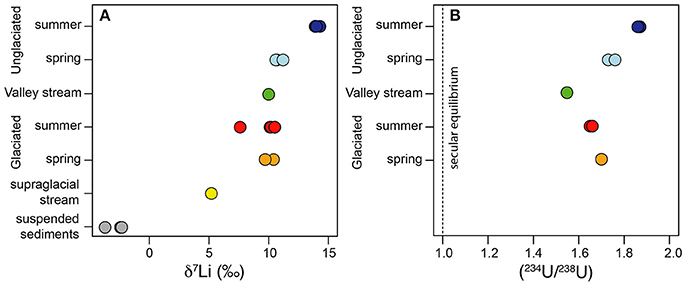
Figure 2. (A) The variation in δ7Li values within the water samples. The suspended sediments are shown for comparison and represent the isotopic composition of the initial Li released during chemical weathering. (B) The variation in (234U/238U) values within the water samples. The dashed line indicates where the solids would be if they were in secular equilibrium i.e., when (234U/238U) = 1.
4.3. Lithium
Lithium concentrations were higher in the unglaciated catchment (1037 ± 394 nmol/L, 2SD) compared to the glaciated catchment (335 ± 125 nmol/L, 2SD). The supraglacial water sample had 47 nmol/L Li (Table 2). These concentrations are comparable to those measured in the Madeira (Amazon) and Mackenzie rivers, which also drain shale (50–1670 nmol/L, Millot et al., 2010; Dellinger et al., 2015). Li isotope values in the glaciated catchment ranged from 7.7 to 10.5‰ with no seasonal variation (Figure 2A, Table 2). In contrast, whilst the spring δ7Li values in the unglaciated catchment (10.8 ± 0.7‰, 2SD) were comparable to the glaciated catchment (10.0 ± 1.8‰, 2SD), the values measured in summer were 3 to 4‰ higher with an average of 14.0‰ (Figure 2A, Table 2). Interestingly, despite the very different lithological and glaciological setting, Wimpenny et al. (2010b) also found a 4‰ difference between glaciated and unglaciated catchments in West Greenland. The supraglacial water sample had a lower Li isotopic composition (5.4‰) compared to both streams.
The average Li concentrations of the bulk solid samples (77 mg/kg) are in the range typical for shale rocks (e.g., Dellinger et al., 2014; Phan et al., 2016) and they are enriched in 6Li (−4.1 to 0.3‰) compared to the water samples (5.4 to 14.3‰, Tables 2, 3). In comparison, stream suspended sediments have higher average Li concentrations (97 mg/kg) and clay samples have the highest average Li concentrations (107 mg/kg). Stream suspended sediment samples were acid treated to remove carbonates, but as Li is not concentrated in carbonates, this leaching step had no effect, within error, on their isotopic composition and a negligible fraction of Li was leached (Table 3). The mean δ7Li value for the stream suspended sediments (leached and unleached) was −2.8 ± 1.5‰ (2SD, n = 9). The δ7Li values of solid samples varied from 0.3‰ in stream sediment (sample O) from the glaciated catchment to −4.1‰ in a shale (sample G). The average value of the solid samples was −2.2± 3.4‰ (2SD, n = 8) in agreement with the average of the stream suspended sediments. The δ7Li values of the clay-sized fraction of bulk samples was 1.8 to 5.7‰ lower than the parent sample in agreement with the known uptake preference for 6Li over 7Li during clay mineral formation (Vigier et al., 2008) and observed in separated clay-size fractions from sediments (Dosseto et al., 2015). The Li isotopic composition of the 0.47 M acetic acid (AA) leach was within error of the bulk rock composition apart from sample G (shale pieces) which was 3.1‰ lower than bulk. In contrast, the Li isotopic composition of the 0.005 M hydroxylamine hydrochloride (HH) leach was markedly lower than the bulk sample, ranging from −7.2 to −14.5‰. The AA leach had higher Li concentrations than the HH leach and the combined fraction of Li leached in the two leaching steps was less than 1.8% of that contained in the bulk.
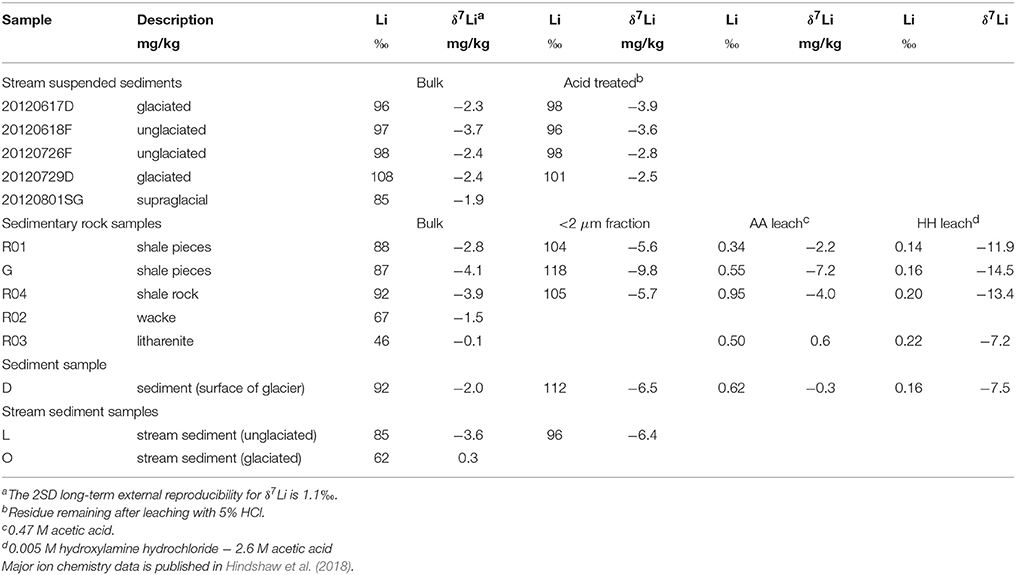
Table 3. Li concentration and isotope data for solid samples.
5. Discussion
5.1. Uranium Activity Ratios in Solution
The rocks in the study area have depositional ages older than 1 Ma (33.9–66 Ma) and are therefore expected to be in secular equilibrium with a (234U/238U) value of 1 (Vigier and Bourdon, 2011). All the water samples measured have (234U/238U) values greater than this (Figure 2, Table 2). There are a number of scenarios which can lead to high U activity ratios in solution: (1) increased physical erosion which decreases the average particle size, increasing the likelihood 234U will be ejected, (2) rapid chemical weathering of lattice-damaged minerals, releasing 234U, and (3) increased water residence time, which increases the time in which 234U can build up in the surrounding solution. We note that these processes are not mutually exclusive and the measured (234U/238U) will reflect the balance between multiple processes.
Physical erosion is greater in glaciated catchments compared to unglaciated catchments (Hallet et al., 1996), resulting in increased (234U/238U) ratios in solution. Exceptions occur in glaciated areas containing volcanic phases which weather rapidly, minimizing disequilibrium (Vigier et al., 2006; Lee et al., 2013). As there are no volcanic rocks present in these catchments, we would expect to measure higher (234U/238U) in Dryadbreen as a result of physical erosion creating small particles, enhancing alpha recoil (Robinson et al., 2004; Andersen et al., 2013). As we do not observe the highest (234U/238U) values in the glaciated catchment, this counts against a direct link between particle size and enhanced release of 234U in these catchments. This may be because the thermal regime of the glacier is either polythermal or cold-based (based on those of similar-sized glaciers in the area, Etzelmüller et al., 2000; Etzelmüller and Hagen, 2005) and, being frozen to the bed, is not actively eroding. Additionally, it is possible that physical erosion in the past could have an impact on the modern U activity ratios measured in the streams. The modern day sediments in the glaciated catchment moraines may have been pre-weathered during earlier times e.g., when the glacier was warm-based, resulting in U activity ratios less than 1 (Vigier et al., 2001). If the sediments in the glaciated catchment have lower U activity ratios than the unglaciated catchment, then this may account for the correspondingly lower (234U/238U) ratios measured in the water samples. Measurements of (234U/238U) values in suspended sediments would be needed in order to test this hypothesis.
Glacial erosion could promote the exposure of fresh mineral surfaces and lead to preferential weathering of trace mineral phases (Tranter, 2003). Carbonates contain high U concentrations and are expected to dissolve rapidly and therefore congruently, with (234U/238U) close to 1 (Andersson et al., 1995; Chabaux et al., 2001; Vigier et al., 2005). Therefore, the weathering of carbonate, which is more abundant in the glaciated catchment (Hindshaw et al., 2016a), could account for the lower (234U/238U) values in the glaciated compared to the unglaciated catchment, despite the higher erosion rate.
Variations in (234U/238U) have been explained by variations in residence time, whereby longer water residence times (e.g., deeper/longer flowpaths) lead to higher (234U/238U)(Riotte and Chabaux, 1999; Riotte et al., 2003; Bagard et al., 2011; Koch et al., 2013; Schaffhauser et al., 2014; Lidman et al., 2016). Water which has had a long residence time (e.g., groundwater) is expected to be more concentrated than water which has had a short residence time. In permafrost, U activity ratios are affected not just by the residence time of the modern water, but by the residence time of the ice itself. The frozen material can accumulate 234U, and this is only released when that material thaws (Tokarev et al., 2005). This process can explain observations of higher (234U/238U) ratios in thaw waters from older permafrost compared to younger permafrost (Ewing et al., 2015). In a reactive transport model, the solute concentration of an initially dilute solution will increase along a flow-path until the solution reaches saturation and secondary phases begin to precipitate (Maher, 2010, 2011). In this scenario, ionic strength, representing the accumulation of solutes, may be used as a proxy for mean water residence time up to the point where the system reaches steady state with respect to concentration. At this point further increases in residence time have only a minor impact on chemical concentrations (Maher, 2011). Freezing will also concentrate solutes and thereby increase ionic strength, but as these pockets of water are unlikely to contribute to the stream chemistry until they are flushed out during spring thaw (Kokelj and Burn, 2005; Lamhonwah et al., 2017; Lehn et al., 2017), they can also be considered to have a long residence time. Consistent with the reactive transport model predictions for dilute waters (Maher, 2011), we observe a positive correlation between ionic strength and (234U/238U) (Figure 3A). This trend implies that the water samples from the unglaciated catchment have the greatest mean water residence times, resulting in the highest ionic strength and (234U/238U) ratios.
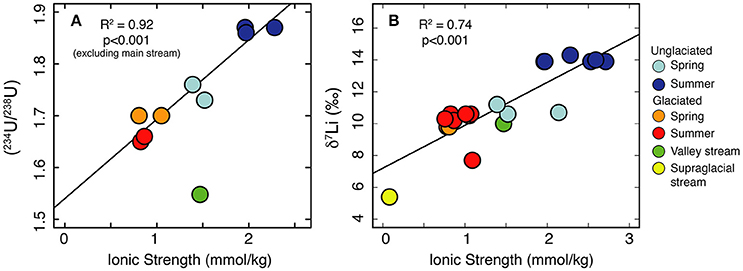
Figure 3. Significant positive trends are observed between ionic strength, a proxy for the amount of chemical weathering, and both (234U/238U) (A) and δ7Li ratios (B).
In the unglaciated catchment, summer (234U/238U) values are greater than in spring (Figure 2B), consistent with seasonal variations observed in the permafrost-affected Kulingdakan stream, Siberia (Bagard et al., 2011). In non-permafrost catchments, seasonal changes in (234U/238U) have been ascribed to a change in the mixing proportion of waters with different residence times (Andersson et al., 1995; Riotte and Chabaux, 1999; Huckle et al., 2016; Arendt et al., 2017). A residence time control would be consistent with the greater depth of the active layer in summer, enabling water to access deeper flowpaths (Koch et al., 2013). Similar to the interpretation of U behavior in an Alaskan permafrost catchment (Koch et al., 2013), the stream water from both catchments can be explained by a mixture of U derived from at least three pools with different residence times (Figure 4). The lower relative proportion of the “long-residence time” end-member (Figure 4), lower ionic strength, lower (234U/238U) ratios and negligible seasonal variation observed in the glaciated catchment (Figures 2B, 3A) can be explained by the layer of dead ice underneath the sandur (glacial outwash plain, Figure 1, Ziaja and Pipała, 2007), which restricts water to the surface and limits water residence time (Figure 5). A similar “blocking” effect was observed in a Canadian high Arctic catchment where ground ice and ice-rich soil prevented active layer deepening and resulted in lower than expected solute fluxes (Lamhonwah et al., 2017).
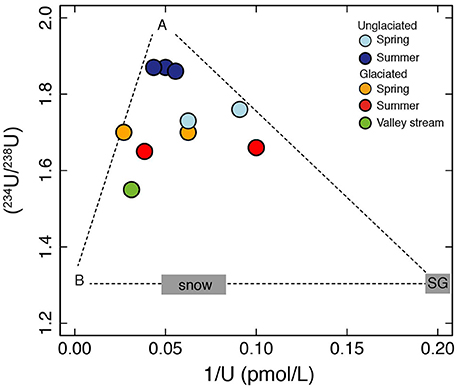
Figure 4. A plot of (234U/238U) vs. 1/U. U concentrations and activity ratios can be explained by mixing between at least three end-members. One end-member (A) has relatively high U concentrations and high activity ratios and is inferred to derive from water with a long residence time. The other two end-members represent short water residence time sources. One is consistent with ice melt with low uranium concentrations represented by the supraglacial sample (SG) and the other (B) has high uranium concentrations and may derive from a mineral source such as carbonate. U from snow or dust contained within snow is an additional end-member and could be important for the spring samples which were collected during snow-melt. We do not have direct measurements of the latter three end-members and plot them using the U activity ratio of 1.3 measured in Siberian snow (Bagard et al., 2011). The lower activity ratio of snow compared to the stream water samples is consistent with short water residence times expected for snow and ice melt (Koch et al., 2013).
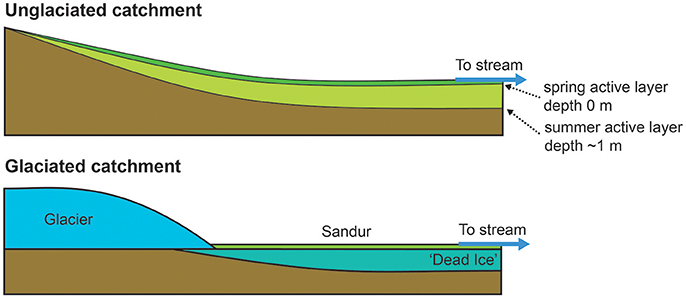
Figure 5. Schematic of the landscape controls on δ7Li and (234U/238U) ratios. Unglaciated: Typical permafrost profile with a seasonal active layer which deepens from spring to summer. Water-rock reaction is low in spring and increases as the melt season progresses. Active layer depths are taken from the Gruvefjellet borehole situated 5 km from this catchment at comparable altitude (Etzelmüller et al., 2011). Glaciated: In contrast to the unglaciated catchment, the dead ice prevents downward expansion of the thawed layer and only a a thin layer of sediment is exposed to weathering. Water-rock reaction time remains low and relatively constant throughout the melt season.
5.2. Li Isotopic Composition of Solid Samples
Previous work in these catchments highlighted the systematic variation of Sr and Nd isotope compositions of rocks and sediments, primarily due to the mixing of sediment populations derived from two different sources (Hindshaw et al., 2018). For Li, the stream suspended sediments from both catchments are, within experimental error, identical despite the differing mineralogy of each catchment. Nevertheless, there is a 4.4‰ variation in δ7Li values in the bulk sedimentary rock and stream sediment samples (Table 3). The bedrock in both catchments is dominantly shale, containing 30–65% clay minerals (Hindshaw et al., 2016a). The δ7Li of the bulk solid samples co-varies with the bulk clay content (Figure 6A) consistent with the preferentially incorporation of 6Li into the clay mineral structure (Vigier et al., 2008). To investigate whether variation in δ7Li values was dependent on the type of clay minerals present, we compared the δ7Li isotope values of the clay-sized fraction with the relative proportions of clay minerals present. For the bulk samples there is an inverse relationship between I/S (an illite-smectite mixed layer phase) and illite (Hindshaw et al., 2018), but the changing relative proportions of these two clay minerals, which have contrasting structures, did not have an obvious effect on δ7Li values (Figure 6B). This finding supports field and modeling studies suggesting that δ7Li ratios in rocks do not show a strong dependence on clay mineralogy, implying that the apparent fractionation factors (Δ7Liclay−water) for different clay minerals are similar (Pogge von Strandmann et al., 2017a; Dupuis et al., 2017).
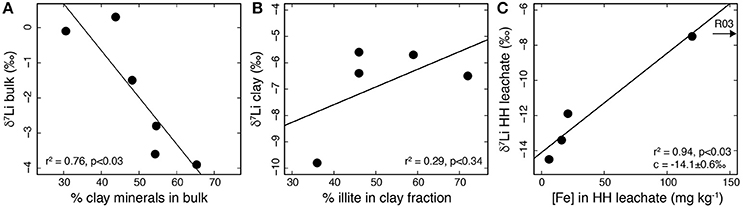
Figure 6. (A) Plot of δ7Libulk vs. the relative proportion of clay minerals in the bulk sample. The proportion of clay minerals was calculated by summing the relative abundances of chlorite, illite/mica, kaolinite and illite/smectite obtained by XRD (Hindshaw et al., 2016a). Higher relative clay mineral abundances are associated with lower δ7Li values. (B) Plot of δ7Liclay vs. the relative proportion of illite in the clay-sized separate (<2 μm). There is no relationship between the relative abundance of illite and δ7Li. (C) Plot of δ7Li vs. Fe concentrations in the HH leachate. The more Fe (assumed to be a proxy for Fe-Mn oxyhydroxide content), the higher the δ7Li value, and presumably a greater fraction of Li removed from solution. Sample R03, which contained 1707 mg kg−1 Fe, was not included in the line fitting. This sample likely has other iron sources in addition to Fe-Mn oxyhydroxides (Hindshaw et al., 2018).
The AA and HH leaching procedure was applied to bulk samples in order to isolate authigenic phases. The AA leach targets carbonates but will also leach out exchangeable and adsorbed ions. As Li is not expected to be concentrated in carbonates (Dellinger et al., 2015), the Li in this leach likely represents exchanged/loosely-bound Li. Being the most readily mobilized fraction of the bulk, it is also expected to represent the isotopic composition of the weathering input to the streams. We note that the Li isotopic composition of this fraction is within uncertainty of the bulk (Table 3) consistent with there being negligible fractionation of Li during the release of Li into water from rock. Similar observations have been made in laboratory dissolution experiments (Pistiner and Henderson, 2003; Wimpenny et al., 2010a), though in the field setting we cannot exclude fractionation into the exchangeable/adsorbed reservoir. The HH leach targets authigenic Fe-Mn oxyhydroxides and the δ7Li isotopic compositions of the HH leaches are low (Table 3). This could be due to adsorption of Li onto Fe-Mn oxyhydroxide surfaces, consistent with previous studies which found that 6Li was preferentially adsorbed onto various Fe and Mn oxides (Pistiner and Henderson, 2003; Chan and Hein, 2007) and the low δ7Li values measured in leachates from a procedure designed to target these phases in river sediments (Wimpenny et al., 2010b). The δ7Li values of the HH leach increase with increasing Fe concentration (Figure 6C), which would be consistent with a greater fraction of Li adsorbing from the initial solution when more Fe-Mn oxyhxdroxides are present.
5.3. Lithium Isotopic Composition of the Dissolved Phase
The dissolved δ7Li values are all greater than those of the stream suspended sediment, consistent with fractionation during secondary mineral formation and/or adsorption (e.g., Huh et al., 2001; Pistiner and Henderson, 2003; Vigier et al., 2008). Element ratios normalize the effect of dilution, allowing samples collected under different discharge conditions to be compared, providing information on sources and processes affecting water chemistry (e.g., Gíslason et al., 1996; Hindshaw et al., 2011). If the normalizing element, X, is fluid mobile and unaffected by secondary processes, then changes in the Li/X ratio between samples reflects the relative amount of Li lost to secondary processes. Often Na is used as the mobile reference element (Millot et al., 2010), but in these catchments Na is predominantly derived from snow-melt and we do not have enough data to be able to accurately calculate the percentage of Na derived from this source. The rocks in these catchments are shales, which contain clay minerals with a high cation exchange capacity compared to silicate minerals contained in granite and basalt, where Na/Li and Mg/Li ratios have been used to calculate the fraction of Li loss (e.g., Dellinger et al., 2015). Adsorption of major cations is likely where clays are present (Huang et al., 2012; Ockert et al., 2013) and these elements may not be fully fluid mobile. As an alternative, we use SO4 as the mobile reference element, as it is less likely than cations to be affected by ion exchange processes due to the predominant negative charge of clay surfaces at the pH of the stream water (6–7.5, Meunier, 2005). Additionally, sulfate forms a major part (up to 86%) of the anion budget (in meq) of the water samples (Hindshaw et al., 2016a) and it is strongly correlated with ionic strength (R2 = 0.99). Li covaries relative to SO4, but with two different slopes (Figure 7A). Assuming SO4 is fully conservative, this implies a process is affecting Li concentrations, resulting in a relative loss of Li from the summer unglaciated catchment samples despite high Li concentrations and ionic strength (Figure 7B). This interpretation is consistent with the isotope data: the samples inferred to have the greatest relative loss of Li also exhibit the greatest δ7Li values (Figure 7B).
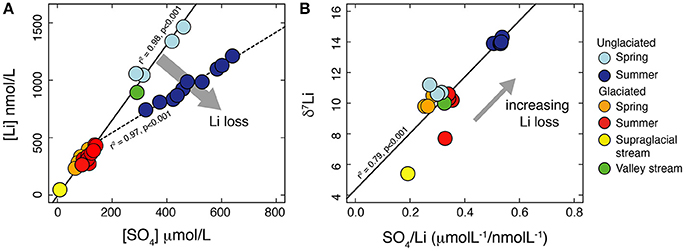
Figure 7. (A) Plot of Li vs. SO4 concentrations highlighting the relative loss of Li from the summer samples collected from Fardalen (unglaciated catchment) compared to all the other samples. (B) Plot of δ7Li vs. SO4/Li. The greater the Li loss from solution relative to SO4, the greater the δ7Li value of the water, consistent with preferential loss of 6Li during removal processes such as adsorption onto Fe-Mn oxyhydroxides.
Whether the loss of Li and associated isotope fractionation is due to adsorption or secondary mineral formation is unclear. Given the high dissolved organic carbon content of the streams (up to 2000 μmol C/L Hindshaw et al., 2016b), lithium may adsorb onto organic molecules. However, work on lithium uptake into plants and phytoplankton suggests that lithium isotope fractionation during complexation with organic molecules is likely to be negligible (Lemarchand et al., 2010; Clergue et al., 2015; Pogge von Strandmann et al., 2016). Small amounts of new clay minerals could be forming but two previous studies on Li isotopes conducted in glaciated and permafrost-dominated catchments (Millot et al., 2010; Wimpenny et al., 2010b) concluded that fractionation was due to adsorption onto particulate Fe/Mn oxyhydroxides. In the Mackenzie Basin, Li isotopes (Millot et al., 2010) could also be consistent with secondary clay formation (Tipper et al., 2012). In contrast to Wimpenny et al. (2010b), who found 60% of Li was released during a 2 M HCl leach of stream suspended sediments, our 5% (~1.4 M) HCl leach of stream suspended sediments did not remove a significant amount of Li (Table 3). Nevertheless, Fe/Mn oxyhydroxides are present in the catchment rocks and sediments and 0.2–0.5% of Li contained in the bulk solids was extracted during the HH leach designed to target these phases. Importantly, these leachates had low δ7Li values compared to bulk samples (−14.5 to −7.2‰, Table 3), supporting the hypothesis that adsorption of Li onto Fe/Mn hydroxides could lead to water enriched in 7Li. Bacterial populations involved in Fe and S cycling were identified in these catchments (Hindshaw et al., 2016a) indicating that a similar reaction mechanism to that observed in Greenland, of sulfide oxidation as a source of Fe-particles to which Li could adsorb (Wimpenny et al., 2010b), could be occurring here. As sulfide weathering increases (higher SO4 concentrations and ionic strength), more Fe oxyhydroxides are likely to form, preferentially removing 6Li from solution via adsorption, increasing stream δ7Li values (Figure 7).
Studies on the seasonal evolution of δ7Li values at a single location invoke mean water residence time as a key parameter. In dilute systems, increased water residence times, as a result of increased ground water contribution or increased water transit time, results in an increased proportion of Li removed into secondary minerals (Lemarchand et al., 2009, 2010; Manaka et al., 2017). Conversely, short water residence times favor greater amounts of dissolution relative to precipitation because the waters are under-saturated, inhibiting 6Li uptake into secondary minerals (Manaka et al., 2017). In permafrost catchments, concentrated solutes (long-residence time) may also be derived from two further processes. On a seasonal timescale, freezing of the active layer during winter results in pockets of water concentrated in solutes, which are flushed out during subsequent thaw (Kokelj and Burn, 2005; Lamhonwah et al., 2017). The highest element concentrations are observed in autumn when the active layer depth is greatest (Lamhonwah et al., 2017). On longer timescales, near-surface permafrost (permafrost immediately below the bottom of the active layer) has been shown to be enriched with solutes, due to the removal of solutes from the active layer by advection and convection along thermal gradients (Kokelj and Burn, 2003, 2005). As for U activity ratios, we suggest that the positive relationship between ionic strength and δ7Li (Figure 3B) arises from a residence time control based on reactive transport models that predict increasing δ7Li with increasing subsurface residence time (Wanner et al., 2014). Compared to spring, the deeper active layer in summer would enable a longer water residence time, promoting increased dissolution (higher ionic load) and increased removal of Li (greater fraction of Li lost). In addition, the summer samples will have a contribution of solutes from the previous season and, if the active layer depth was greater than previous years, then solutes from the near-surface permafrost would also be released. Both of these pools are likely depleted in 6Li due to their long residence time. Combined, these processes result in the greater Li isotope fractionation observed in the unglaciated catchment in summer compared to spring. Like (234U/238U), the δ7Li values for Dryadbreen are remarkably constant and show no seasonal variation. We attribute the lack of variation to the dead-ice underlying the sandur plain which restricts the water to shallow, superficial flow paths (Figure 5).
In summary, the variations in both δ7Li and (234U/238U) values in the two catchments are consistent with residence time control and as a result co-vary with each other (Figure 8A).
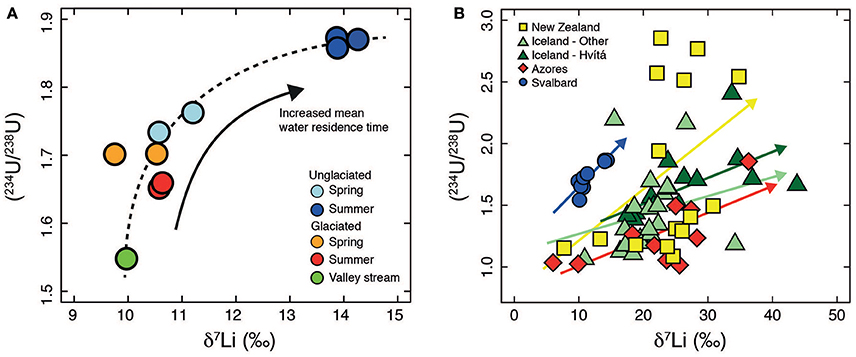
Figure 8. (A) Plot of (234U/238U) vs. δ7Li for Svalbard (this study). The dashed line indicates the overall trend in the data. The positive trend would be consistent with a residence time control. (B) Plot of (234U/238U) vs. δ7Li. Data from New Zealand (Pogge von Strandmann and Henderson, 2015), Iceland (Pogge von Strandmann et al., 2006; Vigier et al., 2006, 2009), Azores (Pogge von Strandmann et al., 2010) and Svalbard (this study). There is no global relationship between (234U/238U) and δ7Li. Relationships are specific to each catchment and are dependent on local environmental conditions and are indicated by the colored arrows.
5.4. Combining Uranium and Lithium
The combination of U and Li isotopes has been proposed as a means to gain simultaneous information on physical and chemical erosion (Pogge von Strandmann and Henderson, 2015) based on the observation of a positive relationship between the two isotope systems, similar to our observations from Svalbard (Figure 8, Pogge von Strandmann et al., 2006; Vigier et al., 2006, 2009; Pogge von Strandmann et al., 2010; Pogge von Strandmann and Henderson, 2015). In the mono-lithological shale catchments investigated in this study we interpret the positive trend to arise from a water residence time control on both δ7Li and U activity ratios. Indeed, the clearest relationships between δ7Li and (234U/238U) occur within single catchments or relatively homogenous areas such as Iceland (a large part of the Iceland data is from a single river catchment, Hvítá), where the positive relationship was interpreted to indicate decreased chemical weathering intensity at high erosion rates (Pogge von Strandmann et al., 2006). Nevertheless on a global scale no overall trend is observed (Figure 8B), but coherency between δ7Li and (234U/238U) is not expected at this scale due to the wide variety of processes which can impact on (234U/238U) (e.g., physical erosion, water residence time, balance between alpha recoil and dissolution, Chabaux et al., 2003) and δ7Li (e.g., weathering intensity, Dellinger et al., 2015) ratios. Both isotope systems also exhibit a range of bedrock compositions precluding coherent relationships over large spatial scales (Riotte and Chabaux, 1999; Chabaux et al., 2001; Teng et al., 2004; Penniston-Dorland et al., 2017). We anticipate that within catchments where the dominant control on water chemistry is residence time, and other factors such as heterogenous lithology and erosion exert a minor influence, then a positive relationship between δ7Li and (234U/238U) specific to that catchment will be observed that would reflect active layer depth in those catchments underlain by continuous permafrost.
5.5. Conclusions and Outlook
We found that both the uranium activity ratio and the Li isotope ratio of stream water samples in two High Arctic catchments were positively related to ionic strength. We interpret this to reflect a key control of mean water (and ice for U) residence time on these two isotope systems in agreement with previous studies (Riotte and Chabaux, 1999; Lemarchand et al., 2010). Increased water residence time, results in increased accumulation of 234U (derived from direct alpha recoil) and an increased proportion of Li removed from solution, either through secondary mineral formation or adsorption.
Time series measurements from the two catchments are consistent with changing water residence times. In the unglaciated, permafrost catchment an increase in both (234U/238U) and δ7Li was observed from spring to summer, consistent with the increase in active layer depth, allowing deeper flowpaths and increased water-rock interaction. Deep thawing of the active layer can additionally release pockets of liquid concentrated in solutes, resulting from solute exclusion during freezing, and melt old ice, which can be enriched in 234U. The lack of seasonal variation in the glaciated catchment is likely due to a layer of dead ice under the sandur, which restricts water flow to a shallow surface layer.
During deglaciation, as glaciers retreat, they are likely to change from a temperate state with bed-wide melting through polythermal to cold-based where limited meltwater reaches the base of the glacier. This transition, which is currently being observed in Svalbard (Nowak and Hodson, 2014), could mean that in high-latitude areas during deglaciation, a decrease in (234U/238U) from glaciated areas due to decreasing physical erosion could be compensated for by increased (234U/238U) from a deepening active layer in unglaciated permafrost areas. Both effects should be considered when calculating how riverine (234U/238U) changes over time.
Measurements of active layer depth are critical for calculating carbon storage and documenting changes at high latitudes (AMAP, 2017). Active layer depth is currently determined by ground-based measurements, which are precise but limited in spatial extent (Brown et al., 2000). Regional mapping can be conducted by extrapolating between ground-based measurements, and there have been recent tests of remote sensing technology to achieve this goal (Liu et al., 2012; Gangodagamage et al., 2014). This study highlights the potential of Li and U isotopes, in combination with standard water chemistry measurements, to provide a complementary method that would enable monitoring of seasonal and inter-annual changes in active layer depth at the catchment-scale. Future research is required to address how, and if, these findings from a small catchment can be scaled up to larger river basins.
Data Availability
All datasets generated for this study are included in the manuscript or available in the referenced companion manuscripts.
Author Contributions
RH designed the project, conducted the fieldwork, and analyzed all samples apart from those for (234U/238U) which SA analyzed. ET helped with Li method development. All authors contributed the interpretation of the results and the writing of the article.
Funding
This project was funded by a Swiss National Science Foundation fellowship for prospective researchers (PBEZP2-137335), a Marie Curie Intra-European Fellowship (PIEF-GA-2012-331501), and NERC Standard Grant NE/M001865/1. Fieldwork was supported by an Arctic Field Grant (219165/E10, The Research Council of Norway).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank the fieldwork team and everyone who made the fieldwork possible, Alex Piotrowski and Christina Larkin for help with the AA/HH leaching procedure, Tim Heaton for performing the stream suspended sediment leaches, Angus Calder for collecting and processing XRD data on clay mineral abundances, Nathalie Vigier for providing the 7Li-N and 6Li-N standard solutions and Sambuddha Misra for help setting up the Li isotope method. We are grateful to Carsten Mueller and Philip Pogge von Strandmann for their constructive reviews of this manuscript.
References
AMAP (2017). Snow, Water, Ice and Permafrost in the Arctic (SWIPA). Oslo: Arctic Monitoring and Assessment Programme (AMAP).
Andersen, M. B., Stirling, C. H., Porcelli, D., Halliday, A. N., Andersson, P. S., and Baskaran, M. (2007). The tracing of riverine U in Arctic seawater with very precise 234U/238U measurements. Earth Planet. Sci. Lett. 259, 171–185. doi: 10.1016/j.epsl.2007.04.051
Andersen, M. B., Vance, D., Keech, A. R., Rickli, J., and Hudson, G. (2013). Estimating U fluxes in a high-latitude, boreal post-glacial setting using U-series isotopes in soils and rivers. Chem. Geol. 354, 22–32. doi: 10.1016/j.chemgeo.2013.06.021
Andersson, P. S., Wasserburg, G. J., Chen, J. H., Papanastassiou, D. A., and Ingri, J. (1995). 238U-234U and 232Th-230Th in the Baltic Sea and in river water. Earth Planet. Sci. Lett. 130, 217–234.
Arendt, C. A., Aciego, S. M., Sims, K. W. W., and Aarons, S. M. (2017). Seasonal progression of uranium series isotopes in subglacial meltwater: Implications for subglacial storage time. Chem. Geol. 467, 42–52. doi: 10.1016/j.chemgeo.2017.07.007
Arendt, C. A., Aciego, S. M., Sims, K. W. W., and Robbins, M. J. (2015). Sequential separation of uranium, hafnium and neodymium from natural waters concentrated by iron coprecipitation. Geostand. Geoanal. Res. 39, 293–303. doi: 10.1111/j.1751-908X.2014.00322.x
Bagard, M.-L., Chabaux, F., Pokrovsky, O. S., Viers, J., Prokushkin, A. S., Stille, P., et al. (2011). Seasonal variability of element fluxes in two Central Siberian rivers draining high latitude permafrost dominated areas. Geochim. Cosmochim. Acta 75, 3335–3357. doi: 10.1016/j.gca.2011.03.024
Bagard, M.-L., Schmitt, A.-D., Chabaux, F., Pokrovsky, O. S., Viers, J., Stille, P., et al. (2013). Biogeochemistry of stable Ca and radiogenic Sr isotopes in a larch-covered permafrost-dominated watershed of Central Siberia. Geochim. Cosmochim. Acta 114, 169–187. doi: 10.1016/j.gca.2013.03.038
Barker, A. J., Douglas, T. A., Jacobson, A. D., McClelland, J. W., Ilgen, A. G., Khosh, M. S., et al. (2014). Late season mobilization of trace metals in two small Alaskan arctic watersheds as a proxy for landscape scale permafrost active layer dynamics. Chem. Geol. 381, 180–193. doi: 10.1016/j.chemgeo.2014.05.012
Bohlin, M. S., Misra, S., Lloyd, N., Elderfield, H., and Bickle, M. J. (2018). High-precision determination of lithium and magnesium isotopes utilising single column separation and multi-collector inductively coupled plasma mass spectrometry. Rapid Commun. Mass Spectrom. 32, 93–104. doi: 10.1002/rcm.8020
Bouchez, J., von Blanckenburg, F., and Schuessler, J. A. (2013). Modeling novel stable isotope ratios in the weathering zone. Am. J. Sci. 313, 267–308. doi: 10.2475/04.2013.01
Bourdon, B., Turner, S., Henderson, G. M., and Lundstrom, C. C. (2003). “Introduction to U-series geochemistry,” in Uranium-Series Geochemistry, Vol. 52, eds B. Bourdon, G. M. Henderson, C. C. Lundstrom, and S. P. Turner (Washington, DC: Mineralogical Society of America), 1–21
Bring, A., Fedorova, I., Dibike, Y., Hinzman, L., Mård, J., Mernild, S. H., et al. (2016). Arctic terrestrial hydrology: A synthesis of processes, regional effects, and research challenges. J. Geophys. Res. Biogeosci. 121, 621–649. doi: 10.1002/2015JG003131
Brown, J., Hinkel, K. M., and Nelson, F. E. (2000). The circumpolar active layer monitoring (CALM) program: Research designs and initial results. Polar Geogr. 24, 166–258. doi: 10.1080/10889370009377698
Carignan, J., Cardinal, D., Eisenhauer, A., Galy, A., Rehkämper, M., Wombacher, F., et al. (2004). A reflection on Mg, Cd, Ca, Li and Si isotopic measurements and related reference materials. Geostand. Geoanal. Res. 28, 139–148. doi: 10.1111/j.1751-908X.2004.tb01050.x
Carignan, J., Vigier, N., and Millot, R. (2007). Three secondary reference materials for lithium isotope measurements: Li7-N, Li6-N and LiCl-N solutions. Geostand. Geoanal. Res. 31, 7–12. doi: 10.1111/j.1751-908X.2007.00833.x
Chabaux, F., Riotte, J., Clauer, N., and France-Lanord, C. (2001). Isotopic tracing of the dissolved U fluxes of Himalayan rivers: implications for present and past U budgets of the Ganges-Brahmaputra system. Geochim. Cosmochim. Acta 65, 3201–3217. doi: 10.1016/S0016-7037(01)00669-
Chabaux, F., Riotte, J., and Dequincey, O. (2003). “U-Th-Ra fractionation during weathering and river transport,” in Uranium-Series Geochemistry, vol. 52, eds B. Bourdon, G. M. Henderson, C. C. Lundstrom, and S. P. Turner (Washington, DC: Mineralogical Society of America), 533–576.
Chadburn, S. E., Burke, E. J., Cox, P. M., Friedlingstein, P., Hugelius, G., and Westermann, S. (2017). An observation-based constraint on permafrost loss as a function of global warming. Nat. Clim. Change 7, 340–344. doi: 10.1038/nclimate3262
Chan, L.-H., and Hein, J. R. (2007). Lithium contents and isotopic compositions of ferromanganese deposits from the global ocean. Deep Sea Res. II 54, 1147–1162. doi: 10.1016/j.dsr2.2007.04.003
Chen, T.-Y., Frank, M., Haley, B. A., Gutjahr, M., and Spielhagen, R. F. (2012). Variations of North Atlantic inflow to the central Arctic Ocean over the last 14 million years inferred from hafnium and neodymium isotopes. Earth Planet. Sci. Lett. 353-354, 82–92. doi: 10.1016/j.epsl.2012.08.012
Cheng, H., Edwards, R. L., Hoff, J., Gallup, C. D., Richards, D. A., and Asmerom, Y. (2000). The half-lives of uranium-234 and thorium-230. Chem. Geol. 169, 17–33. doi: 10.1016/S0009-2541(99)00157-6
Christiansen, H. H., Etzelmüller, B., Isaksen, K., Juliussen, H., Farbrot, H., Humlum, O., et al. (2010). The thermal state of permafrost in the Nordic area during the International Polar Year 2007-2009. Permafrost Periglac. Process. 21, 156–181. doi: 10.1002/ppp.687
Chutcharavan, P. M., Dutton, A., and Ellwood, M. J. (2018). Seawater 234U/238U recorded by modern and fossil corals. Geochim. Cosmochim. Acta 224, 1–17. doi: 10.1016/j.gca.2017.12.017
Clergue, C., Dellinger, M., Buss, H. L., Gaillardet, J., Benedetti, M. F., and Dessert, C. (2015). Influence of atmospheric deposits and secondary minerals on Li isotopes budget in a highly weathered catchment, Guadeloupe (Lesser Antilles). Chem. Geol. 414, 28–41. doi: 10.1016/j.chemgeo.2015.08.015
Dean, J. F., Billett, M. F., Baxter, R., Dinsmore, K. J., Lessels, J. S., Street, L. E., et al. (2016). Biogeochemistry of “pristine” freshwater stream and lake systems in the western Canadian Arctic. Biogeochemistry 130, 191–213. doi: 10.1007/s10533-016-0252-2
Dellinger, M., Gaillardet, J., Bouchez, J., Calmels, D., Galy, V., Hilton, R. G., et al. (2014). Lithium isotopes in large rivers reveal the cannibalistic nature of modern continental weathering and erosion. Earth Planet. Sci. Lett. 401, 359–372. doi: 10.1016/j.epsl.2014.05.061
Dellinger, M., Gaillardet, J., Bouchez, J., Calmels, D., Louvat, P., Dosseto, A., et al. (2015). Riverine Li isotope fractionation in the Amazon River basin controlled by the weathering regimes. Geochim. Cosmochim. Acta 164, 71–93. doi: 10.1016/j.gca.2015.04.042
DePaolo, D. J., Lee, V. E., Christensen, J. N., and Maher, K. (2012). Uranium comminution ages: sediment transport and deposition time scales. C. R. Geosci. 344, 678–687. doi: 10.1016/j.crte.2012.10.014
Dosseto, A., Turner, S. P., and Chappell, J. (2008). The evolution of weathering profiles through time: New insights from uranium-series isotopes. Earth Planet. Sci. Lett. 274, 359–371. doi: 10.1016/j.epsl.2008.07.050
Dosseto, A., Vigier, N., Joannes-Boyau, R., Moffat, I., Singh, T., and Srivastava, P. (2015). Rapid response of silicate weathering rates to climate change in the Himalaya. Geochem. Perspect. Lett. 1, 10–19. doi: 10.7185/geochemlet.1502
Dupuis, R., Benoit, M., Tuckerman, M. E., and Méheut, M. (2017). Importance of a fully anharmonic treatment of equilibrium isotope fractionation properties of dissolved ionic species as evidenced by Li+(aq). Acc. Chem. Res. 50, 1597–1605. doi: 10.1021/acs.accounts.6b00607
Etzelmüller, B., and Hagen, J. O. (2005). “Glacier-permafrost interaction in Arctic and alpine mountain environments with examples from southern Norway and Svalbard,” in Cryospheric Systems: Glaciers and Permafrost, Vol. 242, eds C. Harris and J. B. Murton (London: Geological Society; Special Publications), 11–27.
Etzelmüller, B., Ødegård, R. S., Vatne, G., Mysterud, R. S., Tonning, T., and Sollid, J. L. (2000). Glacier characteristics and sediment transfer system of Longyearbreen and Larsbreen, western Spitsbergen. Norsk Geogr. Tidskr. 54, 157–168. doi: 10.1080/002919500448530
Etzelmüller, B., Schuler, T. V., Isaksen, K., Christiansen, H. H., Farbrot, H., and Benestad, R. (2011). Modeling the temperature evolution of Svalbard permafrost during the 20th and 21st century. Cryosphere 5, 67–79. doi: 10.5194/tc-5-67-2011
Ewing, S. A., O'Donnell, J. A., Aiken, G. R., Butler, K., Butman, D., Windham-Myers, L., et al. (2016). Long-term anoxia and release of ancient, labile carbon upon thaw of Pleistocene permafrost. Geophys. Res. Lett. 42, 10730–10738. doi: 10.1002/2015GL066296
Ewing, S. A., Paces, J. B., O'Donnell, J. A., Jorgenson, M. T., Kanevskiy, M. Z., Aiken, G. R., et al. (2015). Uranium isotopes and dissolved organic carbon in loess permafrost: Modeling the age of ancient ice. Geochim. Cosmochim. Acta 152, 143–165. doi: 10.1016/j.gca.2014.11.008
Fleischer, R. L. (1980). Isotopic disequilibrium of uranium: alpha-recoil damage and preferential solution effects. Science 207, 979–981.
Frey, K. E., and McClelland, J. W. (2009). Impacts of permafrost degradation on arctic river biogeochemistry. Hydrol. Process. 23, 169–182.
Frey, K. E., Siegel, D. I., and Smith, L. C. (2007). Geochemistry of west Siberian streams and their potential response to permafrost degradation. Water Res. Res. 43, W03406. doi: 10.1029/2006WR004902
Gangodagamage, C., Rowland, J. C., Hubbard, S. S., Brumby, S. P., Liljedahl, A. K., Wainwright, H., et al. (2014). Extrapolating active layer thickness measurements across Arctic polygonal terrain using LiDAR and NDVI data sets. Water Res. Res. 50, 6339–6357. doi: 10.1002/2013WR014283
Gíslason, S. R., Arnórsson, S., and Ármannsson, H. (1996). Chemical weathering of basalt in southwest Iceland: effects of runoff, age of rocks and vegetative/glacial cover. Am. J. Sci. 296, 837–907.
Haley, B. A., Frank, M., Spielhagen, R. F., and Eisenhauer, A. (2008). Influence of brine formation on Arctic Ocean circulation over the past 15 million years. Nat. Geosci. 1, 68–72. doi: 10.1038/ngeo.2007.5
Hallet, B., Hunter, L., and Bogen, J. (1996). Rates of erosion and sediment evacuation by glaciers: A review of field data and their implications. Global Planet. Change 12, 213–235.
Hindshaw, R. S., Heaton, T. H. E., Boyd, E. S., Lindsay, M. R., and Tipper, E. T. (2016a). Influence of glaciation on mechanisms of mineral weathering in two high Arctic catchments. Chem. Geol. 420, 37–50. doi: 10.1016/j.chemgeo.2015.11.004
Hindshaw, R. S., Lang, S. Q., Bernasconi, S. M., Heaton, T. H. E., Lindsay, M. R., and Boyd, E. S. (2016b). Origin and temporal variability of unusually low δ13C-DOC values in two High Arctic catchments. J. Geophys. Res. Biogeosci. 121, 1073–1085. doi: 10.1002/2015JG003303
Hindshaw, R. S., Tipper, E. T., Reynolds, B. C., Lemarchand, E., Wiederhold, J. G., Magnusson, J., et al. (2011). Hydrological control of stream water chemistry in a glacial catchment (Damma Glacier, Switzerland). Chem. Geol. 285, 215–230. doi: 10.1016/j.chemgeo.2011.04.012
Hindshaw, R. S., Tosca, N. J., Piotrowski, A. M., and Tipper, E. T. (2018). Clay mineralogy, strontium and neodymium isotope ratios in the sediments of two High Arctic catchments (Svalbard). Earth Surf. Dyn. 6, 141–161. doi: 10.5194/esurf-6-141-2018
Huang, K.-J., Teng, F.-Z., Wei, G.-J., Ma, J.-L., and Bao, Z.-Y. (2012). Adsorption- and desorption-controlled magnesium isotope fractionation during extreme weathering of basalt in Hainan Island, China. Earth Planet. Sci. Lett. 359-360, 73–83. doi: 10.1016/j.epsl.2012.10.007
Huckle, D., Ma, L., McIntosh, J., Vázquez-Ortega, A., Rasmussen, C., and Chorover, J. (2016). U-series isotopic signatures of soils and headwater streams in a semi-arid complex volcanic terrain. Chem. Geol. 445, 68–83. doi: 10.1016/j.chemgeo.2016.04.003
Huh, Y., Chan, L.-H., and Edmond, J. M. (2001). Lithium isotopes as a probe of weathering processes: Orinoco river. Earth Planet. Sci. Lett. 194, 189–199. doi: 10.1016/S0012-821X(01)00523-4
Humlum, O., Instanes, A., and Sollid, J. L. (2003). Permafrost in Svalbard: a review of research history, climatic background and engineering challenges. Polar Res. 22, 191–215. doi: 10.3402/polar.v22i2.6455
Isaksen, K., Benestad, R. E., Harris, C., and Sollid, J. L. (2007a). Recent extreme near-surface permafrost temperatures on Svalbard in relation to future climate scenarios. Geophys. Res. Lett. 34:L17502 doi: 10.1029/2007GL031002
Isaksen, K., Sollid, J. L., Holmlund, P., and Harris, C. (2007b). Recent warming of mountain permafrost in Svalbard and Scandinavia. J. Geophys. Res. 112:F02S04. doi: 10.1029/2006JF000522
James, R. H., and Palmer, M. R. (2000). The lithium isotope composition of international rock standards. Chem. Geol. 166, 319–326. doi: 10.1016/S0009-2541(99)00217-X
Juliussen, H., Christiansen, H. H., Strand, G. S., Iversen, S., Midttømme, K., and Rønning, J. S. (2010). NORPERM, the Norwegian Permafrost Database - a TSP NORWAY IPY legacy. Earth Syst. Sci. Data 2, 235–246. doi: 10.5194/essd-2-235-2010
Keller, K., Blum, J. D., and Kling, G. W. (2010). Stream geochemistry as an indicator of increasing permafrost thaw depth in an arctic watershed. Chem. Geol. 273, 76–81. doi: 10.1016/j.chemgeo.2010.02.013
Kisakürek, B., James, R. H., and Harris, N. B. W. (2005). Li and δ7Li in Himalayan rivers: Proxies for silicate weathering? Earth Planet. Sci. Lett. 237, 387–401. doi: 10.1016/j.epsl.2005.07.019
Koch, J. C., Ewing, S. A., Striegl, R., and McKnight, D. M. (2013). Rapid runoff via shallow throughflow and deeper preferential flow in a boreal catchment underlain by frozen silt (Alaska, USA). Hydrogeol. J. 21, 93–106. doi: 10.1007/s10040-012-0934-3
Kokelj, S. V., and Burn, C. R. (2003). Ground ice and soluble cations in near-surface permafrost, Inuvik, Northwest Territories, Canada. Permafrost Periglac. Process. 14, 275–289. doi: 10.1002/ppp.458
Kokelj, S. V., and Burn, C. R. (2005). Geochemistry of the active layer and near-surface permafrost, Mackenzie delta region, Northwest Territories, Canada. Can J. Earth Sci. 42, 37–48. doi: 10.1139/e04-089
Lamhonwah, D., Lafrenière, M. J., Lamoureux, S. F., and Wolfe, B. B. (2017). Evaluating the hydrological and hydrochemical responses of a High Arctic catchment during an exceptionally warm summer. Hydrol. Process. 31, 2296–2313. doi: 10.1002/hyp.11191
Lee, B., Han, Y., Huh, Y., Lundstrom, C., Siame, L. L., Lee, J. I., et al. (2013). Chemical and physical weathering in south Patagonian rivers: a combined Sr-U-Be isotope approach. Geochim. Cosmochim. Acta 101, 173–190. doi: 10.1016/j.gca.2012.09.054
Lehn, G. O., Jacobson, A. D., Douglas, T. A., McClelland, J. W., Barker, A. J., and Khosh, M. S. (2017). Constraining seasonal active layer dynamics and chemical weathering reactions ocurring in North Slope Alaskan watersheds with major ion and isotope (δ34SSO4, δ13CDIC, 87Sr/86Sr, δ44/40Ca, and δ44/42Ca) measurements. Geochim. Cosmochim. Acta 217, 399–420. doi: 10.1016/j.gca.2017.07.042
Lemarchand, E., Chabaux, F., Vigier, N., Millot, R., and Pierret, M.-C. (2010). Lithium isotope systematics in a forested granitic catchment (Strengbach, Vosges Mountains, France). Geochim. Cosmochim. Acta 74, 4612–4628. doi: 10.1016/j.gca.2010.04.057
Lemarchand, E., Tipper, E. T., Hindshaw, R., Wiederhold, J. G., Reynolds, B. C., Bourdon, B., et al. (2009). Li isotope fractionation in surface waters of an alpine granitic catchment. Geochim. Cosmochim. Acta 73:A742.
Lidman, F., Peralta-Tapia, A., Vesterlund, A., and Laudon, H. (2016). 234U/238U in a boreal stream network - relationship to hydrological events, groundwater and scale. Chem. Geol. 420, 240–250. doi: 10.1016/j.chemgeo.2015.11.014
Liu, L., Schaefer, K., Zhang, T., and Wahr, J. (2012). Estimating 1992-2000 average active layer thickness on the Alaskan North Slope from remotely sensed surface subsidence. J. Geophys. Res. 117:F01005 doi: 10.1029/2011JF002041
Ma, L., Chabaux, F., West, N., Kirby, E., Jin, L., and Brantley, S. (2013). Regolith production and transport in the Susquehanna Shale Hills Critical Zone Observatory, Part 1: Insights from U-series isotopes. J. Geophys. Res. Earth Surf. 118, 722–740. doi: 10.1002/jgrf.20037
Maher, K. (2010). The dependence of chemical weathering rates on fluid residence time. Earth Planet. Sci. Lett. 294, 101–110. doi: 10.1016/j.epsl.2010.03.010
Maher, K. (2011). The role of fluid residence time and topographic scales in determining chemical fluxes from landscapes. Earth Planet. Sci. Lett. 312, 48–58. doi: 10.1016/j.epsl.2011.09.040
Maher, K., DePaolo, D. J., and Christensen, J. N. (2006). U-Sr isotopic speedometer: Fluid flow and chemical weathering rates in aquifers. Geochim. Cosmochim. Acta 70, 4417–4435. doi: 10.1016/j.gca.2006.06.1559
Major, H., Haremo, P., Dallmann, W. K., and Andresen, A. (2000). Geological Map of Svalbard 1:100 000, Sheet C9G Adventdalen, Temakart nr. 31 Tromsø: Norsk Polarinstitutt
Manaka, T., Araoka, D., Yoshimura, T., Hossain, H. M. Z., Nishio, Y., Suzuki, A., et al. (2017). Downstream and seasonal changes of lithium isotope ratios in the Ganges-Brahmaputra river system. Geochem. Geophys. Geosyst. 18, 3003–3015. doi: 10.1002/2016GC006738
Mavromatis, V., Rinder, T., Prokushkin, A. S., Pokrovsky, O. S., Korets, M. A., Chmeleff, J., et al. (2016). The effect of permafrost, vegetation, and lithology on Mg and Si isotope composition of the Yenisey River and its tributaries at the end of the spring flood. Geochim. Cosmochim. Acta 191, 32–46. doi: 10.1016/j.gca.2016.07.003
Millot, R., Vigier, N., and Gaillardet, J. (2010). Behaviour of lithium and its isotopes during weathering in the Mackenzie Basin, Canada. Geochim. Cosmochim. Acta 74, 3897–3912. doi: 10.1016/j.gca.2010.04.025
Moore, D. M., and Reynolds, R. C Jr. (1997). X-ray Diffraction and the Identification and Analysis of Clay Minerals. New York, NY: Oxford University Press.
Nordli, Ø., Przybylak, R., Ogilvie, A. E. J., and Isaksen, K. (2014). Long-term temperature trends and variability on Spitsbergen: the extended Svalbard Airport temperature series, 1898-2012. Polar Res. 33:21349. doi: 10.3402/polar.v33.21349
Nowak, A., and Hodson, A. (2014). Changes in meltwater chemistry over a 20-year period following a thermal regime switch from polythermal to cold-based glaciation at Austre Brøggerbreen, Svalbard. Polar Res. 33:22779. doi: 10.3402/polar.v33.22779
Ockert, C., Gussone, N., Kaufhold, S., and Teichert, B. M. A. (2013). Isotope fractionation during Ca exchange on clay minerals in a marine environment. Geochim. Cosmochim. Acta 112, 374–388. doi: 10.1016/j.gca.2012.09.041
Overland, J. E., Wang, M., Walsh, J. E., and Stroeve, J. C. (2013). Future Arctic climate changes: Adaptation and mitigation time scales. Earths Future 2, 68–74. doi: 10.1002/2013EF000162
Penniston-Dorland, S., Liu, X.-M., and Rudnick, R. L. (2017). Lithium isotope geochemistry. Rev. Miner. Geochem. 82, 165–217. doi: 10.1515/9783110545630-007
Phan, T. T., Capo, R. C., Stewart, B. W., Macpherson, G. L., Rowan, E. L., and Hammack, R. W. (2016). Factors controlling Li concentration and isotopic composition in formation waters and host rocks of Marcellus Shale, Appalachian Basin. Chem. Geol. 420, 162–179. doi: 10.1016/j.chemgeo.2015.11.003
Piotrowski, A. M., Goldstein, S. L., Hemming, S. R., and Fairbanks, R. G. (2004). Intensification and variability of ocean thermohaline circulation through the last deglaciation. Earth Planet. Sci. Lett. 225, 205–220. doi: 10.1016/j.epsl.2004.06.002
Pistiner, J., and Henderson, G. M. (2003). Lithium-isotope fractionation during continental weathering processes. Earth Planet. Sci. Lett. 214, 327–339. doi: 10.1016/S0012-821X(03)00348-0
Pogge von Strandmann, P. A. E., Burton, K. W., James, R. H., van Calsteren, P., and Gíslason, S. R. (2010). Assessing the role of climate on uranium and lithium isotope behaviour in rivers draining a basaltic terrain. Chem. Geol. 270, 227–239. doi: 10.1016/j.chemgeo.2009.12.002
Pogge von Strandmann, P. A. E., Burton, K. W., James, R. H., van Calsteren, P., Gíslason, S. R., and Mokadem, F. (2006). Riverine behaviour of uranium and lithium isotopes in an actively glaciated basaltic terrain. Earth Planet. Sci. Lett. 251, 134–147. doi: 10.1016/j.epsl.2006.09.001
Pogge von Strandmann, P. A. E., Burton, K. W., Opfergelt, S., Eiriksdóttir, E. S., Murphy, M. J., Einarsson, A., et al. (2016). The effect of hydrothermal spring weathering processes and primary productivity on lithium isotopes: Lake Myvatn, Iceland. Chem. Geol. 445, 4–13. doi: 10.1016/j.chemgeo.2016.02.026
Pogge von Strandmann, P. A. E., Desrochers, A., Murphy, M. J., Finlay, A. J., Selby, D., and Lenton, T. M. (2017a). Global climate stabilisation by chemical weathering during the Hirnantian glaciation. Geochem. Perspect. Lett. 3, 230–237. doi: 10.7185/geochemlet.1726
Pogge von Strandmann, P. A. E., Frings, P. J., and Murphy, M. J. (2017b). Lithium isotope behaviour during weathering in the Ganges Alluvial Plain. Geochim. Cosmochim. Acta 198, 17–31. doi: 10.1016/j.gca.2016.11.017
Pogge von Strandmann, P. A. E., and Henderson, G. M. (2015). The Li isotope response to mountain uplift. Geology 43, 67–70. doi: 10.1130/G36162.1
Pokrovsky, O. S., Reynolds, B. C., Prokushkin, A. S., Schott, J., and Viers, J. (2013). Silicon isotope variations in Central Siberian rivers during basalt weathering in permafrost-dominated larch forests. Chem. Geol. 355, 103–116. doi: 10.1016/j.chemgeo.2013.07.016
Riotte, J., and Chabaux, F. (1999). (234U/238U) activity ratios in freshwaters as tracers of hydrological processes: The Strengbach watershed (Vosges, France). Geochim. Cosmochim. Acta 63, 1263–1275.
Riotte, J., Chabaux, F., Benedetti, M., Dia, A., Gérard, M., Boulègue, J., et al. (2003). Uranium colloidal transport and origin of the 234U–238U fractionation in surface waters: new insights from Mount Cameroon. Chem. Geol. 202, 365–381. doi: 10.1016/j.chemgeo.2002.10.002
Robinson, L. F., Henderson, G. M., Hall, L., and Matthews, I. (2004). Climatic control of riverine and seawater uranium-isotope ratios. Science 305, 851–854. doi: 10.1126/science.1099673
Rosner, M., Ball, L., Peucker-Ehrenbrink, B., Blusztajn, J., Bach, W., and Erzinger, J. (2007). A simplified, accurate and fast method for lithium isotope analysis of rocks and fluids, and δ7Li values of seawater and rock reference materials. Geostand. Geoanal. Res. 31, 77–88. doi: 10.1111/j.1751-908X.2007.00843.x
Schaefer, K., Zhang, T., Bruhwiler, L., and Barrett, A. P. (2011). Amount and timing of permafrost carbon release in response to climate warming. Tellus 63B, 165–180. doi: 10.1111/j.1600-0889.2011.00527.x
Schaffhauser, T., Chabaux, F., Ambroise, B., Lucas, Y., Stille, P., Reuschlé, T., et al. (2014). Geochemical and isotopic (U, Sr) tracing of water pathways in the granitic Ringelbach catchment (Vosges Mountains, France). Chem. Geol. 374-375, 117–127. doi: 10.1016/j.chemgeo.2014.02.028
Stumm, W., and Morgan, J. J. (1996). Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd Edn. New York, NY; Chichester; Brisbane, QLD; Toronto, ON; Singapore: Wiley.
Svendsen, J. I., and Mangerud, J. (1997). Holocene glacial and climatic variations on Spitsbergen, Svalbard. Holocene 7, 45–57. doi: 10.1177/095968369700700105
Tank, S. E., Striegl, R. G., McClelland, J. W., and Kokelj, S. V. (2016). Multi-decadal increases in dissolved organic carbon and alkalinity flux from the Mackenzie drainage basin to the Arctic Ocean. Environ. Res. Lett. 11:054015. doi: 10.1088/1748-9326/11/5/054015
Teng, F.-Z., McDonough, W. F., Rudnick, R. L., Dalpé, C., Tomascak, P. B., Chappell, B. W., et al. (2004). Lithium isotopic composition and concentration of the upper continental crust. Geochim. Cosmochim. Acta 68, 4167–4178. doi: 10.1016/j.gca.2004.03.031
Tipper, E. T., Calmels, D., Gaillardet, J., Louvat, P., Campas, F., and Dubacq, B. (2012). Positive correlation between Li and Mg isotope ratios in the river waters of the Mackenzie Basin challenges the interpretation of apparent isotopic fractionation during weathering. Earth Planet. Sci. Lett. 333-334, 35–45. doi: 10.1016/j.epsl.2012.04.023
Tipper, E. T., Louvat, P., Campas, F., Galy, A., and Gaillardet, J. (2008). Accuracy of stable Mg and Ca isotope data obtained by MC-ICP-MS using the standard addition method. Chem. Geol. 257, 65–75. doi: 10.1016/j.chemgeo.2008.08.016
Tokarev, I., Zubkov, A. A., Rumynin, V. G., Polyakov, V. A., Kuznetsov, V. Y., and Maksimov, F. E. (2005). “Origin of high 234U/238U ratio in post-permafrost aquifers,” in Uranium in the Environment: Mining Impact and Consequences, eds B. J. Merkel and A. Hasche-Berger (Berlin; Heidelberg; New York, NY: Springer), 847–856.
Tomascak, P. B., Magna, T., and Dohmen, R. (2016). “The surficial realm: Low temperature geochemistry of lithium,” in Advances in Lithium Isotope Geochemistry, eds P. B. Tomascak, T. Magna, and R. Dohmen (Cham; Heidelberg; New York, NY; Dordrecht; London: Springer), 157–189.
Toohey, R. C., Herman-Mercer, N. M., Schuster, P. F., Mutter, E. A., and Koch, J. C. (2016). Multidecadal increases in the Yukon River Basin of chemical fluxes as indicators of changing flowpaths, groundwater, and permafrost. Geophys. Res. Lett. 43, 12120–12130. doi: 10.1002/2016GL070817
Tranter, M. (2003). “Geochemical weathering in glacial and proglacial environments,” in Treatise on Geochemistry; Surface and Ground Water, Weathering and Soils, Vol. 5, eds H. D. Holland and K. K. Turekian (Amsterdam; Boston, MA; Heidelberg; London; New York, NY; Oxford; Paris; San Diego, CA; San Francisco, CA; Singapore; Sydney, NSW; Tokyo: Elsevier), 189–205.
Vigier, N., and Bourdon, B. (2011). “Constraining rates of chemical and physical erosion using U-series radionuclides,” in Handbook of Environmental Isotope Geochemistry, Advances in Isotope Geochemistry, ed M. Baskaran (Berlin; Heidelberg: Springer), 553–574.
Vigier, N., Bourdon, B., Lewin, E., Dupré, B., Turner, S., Chakrapani, G. J., et al. (2005). Mobility of U-series nuclides during basalt weathering: An example from the Deccan Traps (India). Chem. Geol. 219, 69–91. doi: 10.1016/j.chemgeo.2005.02.010
Vigier, N., Bourdon, B., Turner, S., and Allègre, C. J. (2001). Erosion timescales derived from U-decay series measurements in rivers. Earth Planet. Sci. Lett. 193, 549–563. doi: 10.1016/S0012-821X(01)00510-6
Vigier, N., Burton, K. W., Gislason, S. R., Rogers, N. W., Duchene, S., Thomas, L., et al. (2006). The relationship between riverine U-series disequilibria and erosion rates in a basaltic terrain. Earth Planet. Sci. Lett. 249, 258–273. doi: 10.1016/j.epsl.2006.07.001
Vigier, N., Decarreau, A., Millot, R., Carignan, J., Petit, S., and France-Lanord, C. (2008). Quantifying Li isotope fractionation during smectite formation and implications for the Li cycle. Geochim. Cosmochim. Acta 72, 780–792. doi: 10.1016/j.gca.2007.11.011
Vigier, N., Gislason, S. R., Burton, K. W., Millot, R., and Mokadem, F. (2009). The relationship between riverine lithium isotope composition and silicate weathering rates in Iceland. Earth Planet. Sci. Lett. 287, 434–441. doi: 10.1016/j.epsl.2009.08.026
Wanner, C., Sonnenthal, E. L., and Liu, X.-M. (2014). Seawater δ7Li: A direct proxy for global CO2 consumption by continental silicate weathering? Chem. Geol. 381, 154–167. doi: 10.1016/j.chemgeo.2014.05.005
Weynell, M., Wiechert, U., and Schuessler, J. A. (2017). Lithium isotopes and implications on chemical weathering in the catchment of Lake Donggi Cona, northeastern Tibetan Plateau. Geochim. Cosmochim. Acta 213, 155–177. doi: 10.1016/j.gca.2017.06.026
Wimpenny, J., Gíslason, S. R., James, R. H., Gannoun, A., Pogge von Strandmann, P. A. E., and Burton, K. W. (2010a). The behaviour of Li and Mg isotopes during primary phase dissolution and secondary mineral formation in basalt. Geochim. Cosmochim. Acta 74, 5259–5279. doi: 10.1016/j.gca.2010.06.028
Wimpenny, J., James, R. H., Burton, K. W., Gannoun, A., Mokadem, F., and Gíslason, S. R. (2010b). Glacial effects of weathering processes: New insights from the elemental and lithium isotopic composition of West Greenland rivers. Earth Planet. Sci. Lett. 290, 427–437. doi: 10.1016/j.epsl.2009.12.042
Keywords: permafrost, Arctic, lithium isotopes, uranium isotopes, geochemistry, weathering, active-layer, glacier
Citation: Hindshaw RS, Aciego SM and Tipper ET (2018) Li and U Isotopes as a Potential Tool for Monitoring Active Layer Deepening in Permafrost Dominated Catchments. Front. Earth Sci. 6:102. doi: 10.3389/feart.2018.00102
Received: 19 February 2018; Accepted: 05 July 2018;
Published: 26 July 2018.
Edited by:
Mark Skidmore, Montana State University, United StatesReviewed by:
Philip Pogge von Strandmann, University College London, United KingdomCarsten W. Mueller, Technische Universität München, Germany
Copyright © 2018 Hindshaw, Aciego and Tipper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruth S. Hindshaw, cnV0aC5oaW5kc2hhd0BnbWFpbC5jb20=