 Edith Mollard
Edith Mollard Caroline Martel
Caroline Martel- Université d'Orléans, CNRS/INSU—ISTO—BRGM, UMR 7327, Orléans, France
Magma ascending from the storage region toward the surface may crystallize small and rapidly-grown crystals, designated as microlites. Upon decompression, the rapid changes of the microlite textures, such as number density and crystal size, directly impact the rheology of the magma in the volcanic conduit, and eventually control the effusive vs. explosive style of the subaerial eruption. This is of prime importance for volcanic risk assessment involving highly viscous silica-rich magmas that are prone to fragmentation. To this aim, we present a theoretical modeling of plagioclase nucleation and growth in rhyolitic-like melts, based on previous decompression experiments. Thus, the modeling is valid for plagioclase crystallization in rhyolitic melts decompressed at 875°C from 200 MPa to final pressures of 50, 75, or 100 MPa, which represents effective undercooling (ΔTeff) of 110, 80 and 55°C, respectively. Our results of nucleation-rate calculation using the Classical Nucleation Theory (CNT; case of homogeneous nucleation) and values of crystal-melt interfacial energy (σ) either from literature or empirically calculated, strongly disagree with previously-determined experimental nucleation rates. By inverting the CNT calculation using the experimentally-determined nucleation rates, we propose plagioclase-liquid interfacial energies from 0.041 to 0.059 J.m−2 with ΔTeff increasing from 55 to 110°C, which is about 2–3 times lower than σ determined empirically and macroscopically. We modeled plagioclase growth rates by atom diffusion in melt (following Fick's second law adapted to multicomponent systems), considering a crystal-melt interface advancing over time (as crystal grows) and a chemically-closed finite reservoir (no component supply). The component limiting plagioclase growth has been determined to be CaO, for which we calculated diffusion coefficients from 10−14 to 10−15 m2/s (conditions of a silicic melt, 875°C, and H2O saturation pressures from 50 to 100 MPa). The overall good agreement between the model and the experimental growth laws validates diffusion as the main process controlling isothermal decompression-induced plagioclase growth under moderate ΔTeff < 110°C. Combining both calculations, crystal number density and size, may be relevant to predict plagioclase microlite textures in ascending rhyolitic magmas, which may now be incorporated in conduit flow models to assess the magma rheological behavior that controls eruption dynamics.
Introduction
The complex eruptive behavior of island-arc silicic volcanoes, i.e., effusive vs. explosive eruptions, is commonly attributed to the magma ascent conditions in the volcanic conduit (e.g., Jaupart and Allègre, 1991; Woods and Koyaguchi, 1994; Barclay et al., 1996). In particular, magma ascent rate controls the time available for syn-eruptive degassing and crystallization, so that a magma that has time and capability to outgas commonly erupts effusively whereas magma that still contains overpressurized gases may give rise to explosive eruptions (e.g., Eichelberger et al., 1986; Melnik et al., 2005; Mueller et al., 2005; Martel and Iacono-Marziano, 2015). One of the major consequences of decreasing melt volatile content through degassing is the increase of the liquidus temperature and subsequent melt crystallization (Tuttle and Bowen, 1958). Crystal nucleation and growth modify the flow properties of magma and gases, and may control the eruptive dynamics by posing the limits of a ductile vs. brittle behavior of the magma in response to the imposed stress. Therefore, investigating decompression-induced crystallization may eventually provide links to eruption dynamics.
Numerous studies have been conducted on crystallization kinetics in silicate melts via cooling (Fenn, 1977; Kirkpatrick, 1981; Muncill and Lasaga, 1987, 1988; Lattard and Partzsch, 2001; Zieg and Lofgren, 2006; Iezzi et al., 2008; Pupier et al., 2008) and for compositionally simple systems (e.g., Davis et al., 1997; Potapov et al., 1999). These studies have shown that crystallization kinetics exert the primary control on crystal number density, proportion, size, and shape. However, the main driving force for crystallization in ascending hydrous magmas in volcanic context is more decompression (and volatile loss) than cooling, since temperature is roughly constant upon ascent (Wilson et al., 1980). Such decompression-triggered crystals have common sizes below ~100 μm and are designated as microlites, i.e., small crystals that grow rapidly under thermodynamic disequilibrium.
Several studies have dealt with decompression-induced crystallization in silica-rich melts (dacite to rhyolite), for which the microlite phase is mainly plagioclase, with the aim of understanding the magma ascent conditions in the volcanic conduit and the link with the eruptive behavior. Besides studies of microlite characteristics in natural volcanic pyroclasts (Cashman, 1992; Klug and Cashman, 1994; Wolf and Eichelberger, 1997; Hammer et al., 1999, 2000; Nakada and Motomura, 1999; Clarke et al., 2007; Martel and Poussineau, 2007; Noguchi et al., 2008), experimental simulations of magma decompression have been carried out (Geschwind and Rutherford, 1995; Hammer and Rutherford, 2002; Couch et al., 2003; Martel and Schmidt, 2003; Larsen, 2005; Brugger and Hammer, 2010; Cichy et al., 2011; Martel, 2012; Mollard et al., 2012; Riker et al., 2015; Waters et al., 2015; Befus and Andrews, 2018). On the contrary, theoretical modeling of decompression-induced crystallization is rare. Plagioclase nucleation in a rhyolitic melt has been numerically studied by Hammer (2004). Syn-eruptive crystallization during magma ascent has been considered in numerical models of conduit flow and lava dome extrusion (Melnik and Sparks, 1999; Melnik et al., 2005; De'Michieli Vitturi et al., 2010). These models consider crystallization as a crystal volume fraction at thermodynamic equilibrium, which evolves with time and pressure. One model investigated the effect of crystal growth kinetics on the conduit flow properties via crystal size distributions (Melnik et al., 2011). Theoretical modeling of syn-eruptive crystal nucleation and growth provides a mean to infer the time evolution and extent of groundmass crystallization for a given volcanic system. Predicting groundmass crystallization is a pre-requisite step to further investigate the physicochemical and flow properties of the magma that control the eruptive behavior.
In this paper, we present two theoretical models. The first one concerns the rates of plagioclase homogeneous nucleation, for which the use of the CNT equation is not new [i.e., similar approach to that used in Hammer (2004)], but allows to refine the plagioclase-liquid surface tensions for a new rhyolitic-like composition. The second model concerns plagioclase crystal growth using the equations of diffusion, which first permits the time-evolution assessment of crystal size (and not only bulk volume) in decompressing magmas; note that the simplified melt composition used here (haplotonalite) is an asset for the diffusion modeling purpose. The proposed crystal growth model is thus suitable for decompression-triggered crystallization from a hydrous rhyolitic melt at constant pressure and temperature.
Rapidly-decompressed magmas that crystallize at constant pressure may not be the first scenario invoked to explain explosive eruptions (which are commonly interpreted as being linked to magma ascent rate). However, two main reasons can be highlighted in favor of the interest of such a scenario. First, such a scenario is possible in nature. Indeed, explosive eruptions generated from lava domes, such as blasts (e.g., the eruptions of Montagne Pelée in 1902 and of Mount Saint Helens in 1980), are interpreted to be triggered by gas overpressures in a shallow-level residing magma. Martel (2012) demonstrated experimentally that the blast magmas ascend from the reservoir fast enough (rapid decompression) to prevent crystallization during ascent. Microlite crystallization actually occurs at shallow/dome level, which drastically increases viscosity and has a major control on the degassing process (i.e., capable of generating a second bubble nucleation event). The second reason for considering such a scenario is that it provides the only way to link textural parameters observable in natural erupted products to thermodynamic parameters controlling crystal growth. Crystal growth is driven by undercooling that is the difference between the crystal liquidus temperature (TL) and the real temperature of crystallization. In ascending H2O-saturated silicic magmas, the driving force for crystallization is not cooling, but melt dehydration as pressure decreases. Anhydrous-crystal TL increases with decreasing melt H2O content, so that isothermal decompression of the hydrated magma leads to an increasing difference between TL and magma temperature, referred as to the effective undercooling, ΔTeff (Hammer and Rutherford, 2002). For a given magma temperature, ΔTeff is thus dependent on melt H2O content, here controlled by pressure. The scenario of decompression-induced crystallization at constant pressure is thus ideal to properly assign one ΔTeff to one set of crystal textures (e.g., number density, fraction, size, and morphology). This clearly constitutes a prerequisite at thermodynamic equilibrium to further crystallization modeling in which kinetic factors are involved.
After a brief summary of the experimental results of Mollard et al. (2012), from which the modeling aims at reproducing the empirical laws of plagioclase nucleation and growth, we present below (i) new constraints on the plagioclase-melt interfacial energy that controls the nucleation kinetics and (ii) a model of crystal growth-rate limited by element diffusion that may be used to predict final sizes of plagioclase microlites crystallizing from rhyolitic melts.
Previous Results on Empirical Laws of Plagioclase Crystallization
This section briefly presents the experimentally-determined laws of plagioclase crystallization of Mollard et al. (2012), which the modeling aims at reproducing, in order to validate the processes controlling decompression-induced crystallization in rhyolitic melts.
The experimental data set of the decompression-induced plagioclase crystallization and the experimental and analytical methods are reported in Mollard et al. (2012). The starting glass composition is haplotonalitic (HTND; SiO2: 78.7; Al2O3: 14.1; CaO: 1.8; Na2O: 5.4, in wt %), which represents a simplified analog composition for natural rhyolitic glasses. Such a simplified composition has the advantage to ease modeling. The HTND glass was saturated with H2O at 875°C and 200 MPa (initial pressure, Pi; 6.7±0.3 wt % H2O) and decompressed from these conditions using cold-sealed pressure vessels. The final pressures (Pf) ranged from 160 to 25 MPa and decompression rates were of 1,200, 150, and 30 MPa/h (decompression durations of 0.25, 1.5, and 5 h, respectively). For the purpose of this study, only the Pf of 100, 75, and 50 MPa will be considered. No crystals were observed before and during decompression, thus restricting crystallization, of plagioclase exclusively, to the dwell stage at Pf during time periods up to 17 days. However, degassing was fully completed before the dwell stage at Pf, based on direct measurements of H2O content in the experimental glasses. The driving force for crystallization, ΔTeff, was calculated to 55, 80, and 110°C for Pf of 100, 75, and 50 MPa, respectively. As pointed out in Mollard et al. (2012), the three decompression rates did not lead to systematic differences in phase composition, proportions, and crystallization rate for a given Pf, except for the runs decompressed to 100 MPa at 1,200 MPa/h, which show nucleation difficulties and have thus been ruled out from the calculation. The experimental conditions are reported in Table 1.
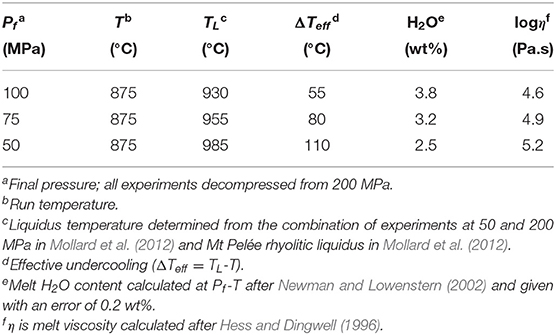
Table 1. Conditions of the decompression-induced crystallization experiments of Mollard et al. (2012).
The area number density (NA), proportion (Φ) and size of the plagioclases were determined by image analysis (using SPO2003 software; Launeau and Robin, 1996) as a function of experimental duration (Table 2). As defined in Mollard et al. (2012), the crystal size was determined on SEM pictures by the ten longest semi-long axes of the crystals (L/2), in order to best approach the true stereological size of the crystals by minimizing cross-cutting effects (Higgins, 1994).
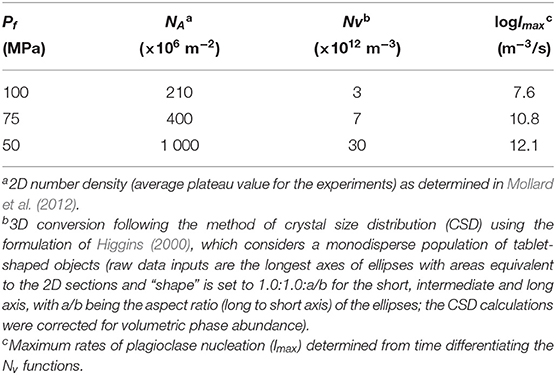
Table 2. Plagioclase number density and maximum nucleation rate.
Methods
Textural and Analytical Data
For the modeling purpose, we had to perform complementary analyses to the experimental study of Mollard et al. (2012).
Regarding plagioclase textures, we had to convert NA into volume number densities (Nv). Because plagioclase microlites show strongly elongated tablet shapes, we determined Nv from the method of crystal size distribution (CSD) using the spreadsheet of Higgins (2000), which considers a monodisperse population of tablet-shaped objects (Table 2). Plagioclase crystals grow anisotropically, but we lack growth rate data for each of the three crystalline axes, so that the crystal size to model was the average of the ten longest semi-lengths of the crystal long axes. The time evolution of plagioclase Nv, Φ, and L/2 are presented in Figure 1. We had also to determine the mean distance between two crystal centers from the SEM images used in Mollard et al. (2012), via the “center-center” function of the SPO2003 software (Launeau and Robin, 1996) (Table 3).
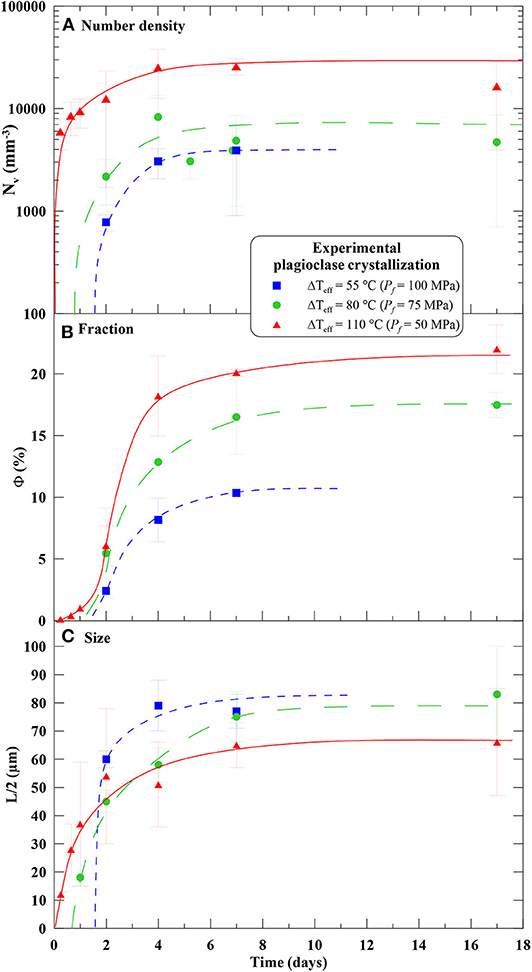
Figure 1. Experimental data of Mollard et al. (2012) showing the time-evolution of the plagioclase crystal (A) volume number density calculated after Higgins (2000) as in Table 2; (B) plagioclase proportion, and (C) average of the ten longest semi-axes of the crystals as defined in Mollard et al. (2012). Temperature was 875°C, initial pressure was 200 MPa, and final pressures were 100, 75, and 50 MPa, corresponding to ΔTeff of 55, 80, and 110°C, respectively. Note that the curves start at different times, corresponding to an increasing delay in plagioclase nucleation with decreasing ΔTeff. However, for growth modeling, 0 time corresponds to the onset of nucleation.
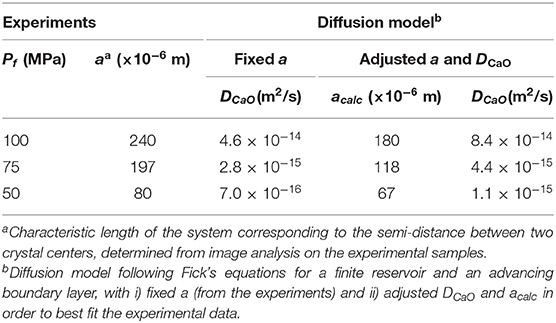
Table 3. Results of the simulations.
Regarding glass compositions, we performed analytical profiles in SiO2, Al2O3, CaO, and Na2O contents from selected decompression run products. The analyses have been performed using a Cameca SX50 electron microprobe (EMP; BRGM-ISTO, Orléans), with the conditions of 15 keV accelerating voltage, 6 nA beam current, 10 s counting time on the element peaks, and a defocused beam (~10 μm in diameter) to minimize Na migration during analysis (Na analyzed first; Pichavant, 1987; Devine et al., 1995). Analytical errors on these analyses were calculated at ~1% (relative) for SiO2, Al2O3 and CaO, and 5% for Na2O.
Classical Nucleation Theory (CNT)
Nucleation may be homogeneous or heterogeneous. Heterogeneous nucleation occurs if any interfaces, such as pre-existing crystals or bubbles, are capable of decreasing the energy barrier required to trigger crystal nucleation. The starting products used in the experiments of Mollard et al. (2012) were aphyric and the SEM images did not show pre-existing gas bubbles that could have acted as preferential sites for plagioclase nucleation (i.e., no systematic crystal attachment on bubbles). Therefore, we assume that nucleation in the experiments was homogeneous.
The CNT has been developed by Volmer and Weber (1926), Farkas (1927), Kaischew and Stranski (1934), Becker and Döring (1935), improved by Turnbull and Fisher (1949), Frenkel (1946), Kashchiev (1969), and compiled in Kirkpatrick (1981) and Fokin et al. (2006). The CNT, for the case of homogeneous nucleation, considers a submicroscopic process in which random fluctuations in a homogeneous one-component liquid held below its liquidus form crystal-like atom clusters, in order to reach thermodynamic stability (low free energy). This results in the creation of a cluster-liquid interface, the maintaining of which causes clusters below a critical size to shrink whereas the largest grow to macroscopic sizes (the critical size decreasing with increasing undercooling). The temperature-dependence of the nucleus size distribution is given by the Boltzmann function:
where n is the number of spherical nuclei, no is the total number of atoms, ΔG is the excess free energy, and kB the Boltzmann's constant. According to the CNT, the steady-state rate of homogeneous nucleation has the following form James (1985):
where Nc = A/Vm, with Nc being the volumetric concentration of reactant atoms, A the Avogadro's constant and Vm the molar volume of the crystallizing phase, h is the Planck's constant, ΔG* is the free energy required to form a spherical critical nucleus having the properties of the macroscopic crystal, and ΔGD is the activation energy required for atom attachment. ΔG* can be expressed as:
where σ is the free energy associated with the crystal-melt interface and ΔGv the free energy change per volume of the transformation that can be defined as ΔGv σ ΔG/Vm.
To compare experimental data with the CNT, several simplifications are necessary. The first one sets that the activation energy of atomic jumps across the nucleus-melt interface equals that of the shear relaxation time of the melt (Stokes-Einstein approximation), allowing ΔGD to be expressed in terms of melt viscosity (η). The second assumption sets that heat capacities of the crystal and melt compare, reducing ΔG to Turnbull's equation:
where ΔH is the enthalpy of formation and ΔT is the undercooling which is replaced by ΔTeff in the present case of decompression-induced isothermal crystallization. Combining (2) and (3), the steady-state nucleation rate becomes:
where ao is the atomic jump distance. This shows that for small ΔTeff, the exponential term controls the shape of the curve and I increases with increasing ΔTeff. In contrast, for high ΔTeff, η increases faster than the exponential term and I passes per a maximum before decreasing. Thus, I should show a bell-shape and an asymmetrical curve around the maximum depending on ΔTeff. To resolve (5), thermochemical and viscosity data are available and I is empirically determined from the crystal number density evolution with time in the experiments, whereas σ remains the most unknown parameter.
Diffusion-Driven Crystal-Growth Theory
A nucleus that reached a critical size starts growing until the thermodynamic equilibrium is reached or the supply in crystal components stops. The theory of crystal growth has been extensively developed by Christian (1965), Dowty (1980), and Kirkpatrick (1981). In principle, crystal growth involves two main mechanisms: (1) atom incorporation at the crystal/melt interface which locally leads to melt depletion in species consumed by the crystal, and thus leads to a concentration gradient that drives (2) transportation of the plagioclase-component atoms from melt to crystal interface. The kinetics of either mechanism control crystal growth. In the following, however, interface processes are not considered as the growth limiting factor, practically meaning that the atoms are assumed to be instantaneously inserted into the crystalline structure. Although this hypothesis should be demonstrated, our first approach of plagioclase growth modeling is to test atom diffusion in melt as the limiting factor on the entire time period of crystal growth.
Adaptation of Fick's Law to Multicomponent Systems
Fick's second law classically describes diffusion in binary aqueous solutions. Therefore, its application to multicomponent silicate melts requires the assumption that the silicate melt can behave as an effective binary system (Cooper, 1968; Zhang, 2010). Given the high silica content of the melt (~80 wt%), we defined SiO2 as the solvent solution, in which solutes of Al, Ca, and Na, diffuse. As the crystal/melt interface processes are not considered, the progression of the crystallization interface only depends on the flux of the various components arriving at the interface. It is reasonable to assume that growth is limited by only one such flux, i.e., the smallest one. Note that in the present case of a finite reservoir (chemically closed system), the flux at the interface depends on the diffusion coefficient of the considered component but also on the concentration gradient at that point. Thus, comparing fluxes does not necessarily mean comparing diffusion coefficients (in contrary to the case of an infinite reservoir with continuous component supply). The species with the lowest diffusion flux may as well be the one with the highest diffusion coefficient, because it starts depleting the reservoir faster than the other species.
Dimensional System
Diffusion in binary aqueous solutions is classically described by Fick's second law (Bird et al., 2007) which gives the solute concentration c(x,t) at distance x from the interface and time t as follows:
where D is the diffusion coefficient of the considered species. At t = 0, the initial concentration is Co and the concentration at the crystal-liquid interface is the equilibrium concentration Ceq (below which the component cannot precipitate anymore into the crystal).
The position of the crystal/melt interface at time t is noted xB(t) and the growth rate vB(t) is its time derivative:
The flux J at the interface is as follows:
The fact that growth rate (i.e., interface velocity) is controlled by diffusion is expressed by the Stefan condition (Ghez, 1980):
with Cs being the species concentration in the crystal. The right-hand term of the equation is the quantity of matter (per unit square per second) that is consumed by the crystal as the interface extends with velocity vB. The left-hand term is the quantity of matter brought at the interface by diffusion. The two are forced to be equal following the principle of mass conservation. Therefore, estimating plagioclase growth velocity is equivalent to computing a diffusive flux at the interface, which requires solving Fick's law (6) with the appropriate boundary conditions.
Considering a finite reservoir requires the determination of the characteristic length of the system (a) over which diffusion operates, which we retrieved from the average semi-distance between neighboring crystal centers (image analysis on the SEM pictures of the experimental samples using the SPO2003 software; Table 3). The position of the interface x verifies 0 < x< a. The boundary conditions are:
(10) postulates that at t = 0 (when nucleation starts), the concentration in the system is equal to the initial concentration co. (13) gives the boundary condition at the interface (at position xB) where the concentration is fixed at the value ceq. (12) is the boundary “closed system” condition at the other end of system, which states that the flux through this extremity is zero. This hypothesis reflects the fact that there is another crystal growing, opposite to this one, on the other side of this boundary. However, because of the diffusion coefficient issue addressed below, we had to circumvent this dimensional system of equation into a non-dimensional one.
Non-dimensional System
To solve (6), one requires knowing D of the growth-limiting component (CaO in our case; see below). Yet, because of the absence of CaO diffusion data relevant to our conditions (see below), we deduce D from the experimental data using a non-dimensional form of (6). The solution is also in a non-dimensional form (i.e., independent of D) and must be transformed back into a dimensional form using an adequate D.
The non-dimensional form of this system is obtained via the change of the following variables:
with their respective differentials (dx'=a dx; dt'=a2/D dt; dc'=(co-ceq) dc), the D-independent form of (6) writes:
with boundary conditions as follows:
This system is solved numerically using a fixed finite difference scheme and a front tracking method to follow the movement of the interface (i.e., giving the position of the interface). The details are available in Crank (1984). The method works as follows:
1. solve the non-dimensional form of Fick's law (16), which returns a D-independent concentration field, a flux at the interface, and an interface velocity [vB (t,D)] Crank (1984);
2. a D-independent interface position is obtained by time integration [xB (t,D)], which is exactly the half-size of the crystal;
3. find D that best fits this computed crystal size with the experimental measurements [crystal size vs. time].
It has to be noted that for comparison, the same procedure was performed using the Fick's equations for a concentric growth [finite reservoir and advancing boundary layer; equations in Crank (1984)]. The results turned out to be very similar to those of the model described above (i.e., planar interface), so that only this latter model is presented here.
Results
Nucleation Rate and Interfacial Energy
The energy required to create interfaces between the solid nucleus and melt (σ) plays a critical role in the crystallization process by drastically impacting the nucleation rate (I), with tiny σ variations generating several orders of magnitude differences in I (Figure 2; Hammer, 2004). Indeed, strong σ forces the magma to become highly supersaturated before crystals can nucleate. Should σ become great enough, crystals would not nucleate, even if the driving force for nucleation, ΔTeff, is high. Solid-liquid σ has been determined independently from the CNT, mostly in pure metals (Howe, 1997) and from indirect estimations in magmatic system (Zanotto, 1987). For instance, macroscopic σ values have been obtained by measurements of crystal-melt wetting angles, giving values of 0.3 J.m−2 for quartz-granite (Laporte and Provost, 2000) and 0.5 J.m−2 for olivine-basalt (Cooper and Kohlstedt, 1982). Thus, we focus below on defining σ for the plagioclase-haplotonalite (rhyolite) system.
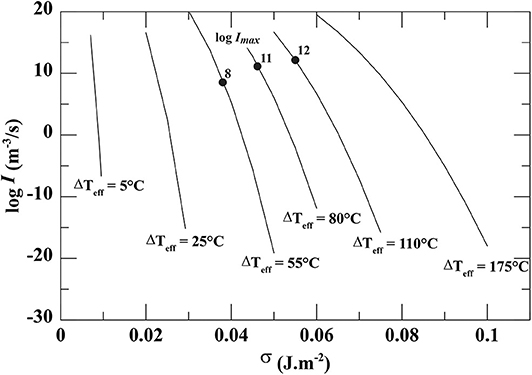
Figure 2. Dependence of the nucleation rate (I) on the interfacial energy (σ) as a function of effective undercooling (ΔTeff), calculated from the Classical Nucleation Theory. The black dots show the empirical Imax determined from the crystallization experiments of Mollard et al. (2012).
The following empirical equation gives macroscopic σ estimations for different compositions (Turnbull, 1950; Kirkpatrick, 1981; James, 1985):
where β is an experimentally-determined constant that ranges from ~0.3 for non-metals to ~0.5 for metals (Matusita and Tashiro, 1973; Neilson and Weiberg, 1979). In the case of our experimental data set, Pi is 200 MPa and the plagioclase composition varies from An27 to An37 depending on Pf (Mollard et al., 2012), so that ΔH varies from 74300 to 79180 J.mol−1 (Burnham and Nekvasil, 1986; Muncill and Lasaga, 1988; Hammer, 2004) and Vm, calculated as a linear albite-anorthite solution and assumed to be pressure independent, is ~1.003 × 10−4 m3.mol−1 (Klein, 1994; Hammer, 2004). From (20) and with β = 0.3, σemp varies from 0.122 J.m−2 for An27 to 0.130 J.m−2 for An37 (Table 4). However, these values may still be overestimated with respect to a microscopic critical nucleus (Fokin et al., 2000).
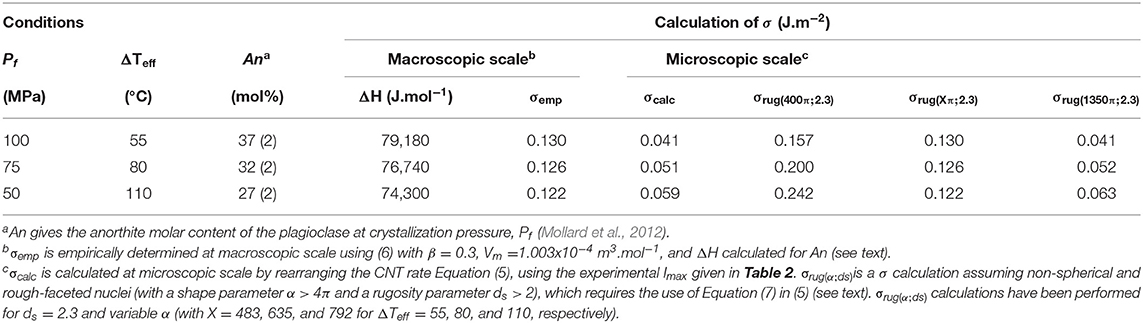
Table 4. Determination of the interfacial energy (σ).
In the absence of additional methods to determine plagioclase-rhyolite σ independently from the nucleation rate data, we calculated I with σ emp (20) using the CNT rate Equation (5), giving logImax below 20 m−3.s−1.
Glass Compositional Gradients
The glass compositions at initial and final conditions are reported in Table 5 and their compositional evolutions as a function of distance from the plagioclase interface for durations of 7 and 17 days at Pf (100, 75, and 50 MPa) are shown in Figure 3.
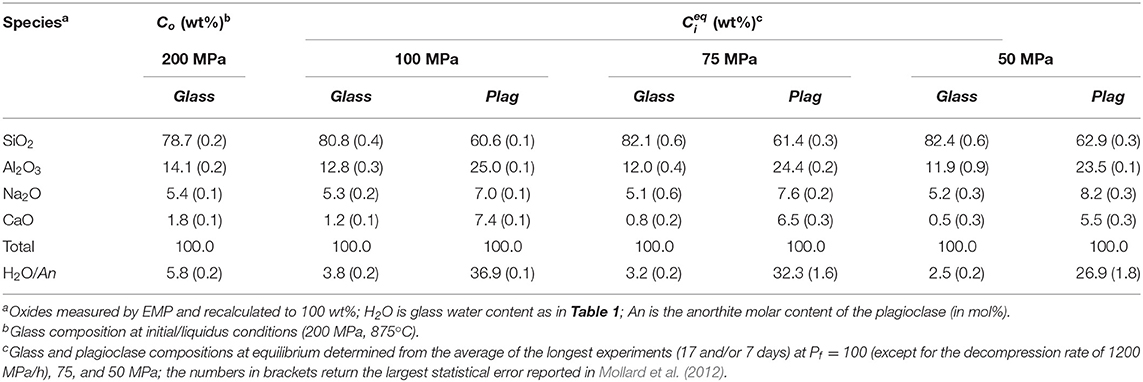
Table 5. Initial and equilibrium compositions of the experimental glasses and plagioclases, from Mollard et al. (2012).
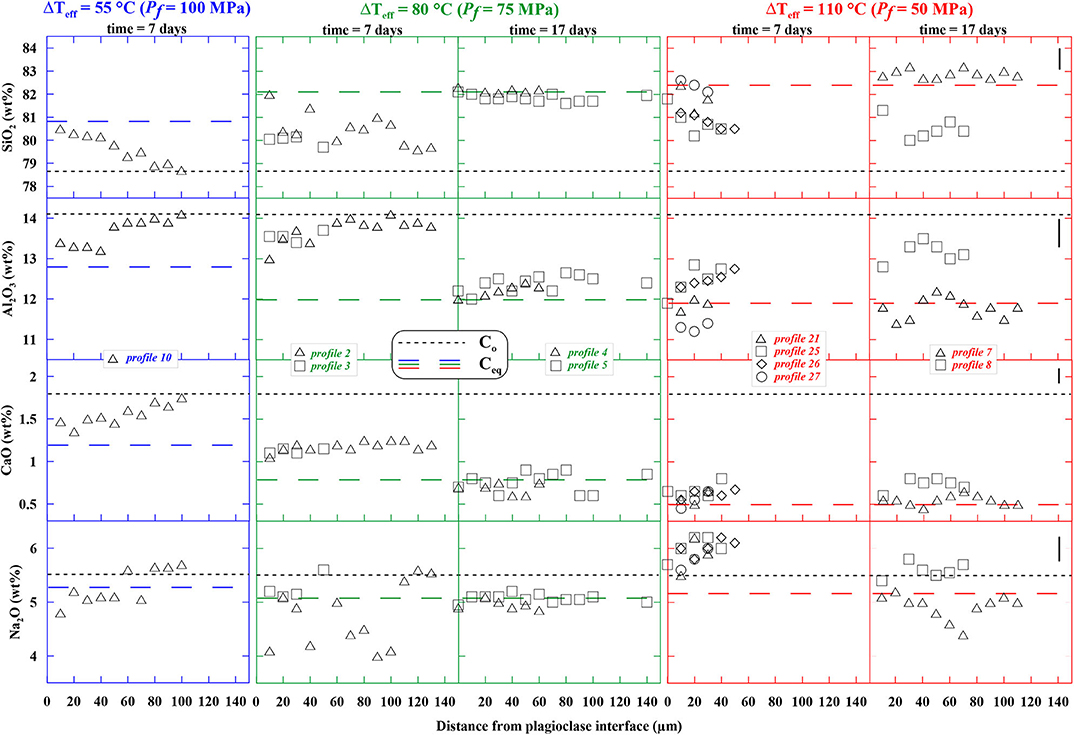
Figure 3. Selected compositional profiles in the glasses from the decompression-induced crystallization experiments of Mollard et al. (2012). Each column corresponds to a given run product at ΔTeff of 55°C (blue), 80°C (green), or 110°C (red), and a crystallization duration of 7 or 17 days. The symbols represent different analytical profiles performed in a given run product. The black dotted lines represent the initial concentrations (Co at 875°C and 200 MPa) and the colored dashed lines give the expected equilibrium melt compositions at crystallization pressure (Ceq at 875°C and Pf) as given in Table 5. The vertical bars on the right give the analytical error.
For ΔTeff = 55°C (Pf of 100 MPa) and a crystallization duration of 7 days, the SiO2 content of the glass next to the plagioclase is similar to the one expected at Pf (i.e., 80.8 wt %) and gradually deviates toward the initial concentration (78.7 wt %). This trend is confirmed by the Al2O3, CaO, and Na2O profiles, which all start at the plagioclase interface close to the value expected at Pf and join the Pi concentration 80–100 μm away from the crystal. The slopes of the Al2O3, CaO, and Na2O profiles are inverted with respect to SiO2, since plagioclase is SiO2-poorer than melt, but richer in Al2O3, CaO, and Na2O. The presence of compositional gradients in the glasses after 7 days suggests that none of the oxides chemically re-equilibrated at 100 MPa over this period of time.
For ΔTeff = 80°C (Pf of 75 MPa) and crystallization duration of 7 days, the SiO2, Al2O3, and Na2O profiles in glasses show similar trends than for ΔTeff = 55°C. After 17 days, all profiles are flat and close to the expected concentrations at Pf, suggesting complete chemical re-equilibration between 7 and 17 days.
For ΔTeff = 110°C and crystallization duration of 7 days, the profiles are close to the expected values at equilibrium, but they show gradients, so that the glass did not fully re-equilibrate. The Na2O profiles at 7 days are richer than the starting glass concentration, an inconsistency that might be attributed to the large analytical errors associated with Na microprobe analysis (although precautions have been taken for the analysis, as mentioned above). It has also to be noted that the small difference between the initial and equilibrium compositions do not favor accurate estimations of the gradients. For crystallization duration of 17 days, the profiles are more or less flat (at least they start and end with similar values), but they show differences in bulk oxide contents. For instance, profile #7 shows flat profiles close to equilibrium compositions, whereas profile #8 shows 2–3 wt% SiO2 less and 1–2 wt% Al2O3 more, that is, closer to the initial concentrations. This suggests that melt areas may be chemically re-equilibrated, whereas others are still in progress after 17 days. Such chemical heterogeneities may result from (i) spatial heterogeneities in plagioclase crystal distribution as described in Mollard et al. (2012), (ii) a nucleation event that is not strictly instantaneous, possibly due to the slow kinetics prevailing at low melt H2O contents; this would result in transient plagioclase crystallization from chemically evolving melts, or (iii) the vicinity of neighboring crystals that contaminate some glass analyses (mixed analyses between glass and crystal).
Crystal-Growth Limiting Component
The component with the smallest flux that limits crystal growth can be identified from comparing the compositional profiles in the experimental melts as a function of distance from plagioclase interface (Figure 3). The source of a given component is assumed to be exhausted when the component profile shows a flat slope at the equilibrium concentration expected at Pf. Al2O3 did not reach equilibrium at Pf after 7 or 17 days (positive slopes and profile #8 far from equilibrium concentration). The Na2O profile looks flat at 17 days, but the large analytical scatter makes it inherently untrustworthy. In contrast, CaO is the only component showing equilibrium concentrations after 7 days (and also after 17 days) at Pf = 75 MPa. CaO is also the component with the lowest concentrations (~1–2 wt%) and the smallest slopes of the profile lines (which give an indication of the magnitude of the flux), explaining why it gets depleted first. Therefore, we chose CaO as the plagioclase growth limiting component.
Thus, solving (6) requires knowing DCaO. Ca diffusivity values are particularly variable in dry rhyolitic melts, with logDCaO ranging from about −11 m2/s at 1,400°C to −13 m2/s at 900°C [data compiled in Zhang (2010)]. In H2O-rich (3.6-6.3 wt%) melts, Baker and Bossanyi (1994) measured logDCaO of the order of −11 to −10 m2/s for temperatures from 1,100 to 1,400°C, highlighting the strong effect of H2O on the diffusion coefficient of CaO. In a Ca-doped (<0.1 wt%) haplogranite, Mungall et al. (1999) measured logDCaO from −14 m2/s at 1,137°C to −11 m2/s at 1,600°C in dry conditions, reaching −11 m2/s at 1,300°C and −10 m2/s at 1,600°C with 3.7 wt% added H2O. In conclusion, none of the above DCaO have been determined for pressure, temperature and H2O content conditions relevant to the experiments of Mollard et al. (2012), Therefore, we deduce D from the experimental data using a non-dimensional form of (6). The solution is also in a non-dimensional form (i.e., independent of D) and must be transformed back into a dimensional form using an adequate D.
Results of the Plagioclase Growth Simulations
Following the methodology given in section § Non-Dimensional System, we performed simulations at Pf = 100, 75, and 50 MPa, the results of which are given in Table 3 and Figure 4. The simulations freely adjust acalc (calculated a) and DCaO in order to best fit the experimental data. For ΔTeff = 55°C, the calculation suggests a couple of acalc and DCaO of 180 μm and 8.4 × 10−14 m2/s, respectively (Figure 4A). For ΔTeff = 80°C, acalc and DCaO are 118 μm and 4.4 × 10−15 m2/s, respectively (Figure 4B). For ΔTeff = 110°C, acalc and DCaO are 67 μm and 1.1 × 10−15 m2/s, respectively (Figure 4C).
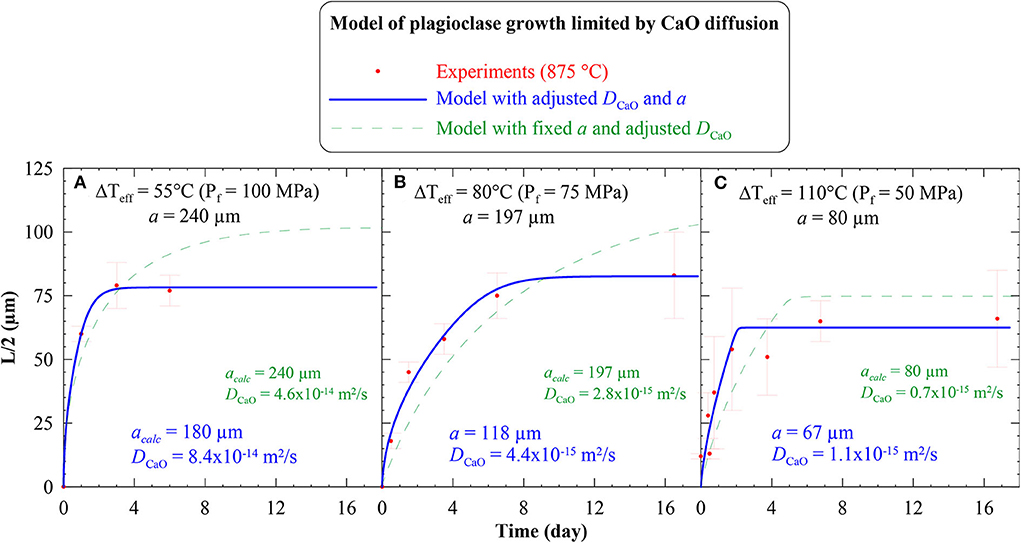
Figure 4. Time evolution of the plagioclase crystal size (half-length of the ten longest long-axes; L/2) for a growth limited by the diffusion of CaO (DCaO), following Fick's diffusion equations considering a time-advancing crystal-melt interface in a finite reservoir. (A) ΔTeff = 55°C; (B) ΔTeff = 80°C, and (C) ΔTeff = 110°C a gives the characteristic length of the system determined from the experimental samples, as given in Table 3. The red dots represent the experimental data, as given in Figure 1C. The plain blue curves show the results for the model adjusting both, the characteristic length of the system (acalc) and DCaO, in order to best fit the experimental data. The dashed green lines show the results for the model adjusting DCaO with a fixed to the experimental value.
Fixing a to the measured values does not allow a good fitting of the experimental data, but the calculated DCaO are of the same order of magnitude. In the worst case, i.e., for ΔTeff of 110°C, the calculation returns DCaO of 0.7 × 10−15 m2/s instead of 1.1 × 10−15 m2/s when calculated for both parameters being adjusted (i.e., about twice as slow; Figure 4).
Discussion
In the section below, we discuss the results of the crystallization modeling in terms of validation and implications for volcanic systems.
New Constraints on the Plagioclase-Rhyolite Interfacial Energy (σ)
σ Values Deduced From the CNT
The calculation of I with σemp (20) using the CNT rate Equation (5) gives extremely low logImax (<20 m−3.s−1), which represents inconsistent values with respect to the experimentally-determined logImax of 8 to 12 m−3.s−1 with ΔTeff increasing from 55 to 110°C (Pf from 100 to 50 MPa, respectively) obtained by time differentiation of the Nv functions (Table 2). This suggests that either the CNT or the calculation of σ is not valid in this case.
Turning things around, we assumed the basic form of the CNT Equation (5) and back-solved for σ using our experimentally-determined nucleation density data (Imax). The calculations suggest σcalc from 0.041 to 0.059 J.m−2 with ΔTeff increasing from 55 to 110°C (Figure 2), which is about 2–3 times lower than σemp (Table 4). Following a similar approach (i.e., assuming the basic form of the CNT and back-solving for σ using the experimental nucleation data of Hammer and Rutherford, 2002), Hammer (2004) suggests σ variations from 0.045 to 0.068 J.m−2 with H2O content decreasing from 3.8 to 2.5 wt% (i.e., Pf from 100 to 50 MPa and ΔTeff from 55 to 110°C). Both results are in very good agreement, giving confidence in the use of such an approach to estimate σ values.
However, these calculated σ may deviate from the real σ because it incorporates all the assumptions inherent in CNT (Hammer, 2004; Shea et al., 2009). In addition, another source of variance is the dwell time during which textural ripening occurs, which may lead to a decrease of the number density during the period between nucleation (first hours to days after perturbation) and the growth of crystals to observable size (7–17 days); such an effect of dwell time may influence the comparison between experimental studies of otherwise similar systems (feldspar in highly evolved silica-rich liquids).
Compositional Dependence of σ
The ΔTeff dependence of σ (Figure 2) is mainly ascribed to the melt H2O content (Hammer, 2004). The decrease of melt H2O content with decompression (increasing ΔTeff) has three main effects. The first one is to increase melt viscosity and thus the kinetic barrier to the atomic attachment (Heslin and Shelby, 1993). The second is the influence of devolatilization on the free energy term in the nucleation rate equation (Hammer, 2004; Shea et al., 2009). The third effect concerns the progressive divergence between the liquid and plagioclase compositions in the course of crystallization that leads to an increase of the crystal-melt σ (Takei and Shimizu, 2003). This effect results from intra-crystalline chemical bonds less energetic than at crystal-liquid interface, so that the crystal-liquid σ will increase as the concentration of the crystal component dissolved in the liquid decreases. In other words, the non-ideality of chemical bonding causes a strong compositional dependence of the crystal-liquid σ (Takei and Shimizu, 2003; Shimizu and Takei, 2005). All three effects may act together to increase σ as the H2O-saturated rhyolitic melt depressurizes and crystallizes plagioclase.
Refinement of σ Through Shape-Dependent Nucleus Parameters
Because the CNT assumes smooth-faceted and spherical nuclei, σcalc may be wrong in case of rough and elongated nuclei. Indeed, a rough surface increases the surface area, which modifies the crystal-melt σ and thus nucleation rate (Sen and Mukerji, 1999). Although the nanometric size of the critical nucleus does not permit determination of its true shape characteristics, an alternative consideration to the CNT is to view the critical nucleus as the unit cell shape of the macroscopic crystal, i.e., triclinic and rough-faceted for plagioclase (Holdren and Speyer, 1987; Brantley and Mellot, 2000). Thus, assuming that the general morphology of the nucleus compares to the one of the macrocrystals, the surface fractal dimension (ds) may be extracted from the surface area (SA), using SA = rds−3, where r is the radius of the nucleus (Mandelbrot, 1982; Van Damme et al., 1987). Measurements on albite and anorthite crystals suggest ds of ~2.2 and ~2.7, respectively (Holdren and Speyer, 1987; Brantley and Mellot, 2000). Assuming an ideal solid solution between albite and anorthite gives ds value of ~2.3 for An30 (average plagioclase composition analyzed in the experiments).
The nucleus shape, expressed as the shape-dependent parameter α, has the value of 4π for a perfect sphere, but there are no α data available for a plagioclase shape. The most relevant ds-α data to our magmatic system concern congruent nucleation in Li2Si2O5 liquids (that have often been taken as melt analogs), which show a positive correlation between ds and α, with α values of ~400π where ds = 2.3 (Sen and Mukerji, 1999). The influence of ds and α on σ is expressed through the following equation:
Assuming ds = 2.3 for An30 and using our experimental Imax, we calculated σ values for rough and non-spherical nuclei, σrug(α;ds), using (5), in which (21) replaces (3), and for α varying up to 2,000 π. Bracketing σrug between σemp and σcalc (calculated for a smooth sphere) suggests α between 400 π and 1,500 π (Figure 5). σemp would be compatible with σrug having ds = 2.3 and α of 483, 635, and 792 π at Pf = 100, 75, and 50 MPa, respectively (Table 4). Although there are no available data on a correlation between α and a given crystal aspect ratio, the elongation and the increasing surface area of the nucleus as ΔTeff increases would agree with the observed evolution of the plagioclase macrocrystal characteristics.
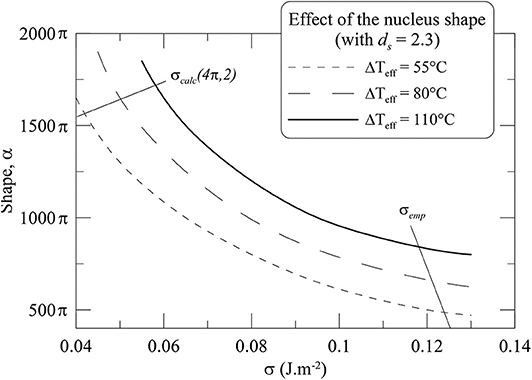
Figure 5. Dependence of the interfacial energy (σ) on the nucleus shape (α) for a roughness ds of 2.3 (An30 plagioclase), as a function of effective undercooling (ΔTeff). σcalc(4π;2) and σemp (as determined in Table 4) define a range of σ bracketed between microscopic and macroscopic values, respectively.
Plagioclase Growth Modeling
Validation of the Process Controlling Crystal Growth
The theoretical results and the experimental decompression-induced plagioclase growth laws agree on three points (Figure 4). First, the experimental data agree with the theoretical curve profiles defined for a finite model with an advancing crystal-melt interface limited by CaO diffusion. Second, the returned acalc, i.e., the distance between two crystals that defines number density, are not far from those measured in the experimental samples, i.e., differing by ~60 μm at Pf = 100 MPa, ~80 μm at Pf = 75 MPa, and ~10 μm at Pf = 50 MPa (Table 5). Third, the returned DCaO from 1.1 × 10−15 to 8.4 × 10−14 m2/s stand in the lower limit of the range of those from the literature (full range of 10−14 to 10−10 m2/s), although none of them were determined for conditions (melt composition, H2O content, temperature, pressure) comparable to those in Mollard et al. (2012).
Therefore, this overall good agreement between the theoretical and empirical models validates CaO diffusion as the main process controlling isothermal decompression-induced plagioclase growth under moderate ΔTeff (<110°C).
Limitations of the Model
Constraining DCaO by an independent method would of course help to refine the model, which, in its present form, possibly compensates different kinds of discrepancies by adjusting DCaO. The discrepancies may come from limitations on Fick's laws and/or crystal-melt interface barriers, as explained below.
First, the aqueous solutions for which the Fick's laws were determined are often chemically dissociated, whereas melts contain polymeric transient units, possibly leading to coupled diffusions of some species. Also, aqueous solutions are dilute, thus permitting to approximate diffusion of particles against a constant background of water, whereas the notion of particles diffusing against a constant background of a silica melt has not been discussed so far. The viscosities in aqueous solutions and melts differ by many orders of magnitude, which may introduce some bias in the formulation. Although these issues need to be addressed in the future, the application of Fick's law to multicomponent systems is a reasonable starting point of work, as already used by many authors willing at determining diffusion coefficients in complex systems such as polymers, glasses, and melts.
Second, plagioclase growth may not be limited by kinetics (diffusion) over the entire crystallization period of time. In particular, Roskosz et al. (2005a,b) demonstrated that at large undercooling, for which melt viscosity is high and the crystal-melt Al/Si ratio is constant, crystal growth is thermodynamically controlled by crystal-melt interface processes. Yet, the calculation of the Al/Si ratios in the experiments used in the present study does not consolidate this hypothesis (Mollard et al., 2012). In the opposite side, interface-controlled growth may be the regime prevailing for small degree of undercooling in case of incongruent crystallization, since plagioclase-melt interfacial energy is high and the component diffusion is often fast compared to the rate of atom attachment. If true for the early crystallization, the starting conditions used for Fick's diffusion modeling may not be correct. However, the equations governing plagioclase growth controlled by interface processes are not time-dependent and consequently do not allow to assess when diffusion actually takes over as the controlling mechanism. Interface-controlled mechanisms may likely affect the early plagioclase crystallization at ΔTeff = 55°C (Pf = 100 MPa), but the overall shape of the experimental plagioclase growth is satisfactorily simulated by diffusion-controlled processes.
Thus, potential biases, such as the application of Fick's law to melts and the possible interface-controlled growth, need further evaluation to fully validate the present model and the CaO diffusion coefficients.
Implication for Volcanic Systems
The present study provides a mean to retrieve crucial parameters for the understanding of syn-eruptive volcanic processes. Indeed, the simulations may be relevant to phenocryst-poor rhyolitic magmas that rapidly ascend from a storage region to a given shallower depth where they crystallize microlites for a given amount of time. Although rapidly-decompressed magmas that crystallize at constant pressure may not be the most common scenario triggering explosive eruptions (more commonly thought to follow polybaric crystallization), dome-generated blasts have been recognized to follow such a decompression-induced isobaric crystallization path (e.g., Castro and Dingwell, 2009; Martel, 2012). The driving force for crystallization, ΔTeff, may be deduced from the plagioclase liquidus for the bulk composition of interest and the crystallization pressure/depth that is either directly determined (e.g., from geophysical measurements) or retrieved post-eruption from the pyroclasts [i.e., from number density, proportion, and shape of the plagioclase microlites as defined in Mollard et al. (2012); their Figure 11]. This study has two major implications for the understanding of the volcanic systems.
Firstly, predicting crystal nucleation rates in volcanology is crucial for two reasons. First, high nucleation rates may lead to an extensive crystallization of microlites at low pressures (<50 MPa), which drastically enhances the risk of dome explosivity (Sparks, 1997; Martel, 2012). Second, nucleation rate directly controls crystal growth kinetics by fixing crystal number density that defines the characteristic distance on which diffusion proceeds between two growing crystals. The shorter the characteristic distance, the quicker the reservoir depletion in the fastest species, and thus the slower the growth rate. Therefore, magmas with high crystal number densities must show slower crystal growth rates than poorly-crystallized magmas, all other conditions being comparable. Our study suggests that homogeneous nucleation rates in rapidly decompressed natural rhyolites may be deduced from the CNT equation, using a plagioclase-melt surface tension from 0.04 to 0.06 J.m−2 with ΔTeff increasing from 55 to 110°C. Yet, natural rhyolitic magmas may contain impurities that could trigger heterogeneous nucleation by lowering crystal-liquid surface tensions and the energetic barrier, for instance on pre-existing crystals or gas bubbles. So far, however, neither experimental or natural products have brought evidence of preferential sites favoring plagioclase nucleation during magma decompression.
Secondly, the size evolution of the plagioclase microlites may be predicted as a function of time at various crystallization pressure (or ΔTeff) by the present model of growth limited by the diffusion of CaO. Our model predicts that a H2O-saturated rhyolitic melt at 875°C that ascends from about 6-8 km deep (~200 MPa) to 1.5–2.0 km (~50 MPa) will nucleate about 1013 m−3 plagioclase microlites (Table 2) with final half-lengths of ~60 μm (Figure 4C). Additional data at lower pressures would be helpful to assess the crystallization conditions at dome level. However, coupling data on crystal number density to crystal size evolution is a key parameter to assess magma flow properties. Indeed, many small needle-shaped crystals do not behave the same as few large crystals under shear. Integrated numerical modeling of magma crystallization during ascent in volcanic conduits (e.g., Melnik and Sparks, 1999; De'Michieli Vitturi et al., 2010) consider crystallization as a volume fraction at thermodynamic equilibrium. Such models would simulate magma rheology more realistically by incorporating time evolutions of textural parameters, such as crystal number density and size.
For a fully confident extrapolation to natural systems, the calculations of the crystallization kinetics still need further investigations. In particular, the chosen composition represents the SiO2-richest end-member of the typical interstitial melts from natural andesites to rhyolites (70–79 wt % of SiO2; Hammer, 2008). A validation of the calculations for SiO2-poorer melts would be necessary, especially to verify whether Fick's diffusion law is still acceptable with a SiO2-poorer background solution. Additionally, the calculations are performed for a four-components simplified analog of a rhyolitic melt. Although such analog compositions have been successfully used to determine various physicochemical properties (including diffusion) of natural magmas, they have to be confronted to more realistic melts, the compositional complexity of which may induce deviations of the crystallization processes and kinetics. Finally, crystallization at fixed ΔTeff needs to be extended to polybaric crystallization, for which crystal nucleation and growth experience time-evolving ΔTeff.
Conclusions
Modeling the experimental results of decompression-induced crystallization of plagioclase from a rhyolite-like melt permits to refine the plagioclase-rhyolite surface tensions that control crystal nucleation rates and validates plagioclase growth limited by diffusion. In particular:
– Plagioclase-rhyolite interfacial energies (σ) have been determined from 0.041 to 0.059 J.m−2 with ΔTeff increasing from 55 to 110°C.
– Considering the shape characteristics of the plagioclase nucleus as similar to those of the macrocrystal, σ can be refined by adjusting parameters of nucleus roughness (ds) of ~2.3 and shape (α) between 500 and 1,350 π depending on ΔTeff.
– Plagioclase growth with time can be numerically simulated by a diffusion-limited model (Fick's second law adapted to multicomponents), considering a (i) finite reservoir, (ii) an advancing crystal-melt interface, and (iii) CaO as the component limiting plagioclase growth.
– The CaO diffusion coefficients, DCaO, that best fit the experimental data range from 8.4 × 10−14 to 1.1 × 10−15 m2/s at 875°C and H2O-saturated pressures from 50 to 100 MPa.
– Predicting crystal nucleation (number density) and growth (time-evolving size) is of prime importance for assessing the rheological properties of magmatic flows that decipher eruptive dynamics, with a direct application to plagioclase microlite crystallization in ascending silica-rich magmas.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Author Contributions
EM participated to the implementation of the model based on her own experimental data. CM participated to the model implementation and overall writing of the manuscript. EL wrote the program using the diffusion equations. GR performed tests using the program. All authors contributed to the article and approved the submitted version.
Funding
This work was part of EM Ph.D. (French MNRT grant) and was funded by the Agence Nationale de la Recherche (contract No. ANR-05-CATT-0004 to CM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank Olivier Rouer and Ida Di Carlo for assistance with the EMP and the SEM analyses. We are grateful to the two reviewers for their fruitful comments and we thank both, Julia Hammer as the guest editor and Valerio Acocella as the chief editor, for the careful handling of the manuscript and their highly valuable contributions to the improvement of the manuscript.
References
Baker, D. R., and Bossanyi, H. (1994). The combined effect of F and H2O on interdiffusion between peralkaline dacitic and rhyolitic melts. Contrib. Mineral. Petrol. 117, 203–214. doi: 10.1007/BF00286843
Barclay, J., Carroll, M. R., Houghton, B. F., and Wilson, C. J. N. (1996). Pre-eruptive volatile content and degassing history of an evolving peralkaline volcano. J. Volcanol. Geother. Res. 74, 75–87. doi: 10.1016/S0377-0273(96)00058-3
Becker, R., and Döring, W. (1935). Kinetische behandlung der keimbuilding in übersättigten dämpfen. Ann. Phys. 24, 719–752. doi: 10.1002/andp.19354160806
Befus, K. S., and Andrews, B. J. (2018). Crystal nucleation and growth produced by continuous decompression of pinatubo magma. Contrib. Mineral. Petrol. 173, 92–112. doi: 10.1007/s00410-018-1519-5
Bird, R. B., Stewart, W. E., and Lightfoot, E. N. (2007). Transport Phenomena, 2nd revised Edn. New York, NY: John Wiley & Sons, 780.
Brantley, S. L., and Mellot, N. P. (2000). Surface area and porosity of primary silicate minerals. Am. Mineral. 85, 1767–1783. doi: 10.2138/am-2000-11-1220
Brugger, C. R., and Hammer, J. E. (2010). Crystallization kinetics in continuous decompression experiments: implications for interpreting natural magma ascent processes. J. Petrol. 51, 1941–1965. doi: 10.1093/petrology/egq044
Burnham, C. W., and Nekvasil, H. (1986). Equilibrium properties of granite pegmatite magmas. Am. Mineral. 71, 239–263.
Cashman, K. V. (1992). Groundmass crystallization of mount st. helens dacite 1980-1986: a tool for interpreting shallow magmatic processes. Contrib. Mineral. Petrol. 109, 431–449. doi: 10.1007/BF00306547
Castro, J. M., and Dingwell, D. B. (2009). Rapid ascent of rhyolitic magma at Chaiten volcano, Chile. Nature 461, 780–783. doi: 10.1038/nature08458
Christian, J. W. (1965). The Theory of Transformations in Metals And Alloys. Oxford: Pergamon Press, 1200.
Cichy, S. B., Botcharnikov, R. E., Holtz, F., and Behrens, H. (2011). Vesiculation and microlite crystallization induced by decompression: a case study of the 1991-1995 mt unzen eruption (Japan). J. Petrol. 52, 1469–1492. doi: 10.1093/petrology/egq072
Clarke, A. B., Stephens, S., Teasdale, R., Sparks, R. S. J., and Diller, K. (2007). Petrologic constraints on the decompression history of magma prior to vulcanian explosions at the Soufrière hills volcano, montserrat. J. Volcanol. Geother. Res. 161, 261–274. doi: 10.1016/j.jvolgeores.2006.11.007
Cooper, A. R. (1968). “The use and limitations of the concept of an effective binary diffusion coefficient for multicomponent diffusion,” in Mass Transport in Oxides, eds J. B. Wachma and A. D. Franklin (Washington, DC: National Bureau of Standards Special Publications), 79–84.
Cooper, R. F., and Kohlstedt, D. L. (1982). “Interfacial energies in the olivine-basalt system,” in High Pressure Research in Geophysics, eds S. Akimoto and M. H. Manghani (Tokyo: Center for academic publications), 217–228. doi: 10.1007/978-94-009-7867-6_17
Couch, S., Sparks, R. S. J., and Carroll, M. R. (2003). The kinetics of degassing-induced crystallization at soufrière hills volcano, montserrat. J. Petrol. 44, 1477–1502. doi: 10.1093/petrology/44.8.1477
Davis, M. J., Ihinger, P. D., and Lasaga, A. C. (1997). Influence of water on nucleation kinetics in silicate melts. J. Non Cryst. Solids 219, 62–69. doi: 10.1016/S0022-3093(97)00252-4
De'Michieli Vitturi, M., Clarke, A. B., Neri, A., and Voight, B. (2010). Transient effects of magma ascent dynamics along a geometrically variable dome-feeding conduit. Earth Planetary Sci. Lett. 295, 3–4, 541–553. doi: 10.1016/j.epsl.2010.04.029
Devine, J. D., Gardner, J. E., Brack, H. P., Layne, G. D., and Rutherford, M. J. (1995). Comparison of microanalytical methods for estimating H2O contents of silicic volcanic glasses. Am. Mineral. 80, 319–328. doi: 10.2138/am-1995-3-413
Dowty, E. K. (1980). “Crystal growth and nucleation theory and numerical simulation of igneous crystallization,” in Physics of magmatic processes, ed R. B. Hargraves (New York, NY: Princeton University Press), 385–417. doi: 10.1515/9781400854493.419
Eichelberger, J. C., Carrigan, C. R., Westrich, H. R., and Price, R. H. (1986). Non-explosive silicic volcanism. Nature 323, 598–602. doi: 10.1038/323598a0
Farkas, L. (1927). Keimbildungsgeschwindigkeit in übersättigten dämpfen. Zeitschrift für Physikalischee Chemie – Abteilung B – Chemie der Elementarprozesse Aufbau der Materie 125, 236–242. doi: 10.1515/zpch-1927-12513
Fenn, P. M. (1977). The nucleation and growth of alkali feldspars from hydrous melts. Can. Mineral. 15, 135–161.
Fokin, V. M., Zanotto, E. D., and Schmelzer, J. W. P. (2000). Method to estimate crystal/liquid surface energy by dissolution of subcritical nuclei. J. Non Cryst. Solids 278, 24–34. doi: 10.1016/S0022-3093(00)00335-5
Fokin, V. M., Zanotto, E. D., Yuritsyn, N. S., and Schmelzer, J. W. P. (2006). Homogeneous crystal nucleation in silicate glasses: a 40 years perspective. J. Non Cryst. Solids 352, 2681–2714. doi: 10.1016/j.jnoncrysol.2006.02.074
Geschwind, C. H., and Rutherford, M. J. (1995). Crystallization of microlites during magma ascent: the fluid mechanics of 1980-1986 eruptions of mt st helens. Bull. Volcanol. 57, 356–370. doi: 10.1007/BF00301293
Ghez, R. (1980). Expansions in time for the solution of one-dimensional Stefan problems of crystal growth. Int. J. Heat Mass Transf. 23, 425–432. doi: 10.1016/0017-9310(80)90083-6
Hammer, J. E. (2004). Crystal nucleation in hydrous rhyolite: experimental data applied to classical theory. Am. Mineral. 89, 1673–1679. doi: 10.2138/am-2004-11-1212
Hammer, J. E. (2008). “Experimental studies of the kinetics and energetics of magma crystallization” in Minerals, Inclusions, and Volcanic Processes, eds K. D. Putirka, and F. J. Tepley (III Reviews in Mineralogy and Geochemistry) 69, 9–59. doi: 10.1515/9781501508486-003
Hammer, J. E., Cashman, K. V., Hoblitt, R. P., and Newman, S. (1999). Degassing and microlite crystallization during pre-climactic events of the 1991 eruption of Mt. Pinatubo, Philippines. Bull. Volcanol. 60, 355–380. doi: 10.1007/s004450050238
Hammer, J. E., Cashman, K. V., and Voight, B. (2000). Magmatic processes revealed by textural and compositional trends in merapi dome lavas. J. Volcanol. Geother. Res. 100, 165–192. doi: 10.1016/S0377-0273(00)00136-0
Hammer, J. E., and Rutherford, M. J. (2002). An experimental study of the kinetics of decompression-induced crystallization in silicic melts. J. Geophys. Res 107, 1–23. doi: 10.1029/2001JB000281
Heslin, M. R., and Shelby, J. E. (1993). Effect of hydroxyl content on the nucleation and crystallization of lithium disilicate glass. Ceram. Trans. 30, 189–196.
Hess, K. U., and Dingwell, D. B. (1996). Viscosities of hydrous leucogranitic melts: non-arrhenian model. Am. Mineral. 81, 1297–1300.
Higgins, M. D. (1994). Numerical modelling of crystal shapes in thin-sections: estimation of crystal habit and true size. Am. Mineral. 79, 113–119.
Higgins, M. D. (2000). Measurement of crystal size distributions. Am. Mineral. 85, 1105–1116. doi: 10.2138/am-2000-8-901
Holdren, G. R., and Speyer, P. M. (1987). Reaction rate-surface area relationships during the early stages of weathering. II Data on eight additional feldspars. Geochim. Cosmochim. Acta 51, 2311–2318. doi: 10.1016/0016-7037(87)90284-5
Howe, J. M. (1997). Interfaces in Materials: Atomic Structure, Kinetics and Thermodynamics of Solid-Vapor, Solid-Liquid, and Solid-Solid Interfaces. New York, NY: John Wiley & Sons, 505.
Iezzi, G., Mollo, S., Ventura, G., Cavallo, A., and Romano, C. (2008). Experimental solidification of anhydrous latitic and trachytic melts at different cooling rates: the role of nucleation kinetics. Chem. Geol. 253, 91–101. doi: 10.1016/j.chemgeo.2008.04.008
James, P. F. (1985). Kinetics of crystal nucleation in silicate glasses. J. Non Cryst. Solids 73, 517–540. doi: 10.1016/0022-3093(85)90372-2
Jaupart, C., and Allègre, C. J. (1991). Gas content, eruption rate and instabilities of eruption regime in silicic volcanoes. Earth Planet. Sci. Lett. 102, 413–429. doi: 10.1016/0012-821X(91)90032-D
Kaischew, R., and Stranski, I. N. (1934). On the kinetic deflection rate of germ formation. Zeitschrift für Physikalischee Chemie – Abteilung B – Chemie der Elementarprozesse Aufbau der Materie 26, 317–236.
Kashchiev, D. (1969). Solution of the non-steady state problem in nucleation kinetics. Surf. Sci. 14, 209–220. doi: 10.1016/0039-6028(69)90055-7
Kirkpatrick, R. J. (1981). “Kinetics of crystallization of igneous rocks,” in Kinetics of Geochemical Processes, eds A. C. Lasaga and R. J. Kirkpatrick (Mineralogical Society of America), 321–398. doi: 10.1515/9781501508233-012
Klug, C., and Cashman, K. V. (1994). Vesiculation of May 18, 1980, Mount St. Helens magma. Geology 22, 468–472. doi: 10.1130/0091-7613(1994)022<0468:VOMMSH>2.3.CO;2
Laporte, D., and Provost, A. (2000). “The grain-scale distribution of silicate, carbonate, and metallosulfide partial melts: a review of theory and experiments,” in Physics and Chemistry of Partially Molten Rocks, eds N. Bagdassarov, D. Laporte, and A. B. Thompson (Kluwer Academic Publishers). 93–140. doi: 10.1007/978-94-011-4016-4_4
Larsen, J. F. (2005). Experimental study of plagioclase rim growth around anorthite seed crystals in rhyodacitic melt. Am. Mineral. 90, 417–427. doi: 10.2138/am.2005.1456
Lattard, D., and Partzsch, G. (2001). Magmatic crystallization at 1 bar in systems close to oxygen: a new/old experimental approach. Eur. J. Mineral. 13, 467–478. doi: 10.1127/0935-1221/2001/0013-0467
Launeau, P., and Robin, P. Y. (1996). Fabric analysis using intercept method. Tectonophysics 267, 91–119. doi: 10.1016/S0040-1951(96)00091-1
Martel, C. (2012). Eruption dynamics inferred from microlite crystallization experiments: application to plinian and dome-forming eruptions of Mt. Pelée (Martinique, Lesser Antilles). J. Petrol. 53, 699–725. doi: 10.1093/petrology/egr076
Martel, C., and Iacono-Marziano, G. (2015). Bubble coalescence, outgassing, and foam collapsing in decompressed rhyolitic melts. Earth Planetary Sci. Lett. 412, 173–185. doi: 10.1016/j.epsl.2014.12.010
Martel, C., and Poussineau, S. (2007). Diversity of eruptive style inferred from the microlites of Mt. Pelée andesite (Martinique, Lesser Antilles). J. Volcanol. Geother. Res. 166, 233–254. doi: 10.1016/j.jvolgeores.2007.08.003
Martel, C., and Schmidt, B. C. (2003). Decompression experiments as an insight into ascent rates of silicic magmas. Contrib. Mineral. Petrol. 144, 397–415. doi: 10.1007/s00410-002-0404-3
Matusita, K., and Tashiro, M. (1973). Rate of homogeneous nucleation in alkali disilicate glasses. J. Non Cryst. Solids 11, 471–484. doi: 10.1016/0022-3093(73)90092-6
Melnik, O., Barmin, A. A., and Sparks, R. S. J. (2005). Dynamics of magma flow inside volcanic conduits with bubble overpressure buildup and gas loss through permeable magma. J. Volcanol. Geother. Res. 143, 53–68. doi: 10.1016/j.jvolgeores.2004.09.010
Melnik, O., Blundy, J. D., Rust, A. C., and Muir, D. D. (2011). Subvolcanic plumbing systems imaged through crystal size distributions. Geology 39, 403–406. doi: 10.1130/G31691.1
Melnik, O., and Sparks, R. S. J. (1999). Nonlinear dynamics of lava dome extrusion. Nature 402, 37–41. doi: 10.1038/46950
Mollard, E., Martel, C., and Bourdier, J.-L. (2012). Decompression-induced experimental crystallization in hydrated silica-rich melts: I. Empirical models of plagioclase nucleation and growth kinetics. J. Petrol. 53, 1743–1766. doi: 10.1093/petrology/egs031
Mueller, S., Melnik, O., Spieler, O., Scheu, B., and Dingwell, D. B. (2005). Permeability and degassing of dome lavas undergoing rapid decompression: an experimental determination. Bull. Volcanol. 67, 526–538. doi: 10.1007/s00445-004-0392-4
Muncill, G. E., and Lasaga, A. C. (1987). Crystal-growth kinetics of plagioclase in igneous systems: one-atmosphere experiments and application of a simplified growth model. Am. Mineral. 72, 299–311.
Muncill, G. E., and Lasaga, A. C. (1988). Crystal-growth kinetics of plagioclase in igneous systems: isothermal H2O-saturated experiments and extension of growth model to complex silicate melts. Am. Mineral. 73, 982–992.
Mungall, J. E., Dingwell, D. B., and Chaussidon, M. (1999). Chemical diffusivities of 18 trace elements in granitoid melts. Geochim. Cosmochim. Acta 63, 2599–2610. doi: 10.1016/S0016-7037(99)00209-4
Nakada, S., and Motomura, Y. (1999). Petrology of the 1990-1995 eruption at Unzen: effusion pulsation and groundmass crystallization. J. Volcanol. Geother. Res. 89, 173–196. doi: 10.1016/S0377-0273(98)00131-0
Neilson, G. F., and Weiberg, M. C. (1979). Test of classical nucleation theory – crystal nucleation of lithium disilicate glass. J. Non Cryst. Solids 34, 137–147. doi: 10.1016/0022-3093(79)90013-9
Newman, S., and Lowenstern, J. B. (2002). VOLATILECALC: a silicate melt-H2O-CO2 solution model written in Visual Basic for excel. Comput. Geosci. 28, 597–604.
Noguchi, S., Toramaru, A., and Nakada, S. (2008). Relation between microlite textures and discharge rate during the 1991-1995 eruptions at Unzen, Japan. J. Volcanol. Geother. Res. 175, 141–155. doi: 10.1016/j.jvolgeores.2008.03.025
Pichavant, M. (1987). Effect of B and H2O on liquidus phase-relations in the haplogranite system at 1-kbar. Am. Mineral. 72, 1056–1070.
Potapov, O., Fokin, V., and Filipovich, V. (1999). Nucleation and crystal growth in water containing soda-lime silica glasses. J. Non Cryst. Solids 247, 74–78. doi: 10.1016/S0022-3093(99)00037-X
Pupier, E., Duchene, S., and Toplis, M. J. (2008). Experimental quantification of plagioclase crystal size distribution during cooling of a basaltic liquid. Contrib. Mineral. Petrol. 155, 555–570. doi: 10.1007/s00410-007-0258-9
Riker, J. M., Cashman, K. V., Rust, A. C., and Blundy, J. D. (2015). Experimental constraints on plagioclase crystallization during H2O- and H2O–CO2- saturated magma decompression. J. Petrol. 56, 1967–1998. doi: 10.1093/petrology/egv059
Roskosz, M., Toplis, M. J., Besson, P., and Richet, P. (2005a). Nucleation mechanisms: a crystal-chemical investigation of phases forming in highly supercooled aluminosilicate liquids. J. Non Cryst. Solids 351, 1266–1282. doi: 10.1016/j.jnoncrysol.2005.02.021
Roskosz, M., Toplis, M. J., and Richet, P. (2005b). Experimental determination of crystal growth rates in highly supercooled aluminosilicate liquids: Implications for rate-controlling processes. Am. Mineral. 90, 1146–1156. doi: 10.2138/am.2005.1627
Sen, S., and Mukerji, T. (1999). A generalized classical nucleation theory for rough interfaces: application in the analysis of homogeneous nucleation in silicate liquids. J. Non Cryst. Solids 246, 229–239. doi: 10.1016/S0022-3093(99)00093-9
Shea, T., Larsen, J. F., Gurioli, L., Hammer, J. E., Houghton, B. F., and Cioni, R. (2009). Leucite crystals: Surviving witnesses of magmatic processes preceding the 79AD eruption at Vesuvius, Italy. Earth Planet. Sci. Lett. 281, 88–98. doi: 10.1016/j.epsl.2009.02.014
Shimizu, I., and Takei, Y. (2005). Temperature and compositional dependence of solid-liquid interfacial energy: application of the cahn-hilliard theory. Phys. B Condensed Matter. 362, 169–179. doi: 10.1016/j.physb.2005.02.008
Sparks, R. S. J. (1997). Causes and consequences of pressurisation in lava dome eruptions. Earth Planet. Sci. Lett. 150, 177–189. doi: 10.1016/S0012-821X(97)00109-X
Takei, Y., and Shimizu, I. (2003). The effet of liquid composition, temperature, and pressure on the equilibrium dihedral angles of binary solid-liquid systems inferred from a lattice-like model. Phys. Earth Planetary Inter. 139, 225–242. doi: 10.1016/j.pepi.2003.08.004
Turnbull, D. (1950). Formation of crystal nuclei in liquid metals. J. Appl. Phys. 21, 1022–1028. doi: 10.1063/1.1699435
Turnbull, D., and Fisher, J. C. (1949). Rates of nucleation in condensed systems. J. Chem. Phys. 17, 71–73. doi: 10.1063/1.1747055
Tuttle, O. F., and Bowen, N. L. (1958). Origin of granite in the light of experimental study in the system NaAlSi3O8-KAlSi3O8-SiO2-H2O. Geol. Soc. Am. Mem. 74. doi: 10.1130/MEM74-p1
Van Damme, H., Levitz, P., Gatineau, L., Alcover, J. F., and Fripiat, J. J. (1987). On the determination of the surface fractal dimension of powders by granulometric analysis. J. Colloid Interface Sci. 122, 1–8. doi: 10.1016/0021-9797(88)90281-0
Volmer, M., and Weber, A. (1926). Germ-formation in oversaturated figures. Zeitschrift für Physicalische Chemie – Stochiometrie und Verwandtschaftslehre 119, 227–301. doi: 10.1515/zpch-1926-11927
Waters, L. E., Andrews, B. J., and Lange, R. A. (2015). Rapid crystallization of plagioclase phenocrysts in silicic melts during fluid-saturated ascent: phase equilibrium and decompression experiments. J. Petrol. 56, 981–1006. doi: 10.1093/petrology/egv025
Wilson, L., Sparks, R. S. J., and Walker, G. P. L. (1980). Explosive volcanic eruptions- IV. The control of magma properties and conduit geometry on eruption column behaviour. Geophys. J. Roy. Astronom. Soc. 63, 117–148. doi: 10.1111/j.1365-246X.1980.tb02613.x
Wolf, K. J., and Eichelberger, J. C. (1997). Syn-eruptive mixing, degassing, and crystallization at redoubt volcano, eruption of December, 1989 to May 1990. J. Volcanol. Geother. Res. 75, 19–37. doi: 10.1016/S0377-0273(96)00055-8
Woods, A. W., and Koyaguchi, T. (1994). Transitions between explosive and effusive eruptions of silicic magmas. Nature 370, 641–644. doi: 10.1038/370641a0
Zanotto, E. D. (1987). Isothermal and adiabatic nucleation in glass. J. Non Cryst. Solids 89, 361–370. doi: 10.1016/S0022-3093(87)80278-8
Zhang, Y. X. (2010). “Diffusion in minerals and melts, theoretical background,” in Diffusion in minerals and melts. eds Y. X. Zhang and D. J. Cherniak (Mineralogical Society of America). 5–57. doi: 10.1515/9781501508394-003
Keywords: plagioclase, undercooling, nucleation and growth, diffusion, modeling
Citation: Mollard E, Martel C, Le Trong E and Rogerie G (2020) Theoretical Models of Decompression-Induced Plagioclase Nucleation and Growth in Hydrated Silica-Rich Melts. Front. Earth Sci. 8:203. doi: 10.3389/feart.2020.00203
Received: 05 December 2019; Accepted: 18 May 2020;
Published: 19 June 2020.
Edited by:
Julia E. Hammer, University of Hawaii at Manoa, United StatesReviewed by:
Leonid Danyushevsky, University of Tasmania, AustraliaMichael Robert Carroll, University of Camerino, Italy
Copyright © 2020 Mollard, Martel, Le Trong and Rogerie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Martel, Y2Fyb2xpbmUubWFydGVsQGNucnMtb3JsZWFucy5mcg==