 Robert Raiswell
Robert Raiswell Jon Hawkings
Jon Hawkings Amira Elsenousy5
Amira Elsenousy5 Ros Death
Ros Death- 1Cohen Biogeochemistry Laboratories, School of Earth and Environment, Leeds University, Leeds, United Kingdom
- 2Bristol Glaciology Centre, School of Geographical Sciences, Bristol University, Bristol, United Kingdom
- 3National High Magnetic Field Lab and Earth, Ocean and Atmospheric Sciences, Florida State Universtiy, Tallahassee, FL, United States
- 4German Research Centre for Geosciences GFZ, Potsdam, Germany
- 5Department of Earth Sciences, University of California, Riverside, Riverside, CA, United States
A more insightful view of iron in glacial systems requires consideration of iron speciation and mineralogy, the potential for iron minerals to undergo weathering in ice-water environments, the impact of freezing on concentration and speciation, and potential for glacial delivery to undergo alteration during transport into the ocean. A size fractionation approach improves recognition of iron speciation by separating dissolved Fe (<0.2 or <0.45 μm) into soluble Fe (<0.02 μm) and colloidal/nanoparticulate Fe (0.02 to 0.2 or 0.45 μm). The ranges of soluble Fe concentrations in icebergs and meltwaters are similar (tens of nanomolar). The range of colloidal/nanoparticulate Fe concentrations in icebergs are an order of magnitude higher (hundreds of nanomolar) and up to thousands of nanomolar in meltwaters. The importance of particulate iron speciation in glacial sediments is also recognized by using carefully calibrated sequential extractions with ascorbic acid (FeA comprising fresh ferrihydrite which is potentially bioavailable) and dithionite (FeD comprising all remaining (oxyhydr)oxide Fe). Iceberg and glacier sediments contain lower concentrations of FeA (0.032 ± 0.024 and 0.042 ± 0.059 wt. %) than meltwater suspended sediments (FeA 0.12 ± 0.09 wt. %). Glacier sediments also contain low concentrations of FeD (0.060 ± 0.036) but concentrations of FeD are comparable in iceberg and meltwater sediments (0.38 ± 0.24 wt. % compared to 0.31 ± 0.09 wt.%). Reactions in ice-water systems produce potentially bioavailable Fe(II) and ferrihydrite by pyrite oxidation, iron mineral dissolution (aided by low pH and organic complexes) and reduction (aided by UV radiation). Some icebergs contain high concentrations of FeA (>0.1 wt. %) which represent samples in which the on-going transformation of ferrihydrite to goethite/hematite is incomplete. Numerical models of freezing in subglacial systems show that the nanomolar levels of soluble Fe in icebergs cannot be achieved solely by freezing, and must indicate the presence of nanoparticulate Fe and/or iron desorbed from ice or sediments during melting. Models of freezing effects in sea ice show that nanomolar levels of soluble Fe are achievable because high concentrations of hydroxide and chloride ions maintain dissolved iron as soluble complexes. Delivery of iron through fjords is temporally and spatially variable due to circulation patterns, mixing of different sources, and aggregation through salinity gradients.
Introduction
Recent studies of the iron cycle in the Southern Ocean acknowledge a range of sources, although the primary focus remains on iron sourced from atmospheric dust, continental shelf sediments and hydrothermal inputs (Tagliabue et al., 2016). Many publications also show the Antarctic Ice Sheet is a potential iron source, both to the open ocean and the shelf. Thus studies of iceberg Fe delivery to the Southern Ocean by Smith et al. (2007, 2013); Raiswell et al. (2008, 2016); Shaw et al. (2011); Death et al. (2014); Duprat et al. (2016) and Herraiz-Borreguero et al. (2016) are supported by many others showing dissolved and particulate Fe delivery from a variety of different localized glacial sources (Lannuzel et al., 2008, 2014; Gerringa et al., 2012; De Jong et al., 2013, 2015; Annett et al., 2015, 2017; Lyons et al., 2015; Monien et al., 2017). These publications demonstrate a significant role for glacial processes in delivering potentially bioavailable Fe to the Southern Ocean.
Here we will construct five working hypotheses in relation to iron behavior in glacial systems and examine the status of each in the light of new and/or recent developments. We contend that; (1) A more detailed approach to iron speciation and mineralogy will improve our understanding of iron behavior and potential bioavailability, (2) Iron minerals stored in ice can undergo a variety of reactions that may enhance or diminish potential bioavailability, (3) Freezing iron-subglacial water systems may concentrate potentially bioavailable species, (4) Freezing iron-seawater systems may concentrate potentially bioavailable species, (5) The delivery of dissolved iron may undergo significant alteration during post-melt transport. Our examples mainly focus on the Arctic but these approaches should also be applied to the Southern Ocean iron cycle. However, we exclude any detailed discussion of the influence glacial sources might have on productivity, as there are no areas in the Arctic Ocean which are currently known to be persistently iron-limited.
A primary goal of research in the iron cycle is to identify and measure those species of iron that are capable of enhancing productivity when delivered to High Nutrient-Low Chlorophyll waters. However, different organisms have widely varying abilities to transform different Fe species into a form that can be assimilated (Shaked and Lis, 2012). Furthermore bioavailability can be influenced by a wide variety of factors external to the organism (e.g., ligand concentrations, photochemistry, biological reduction, and enzymatic reduction at the cell surface). Hence there is no simple definition of bioavailability. Aqueous (< 0.02 μm) inorganic and organically-bound iron are considered the most bioavailable species (Shaked and Lis, 2012; Tagliabue et al., 2017). Fe(II) is regarded as being more readily bioavailable (von der Heyden et al., 2012; Shoenfelt et al., 2017) than Fe(III), which is poorly soluble in seawater. Fresh amorphous iron (III) (oxyhydr)oxides are also considered a better source of bioavailable Fe than aged or more crystalline minerals (e.g., Yoshida et al., 2006). Clearly iron measurements in biogeochemical studies should to be related to those species that are potentially bioavailable, including aqueous Fe, and fresh amorphous iron (oxyhydr)oxides.
Detailed Approaches to Iron Speciation And Mineralogy
Background
Wells and Goldberg (1994) have observed that “distinguishing between soluble, colloidal and particulate phases will be essential if we are to understand the cycling of matter in the ocean.” Here we evaluate current practice in this context, identify improved approaches to determine speciation and mineralogy, and evaluate results obtained using these approaches.
Dissolved Iron
Measurements of dissolved Fe (dFe) are routinely made by filtration through 0.45 or 0.2 μm filters and thus these measurements are based on size, not speciation. Size fractions only crudely approximate to speciation; the fraction >1.0 μm is particulate, that between 0.1 and 1.0 μm is colloidal and that between 0.02 and 0.1 μm is nanoparticulate. Only the <0.02 μm fraction contains any significant concentration of aqueous species.
We will use the following convention (Table 1) based on Hawkings et al. (2014) and Von der Heyden and Roychoudhury (2015), as described below.
a) Aqueous Fe (aqFe) comprises all truly aqueous Fe, including the ionic species Fe3+ and Fe2+and all their complexes with inorganic and organic ligands,
b) Soluble Fe (sFe) includes all aqFe plus those nanoparticles that pass through a 0.02 μm filter,
c) Colloidal/nanoparticulate Fe (CNFe) includes all Fe between 0.02 and 0.2 or 0.45 μm,
d) Dissolved Fe (dFe) = sFe + CNFe,
e) Particulate Fe (pFe) is all Fe extracted from material > 0.2 or 0.45 μm.

Table 1. Explanation of the terminology used.
The sFe fraction comprises ~30–40% of dFe in the open ocean whereas the colloidal/nanoparticulate/ fraction comprises at least 60–70% (Boye et al., 2010; Von der Heyden and Roychoudhury, 2015). Few measurements of sFe have been made in glacial systems although sFe is probably the best empirical measure of potentially bioavailable aqueous iron.
It is important to recognize that the CNFe fraction includes solid and amorphous phases and comprises complex mixtures of organic and inorganic materials. The inorganic materials are minerals (or mineral aggregates) whose properties are modified by size and by the nature of the organic admixtures and/or complexes. There are a variety of techniques (Shi et al., 2012; Raiswell et al., 2017) available to study the CNFe fraction. These techniques include; scanning electron microscopy (SEM), high resolution transmission electron microscopy (HR-TEM), energy dispersive X-ray spectrometry (EDX) and selective area X-ray diffraction (SAED), all of which provide compositional, structural, and mineralogical information down to nanometre scales. Synchroton-based spectroscopy data (e.g., X-ray absorption near-edge structure or XANES) can also be used to identify Fe valency at high resolution. The above techniques have been used on glacial sediments to determine mineralogy and Fe valency, most recently by von der Heyden et al., 2012, Hawkings et al. (2014, 2018) and Shoenfelt et al. (2017, 2018). Their valuable insights are discussed below.
Particulate Fe
The particulate fraction is commonly defined in terms of fractional solubility which is the percentage of total Fe that passes through a 0.2, 0.4, or 0.45 μm filter after treatment with a specified extractant (Meskhidze et al., 2016; Raiswell et al., 2017). Unfortunately a wide variety of extraction techniques are commonly used such that fractional solubilities vary widely, e.g., the fractional solubility of aeolian dusts ranges from 0.001 to 80% (Jickells and Spokes, 2001; Baker and Croot, 2010). Many modeling studies simply use a solubility value of 1–2% for aeolian dusts although the extraction data of Jickells and Spokes, 2001 indicate that this choice is arbitrary (Boyd et al., 2010). However this choice is supported ascorbic acid extractions of iron from aeolian dusts, which correspond to a fractional solubility of ~1% (see paragraph below and Raiswell et al., 2016).
Many extraction methodologies provide only qualitative statements on mineral extraction behavior and few of these distinguish between the different Fe (oxyhydr)oxides (ferrihydrite, lepidocrocite, goethite, and hematite) although the optimum target is ferrihydrite. Baker et al. (2006) use a pH 4.7 acetate extraction which Poulton and Canfield (2005) found to dissolve carbonate Fe but only negligible concentrations of ferrihydrite, lepidocrocite, goethite, and hematite. It is likely that this extraction also dissolves weakly-bound Fe (e.g., adsorbed or surface active) non-carbonate Fe. Chen et al. (2003) use a pH 4.5 formate-acetate extraction which was stated to dissolve Fe (oxyhydr)oxides (mineralogy unspecified). Winton et al. (2015) report that pH 2 hydroxylamine hydrochloride dissolves Fe from biogenic material, Fe adsorbed to clay minerals and Fe as (oxyhydr)oxides (but only from ferrihydrite and lepidocrocite; see Poulton and Canfield, 2005). The distilled water leach technique of Buck et al. (2006, 2010) is believed to extract Fe-rich nanoparticles, Fe (oxyhydr)oxides and Fe in aluminosilicate minerals. Raiswell et al. (2010) use a pH 7.5 ascorbic acid extraction that is highly selective for fresh ferrihydrite and which dissolves only very small concentrations of iron from aged ferrihydrite, lepidocrocite, goethite, hematite, and aluminosilicate minerals. Following the extraction of FeA, the residual, more crystalline iron (oxyhydr)oxides can be removed by a dithionite extraction as FeD (Raiswell et al., 1994). The significance of ferrihydrite is that the pure, freshly precipitated mineral phase (commonly termed HFO or Hydrous Ferric Oxide) has been found experimentally to be bioavailable (Wells et al., 1983; Rich and Morel, 1990; Kuma and Matsunaga, 1995; Nodwell and Price, 2001). Hence the quantitative extraction of fresh ferrihydrite by ascorbic acid represents a rough measure of potentially bioavailable particulate Fe.
New Insights
Dissolved Iron
Here we present speciation data for dissolved iron in icebergs and meltwaters. Iceberg sFe and CNFe data are available only for icebergs sampled from Greenland and Svalbard from Hawkings (2015). Icebergs from Greenland (7 samples) have range of sFe from below detection limit to 11 nM and CNFe from 7 to 182 nM. In Svalbard icebergs (8 samples) the sFe ranges from below detection limit to 20 nM and CNFe ranges from below detection limit to 550 nM. These two data set provide a mean for sFe of 5.7 ± 6.6 nM and for CNFe of 104 ± 160 nM. Data for CNFe only are available from Hopwood et al. (2016) who measure concentrations of 3–300 nM in Greenlandic icebergs, Table 2 presents an estimated range of <1 to 20 nM for sFe and <1 to 550 nM for CNFe.
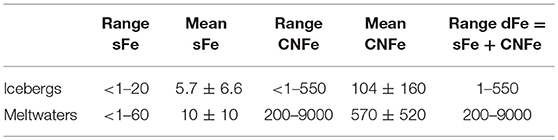
Table 2. Means and ranges of sFe and CNFe in icebergs and meltwaters (nM).
Table 2 shows data for glacial meltwaters draining from the Greenland Ice Sheet for which there are measurements of sFe and CNFe. Hawkings et al. (2014) published analytical data for more than 60 samples of meltwater from the Leverett Glacier that show sFe ranging < 1 to 37 nM. Rather higher sFe data have been found by Statham et al. (2008) for the Bristol Glacier (range 6 to 51 nM) and the Bath Glacier (range 10 to 60 nM) and together these two data sets suggest a range of sFe from < 1 to 60 nM with a mean of 10 ± 10 nM (Table 2). Hawkings et al. (2014) found CNFe ranging from 230 to 4,700 nM for the Leverett meltwaters but Statham et al. (2008) found much lower CNFe measurements (Bristol Glacier 2 to 86 nM; Bath Glacier 10 to 117 nM). Together these data suggest a range of CNFe of 2–4700 nM and a mean of 570 ± 520 nM. This range is rather lower than the range of dFe (essentially equivalent to CNFe, see above) found by Bhatia et al. (2013) for runoff sampled close to three Greenlandic glaciers (range 2200 to 9300 nM). However the data are few and are here used uncritically in Table 2 which estimates the range for CNFe in meltwaters as 200 to 9000 nM.
Particulate Fe Mineralogy
von der Heyden et al. (2012) studied the >0.2 μm fraction of particles collected from the Southern Ocean which mainly comprised discrete 20–700 nm nanoparticulate aggregates. XANES spectra of glacially-influenced samples from the Weddell Sea gyre contained Fe(II)-rich nanoparticles closely associated with organic C. In the Southern Ocean beyond the Weddell Sea Fe(III)-rich particles were found with X-ray spectral features more characteristic of Fe(III) (oxyhydr)oxides. Glacial and iceberg sediments have also been shown to contain Fe(II) in ferrihydrite-like phases (Hawkings et al., 2018). XANES spectra were used to study amorphous Fe nanophases (previously identified as ferrihydrite by Hawkings et al., 2014) in iceberg and meltwater suspended sediment particulates. The iceberg particles comprised mixtures of Fe(III) and Fe(II) containing up to ~40% Fe(II). Shoenfelt et al. (2017) used X-ray absorption spectroscopy to show that glacial sediments contained Fe(II)-bearing minerals (with spectra corresponding to silicates) were more bioavailable than non-glacial sediments containing mainly Fe(III) silicates and (oxyhydr)oxides. These studies suggest that determining the Fe(II) content of CNFe should be an important research target, with the presence of Fe(II) in ferrihydrite and/or silicate minerals representing a powerful argument for the bioavailability of iceberg sediments and meltwater suspended sediments.
Hitherto fresh ferrihydrite has been regarded as the most likely mineral source of bioavailable Fe (see above). However, Shoenfelt et al. (2017, 2018) show that Fe(II) derived from aluminosilicates (thought to be surface-active and weakly-bound in glacial sediments) is also bioavailable. This surface Fe(II) will be dissolved by ascorbic acid because the mechanism of ascorbic acid dissolution (Banwart et al., 1989) is initiated by the reduction of Fe(III), with surface (Fe(II) then being more readily detached than the residual mineral Fe(III). Hence FeA will comprise both Fe reduced from fresh Fe(III) as well any mineral surface Fe(II). Concentrations of FeA extracted from Fe(II)-bearing silicates range from 0.03 to 0.05 wt. % (Raiswell et al., 2010) which are capable of producing ~100 nM concentrations of Fe where iron silicate loadings are ~10 mg/liter (which represents 10% of a sediment load of 0.1 g/liter). These concentrations of FeA derived from Fe silicates are comparable to the range of sFe concentrations in icebergs and meltwaters (< 1–60 nM; Table 2). Trace amounts of Fe(II), whatever their mineralogy, in glacial sediments can clearly be a source of bioavailable Fe. The ascorbic acid extraction is therefore able to provide the best estimate of potential bioavailable Fe from both aluminosilicates or fresh ferrihydrite.
Particulate Fe Speciation
Data for iron present as ferrihydrite (FeA) in sediment-laden ice from glaciers are currently available from Raiswell et al. (2016) and Hopwood et al. (2017). Here we derive a combined glacial data base (Table 3) using the 8 samples collected from the Russell and Mittivakkt Glaciers (Greenland), Finsterwalderbreen (Svalbard), and Engabreen (Norway), as described by Raiswell et al. (2016). These data are combined with 12 samples (those embedded in ice) from the Midtre Lovenbreen and Kongsvegen Glaciers (Greenland) from Hopwood et al. (2017). Surface glacial samples are excluded due to their potential for alteration during sub-aerial exposure. The small 8 sample data set from Raiswell et al. (2016) deviated from normality and were treated as log normal but this data set, when combined with the Hopwood et al. (2017) data, more closely approaches normality and arithmetic means and standard deviations can be used with caution, as is the case for subsequent data compilations. These data (from 6 different glaciers) have mean of 0.042 ± 0.059 wt. %.
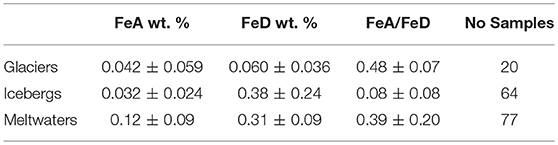
Table 3. Means and standard deviations of FeA and FeD in sediments from glaciers, icebergs and meltwaters.
The iceberg sediment FeA content is recorded in Table 3. There are two sets of Arctic iceberg data. One set utilizes 30 iceberg samples from Greenland (Sermilik and Tunulliarfik fjords) and Svalbard (Kongsfjorden, Liefdenfjorden, and Wallensbergfjorden) from Raiswell et al. (2016) but excludes samples with >0.1 wt. % FeA (as discussed in Particulate iron speciation controls). The other data set uses 34 samples (those embedded in ice) from icebergs in Kongsfjorden (Svalbard; Hopwood et al., 2017). These two iceberg data sets have different mean FeA contents (0.047 ± 0.020 and 0.019 ± 0.020 wt. %) due sampling methodology and sample heterogeneity as well as differences in bedrock geology (see Hopwood et al., 2017). However there is overlap in the two means and standard deviations and combining the two data sets produces a mean of 0.032 ± 0.024 wt. % FeA and 0.38 ± 0.24 wt % FeD.
The Greenlandic meltwater sediment compositions in Table 3 (Hawkings, 2015) are based on analyses of 33 samples taken from Leverett Glacier in 2012, 28 samples taken from Kiattuut Sermiat in 2013 and a further 16 samples taken from Leverett Glacier in 2015. Meltwater suspended sediment concentrations are variable seasonally and spatially (between localities). The samples taken from Leverett Glacier in years 2012 and 2015 are not significantly different (FeA 0.15 ± 0.02 wt. % and 0.13 ± 0.07 wt.%; FeD 0.38 ± 0.08 wt.% and 0.31 ± 0.07 wt. %) but the samples from Kiattuut Sermiat have significantly lower concentrations of FeA (0.074 ± 0.018 wt.%) and FeD (0.22 ± 0.05 wt.%). These data provide a mean FeA content of 0.12 ± 0.09 and mean FeD content of 0.31 ± 0.09 wt. %. A discharge-weighted mean would provide slightly higher mean values for FeA and FeD as the annual discharge at Kiattuut Sermiat is an order of magnitude lower than that at Leverett Glacier (Hawkings et al., 2016). Interestingly the ratios of FeA/FeD are not significantly different between the three sample sets (Leverett 2012 is 0.39 ± 0.08; Kiatuut 2013 is 0.34 ± 0.11 and Leverett 2015 is 0.43 ± 0.25) and the mean FeA/FeD ratio is 0.39 ± 0.20.
Synthesis
Some preliminary conclusions can be drawn from Tables 2, 3 but these require further validation as the data sets are small and representivity is low.
(i) The ranges of iceberg and meltwater sFe are comparable but values are considerably in excess of the saturation levels with respect to iron (oxyhydr)oxides (see Synthesis in following section). A variety of factors may be responsible; kinetic effects may inhibit the precipitation of iron (oxyhydr)oxides, sFe may contain large concentrations of nanoparticles < 0.02 μm, organic complexes may stabilize aqFe in solution (see Complexing and adsorption) and sFe may have been de-sorbed from ice or sediment surfaces during melting.
(ii) Icebergs contain an order magnitude more CNFe than sFe, but concentrations are comparable with CNFe (= dFe) in mean river water (740 nM; De Baar and de Jong, 2001). Meltwaters sampled near the glacier portal may have higher CNFe concentrations than rivers due to turbulence and insufficient time for aggregation and settling. Potentially bioavailable Fe found in CNFe includes ferrihydrite and surface-active Fe in silicates.
(iii) Mean concentrations of potentially bioavailable FeA (representing fresh ferrihydrite and surface extracted Fe) are comparable in glaciers (0.042 wt. %), icebergs (0.032 wt. %) but much higher in meltwater suspended sediments (0.12 wt. %).
Iron Mineral Reactions In Ice-Water Systems
Background
A variety of complex reactions may occur in ice-hosted, sediment-bearing layers or cavities, the inter-play between which depends primarily on sediment and water composition. These reactions have the potential to greatly enhance the Fe content of ice (as sFe, CNFe and pFe). The following discussion uses a solubility product of ferrihydrite, (Ksp ~10−39 mol−4 l−4; Stumm and Morgan, 1981) here defined qualitatively in concentration terms (i.e. for an ideal solution) as;
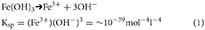
Solutions with ion product levels that exceed Ksp are saturated with ferrihydrite and further dissolution cannot occur. However, the Ksp is defined in terms of the Fe3+ ion only, and thus complexes with the Fe3+ ion do not contribute to saturation levels (see Complexing and adsorption).
Oxidation of Pyrite and Fe(II) Silicates
Sediments may remain encased in ice for periods of time that range from a few years for sea ice, up to thousands of years for subglacial ice. However, the kinetics of most mineral reactions are slow at freezing temperatures, and only rapid reactions are likely to be significant, particularly on the shorter timescales. In oxic microenvironments the most rapid iron mineral reactions involve the oxidation of pyrite and/or other Fe(II) minerals (Tranter et al., 2002; Wadham et al., 2010). The identification of schwertmannite in iceberg sediments specifically implicates pyrite oxidation (Raiswell et al., 2009) which produces iron (oxyhydr)oxides (written here as Fe(OH)3) and acidic species (Nordstrom, 2011);
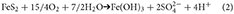
Iron oxidizing bacteria are important in catalyzing this reaction and the resulting Fe3+, formed from the dissociation of Fe(OH)3 in acidic conditions, oxidizes pyrite more rapidly than oxygen (Moses et al., 1987). Thus reaction (2) can be written as;
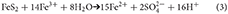
and the resulting acidity produces proton-assisted dissolution (see Dissolution and reduction of Fe(III) oxides) unless buffered by carbonate minerals. Pyrite oxidation proceeds very rapidly because the oxidation of Fe2+ to Fe3+ is biologically mediated by iron oxidizing bacteria whereas the abiotic reaction of Fe2+ with dissolved oxygen is slower. Nordstrom (1982) concluded that oxidation via Fe3+ (with a half -life ~1.5 to 15 days) predominates at pH < 8. Above pH 8, Fe3+ hydrolyses, and precipitates as Fe(OH)3 and oxygen becomes the most important oxidant (and the half -life for oxidation increases to about a year). Both biotic and biotic oxidation have been observed down to −12°C (Elberling, 2005).
Similarly rapid oxidation rates are found in seawater. Gartman and Luther (2014) have conducted experiments in oxygenated seawater at pH 8 and 25°C using synthesized pyrite (faster rates are found using crushed mineral pyrite). These experiments found half-lives for initial rates of oxidation ranging from 13 to 356 days, depending on the initial concentrations of the reactants, but not on particle size or surface area. Rates were found to be ~3x faster in diluted (half strength) seawater but ~3x slower at 9°C. These data suggest that oxidation rates will be slower in sea ice (where temperatures are typically sub-zero and solutes are concentrated by freezing).
The oxidation of Fe(II) in silicates or carbonates occurs less rapidly because insufficient acidity is generated to solubilise the Fe3+needed to catalyze rapid microbial oxidation. These reactions also produce iron (oxyhydr)oxides but are only likely to be more important in older ice.
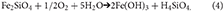
Reactions 2 to 4 all consume oxygen and may thus produce dysoxic or anoxic microenvironments in which pyrite can be oxidized using nitrate (e.g., Bosch et al., 2012). In summary, the rapid weathering of Fe(II)-bearing minerals in oxic conditions produces a high degree of supersaturation that favors the formation of nanoparticles of ferrihydrite (Waychunas et al., 2005).
Dissolution and Reduction of Fe(III) Oxides
Dissolution of ferrihydrite (and other iron (oxyhydr)oxides) can occur in aqueous, ice-enclosed cavities and produces Fe3+as in equation (1). The slightly acidic pH (5.5-6.0) of snow and glacial ice melt (see New Insights in following section and Tranter and Jones, 2001) expedites proton-assisted dissolution of iron minerals. Dissolution in glacial and sea ice is also enhanced because freezing concentrates particulate iron and protons into liquid at ice grain boundaries (Kim et al., 2010). In a closed system the accumulation of aqueous Fe3+ rapidly produces saturation with respect to ferrihydrite and dissolution stops when the solubility product (~10−39) is reached, unless the pH becomes more acidic (consuming OH−) and/or unless complexing and adsorption occur (which decrease the concentration of Fe3+).
Reducing environments can result where closed system pyrite oxidation is able to consume the available oxygen. Iron (oxyhydr)oxides may then be microbially reduced in anaerobic subglacial environments that are able to host viable iron reducing bacteria (Nixon et al., 2017). Fe3+can also be abiotically reduced to ferrous iron in near-surface ice by UV (shown as hv) radiation (Kim et al., 2010).

UV radiation can penetrate to depths of 5–10 cm into ice depending on ice crystallinity, sediment content etc. (Rowland and Grammas, 2011). The resulting ferrous iron may be incorporated into ice during freeze/thaw cycles (see Freeze-thaw reactions in ice) although ferrous iron at shallow depths in ice will be rapidly re-oxidized to (oxyhydr)oxide minerals where ice becomes permeable or melts.
Complexing and Adsorption
Complexing plays an important role by removing the Fe3+ formed in equation (1) into species that do not influence the solubility product. However, complexed species still contribute to aqFe and sFe. The most abundant inorganic species that have the ability to complex Fe3+are the hydroxide and chloride ions (see Geochemical modeling sections). Organic complexes (Lannuzel et al., 2014) are important in sea ice and the presence of EPS (extracellular polymeric substances) both aids dissolution and converts Fe3+ into a complex which has no influence on the solubility product.
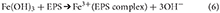
Adsorption of multi-valent ions may also occur to ice surfaces (Lannuzel et al., 2014) and to negatively charged species such as clay minerals. The removal of Fe3+ by adsorption thus diminishes the contribution of this ion to the solubility product but the adsorped species may still be measured as sFe and CNFe (when released from melted ice) or pFe.
New Insights
Freeze-Thaw Reactions in Ice
Freezing concentrates water and solutes into microenvironments at grain boundaries (Kim et al., 2010). Freezing may also conserve gases hence creating oxic environments where the rapid oxidation of pyrite and other Fe(II) minerals may occur (see Oxidation of pyrite and Fe(II) silicates). Figure 1 summarizes the potential reactions that could occur in a sediment-rich, watery layer or cavity enclosed in ice, which is close enough to the ice surface to allow the transmission of UV radiation and to allow temperature fluctuations due to solar heating and freezing.Temperature changes arise because solar energy is absorbed more readily in sediments than in the enclosing ice (which conducts heat less readily). Weathering reactions slow or stop when solar radiation diminishes and temperatures fall to freezing.
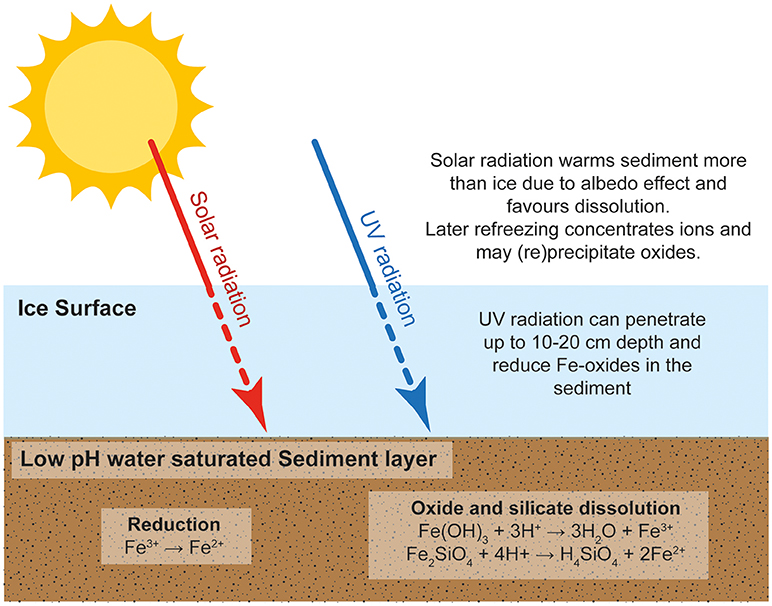
Figure 1. Schematic representation of the redox and dissolution/precipitation cycles that occur in an ice-hosted sediment layer near the ice surface.
Warmer intervals initiate thaw cycles and iron mineral reactions (equations 1, 3, and 4) occur. However, Fe3+ addition to the enclosed water may be limited by saturation with ferrihydrite where oxic conditions exist. As freezing is initiated, so concentrations of dissolved iron increase, and Fe3+ may be adsorbed to ice surfaces, precipitated as ferrihydrite (or with other minerals) and aggregated into CNFe. This removal of Fe3+ into sediment and/or ice may not be fully reversed when ice melts and undersaturation with respect to ferrihydrite then exists. Hence Fe3+ is then below saturation levels and further dissolution can occur. Repetitive freeze/thaw cycles can thus continue to add Fe to ice, water and/or sediment.
There are two sets of Arctic iceberg data (see Particulate Fe speciation). One set utilizes 30 iceberg samples from Greenland (Raiswell et al., 2016) but excludes samples with >0.1 wt. % FeA (see Particulate iron speciation controls in icebergs). The other data set uses 34 samples (those embedded in ice) from icebergs in Kongsfjorden (Svalbard; Hopwood et al., 2017). These two iceberg data sets have different mean FeA contents but combining the two data sets produces a mean of 0.032 ± 0.024 wt. %. Including the samples from Raiswell et al. (2016) with >0.1% FeA produces a higher mean of 0.059 ± 0.085 wt. %. Neither estimate is significantly different from the FeA content of sediment entombed in glaciers.
Particulate Iron Speciation Controls in Icebergs
Pyrite is present in a wide variety of sedimentary, igneous, and metamorphic rocks although average crustal S contents are low (0.02 to 0.24 wt. %; Faure, 1998). Hence ferrihydrite should be a common subglacial weathering product, irrespective of bedrock geology. However, ferrihydrite (measured as FeA) is not found in large concentrations in ice-hosted sediments (Table 3). There could be several reasons for this; the initial pyrite concentrations in the glacial bedrock might have been low and entombment in ice may have minimized water contact (and thus weathering and alteration). However, Schwertmann et al. (2004) have shown that the transformation of ferrihydrite (FeA) to goethite/hematite (FeD) occurs at low temperatures (half-lives at 4°C range from ~30 year at pH 6 to ~3 year at pH 8). Transformation can be prevented by adsorption of Si and organic matter (Jones et al., 2009). However, the ferrihydrite formed in a subglacial environment (where contact with organic matter is limited and where the alteration kinetics are slow) may be preserved in ice and may remain potentially bioavailable.
Raiswell et al. (2016) found that the FeA contents of Arctic and Antarctic samples of iceberg sediments were significantly higher than glacier-hosted sediments. The icebergs were not all calved from the land-based glaciers that were sampled but it was hypothesized that the iceberg sediments had been weathered and altered during post-calving transport due to interactions with seawater and temperature fluctuations that induced melting, weathering and re-freezing (see Freeze-thaw reactions in ice).The difference between sediments in glaciers and icebergs that Raiswell et al. (2016) identified is here re-examined using the databases shown in Table 3. The new means and standard deviations for the sediments from icebergs (0.032 ± 0.024 wt. %) and glaciers (0.042 ± 0.059 wt. %) are not significantly different. This contradiction with the Raiswell et al. (2016) conclusion mainly arises from separating out the samples >0.1% FeA from the iceberg data. Plots of FeA against FeD (Figure 2) show two different relationships between FeA and FeD. One relationship shows persistently low concentrations of FeA (< 0.1 wt. %) over relatively large concentrations of FeD (ranging from 0.1 to 1.2 wt. %). There are several explanations for the low variations in FeA over a large range in FeD; these samples may originally have contained little pyrite, or may have been little weathered, or sufficient time may have passed for all FeA to have been altered to FeD. These circumstances will all produce sediments with low values of FeA associated with high values of FeD.
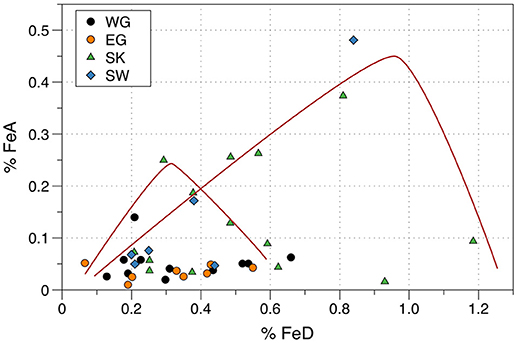
Figure 2. Relationship between FeA wt. % and FeD wt. % for Arctic iceberg sediments. Data from Raiswell et al. (2016). Localities are Wallensbergfjorden (Svalbard, SW), Kongsfjord (Svalbard, SK), Sermilk (East Greenland, EG), and Narsarsuaq (West Greenland, WG). Red lines represent possible pathways of FeA formation by weathering and loss by transformation in different iceberg microenvironments.
However, Figure 2 also shows that nine iceberg sediments have FeA > 0.1 wt. % (and up to 0.5 wt. %) over a wide range of FeD (0.2 to 1.2 wt. %). These samples crudely fit a series of curves, each with a limb of rising FeA that reaches a peak and then declines as FeD continues to increase. Such curves would be expected in a system where pyrite initially weathers to form FeA faster than FeA alters to FeD. No single curve would be expected as each ice-enclosed microenvironment will evolve over a different pathway depending on the amount of pyrite, the rate of weathering and the rate of alteration to goethite/hematite. Figure 2 shows that these temporarily enhanced proportions of FeA can produce relatively high ratios of FeA/FeD. Short transport times of icebergs to the marine environment will favor the preservation of FeA and thus some iceberg sediments with high ratios of FeA/FeD may be released to the open ocean.
Synthesis
Sediment Fe speciation data suggest the following controls on composition, which require validation.
(i) The presence of schwertmannite demonstrates that pyrite oxidation occurs in subglacial systems and produces potentially bioavailable ferrihydrite measured as FeA. Concentrations of FeA in ice are low, either because glacial bedrock contains little pyrite, or oxidative weathering of pyrite was limited.
(ii) Ferrihydrite entombed in ice transforms slowly to goethite/hematite (measured as FeD). Measurements of FeA and FeD in icebergs are consistent with a model where temporarily high concentrations of FeA exist prior to their slow transformation to FeD. Further studies of icebergs are required to confirm this model, which suggests that models of FeA release by iceberg melting may need to incorporate a time-decay function based on the kinetics of ferrihydrite conversion to goethite/hematite.
Iron Behavior During Subglacial Freezing
Background
Complex interactions occur between ions, gases, ice and water during freezing. The major ions are not readily incorporated into the ice lattice and the remaining unfrozen water may thus become highly concentrated in solutes. Consistent with this, observations of natural systems show that the water first released from melting ice is enriched in the major cations by 3–5 times compared to the initial fluids (producing an “ionic pulse”; e.g., Lilbaek and Pomeroy, 2008; Telling et al., 2014). Rates of solute exclusion are influenced by the rate of freezing and the morphology of the ice-water interface. Fast rates of freezing produce solute-rich boundary layers at the ice-water interface that favor incorporation into ice, unless the layers are removed by diffusion, turbulence or convection (Killawee et al., 1998).
Experimental work with cations has mainly focused on Ca2+-HCO waters (Hallet, 1976; Killawee et al., 1998). There are no studies that consider the behavior of Fe during subglacial freezing but important clues are provided by Killawee et al. (1998), who conducted freezing experiments with varying initial CaCO3 saturation states and varying concentrations of the major cations. No attempt was made to remove boundary layer effects. All the experiments showed CaCO3 precipitation and steady increases in Ca, Mg, Sr and Na as freezing progressed. The combination of calcite precipitation plus freezing caused the pCO2 to rise steadily up to values of ~10−2.5 atm. The concentration increases for Na (~3.5×) were larger than those for Ca, Mg, and Sr (which were being removed into the precipitating calcite), nevertheless Ca, Mg, and Sr were still enriched in the final solution by a factor of ~2.5×. Relatively low rates of freezing reduced boundary layer effects (and thus incorporation into ice) and allowed ions at the ice interface to diffuse away into the residual solution. Hence the rate of freezing exerts a significant influence on the extent of concentration (Killawee et al., 1998). These experiments show that progressive freezing increases concentrations in residual water but even ions removed by mineral precipitation continued to increase in the residual, freezing water.
New Insights
In this section the Geochemists Workbench is used to explore the behavior of iron during freezing. The Geochemists Workbench uses Pitzer equations to derive the ion interaction parameters in concentrated solutions. The models envisage a water-filled cavity enclosed in ice with no connections to external fluids or the atmosphere (a closed system), from which water is progressively removed (following the approach of Elsenousy et al., 2015). It is assumed that;
(a) The water is maintained at 0°C throughout by slow freezing (no boundary layer effects),
(b) Closed system freezing removes only water (gases and ions are conserved in the residual water, except where mineral dissolution or precipitation occurs as described below),
(c) Excess ferrihydrite is always present and the waters are maintained in equilibrium with ferrihydrite throughout freezing.
The absence of boundary layer effects in (a) minimizes the concentration of aqFe, whereas the presence of a closed system in (b) maximizes the concentration of aqFe in the freezing water whilst (c) limits the accumulation of aqFe by maintaining saturation with respect to ferrihydrite. The models take no account of kinetics and kinetic inhibition of precipitation is likely to cause species concentrations to be higher than plotted in Figures 3–10. However, many different models could be constructed using different assumptions (temperature and water composition etc) and the present models should be regarded as only exploratory. Our purpose here is simply to investigate the aqFe concentrations that could be reasonably be achieved during subglacial freezing in the absence of organic complexes and Fe mineral dissolution (except as required to maintain equilibrium with ferrihydrite). Note that the initial composition is based on an unpolluted rainwater (Table 4) that includes only the major inorganic cations and anions. The models examine how freezing influences the concentration of aqFe species. Some proportion of aqFe may also be removed by aggregation to CNFe and/or pFe (but quantifying these contributions is impossible).
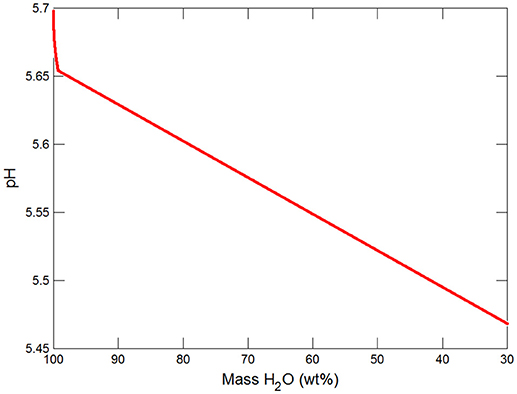
Figure 3. Variations in pH during freezing of glacial water.

Table 4. Initial water compositions for freeze models (from Berner and Berner, 1987).
Figures 3, 4 show the first changes produce vertical trends in pH and dissolved pCO2 as the carbonate system in the water equilibriates with the atmosphere at 0°C. Carbonates precipitate (mainly CaCO3 plus a lesser amount of dolomite) which produce the sharp linear increases in pCO2 and decreases in Ca2+ (Figure 5) and pH. When ~50% of the water has been lost and the pH is ~5.55, the dominant dissolved Ca species is the CaHCO complex (Figure 5), the concentration of which continues to increase through the remaining water loss by freezing. Carbonate precipitation continues but the rates of Ca removal by precipitation, and additions by freezing, are similar and thus the concentration of Ca remains uniform. The lowest pH reached is ~5.47 after the removal of ~70% of the water. Figure 6 shows that Fe(OH)2+and Fe(OH)+ complex ions are the dominant dissolved iron species over the pH range from 5.7 to 5.47. Iron chloride complexes increase during freezing as chloride concentrations increase, but these species never become quantitatively significant.
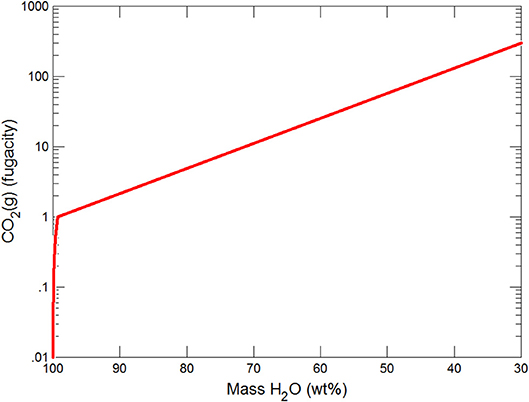
Figure 4. Variations in pCO2 during freezing of glacial water.
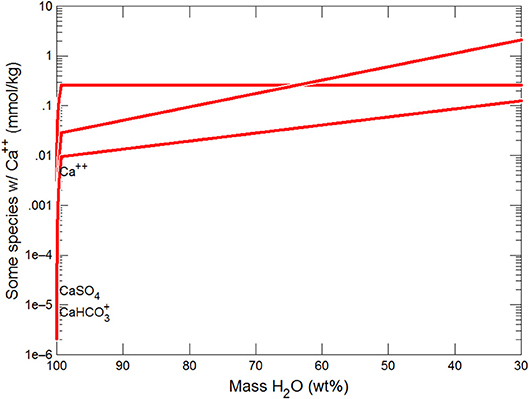
Figure 5. Variations in Ca species during freezing of glacial water. Final solution has Ca bicarbonate>Ca>Ca sulfate.
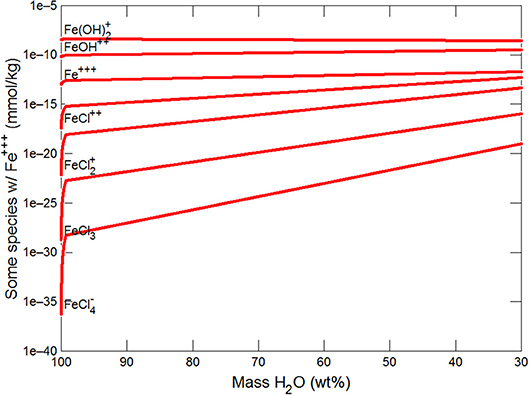
Figure 6. Variations in Fe species during freezing of glacial water.
Synthesis
The models require further validation but are broadly consistent with the observations of natural and experimental systems in that;
(i) Significant solute concentration increases can be achieved during subglacial freezing and are accompanied by decreases in pH and increases in pCO2.
(ii) The models predict aqFe concentrations in this subglacial system, in equilibrium with ferrihydrite, that are maintained at ~4 × 10−8 mM or ~ 0.04 nM throughout freezing. However, the nanomolar levels of sFe in ice (<1 to 20 nM; Table 2) are many orders of magnitude higher than the model aqFe predictions and appear too high to be reached by simple freezing.
(iii) Higher aqFe values would result if high concentrations of complexing species (chloride, EPS etc) were present (as in the seawater models, see New Insights in following section) and/or continued dissolution of Fe-bearing minerals occurred (especially in anoxic and/or acidic conditions), assisted by UV radiation.
(iv) Alternatively high sFe values from ice may result the measurement methodology which includes nanoparticulate Fe < 0.02 μm and Fe species desorbed from sediment during melting.
(v) High CNFe values in ice and meltwaters (200–9000 nM) are considerably in excess of ferrihydrite equilibrium levels and must be mainly present as nanoparticles.
Iron Behavior During Seawater Freezing
Background
Sea ice exerts an important role in the iron cycle in the polar regions (Vancoppenolle et al., 2013; Wang et al., 2014; Lannuzel et al., 2016). There are many more measurements of sea ice Fe in the Antarctic than the Arctic but measurements in both regions are too few to characterize adequately a system which is highly variable, both temporally and spatially. In general, however; (i) measurements of dFe and pFe in land-fast sea ice are higher than in pack ice due to the increased opportunity for sedimentary inputs, (ii) concentrations of dFe are higher toward the bottom of the ice because sea ice algae scavenge Fe from seawater and Fe can be transported vertically downwards by brine drainage, and (iii) the concentration of dFe in sea ice is several times higher than in the seawater under the ice (Lannuzel et al., 2016). Thus the seasonal release of potentially bioavailable dFe is able to relieve iron limitation and produce phytoplankton blooms at melting sea ice margins (Vancoppenolle et al., 2013; Wang et al., 2014; Lannuzel et al., 2016).
However, the processes by which Fe is incorporated into sea ice are not well-known. Wang et al. (2014) suggest three potential mechanisms; (1) dissolved and particulate Fe can be removed from seawater by entrainment during ice formation, (2) Fe-rich particles, colloids, and organic matter are harvested during winter by ice crystals that rise through the water column toward the ice base, forming an iron-rich substrate, and (3) re-suspended shelf sediments can also be added to the underlying ice surface and aeolian dust can be deposited on the ice surface and incorporated into the ice.
Subsequent within-ice processes are also poorly understood. Observations from melted ice sections indicate that dFe is excluded from ice into brine-rich microenvironments (created by freezing seawater) which are trapped within relatively impermeable winter ice. In spring, sea ice becomes more permeable and dFe-rich brines can then drain into the underlying seawater (Lannuzel et al., 2007, 2014). Both dFe and pFe are believed to exchange with each other within sea ice. Algae and organic compounds incorporated within sea ice have a high affinity for dFe which is thus removed into pFe (Hassler and Schoemann, 2009). Conversely some pFe goes into dFe due to cell lysis, heterotrophic activity and photo-oxidation. Heterotrophic reduction of Fe (oxyhydr)oxides produces ferrous iron, which can be stabilized by EPS complexes. Leakage of complexed Fe from brine inclusions delivers dFe to seawater as the ice becomes more permeable during melting.
Experimental and computer modeling studies have been aimed at understanding the sequential chemical changes that occur as seawater is progressively frozen in sea ice. Two main pathways have been identified, depending on whether precipitated minerals are removed from the residual brines (the Gitterman pathway) or are allowed to interact with the brines (the Ringer-Nelson-Thompson or R-N-T pathway). Both pathways sequentially produce mirabilite (Na2SO4), hydrohalite (NaCl.2H2O), sylvite (KCl), and finally MgCl2.12H2O. The main differences between the two pathways are the non-appearance of gypsum in the R-N-T pathway and minor differences in precipitation temperatures. Subsequent studies of natural and experimental systems (Marion et al., 1999; Geilfus et al., 2013; Butler and Kennedy, 2015) confirm the formation of gypsum and thus favor the Gitterman pathway.
Sea ice is a very heterogeneous medium. Delille et al. (2014) envisage that vertical heterogeneity produces an upper layer with dense ice which is also porous, and a lower layer that contains pockets and channels that may be temporarily or permanently open to seawater. The upper layer can be considered as a closed system where carbonate precipitation occurs and the pCO2 increases due to precipitation and freezing (Delille et al., 2014). Temperatures in the upper layer may reach −20 to −30°C but the lower layer in contact with seawater is much warmer (−2 to −5°C; Delille et al., 2014; Lannuzel et al., 2016). The models explored here apply only to the upper, closed system layer and examine the potential for aqFe concentrations to be increased by freezing removal of water.
New Insights
This section follows the modeling approach developed in the section on modeling subglacial waters using Geochemists Workbench. The seawater models use the same initial assumptions as the glacial model but with an initial composition corresponding to average seawater (Table 4). Freezing initially produces precipitation of CaCO3 (with a small amount of dolomite) which together increase pCO2 and dramatically decrease pH (Figures 7, 8). When the water mass has declined to ~80%, the Na+ and sulfate become concentrated to the point where mirabilite precipitates (Figure 9). Figure 9 shows that the concentration of Na+ continues to increase due to freezing (even whilst mirabilite precipitates), until the water mass declines to ~55% of the initial value. Now hydrohalite is supersaturated and Na+ concentrations rapidly decline. As the water mass continues to decline, magnesite precipitates (at ~50%), and then sylvite (at 37%), by which point the pH has decreased to ~4.7. These changes broadly correspond to those observed in the seawater freezing experiments described above. The increasing chloride concentrations enable high concentrations of aqFe to exist as the complexes FeCl and FeCl. Figure 10 shows that FeCl constitutes ~10−6 mM or ~ 1 nM when the water mass has been reduced to ~25%. Note that these concentration levels represent aqFe, which will be lower than sFe (as in the glacial model, see Synthesis in previous section) and substantially less than dFe.
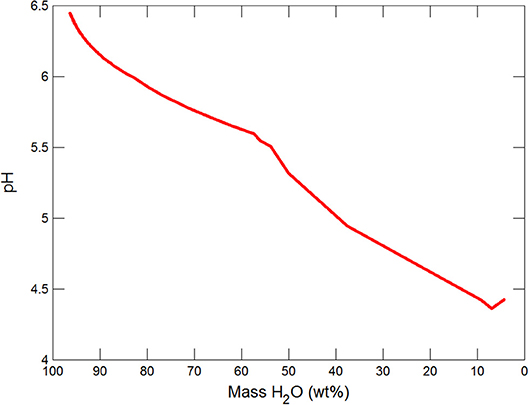
Figure 7. Variations in pH during freezing of seawater.
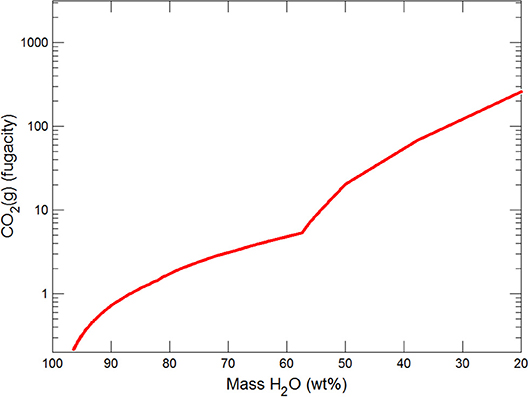
Figure 8. Variations in pCO2 during freezing of seawater.
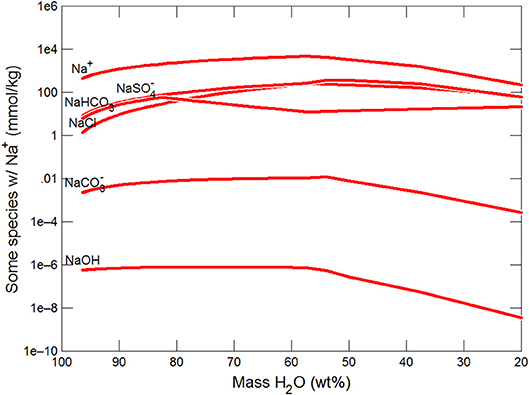
Figure 9. Variations in Na species during freezing of seawater.
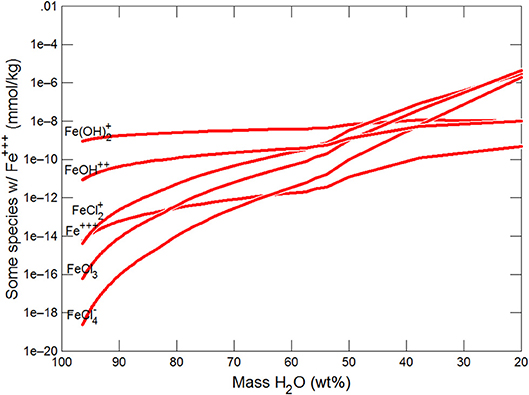
Figure 10. Variations in Fe species during freezing of seawater.
Synthesis
The model predictions of mineral behavior are broadly consistent with the experimental behavior but modeling has produced some important additional insights that should be re-evaluated. In respect of Fe the models predict that;
(i) High concentrations of aqFe (~ 1 nM) can be achieved during seawater freezing due to increasing concentrations of chloride which complex Fe and retain aqFe in solution. These aqFe concentrations are at the upper end of the observed concentrations of dFe in Arctic and Southern Oceans (typically ~0.1 to 1.0 nM; Wang et al., 2014).
(ii) Higher aqFe concentrations could be achieved by invoking within-ice biological processes and continued mineral-water reactions and complexation with EPS and/or other organic species (see Lannuzel et al., 2015).
(iii) Fe enrichment would be confined to brine pockets or hotspots that would be irregularly distributed within the upper ice layers. Drainage of these hotspots produces pulses of Fe-enriched water that may be transferred to sea water as the ice melts, becomes more permeable and drains through channels into seawater.
Modification of Glacial Sediments During Transport
Background
Glacial sediment sources may undergo substantial losses and/or modification during transport into the marine environment. Sediments produced from land-terminating glaciers discharging into meltwater rivers may be altered during passage through the pro-glacial environment, especially where there is storage in pro-glacial lakes (Bhatia et al., 2013; Zhang et al., 2015). However two extreme situations exist for the transport of ice-hosted sediment; one represents glaciers where the main ice mass loss is directly into the ocean, and the other occurs where glaciers enter long fjords (up to 150 km in length) where the main ice mass loss is by melting into the fjord (Hopwood et al., 2015; Straneo and Cenedes, 2015). In this latter case, two main processes have been identified that have the potential to alter the speciation of iron and/or its delivery into the marine environment; (a) circulation patterns in fjords may diminish, or even prevent, the export of ice-hosted iron species into coastal waters (e.g., Hopwood et al., 2016, 2017), and (b) passage through a salinity gradient assembles meltwater sFe and CNFe into aggregates that can slowly sediment out before entry into coastal waters (e.g., Hawkings et al., 2014; Schroth et al., 2014).
Some Challenging Problems
Many glaciers in the Arctic terminate in fjords, which can be up to 150 km in length (Straneo and Cenedes, 2015). Seasonal discharge from the Greenland Ice Sheet creates strong lateral and vertical gradients in the physical and chemical properties of water along these fjords. Hence icebergs and meltwaters (and their associated sediments) derived from fjord-terminating glaciers will be affected by complex interactions between tides, density driven seawater inflows, wind patterns and freshwater glacial melt (Meire et al., 2016).
Fjord-terminating glaciers supply meltwater in two ways; supraglacial drainage off the glacier forms a fresh water surface layer (which is a relatively minor contribution) whilst most melting produces a buoyant plume of subglacial meltwater that entrains and mixes with the ambient fjord water as it rises near the glacier front. These plumes can either reach the surface water or travel below the surface at their neutral buoyancy level (Straneo and Cenedes, 2015). In Greenland, highly turbid plumes reaching the surface have been observed at some tidewater glaciers, at least for some part of the melt season (Meire et al., 2017; Kanna et al., 2018). However other studies have found only mid-depth intrusions of glacially modified water (Mortensen et al., 2011) and the impact of this on the transport of Fe is currently unknown. Plume behavior is likely to be temporally variable with buoyancy effects controlled by the magnitude of the subglacial discharge and the salinity and temperature of the fjord water at depth. A strong vertical stratification may prevent rising sub-glacial meltwater (and the associated Fe) from mixing into surface water. These complexities are illustrated in the example discussed below.
Godthabsfjord (SW Greenland)
Cold subglacial discharge in Godthabsfjord has been observed rising from < 100 m to mix with fjord water that became trapped beneath a low salinity surface layer (Mortensen et al., 2013). However, surface waters in Godthabsfjord do transport glacial particulate material (and associated Fe) out of the fjord although the precise origin these sediments is not clearly established. Figure 11 shows data from Hopwood et al. (2016) who measured DFe (filtered < 0.2 μm; comprising sFe plus CNFe, see Table 1) and TDFe (unfiltered and acidified to pH 2; comprising sFe, CNFe and some leached particulate Fe, see Table 1). This data allows particulate Fe to be estimated as (TDFe—DFe), which can be plotted against turbidity (measured by Optical Back Scattering or OBS). The LT samples are from downstream of Lake Tasersuaq and have low ratios of (TDFe—DFe)/OBS due to high turbidity arising from a particle plume. This plume was interpreted by Hopwood et al. (2016) as resulting from re-suspended mud flat pro-glacial sediments. However, headwaters with high particulate loads also enter Lake Tasersuaq to the north of where the LT samples were taken, sourced from a large land-terminating glacier. Furthermore, Meire et al. (2017) have observed surface water samples up-fjord from LT which have high particulate loads derived from the Kangiata Nunaata Sermai and Akullersup Sermia glaciers. These glaciers may thus be the source of some, or all, of the high Fe particulate material in the LT samples.
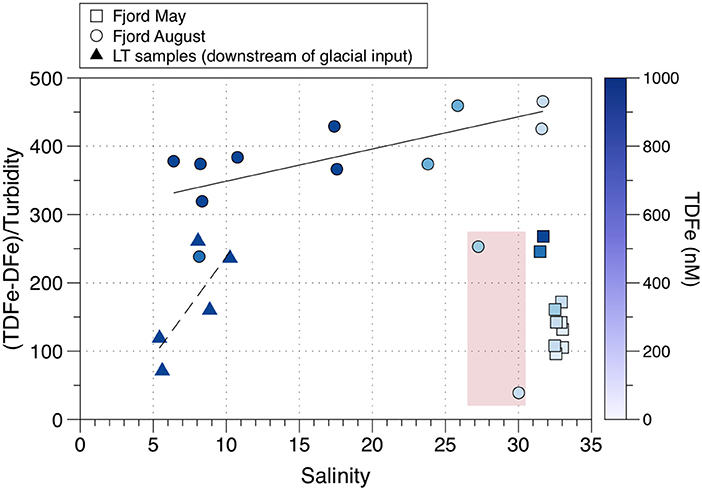
Figure 11. Variations in the ratio of (TDFe-DFe)/Turbidity (measured as OBS or Optical Back Scattering) with salinity in Godthabsfjord (Greenland). The dash lined indicates a linear best fit of LT samples (low salinity, high turbidity meltwater plume), and the solid line indicates a best fit of other August fjord samples (excluding outlier stations GF4 and GF5, highlighted in red). Data from Hopwood et al. (2016).
Whatever their source, the plot of the LT samples trends up to the low salinity GF samples defining a rise in (TDFe—DFe)/OBS that reflects declining OBS, due to the sedimentation of coarser material, which produces particulate iron enrichment. The remaining GF samples show a uniform trend in (TDFe—DFe)/OBS that runs from ~5–32 ppt salinity (with no evidence for Fe losses through the salinity gradient, see below). Interestingly, there is a sharp anomalous decline in this relationship between a salinity of 27–30 ppt. The reasons for this are unknown, and the trend over a salinity of 30 ppt follows that below 27 ppt. These two stations (GF4, GF5) are close to the Greenland capital city of Nuuk and therefore there could be a localized higher turbidity influence from the settlement. In the present context, Figure 11 clearly shows that the surface waters in Godthabsfjord can transport fine, iron-enriched glacially-derived particulates out of the fjord. However, dissolved and particulate glacial Fe export may be impeded when deep turbid, subglacial melt cannot reach the surface waters and when dominant southerly winds and warm surface waters ensure that most ice is trapped, and melts, within the fjord surface waters (Hopwood et al., 2016). The variable role of subsurface upwelled glacial water in supplying nutrients to the fjord surface waters requires further study.
The Influence of Salinity Gradients
Meltwaters emerging into seawater pass through a salinity gradient where aggregation removes a significant proportion of dFe. Zhang et al. (2015) found losses of ~90% for dFe in meltwaters passing through the Kongsfjorden estuary, with only 4.7 nM of dFe (stabilized by organic complexes) actually reaching seawater. Hence losses through estuaries are commonly assumed to be ~90% for dFe in meltwater (Hawkings et al., 2014; Schroth et al., 2014). The removal of dFe from river water varies widely from 75 to >95% but aggregation may only affect CNFe and not aqFe. Dai and Martin (1995) have shown that aqueous Fe (< 3 nm) shows a linear, conservative mixing trend with increasing salinity whereas their colloidal/nanoparticulate fraction (3 nm to 0.4 μm) shows a concave, non-conservative trend of flocculation and removal with increasing salinity.
The removal of dFe (mainly CNFe) will be influenced by compositional differences such as particle composition and ligand availability, but it has also been suggested that aggregation is more efficient in summer than winter (Mayer, 1982). Laboratory studies (Markussen et al., 2016) also show that the kinetics of the flocculation and aggregation process are linked to the labile Fe content of the particles (as measured by extraction with ammonium oxalate at pH 3; see Particulate Fe). Increased levels of dFe increase the surface area of the flocs and decreases their density. In situ observations in Disko Fjord showed that the horizontal transport of particulate labile Fe occurs partly because μM levels of dFe (essentially all CNFe; see Table 1) produce particles as larger flocs but with a lower density that sediment more slowly (Markussen et al., 2016). Such a mechanism might produce the Fe-enriched particles observed by Hopwood et al. (2016) at high salinities in Godthapsfjord. Further studies of particulate Fe measurements in relation to particulate concentration (mass/volume, turbidity) are needed to ascertain the influence of the density alterations observed by Markussen et al. (2016) in other fjord systems.
Synthesis
The temporal and spatial variations in fjords present challenging problems for recognizing and quantifying glacial ice-hosted Fe sources, which will require that mineralogy and speciation are considered within a rigorous oceanographic framework. Complications may also arise from the addition of multiple dFe sources, including pro-glacial drainage to surface fjord waters and benthic recycling to bottom waters (e.g., Wehrmann et al., 2014). Removal processes through salinity gradients also need to pay more attention to speciation to distinguish between sFe and CNFe and examine their different behavior through a salinity gradient.
Conclusions
Accurate estimates of bioavailable Fe are at present impossible but the optimum measure of bioavailable Fe is sFe (the <0.02 μm fraction of dFe). However, the larger CNFe fraction contains ferrihydrite that has been shown experimentally to be bioavailable. In general the CNFe fraction is poorly characterized and the routine use of high resolution microscopy techniques would provide valuable information about the form of CNFe and its potential bioavailability. At present the optimum bioavailability targets are weakly-bound, surface Fe(II) in aluminosilcates (and other minerals), and Fe(II) or Fe(III) in ferrihydrite. These potentially bioavailable species can be extracted by ascorbic acid (as FeA), thus we recommend more widespread use of this procedure.
Improved speciation data obtained here show that the ranges of iceberg and meltwater sFe are comparable (tens of nanomolar) but icebergs contain an order magnitude more CNFe (hundreds of nanomolar), as compared to thousands of nanomolar in meltwaters. Meltwater suspended sediments also contain an order of magnitude more FeA than iceberg (or glacier) sediments but the FeD contents of iceberg sediment and suspended sediments are not significantly different.
Iron minerals encased in ice are not inert but reaction kinetics are slow. The most rapid reactions involve the oxidation of pyrite to form iron (oxyhydr)oxides, including nanoparticulate ferrihydrite. The iron (oxyhydr)oxides in ice-hosted sediments can also be reduced by UV radiation, which can penetrate up to 10 cm in ice. Near surface sediment layers in ice are also warmed by solar radiation and freeze-thaw cycles are able to facilitate dissolution of iron (oxyhydr)oxides and transfer sFe and CNFe to ice. The ferrihydrite produced by weathering in ice alters slowly to goethite/hematite, so decreasing FeA and increasing FeD. The ratio FeA/FeD thus increases during the early stages of ice processing, then declines as FeA is altered to goethite/hematite. The delivery of FeA from icebergs is therefore determined by the rate of alteration of FeA to FeD in relation to the transit time of icebergs.
The reactions occurring as water is frozen have been modeled in subglacial environments, assuming an initial rainwater composition which is maintained at saturation with ferrihydrite. After removal of ~70% of water the pH has declined to ~5.5 and aqFe concentrations are substantially increased up to ~0.04 nM. AqFe concentrations are not directly comparable to measurements of sFe in ice (tens on nanomolar) which include some nanoparticulate Fe as well aqFe. Nevertheless, the modeled values are so far below ice sFe concentrations as to indicate that simple freezing in subglacial systems cannot produce the observed sFe values. The models, therefore, point to the involvement of redox and/or dissolution reactions with iron (oxyhydr)oxides and the presence of species that could complex Fe, and retain it in solution.
The same approach was used to model the reactions that occur in sea ice, using a starting composition of seawater that is maintained at saturation with ferrihydrite. After the removal of ~70% of water the residual brines have declined to a pH ~4.7 and there are substantial increases in aqFe to ~1 nM, due to the stabilization of aqFe as chloride complexes. These aqFe values are numerically comparable to dFe values in surface seawater in the polar regions, although aqFe values do not include the CNFe measured as dFe. Freezing of seawater can clearly play an important role in generating concentrations of dFe that are able to fuel productivity increases when Fe is released by melting at sea ice margins.
The delivery of ice-hosted Fe from glacial and iceberg sediments to the open ocean is considerably dampened where glaciers terminate in long fjords. Circulation patterns may hamper the incorporation of sediments into surface outflows of water and particulate material is removed from the water column. Glaciers terminating in, or close to, coastal waters also lose CNFe during passage through a salinity gradient. Estimates of losses (75–95%) are based on river systems may not be appropriate analogs for meltwaters in fjords. Aggregation losses may be lower due to colder temperatures and changes in particle shape and density may slow sedimentation losses. Fjord systems need intensive study to clarify how mixing and circulation systems control the delivery of sediment Fe and dFe to the coastal ocean.
Sea ice may accumulate substantial concentrations of aeolian dust during glacial intervals (such as the Last Glacial Maximum), when ice coverage persisted for prolonged periods and dust fluxes were high (Maher et al., 2010). Ice processing redox and dissolution reactions, augmented by freezing effects, will occur during storage producing dFe which can be released at the melting sea ice margins to stimulate productivity enhancements. The role of sea ice in storing and redistributing iron is rarely considered in most modeling studies of the Southern or Arctic Oceans (2016), with two exceptions (Lancelot et al., 2009; Wang et al., 2014 for the Southern Ocean). The freezing model presented here shows that the formation of sea ice has the potential to stabilize Fe, mainly through chloride complexes. The effects of freezing may be enhanced by the presence of organic ligands and by redox and dissolution reactions. The detailed investigation of sea incorporation mechanisms, and within-ice processes, should be a research priority.
Author Contributions
RR, JH, and MT wrote the initial draft of the paper and all authors contributed to the subsequent and final drafts. JH, JW, and RR collected most of the data. AE provided the freezing models and RD provided input for literature Fe model discussion.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JH and JW were supported by NERC funded PISCES (NE/P003133/1) and a Leverhulme Trust Research Grant (RPG-2016-439). JH was additionally supported by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions fellowship ICICLES (grant agreement #793962). MT wishes to acknowledge NERC support (via grant number GR3/11689). This material is based upon work supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under Cooperative Agreement No. NNA15BB03A issued through the Science Mission Directorate. Two reviewers and Andy Mitchell (as editor) have substantially improved the manuscript and their help is much appreciated.
References
Annett, A., Fitzsimmons, J. N., Seguret, M. J. M., Lagerstrom, M., Meredith, M. P., Scholfield, O., et al. (2017). Controls on dissolved and particulate iron distributions in surface waters of the Western Antarctic shelf. Mar. Chem.196, 81–97. doi: 10.1016/j.marchem.2017.06.004
Annett, A., Skiba, M., Henley, S. F., Venables, H. J., Meredith, M. P., Statham, P. J., et al. (2015). Comparitive roles of upwelling and glacial iron sources in Ryder Bay, coastal western Antarctic Peninsula. Mar. Chem. 176, 21–33. doi: 10.1016/j.marchem.2015.06.017
Baker, A. R., and Croot, P. L. (2010). Atmospheric and marine controls on aerosol solubility in seawater. Mar. Chem. 120, 4–13. doi: 10.1016/j.marchem.2008.09.003
Baker, A. R., Jickells, T. D., Witt, M., and Linge, K. L. (2006). Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean. Mar. Chem. 98, 43–58. doi: 10.1016/j.marchem.2005.06.004
Banwart, S., Davies, S., and Stumm, W. (1989). The role of oxalate in accelerating the reductive dissolution of hematite (α-Fe2O3) by ascorbate. Coll. Surf. 39, 303–309. doi: 10.1016/0166-6622(89)80281-1
Bhatia, M. P., Kujawinski, E. B., Das, S. B., Breier, C. F., Henderson, P. B., and Charette, M. A. (2013). Greenland meltwater as a significant and potentially bioavailable source of iron to the ocean. Nat. Geosci. 6, 274–278. doi: 10.1038/ngeo1746
Bosch, J., Lee, K. Y., Jordan, G., Kim, K. W., and Neckenstock, R. U. (2012).Anaerobic, nitrate-dependent oxidation of pyrite nanoparticles by Thiobaccillus denitrificans. Environ. Sci. Tech. 46, 2095–2101. doi: 10.1021/es2022329
Boyd, P. W., Mackie, D. S., and Hunter, K. A. (2010). Aerosol iron deposition to the surface ocean- Modes of iron supply and biological responses. Mar. Chem. 120, 128–143. doi: 10.1016/j.marchem.2009.01.008
Boye, M., Nishioka, J., Croot, P., Lann, P., Timmermans, K. R., Strass, V. H., et al. (2010). Significant proportion of dissolved organic Fe complexes in fact is Fe colloids. Mar. Chem. 122, 20–27. doi: 10.1016/j.marchem.2010.09.001
Buck, C. S., Landing, W. M., and Resing, J. A. (2010). Particle size and aerosol iron solubility: a high-resolution analysis of Atlantic aerosols. Mar. Chem. 120, 14–24. doi: 10.1016/j.marchem.2008.11.002
Buck, C. S., Landing, W. M., Resing, J. A., and Lebon, G. T. (2006). Aerosol iron and aluminium solubility in the northwest Pacific Ocean: results from the 2002 IOC cruise. Geochem. Geophys. Geosyst. 7, 1–10. doi: 10.1029/2005GC000977
Butler, B. M., and Kennedy, H. (2015). An investigation of mineral dynamics in frozen seawater brines by direct measurement with synchrotron X-ray powder diffraction. J. Geophys. Res. 120, 5686–5697. doi: 10.1002/2015JC011032
Chen, Y., and, R. L., and Siefert, R.L. (2003). Determination of different types of labile atmospheric iron over remote oceans. J. Geophys. Res. 108, D24, 4774, doi: 10.1029/2003JD003515
Dai, M.-H., and Martin, J.-M. (1995). First data on trace metal level and behavior in two major Arctic river-estuarine systems (Ob and Yenisey) and in the adjacent Kara Sea, Russia. Earth Planet. Sci. Lett. 131, 127–141. doi: 10.1016/0012-821X(95)00021-4
De Baar, H. J. W., and de Jong, J. T. M. (2001). Distribution, sources and sinks of iron in seawater in The Biogeochemistry of Iron in Seawater, eds D. R. Turner and K. A. Hunter (New York, NY: Wiley), 123–253.
De Jong, J. T. M., Schoemann, V., Maricq, N., Mattielli, N., Langhorne, P., Haskell, T., et al. (2013). Iron in land-fast sea ice of McMurdo Sound derived from sediment re-suspension and wind-blown dust attributes to primary productivity in the Ross Sea, Antarctica. Mar. Chem. 157, 24–43. doi: 10.1016/j.marchem.2013.07.001
De Jong, J. T. M., Stammerjohn, S. E., Ackley, S. F., Tilson, J.-L., Mattielli, N., and Schoemann, V. (2015). Sources and fluxes of dissolved iron in the Bellinghausen Sea (West Antarctic): the importance of sea ice, icebergs and the continental margin. Mar. Chem. 177, 518–535. doi: 10.1016/j.marchem.2015.08.004
Death, R., Wadham, J. L., Monteiro, F., Le Brocq, A. M., Tranter, M., Ridgewell, A., et al. (2014). Antarctic ice sheet fertilizes the Southern Ocean. Biogeosciences 10, 12551–12570. doi: 10.5194/bgd-10-12551-2013
Delille, B., Vancoppenolle, M., Geilfus, N.-X., Tilbrook, B., Lannuzel, D., Schoemann, V., et al. (2014). Southern Ocean CO2 sink: the contribution of sea ice. J. Geophys. Res. 110, 6340–6355. doi: 10.1002/2014JC009941
Duprat, L. P. A. M., Bigg, G. R., and Wilton, D. J. (2016). Giant icebergs significantly enhance the marine productivity of the Southern Ocean. Nat. Geosci. 9, 219–221, doi: 10.1038NGEO2533
Elberling, B. (2005). Temperature and oxygen control on pyrite oxidation in frozen mine tailings. Cold Reg. Sci. Tech. 41, 121–133. doi: 10.1016/j.coldregions.2004.09.004
Elsenousy, A., Hanley, J., and Chevrier, V. A. (2015). Effect of evaporation and freezing on the salt paragenesis and habitability of brines at the Phoenix landing site. Earth Planet. Sci. Lett. 421, 39–46. doi: 10.1016/j.epsl.2015.03.047
Faure, G. (1998). Principles and Applications of Geochemistry, 2nd Edn. New Jersey, NJ: Prentice Hall.
Gartman, A., and Luther, G. W. I. I. I. (2014). Oxidation of synthesized sub-micron pyrite (FeS2) in seawater. Geochim. Cosmochim. Acta 144, 96–108. doi: 10.1016/j.gca.2014.08.022
Geilfus, N.-X., Galley, R. J., Cooper, M., Halden, N., Hare, A., Wang, F., et al. (2013). Gypsum crystals observed in experimental and natural sea ice. Geophys. Res. Lett. 40, 1–6. doi: 10.1002/2013GL058479
Gerringa, L. J. A., Aldercamp, A.-C., Laan, P., Thuroczy, C.-E., de Baar, H. J. W., Mills, M. M., et al. (2012). Iron from melting glaciers fuels the phytoplankton blooms in Amundsen Sea (Southern Ocean): iron biogeochemistry. Deep Sea Res. 71–76, 16–31. doi: 10.1016/j.dsr2.2012.03.007
Hallet, B. (1976). Deposits formed by the subglacial precipitation of CaCO3. Geol. Soc. Am. Bull. 87, 1003–1015. doi: 10.1130/0016-7606(1976)87<1003:DFBSPO>2.0.CO;2
Hassler, C. S., and Schoemann, V. (2009). Bioavailability of organically bound Fe to model phytoplankton of the Southern Ocean. Biogeosciences 6, 2281–2296. doi: 10.5194/bg-6-2281-2009
Hawkings, J. (2015). An Investigation into the Production and Export of Nutrients from Glaciers, Ph.D thesis, University of Bristol, Bristol, 220.
Hawkings, J. R., Benning, L. G., Raiswell, R., Kaulich, B., Araki, T., Abyaneh, M., et al. (2018). Biolabile ferrous iron nanoparticles in glacial sediments. Earth Planet. Sci. Lett. 493, 92–101 doi: 10.1016/j.epsl.2018.04.022
Hawkings, J. R., Wadham, J., Tranter, M., Telling, J., Bagshaw, E., Beaton, A., et al. (2016). The Greenland Ice sheet as a hotspot of phosphorus weathering and export in the Arctic. Global Biogeochem. Cycles 30, 191–210. doi: 10.1002/2015GB005237
Hawkings, J. R., Wadham, J. L., Tranter, M., Raiswell, R., Benning, L. G., Statham, P. J., et al. (2014). Ice sheets as a significant source of highly reactive nanoparticulate iron to the oceans. Nat. Commun. 5:3929. doi: 10.1038/ncomms4929
Herraiz-Borreguero, L., Lannuzel, D., van der Merwe, P., Treverrow, A., and Pedro, J.B. (2016). Large flux of iron from the Amery Ice Shelf marine ice to Prydz Bay, East Antarctica. J. Geophys. Res. 121, 6009–6020. doi: 10.1002/2016JC011687
Hopwood, M. J., Bacon, S., Arendt, K., Conneelly, D. P., and Statham, P. J. (2015). Glacial meltwater from Greenland is not likely to be an important source of Fe to the North Atlantic. Biogeochemistry 124, 1–11. doi: 10.1007/s10533-015-0091-6
Hopwood, M. J., Cantoni, C., Clarke, J. S., Cozzi, S., and Achterburg, E. P. (2017). The heterogeneous nature of Fe delivery from melting icebergs. Geochem. Perspect. Lett. 3, 200–209. doi: 10.7185/geochemlet.1723
Hopwood, M. J., Connelly, D. P., Arendt, K. E., Juul-Pedersen, T., Stichcombe, M., Meire, L., et al. (2016). Seasonal changes in Fe along a glaciated Greenlandic fjord. Front. Earth Sci. 4:15. doi: 10.3389/feart.2016.00015
Jickells, T. D., and Spokes, L. J. (2001), Atmospheric inputs to the ocean, in The Biogeochemistry of Iron in Seawater, eds D. R. Turner K. A. Hunter (New York, NY: Wiley), 123–251.
Jones, A. M., Collins, R. N., Rose, J., and Waite, T. D. (2009). The effect of silica and natural organic matter on the Fe(II)-catalysed transformation and reactivity of Fe(III) minerals. Geochim. Cosmochim. Acta 73, 4009–4433. doi: 10.1016/j.gca.2009.04.025
Kanna, N., Sugiyama, S., Ohashi, Y., Sakakibara, D., Fukamachi, Y., and Nomura, D. (2018). Upwelling of macronutrients and dissolved inorganic carbon by a subglacial freshwater driven plume in Bowdoin Fjord, northwestern Greenland. J. Geophys. Res. Biogeosci. 123:1666–1682. doi: 10.1029/2017JG004248
Killawee, J. A., Farichild, I. J., Tison, J.-L., Janssens, L., and Lorrain, R. (1998). Segregation of solutes and gases in experimental freezing of dilute solutions: Implications for natural glacial systems. Geochim. Cosmochim. Acta 62, 3637–3655. doi: 10.1016/S0016-7037(98)00268-3
Kim, K., Choi, W., Hoffmann, M. R., Yoon, H. I., and Park, B. K. (2010). Photoreductive dissolution of iron oxides trapped in ice and its environmental implications. Environ. Sci. Tech.44, 4142–4148. doi: 10.1021/es9037808
Kuma, K., and Matsunaga, K. (1995).Availability of colloidal ferric oxides to coastal marine phytoplankton. Mar. Biol. 122, 1–11. doi: 10.1007/BF00349272
Lancelot, C., de Montey, A., Goose, H., Becquefort, S., Schoemann, V., Basquer, B., et al. (2009). Spatial distribution of the iron supply to phytoplankton in the Southern Ocean: a model study. Biogeosciences 6, 2861–2878. doi: 10.5194/bg-6-2861-2009
Lannuzel, D., Grotti, M., Albelmoschi, M. L., and van der Merwe, P. C. (2015). Organic ligands control the concentrations of dissolved iron in Antarctic sea ice. Mar. Chem. 174, 120–130. doi: 10.1016/j.marchem.2015.05.005
Lannuzel, D., Schoemann, V., de Jong, J., Chou, L., Delille, B., Becquevort, S., et al. (2008). Iron study in during a time series in the western Weddell Sea pack ice. Mar. Chem. 108, 85–95. doi: 10.1016/j.marchem.2007.10.006
Lannuzel, D., Schoemann, V., de Jong, J., Tison, J.-L., and Chou, L. (2007). Distribution and biogeochemical behavior of iron in the East Antarctic sea ice. Mar. Chem. 106, 18–32. doi: 10.1016/j.marchem.2006.06.010
Lannuzel, D., van der Merwe, P. C., Townsend, A. T., and Bowie, A. R. (2014). Size fractionation of iron, manganese and aluminium in Antarctic fast ice reveals a lithogenic origin and low iron solubility. Mar. Chem. 161, 47–56. doi: 10.1016/j.marchem.2014.02.006
Lannuzel, D., Vancoppenolle, M., van der Merwe, P. C., de Jong, J., Meiners, K. M., Grotti, M., et al. (2016). Iron in sea ice: Review and new insights. Elementa 4:000138. doi: 10.1/12952/journal.elementa.00130
Lilbaek, G., and Pomeroy, J. W. (2008). Ion enrichment of snow melt runoff water caused by basal ice formation. Hydrol. Process. 22, 2758–2766. doi: 10.1002/hyp.7028
Lyons, W. B., Dailey, K. R., Welch, K. A., Deuerling, K. M., Welch, S. A., and McKnight, D. M. (2015). Antarctic streams as a potential source of iron for the Southern Ocean. Geology 43, 1003–1006. doi: 10.1130/G36989.1
Maher, B. A., Prospero, J. M., Mackie, D., Gaiero, D., Hesse, P., and Balkanski, Y. (2010). Global connections between desert dust, ocean biogeochemistry and climate. Earth Sci. Rev. 99, 61–97. doi: 10.1016/j.earscirev.2009.12.001
Marion, G. M., Farren, R. E., and Komrowski, A. J. (1999). Alternative pathways for seawater freezing. Cold Regions Science and Technology 29, 259–266. doi: 10.1016/S0165-232X(99)00033-6
Markussen, T. N., Elberling, B., Winter, C., and Andersen, T. J. (2016). Flocculated meltwater particles control Arctic land-sea fluxes. Sci. Rep. 6:24033. doi: 10.1038/srep24033
Mayer, L. M. (1982). Aggregation of colloidal iron during estuarine mixing-kinetics, mechanism and seasonality. Geochim. Cosmochim. Acta 46, 1003–1009. doi: 10.1016/0016-7037(82)90055-2
Meire, L., Mortensen, J., Meire, P., Juul-Pedersen, T., Sejr, M. K., Rysgaard, S., et al. (2017). Marine-terminating glaciers sustain high productivity in Grennland fjords. Glob. Chang. Biol. 23, 5344–5357. doi: 10.1111/gcb.13801
Meire, L., Mortensen, J., Rysgaard, S., Bendtsen, J., Boone, W., Meire, P., et al. (2016). Spring bloom dynamics in a subarctic fjord influences by tidewater outlet glaciers. J. Geophys. Res. 121, 1581–1592. doi: 10.1002/2015JG003240
Meskhidze, N., Johnson, M. S., Hurley, D., and Dawson, K. (2016). Influence of measurement uncertainties on fractional solubility of iron mineral aerosols over the oceans. Aeolian Res. 22, 85–92. doi: 10.1016/j.aeolia.2016.07.002
Monien, D., Monien, P., Brunjes, R., Widmer, T., Kappenberg, A., Busso, A. A. S., et al. (2017). Meltwater as a source of potentially bioavailable iron to Antarctica waters. Antar. Sci. 29, 277–291. doi: 10.1017/S095410201600064X
Mortensen, J., Bendtsen, J., Moytka, R. J., Lennert, K., Truffer, M., Fahenstock, M., et al. (2013). On the seasonal freshwater stratification in the proximity of fast-flowing tidewater outlet glaciers in a sub-Arctic fjord. J. Geophys. Res. 118, 1382–1395. doi: 10.1002/jgrc.20134
Mortensen, J., Lennert, K., Bendtsen, J., and Rysgaard, S. (2011). Heat sources for glacial melt in a sub-arctic fjord (Godthabsfjord) in contact with the Greenland Ice Sheet. J. Geophys. Res. 116:C01013, doi: 10.1029/2010/C006528
Moses, C. O., Nordstrom, D. K., Herman, J. S., and Mills, A. L. (1987). Aqueous pyrite oxidation by dissolved oxygen and by ferric iron. Geochim. Cosmochim. Acta 51, 1561–1571. doi: 10.1016/0016-7037(87)90337-1
Nixon, S. L., Telling, J. P., Wadham, J. L., and Cockell, C. (2017). Viable cold-tolerant iron-reducing microorganisms in geographically diverse subglacial environments. Biogeosciences 14, 1445–1455. doi: 10.5194/bg-14-1445-2017
Nodwell, L. M., and Price, N. M. (2001).Direct use of inorganic colloidal iron by marine thixotrophic phytoplankton. Limnol. Oceanogr. 46, 765–777. doi: 10.4319/lo.2001.46.4.0765
Nordstrom, D. K. (1982). Aqueous pyrite oxidation and the formation of secondary iron minerals in Acid Sulfate Weathering, eds J. A. Kittrick and D. S. Fanning (Hosner, LR: Soil Science Society of America), 37–56.
Nordstrom, D. K. (2011). Mine wastes: acidic to circumneutral. Elements 7, 393–398. doi: 10.2113/gselements.7.6.393
Poulton, S. W., and Canfield, D. E. (2005). Development of a sequential extraction procedure for iron; implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221. doi: 10.1016/j.chemgeo.2004.09.003
Raiswell, R., Benning, L. G., Davidson, L., Tranter, M., and Tulaczyk, S. (2009). Schwertmannite in wet, acid, and oxic microenvironments beneath polar and polythermal glaciers. Geology 37, 431–434. doi: 10.1130/G25350A.1
Raiswell, R., Benning, L. G., Tranter, M., and Tulaczyk, S. (2008), Bioavailable iron in the Southern Ocean: the significance of the iceberg conveyor belt. Geochem. Trans. 9:7. doi: 10.1186/1467-4866-9-7
Raiswell, R., and Berner, R.A. (1994), A comparison of iron extraction methods for the determination of degree of pyritization recognition of iron-limited pyrite formation. Chem. Geol. 111, 101–111. doi: 10.1016/0009-2541(94)90084-1
Raiswell, R., Hawkings, J. R., Benning, L. G., Albani, S., and Mahowald, N. (2017). Comments on ‘Influence of measurement uncertainties on fractional solubility of iron mineral aerosols over the oceans. Aeolian Research 25, 123–125. doi: 10.1016/j.aeolia.2017.03.003
Raiswell, R., Hawkings, J. R., Benning, L. G., Baker, A. R., Death, R., Albani, S., et al. (2016). Potentially bioavailable iron delivery by iceberg-hosted sediments and atmospheric dust to the polar oceans. Biogeosciences 13, 1–14. doi: 10.5194/bg-13-3887-2016
Raiswell, R., Vu, H. P., Brinza, L., and Benning, L. G. (2010). The determination of Fe in ferrihydrite by ascorbic acid extraction: methodology, dissolution kinetics and loss of solubility with age and de-watering. Chem. Geol. 278, 70–79. doi: 10.1016/j.chemgeo.2010.09.002
Rich, H. W., and Morel, F. M. M. (1990). Availability of well-defined iron colloids to the marine diatom Thalassiosiraweissflogii. Limnol. Oceanogr. 35, 652–662. doi: 10.4319/lo.1990.35.3.0652
Rowland, G. A., and Grammas, A. M. (2011). A solid-phase chemical actinometer film for measurement of solar UV penetration into snow pack. Cold Reg. Sci. Techn. 66, 75–83. doi: 10.1016/j.coldregions.2011.01.009
Schroth, A. W., Crusius, J., Campbell, R. W., and Hoyer, I. (2014). Estuarine removal of glacial iron and implications for iron fluxes to the ocean. Geophys. Res. Lett. 41, 3951–3958. doi: 10.1002/2014GL060199
Schwertmann, U., Stanjek, H., and Becher, H.-H (2004). Long term in vitriol transformation of 2-line ferrihydrite to goethite/hematite at 4, 10, 15 and 25°C. Clay Miner. 39, 433–438. doi: 10.1180/0009855043940145
Shaked, Y., and Lis, H. (2012). Disassembling iron availability to phytoplankton. Front. Microbiol. 123, 1–26. doi: 10.3389/fmicb.2012.00123
Shaw, T. J., Raiswell, R., Hexel, C. R., Vu, H. P., Moore, W. S., Dudgeon, R., et al. (2011). Input, composition and potential impact of terrigenous material from free-drifting icebergs in the Weddell Sea. Deep Sea Res. II 58, 1376–1383. doi: 10.1016/j.dsr2.2010.11.012
Shi, Z., Krom, M. D., Jickells, T. J., Bonneville, S., Carlson, K. S., Mihalopoulos, N., et al. (2012). Impacts on iron solubility in the mineral dust by processes in the source region and the atmosphere: a review. Aeol. Res. 5, 21–42. doi: 10.1016/j.aeolia.2012.03.001
Shoenfelt, E. M., Sun, J., Winckler, G., Kaplan, M. R., Borunda, A. L., Farrel, K. R., et al. (2017). High particulate Fe(II) content in glacially sourced dusts enhances productivity of a model diatom. Sci. Adv. 3:e 1700314. doi: 10.1126/sciadv.1700314
Shoenfelt, E. M., Winckler, G., Lamy, F., Anderson, R. F., and Bostick, B. C. (2018). Highly bioavailable dust-borne iron delivered to the southern Ocean during glacial periods. Proc. Natl. Acad. Sci. U.S.A. 115, 11180–11185. doi: 10.1073/pnas.1809755115
Smith, K. L., Robison, B. H., Helly, J. J., Kaufmann, R. S., Ruhl, H. A., Shaw, T. J., et al. (2007). Free-drifting icebergs: Hot spots of chemical and biological enrichment in the Weddell Sea. Science 317, 478–483. doi: 10.1126/science.1142834
Smith, K. L., Sherman, A. D., Shaw, T. J., and Springall, J. (2013). Icebergs as unique Lagrangrian ecosystems in polar seas. Ann. Rev. Mar. Sci. 5, 269–287. doi: 10.1146/annurev-marine-121211-172317
Statham, P. J., Skidmore, M.M., and Tranter, M. (2008). Inputs of glacially derived dissolved and colloidal iron to the coastal ocean and implications for primary productivity. Glob. Biogeochem. Cycles 22:GB3013. doi: 10.1029/2007GB003106
Straneo, F., and Cenedes, C. (2015). The dynamics of Greenland's glacial fjords and their role in climate Ann. Rev. Mar. Sci. 7, 89–112. doi: 10.1146/annurev-marine-010213-135133
Tagliabue, A., Aumont, O., Death, R., Dunne, J. P., Dutkiewicz, S., Galbraith, E., et al. (2016). How well do global ocean biogeochemistry models simulate dissolved iron distributions? Global Biogeochem. Cycles 23, 17764–17774. doi: 10.1002/2015GB00528
Tagliabue, A., Bowie, A. R., Boyd, P. W., Buck, K. N., Johnson, K. S., and Sato, M. A. (2017). The integral role of iron in ocean chemistry. Nature 543, 51–59. doi: 10.1038/nature21058
Telling, J., Anesio, A. M., Tranter, M., Fountain, A. G., Nylen, T., Hawkings, J., et al. (2014).Spring thaw ionic pulses boost nutrient availability and microbial growth in entombed Antarctic Dry Valley cryoconite holes. Front. Microbiol. 5:694. doi: 10.3339/fmicb.2014.0694
Tranter, M., Huybrechts, P., Munhoven, G., Sharp, M. J., Brown, G. H., Jones, J. W., et al. (2002). Glacial bicarbonate, sulphate and base cation fluxes during the last glacial cycle, and their potential impact on atmospheric CO2. Chem. Geol. 190, 33–44. doi: 10.1016/S0009-2541(02)00109-2
Tranter, M., and Jones, H. G. (2001). The chemistry of snow: processes and nutrient recycling in Snow Ecology, eds H. G. Jones, J. W. Pomeroy, D. A. Walker, and R. Hoham (Cambridge: Cambridge University Press), 127–167.
Vancoppenolle, M., Meiners, K. M., Michel, C., Bopp, L., Brabant, F., Carnat, G., et al. (2013). Role of sea ice in global biogeochemical cycles: emerging views and challenges. Quat. Sci. Rev. 79, 207–230. doi: 10.1016/j.quascirev.2013.04.011
Von der Heyden, B. P., and Roychoudhury, A. N. (2015). A review of colloidal iron partitioning and distribution in the open ocean. Mar. Chem. 177, 9–19. doi: 10.1016/j.marchem.2015.05.010
von der Heyden, B. P., Roychoudhury, A. N., Mtshall, T. N., and Tyliszcak, S. C. B. (2012). Chemically and geographically distinct solid-phase iron pools in the Southern Ocean. Science 338, 1199–1201. doi: 10.1126/science.1227504
Wadham, J. L., Tranter, M., Skidmore, M., Hodson, A. J., Priscu, J., Lyons, W. B., et al. (2010). Bigeochemical weathering under ice: size matters. Glob. Biogeochem. Cycles 24:GB3025. doi: 10.1029/2009GB003688
Wang, S., Lindsay, K., Moore, J. K., and Holland, M. (2014), Impact of sea ice on the marine iron cycle phytoplankton productivity. Biogeosciences 11, 4713–4731. doi: 10.5194/bg-11-4713-2014
Waychunas, G. A., Kim, C. S., and Banfield, J. F. (2005). Nanoparticulate iron oxide minerals in soils and sediments: unique properties and contaminant scavenging mechanisms. J. Nanopart. Res. 7, 409–433. doi: 10.1007/s11051-005-6931-x
Wehrmann, L. M., Formolo, M. J., Owens, J. D., Raiswell, R., Ferdelman, T. G., Riedinger, N., et al. (2014). Iron and manganese speciation and cycling in glacially-influenced high-latitude fjord sediments (West Spitzbergen, Svalbard): evidence for a benthic recycling-transport mechanism. Geochim. Cosmochim. Acta 141, 628–655. doi: 10.1016/j.gca.2014.06.007
Wells, M. J., and Goldberg, E. D. (1994). Marine sub-micron particles. Mar. Chem. 40 5–18. doi: 10.1016/0304-4203(92)90045-C
Wells, M. L., Zorkin, N. G., and Lewis, A. G. (1983). The role of colloid chemistry in providing a source of iron to phytoplankton. J. Mar. Res. 41, 731–746. doi: 10.1357/002224083788520478
Winton, V. H. L., Bowie, A. R., Edwards, R., Keywood, M., Townsend, A. T., van der Merwe, P., et al. (2015). Fractional solubility of atmospheric iron inputs to the Southern Ocean. Mar. Chem. 177, 20–32. doi: 10.1016/j.marchem.2015.06.006
Yoshida, M., Kuma, K., Iwade, S., Isoda, Y., Takata, H., and Yamada, M. (2006). Effect of aging time on the availability of freshly precipitated ferric hydroxide to coastal marine diatoms. Mar. Biol. 149, 379–392. doi: 10.1007/s00227-005-0187-y
Keywords: iron, speciation, icebergs, meltwaters, freezing
Citation: Raiswell R, Hawkings J, Elsenousy A, Death R, Tranter M and Wadham J (2018) Iron in Glacial Systems: Speciation, Reactivity, Freezing Behavior, and Alteration During Transport. Front. Earth Sci. 6:222. doi: 10.3389/feart.2018.00222
Received: 18 July 2018; Accepted: 21 November 2018;
Published: 11 December 2018.
Edited by:
Andrew C. Mitchell, Aberystwyth University, United KingdomReviewed by:
Gilad Antler, Ben-Gurion University of the Negev, IsraelZiming Yang, Oakland University, United States
Copyright © 2018 Raiswell, Hawkings, Elsenousy, Death, Tranter and Wadham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon Hawkings, am9uLmhhd2tpbmdzQGdtYWlsLmNvbQ==