 Alan J. Stephen
Alan J. Stephen Neil V. Rees1
Neil V. Rees1 Iryna Mikheenko
Iryna Mikheenko Lynne E. Macaskie
Lynne E. Macaskie- 1School of Chemical Engineering, University of Birmingham, Birmingham, United Kingdom
- 2School of Biosciences, University of Birmingham, Birmingham, United Kingdom
A hydrogen economy powered by fuel cells is emerging as an alternative to the current fossil-fuel based energy system where hydrogen, produced through renewable sources, is used to generate electricity via fuel cells. A commonly investigated fuel cell, the Polymer Electrolyte Fuel Cell (PEMFC), usually uses platinum and other platinum group metal nanomaterials to catalyze the rate limiting Oxygen Reduction Reaction (ORR) of this process. The high prices and durability limitations of these catalysts have prevented their mass commercialization. Biosynthesis of nanomaterials has emerged as a potentially attractive “eco-friendly” alternative to conventional chemical synthesis methods. Various attempts have been made to biosynthesize nanoparticles for use in fuel cells. However, the processing methods used during and post synthesis increase their costs and limit their overall efficacies. We report bimetallic Pt/Pd nanoparticles (NPs) biosynthesized by E. coli [E. coli-Pt/Pd (10 wt%:10 wt%)] that shows promise for direct use as a PEMFC catalyst. This catalyst outperformed single metal versions of the same, i.e., E. coli-Pt (20 wt%) and E. coli-Pd (20 wt%) when tested as an electrocatalyst ex-situ. Direct use of E. coli-synthesized nanoparticles in PEMFCs is limited by the inherent resistances of the bacteria and the internal localization of nanoparticles. Transmission electron microscopy images and impedances (resistivity) tests showed that by initially synthesizing Pd nanoparticles on the E. coli cells, followed by Pt, gave a cell surface-localized metallic shell that improved conductivity of the catalyst. Catalyst Pt synthesis is likely mediated by initially-formed Pd-NPs reducing Pt (IV) under H2 resulting in alloying; this was evidenced by XRD data that showed XRD peaks for E. coli-Pt/Pd (10%:10%) which lie in between XRD peaks for Pt-only and Pd-only nanoparticles on the same planes. Protrusions of agglomerated nanoparticles were seen on the cell surface to form sites for catalytic activity. The catalyst, used in the ORR without optimization, performed significantly worse (~100 times) than a commercial catalyst (extensively developed for purpose) but which contained 5 times as much Pt. This serves as a starting point for a more engineered approach to bio-synthesizing nanoparticles for PEMFC catalysts.
Introduction
With the effects of climate change being increasingly apparent, research focus has shifted to non-carbon energy systems. An emerging concept is a hydrogen economy where renewable energy produced is stored as H2, an energy vector (Dutta, 2014). This can be made sustainably via electrolysis of water, driven by renewable technologies like solar or wind (Dincer and Acar, 2015), or via algal or bacterial photosynthesis (Stephen et al., 2017). H2 produced can be combusted, used in nuclear fusion (as deuterium) or, with available technology, used to generate electricity via fuel cells (Zhang et al., 2013a). Comparable to batteries, fuel cells are electrochemical cells that convert chemical energy into electricity but are distinct from batteries by requiring a continuous supply of fuel to sustain the chemical reaction (Zhang et al., 2013a). Figure 1 shows a general scheme for the polymer electrolyte fuel cell (PEMFC). Here, H2 is supplied to the anode where it dissociates into protons and electrons. The electrons travel through the circuit to the cathode, while the protons travel through the polymer electrolyte (often Nafion®) and meet the electrons to reduce oxygen at the cathode; this oxygen reduction reaction (ORR) at the cathode is the rate-determining step of the entire electrochemical reaction (Zhang et al., 2013b).
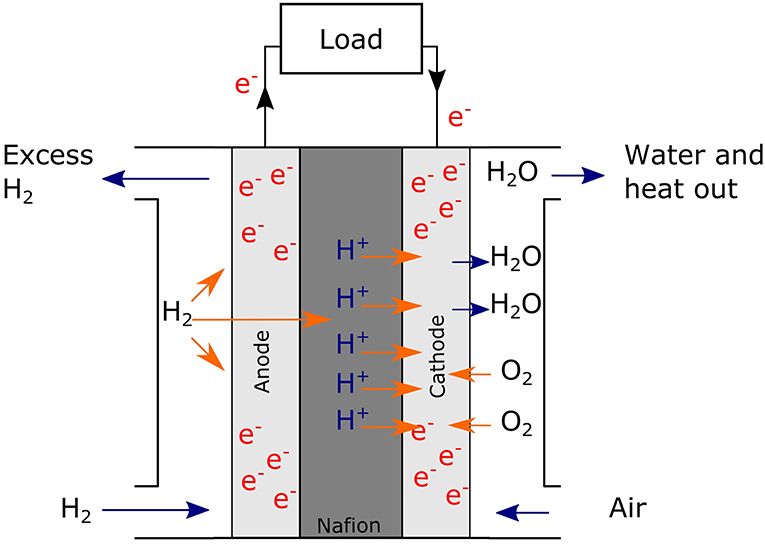
Figure 1. A general schematic of a PEMFC. H2 (fuel) flows in to the anodic side where H2 dissociates into protons and electrons. The electrons flow through the circuit while the protons are transferred via the electrolyte (Nafion®) to the cathodic side. Here they meet O2 at the cathode and combine to form H2O.
The Sabatier principle, which describes the ideal interaction between a substrate and a catalyst, was used to identify the most efficient ORR catalyst (Nørskov et al., 2004). From this it was identified that Pt, followed by Pd, were the most efficient of all bulk metals (Holton and Stevenson, 2013). The high prices of platinum group metals (PGM), combined with a major competing global demand for use in automotive catalysts (Zhang et al., 2017), has driven research into decreasing the amount of Pt (loading) used through Pt-alloyed nanomaterials (Zhu et al., 2015). Such metal “thrifting” is used in automotive catalysts (Cooper and Beecham, 2013); the PGM are often substitutable but the degree of success may be application-specific. Many groups have researched non-PGM ORR catalysts with reasonable success, although none have become economically viable (Banham and Ye, 2017). Hence, decreasing Pt loading and/or developing cheaper Pt catalytic nanoparticle (NP) synthesis remains a priority. The average life of an automotive catalyst is about 100,000 miles, whereas durability is a major limitation of fuel cells. Durability targets, set by the US Department of Energy and the Japanese New Energy and Industrial Development Organization are 5,000 h of operation for automotive application and 40,000 h for stationary FCs over 10 years (Rice et al., 2015). By way of illustration, in the 2000s the US Department of Defense installed 5 kW Plug Power Combined Heat and Power (CHP) systems (PEM fuel cells) in ~40 military bases at a cost of more than $100,000 per system. With a lifespan of only 500 h in operation these systems had to be overhauled once a year which was not cost-effective (Staffell et al., 2015). The need for cheaper, effective catalysts still limits widespread FC adoption.
The fluctuating market price of individual PGM is also a contributory factor. Pd is usually cheaper than Pt (typically by >3-fold) but periodically parity is achieved, e.g., in 2000 and again in 2018 1; (currently ~$1,000/oz). The price of Pt was stable between ~1980 and 2000 at ~$500/oz, but reached $1,500/oz periodically from 2008 to 2014. Thrifting within the PGM would offer the same flexibility as for automotive catalysts, “buying time” for alternative FC catalysts to be developed, in the context of the urgency for implementation of zero carbon energy.
Recently bio-synthesis of nanoparticles (NPs) has emerged as an attractive and “eco-friendly” alternative to conventional chemical synthesis (Schröfel et al., 2014) including the use of bio-NPs as fuel cell catalysts (Macaskie et al., 2017). This approach entails the use of biological materials to promote NP synthesis from aqueous solutions of metal salts. Plants, fungi, yeasts, bacteria, and biomolecules obtained from these can be used to synthesize NPs (Schröfel et al., 2014). While the majority of work in this field has focused on gold and silver NPs with size and shape control, biosynthesis of Pt and Pd–NPs (and also bimetallics) has been achieved using a variety of bacteria at the expense of various electron donors for bio-reduction of soluble metal ions into metallic NPs (Singh et al., 2015; Macaskie et al., 2017; Murray et al., 2018). Furthermore some of these have been used with reasonable success as anodic catalysts in fuel cells (Yong et al., 2007, 2010; Xiong et al., 2015; Liu et al., 2016; Macaskie et al., 2017) while electrochemical activity of “Bio-Pt” in the cathodic reaction was shown as comparable to a commercial Pt on carbon FC cell catalyst (TKK: Williams, 2015). Bio-manufacturing of catalytic NPs has additional clear advantages such as delivering a cheap, eco-friendly synthesis method via bio-recovery of metal from waste streams (Yong et al., 2010; Murray et al., 2017) (bringing significant savings in the CO2 cost of extraction from primary ores) alongside unexpected benefits such as an increased durability relative to commercial catalysts (BioNP had the smallest decrease in stability (9%) after accelerated stress tests relative to various commercial catalysts (Pt/C at 12% decrease or higher) (Xiong et al., 2015; Liu et al., 2016). As such, this approach warrants further research.
In engineering biosynthesized nanoparticles (Bio-NPs) as FC catalysts, a key factor to consider is the microenvironment in which the NP sits. As shown in Figure 2, the ORR occurs at sites where the electrolyte, the gaseous oxidant and the catalyst supported on a conductive material interact, known as the triple phase boundary (TPB). Thus, it is essential to recreate the TPB using Bio-NPs and this can prove challenging. Bacteria are inherently non-conductive and the nanoparticles synthesized are often localized both on the cell surface and also intracellularly (Omajali et al., 2015) and thus are in poor contact with both electrolyte and conductive support. Various strategies have been previously used to alleviate these problems (Yong et al., 2010; Attard et al., 2012; Xiong et al., 2015; Liu et al., 2016), however none of the resulting materials are directly applicable as potential commercial catalysts.
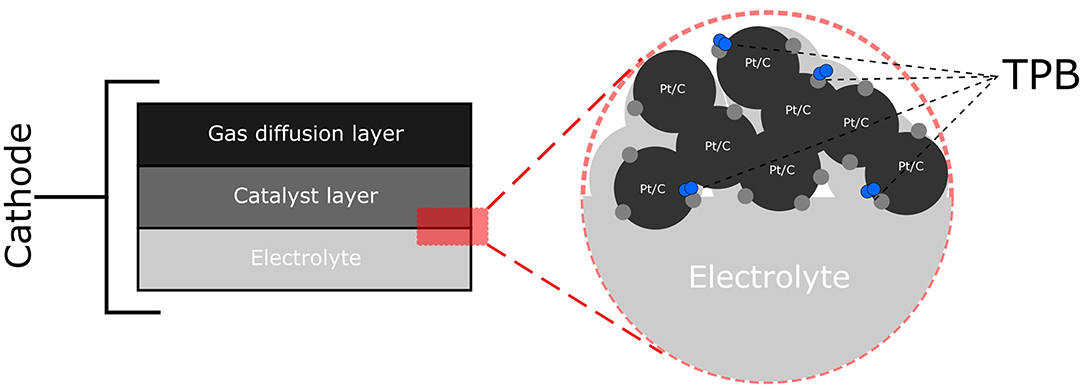
Figure 2. Schematic diagram of the cathode of a PEMFC showing the gas diffusion layer through which oxidant flows and the catalyst layer in contact with the electrolyte where the oxygen reduction reaction (ORR) occurs. This is shown in further detail where the electrolyte is contact with the catalyst (Pt/C). The ORR occurs at the triple phase boundary (TPB) where the electrolyte, the catalyst (Pt/C) and O2 meet.
To utilize Bio-NPs, the various described methods adopted destructive high temperature, energy- consuming methods to carbonize the material (Yong et al., 2007, 2010; Xiong et al., 2015; Liu et al., 2016), or aggressive chemical treatment (Attard et al., 2012; Williams, 2015) to clean and access the nanoparticles. While carbonizing the bacterial support makes it conductive, the temperatures used are likely to result in coarsening and agglomeration of nanoparticles which can further result in the loss of electrochemically active surface areas (ECSAs) and lower performance resulting from fewer sites available for catalysis (Simonsen et al., 2012). Furthermore, carbonized bacterial supports would be subject to similar carbon corrosion effects faced by commercial catalysts (Yousfi-Steiner et al., 2009). Chemical treatment with NaOH released NPs with high catalytic activity but the method is not applicable to commercial synthesis, requiring several weeks of digestion to obtain a suitable electrochemically-active surface (Attard et al., 2012; Williams, 2015). It is timely to develop further the direct use of bacteria as a NP-support for a possible high electrochemical activity coupled with enhanced durability. In addition, it is important to limit costs in synthesis to remain competitive as a catalyst; the incorporation of additional components such as reduced graphene oxide (rGO) (Liu et al., 2016) to improve conductivity and prevent NP agglomeration, may undermine the advantages delivered by this method; the synergistic co-use of E. coli and rGO was previously shown to make a potentially useful material but this was not tested operationally (Priestley et al., 2015) against chemical comparators. Finally, the bacterial strain used affects the nanoparticle localization within the cell and hence bacterial strain choice is an important consideration. While Yong et al. (2007) utilized ubiquitous E. coli (the “laboratory workhorse”) for NP biosynthesis, the research was done using non-industrial strains, the “second life” use of which would reduce cost and make the process more readily scalable. E. coli has data available on the localization of both Pt and Pd nanoparticles (Deplanche et al., 2010; Williams, 2015) and the wealth of genetic information available, as well as the widespread use of this organism for synthetic biology, could aid a more engineered approach to “tailored” Bio NP synthesis using the orthogonality afforded by synthetic biology, which cannot be achieved in a purely chemical system.
An alternative strategy toward sustainable development could use waste E. coli bacteria left over from other primary industrial processes, valorizing waste into high value material while mitigating waste disposal costs (Macaskie et al., 2017). Such “second life” use of E. coli bacteria to make Bio-NP catalyst was shown in chemical catalysis (Zhu et al., 2016) while Orozco et al. (2010) showed use of E. coli left over from a biohydrogen process to make a fuel cell catalyst to convert the biogenic H2 (which is free of the catalyst poisons that require extensive purification of petrochemically-derived H2) into power. Indeed, E. coli is widely used industrially (e.g., for pharmaceuticals production) and hence is potentially available as a low cost resource (Baeshen et al., 2015). However, regardless of bacteria, all approaches would require bound NPs to maintain contact with both a conductive material and the electrolyte to form a TPB. Courtney et al. (2016) showed that E. coli-Pd at high loading (20% metal) is conductive and this could be attributed to a Pd shell-layer at the cell surface; such an approach could possibly alleviate the conductance problem. The Bio-Pd catalyst, however, showed low performance which is expected due to the loss of ECSA resulting from the inaccessible intracellular Pd, which formed a significant proportion of the cellular Pd-NPs.
The objective of the current study is to show that this power loss can be mitigated by the combined synthesis of Pt and Pd nanoparticles on E. coli. As well as giving the flexibility to thrift metal usage according to PGM price and availability, use of a mixed metal catalyst is unavoidable when made from a waste. The metal composition of the bio-NPs reflects the composition of the original waste solution (Mabbett et al., 2006) and the metallic NPs are “diluted out” by additional metals that may not contribute to catalysis but may aid in conductivity. Such a PEMFC anode bio-NP catalyst made from an example industrial waste in a proof of concept study (here sintered to carbonize the biomass) generated ~ half as much power in a PEMFC as compared to pure Bio-Pd(0) (Yong et al., 2010) but no attempt was made to optimize this. Also, as described above, since the anodic oxidation reaction is not rate-limiting, a more realistic assessment would examine the ORR, which is the electrochemical reaction addressed in the current work.
Materials and Methods
Bacterial Growth and Biosynthesis of Nanoparticles
E. coli MC4100 was provided by Dr. D. Linke (University of Oslo). Cells were maintained aerobically at 30°C on nutrient agar plates. Cultures were first grown aerobically in nutrient broth no. 2 (NB 2, Oxoid) at 37°C to the mid-exponential phase and a 10% v/v inoculum was used to inoculate 200 mL of anaerobic growth medium [NB 2 with 0.5% (vol/vol) glycerol and 0.4% fumarate (wt/vol) under oxygen free N2 (OFN)] at 37°C and left to grow overnight. At mid-exponential phase, this was used to inoculate 2,000 mL of the same anaerobic medium and cells were harvested in mid exponential phase and concentrated via centrifugation (13,720 g, 15 min). These were then washed three times and re-suspended in 20 mM MOPS-NaOH buffer (buffer pH 7) to 25–30 mg/mL dry weight of cells and stored under OFN at 4°C until use, usually the next day.
For nanoparticle synthesis, 2 mM salt solutions of K2PtCl6 and Na2PdCl4 in 0.01 M HNO3 were used as precursors. For single metal NP synthesis: To a specified volume of metal salt solution, an appropriate amount of E. coli suspension was added for a final dry weight ratio of metal:cells of 1:9 (for 10% metal loading, or as otherwise stated). The metal/cell mixture was left to stand at (30°C, 30 min) for metal biosorption and then sparged with H2 (10 min) to promote metal ion reduction into nanoparticles. Metal uptake from solution was monitored using a tin chloride assay as described in Deplanche et al. (2010). Once nanoparticle synthesis had reached completion, the “as made” E. coli-NP preparation was centrifuged (13,720 g, 15 min), washed three times and re-suspended in ultrapure water.
For bimetallic nanoparticle synthesis, E. coli-Pd was first made as above (10 wt% Pd) and to this an appropriate volume of Pt solution was added for a final Pd:Pt:cells (w/w/w) ratio of 1:1:8 (for a 10%Pt:10%Pd loading) and sparged with H2 for a further 20 min. As with Pd, Pt uptake was monitored using a tin chloride assay. Once nanoparticle synthesis was complete, the “as made” E. coli-Pt/Pd was similarly washed and re-suspended in ultrapure water.
Characterization of Samples
Samples were harvested and prepared for TEM visualization (Supplementary Information, S1). Sections (100–150 nm) were placed onto a copper grid and routinely viewed with JEOL 1200CX2 TEM with an accelerating voltage of 80 kV. For better visualization of small bio-Pd NPs the samples were examined using scanning transmission electron microscopy using a FEI image Cs-corrector configuration TitanTM G2 60–300 STEM microscope, accelerating voltage of 300 kV.
Catalyst was dried on a filter paper overnight (105°C), ground to a fine powder using an agate mortar and X-ray powder diffraction patterns were obtained using a Bruker D8 Advance Diffractometer using a Cu Kα x-ray source. Scans were taken between 2θ values 20 and 80°. XRD analysis used the principal (111) peak to identify the mean particle size using the Scherrer equation where FWHM is the full width at half-maximum of the principal diffraction peak, k is the shape constant, D is the crystallite size and θ is the Bragg angle. All peaks were used to identify elemental mol-fractions of Pd and Pt in the materials using Vergard's equation: dPtPd = XdPt+ (1−X)dPd where d is the lattice parameters of the corresponding nanoparticle and X is the mol fraction of Pd from each peak. The final mol fraction was taken as an average of mol fractions from all three peaks.
Inductively coupled plasma mass spectrometry (ICP-MS) was used to confirm the actual metal loading on the cells (metal weight/total dry weight of catalyst) (Supplementary Information, S2).
Electrochemical Testing
All catalysts were electrochemically tested ex-situ in a three electrode half-cell against an Ag/AgCl reference electrode in 0.1 M HClO4 electrolyte (Supplementary Information, S3). The electrodes were connected to an Autolab (PGStat302N, Metrohm-Autolab, Utrecht, The Netherlands) potentiostat/galvanostat through a Pine modulate speed rotator (AFMSRCE Pine research instrumentation, 5 mm).
For cyclic voltammetry (CV) tests, the electrolyte was first purged with N2 (20 min). Prior to electrochemical analysis, the electrode was cleaned by cycling the applied potential between −0.235 and 0.9V vs. Ag/AgCl at a scan rate of 0.1 V/s for 50 scans. CVs were then taken at scan rates of 0.1V/s. Electrode chemical surface areas (ECSAs) were calculated from the CV data as described by Garsany et al. (2010). While ECSA for palladium nanoparticles are usually calculated from CO stripping experiments, this method was shown to be unusable here as the biomass adsorption of CO results in an overestimated result (A. J. Stephen, unpublished work). For ORR kinetics tests the electrolyte was saturated with O2 and linear sweep voltammetries (LSV) were taken in the same potential window at rotation speeds of 0, 400, 800, 1,200, and 1,600 RPM and scan rate of 0.025/s. From this mass activities were calculated as described by Garsany et al. (2010). Impedances of the catalysts were measured at a voltage of 0.6 V at a frequency range of 105-10−1 Hz after the electrolyte was saturated with O2. From this Nyquist plots were made to show catalyst impedances where the size of the semicircle obtained is related to the impedance (resistance) of the catalyst (Zhang et al., 2013c).
Results and Discussion
Formation of Metallic Nanoparticles on E. coli
Initially Bio-Pd was made on E. coli at 5 and 20 wt% Pd. Non-metallized cells appeared gray with no electron opaque deposits (Figure 3A). Cells palladized to 5 wt% Pd (~equivalent to 10 wt% Pt on an atom-loading basis; respective atomic weights of Pd and Pt are 106.4 and 195.1 Da) showed numerous small NPs located at the cell surface and intracellularly, with occasional larger deposits (Figure 3B). The large distance between NPs suggests that there would be little conductivity between them; Orozco et al. (2010) confirmed that, even after sintering, 5 wt% Pd-E coli gave poor activity in a PEMFC. On palladizing the cells to 20 wt% larger NPs were visible, with evidence of contact between them and with electrocatalytic activity reported previously (Figure 3C; Courtney et al., 2016).
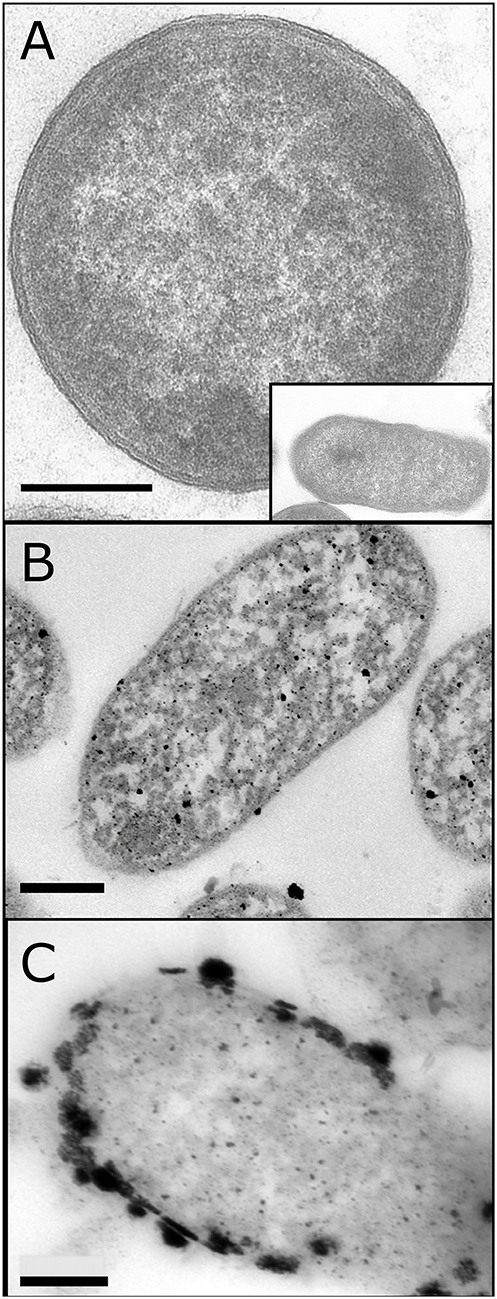
Figure 3. TEM images of E. coli cells without Pd (A) (main figure shows vertical cross section of cell; inset and following images show horizontal cross section of the cell) and loaded to 5 wt% (B) and 20% (C) of the cell dry weight. (B) was provided by Mr. J. Gomez-Bolivar and (A,C) are adapted from Courtney et al. (2016). Bars are as shown (0.2 μm).
E. coli-Pt/Pd (10%:10%) was made by synthesizing Pd nanoparticles on E. coli under H2 followed by Pt reduction as described above. Figure 4A shows a TEM image of E. coli-Pt 10%. The latter made highly agglomerated nanoparticles (black arrow) at the surface of the bacteria with no apparent intracellular NPs. Figures 4B,C show the bimetallic E. coli-Pt/Pd (10%:10%) with Figure 4C showing a clear nanoparticle metal “shell” at the cell surface. Some intracellular NPs can be seen (circled) but these appear to be few in number compared to the surface-localized material.
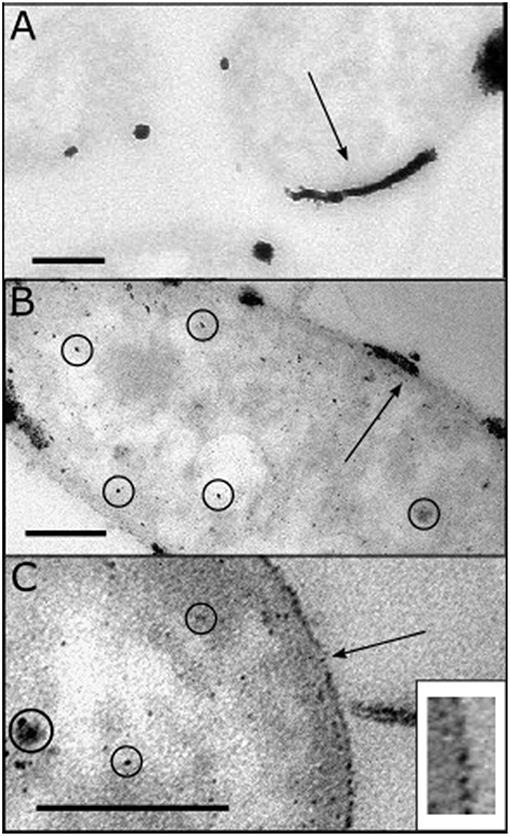
Figure 4. TEM images taken of E. coli-NP catalysts fixed and sectioned as detailed in Materials and Methods. (A) adapted from Williams (2015), shows E. coli-Pt (10%) where agglomerated periplasmic Pt NPs are identified with a black arrow (c.f. 5 wt% Bio-Pd in Figure 3B); note lack of intracellular NPs using equimolar Pt. (B,C) show E. coli-Pt/Pd (10%:10%) where the black arrow shows a degree of agglomeration at the periplasm, while intracellular nanoparticles can be seen (circled). The higher magnification image (C) shows a “nanoparticle shell” in the periplasm as indicated by the black arrow. Bars are 200 nm. Inset: expanded region of the cell surface of the cell shown in (C), showing discrete supported NPs.
Examination of Bio-Nanomaterial by X-Ray Powder Diffraction
The catalysts were dried to a powder, ground and analyzed under XRD (Figure 5) E. coli-Pt/Pd (10%:10%) and E. coli-Pd (20%) both had crystalline components showing FCC crystal peaks. The bimetallic catalyst showed (Figure 5A) peaks at 2θ values of 39.83 (111), 46.36(200), and 67.81(220). Furthermore, as shown in Figure 5A, these peaks have shifted to lower angles relative to the reference of E. coli-Pd (20%) which can be interpreted as evidence of alloying (Butera and Waldeck, 1997).
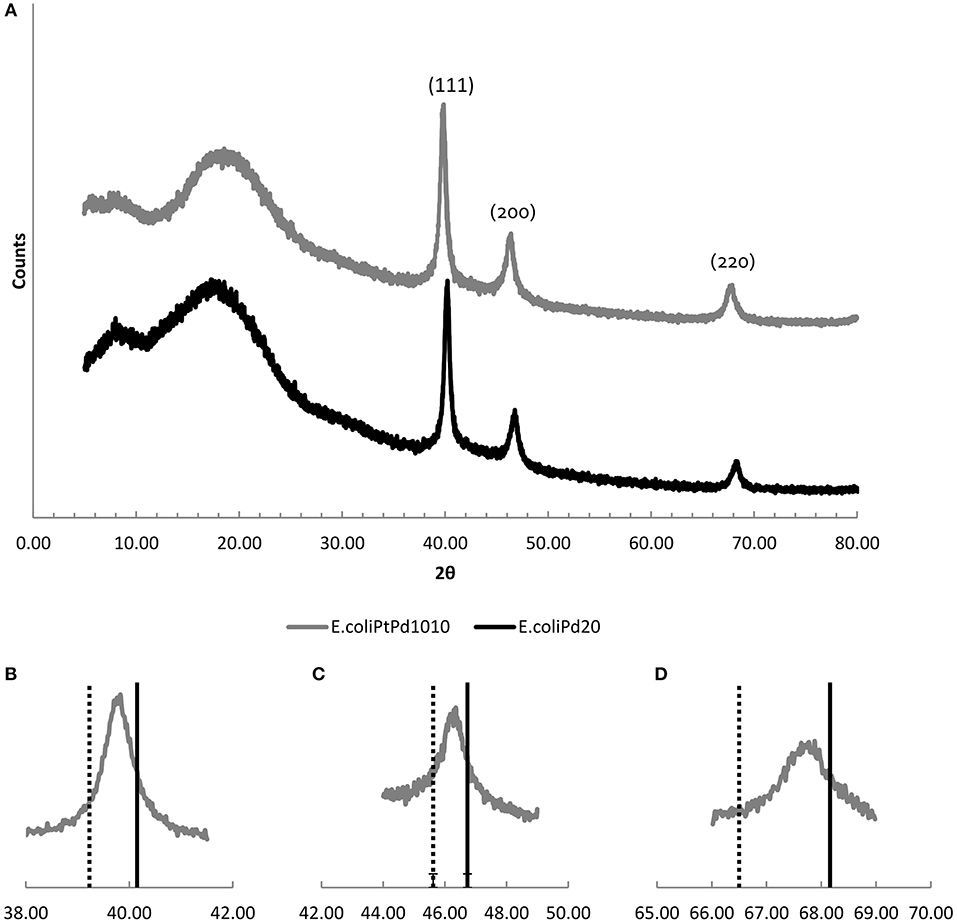
Figure 5. (A) XRD patters of E. coli-Pt/Pd (10%:10%; top, gray) compared to E. coli-Pd (20%; bottom, black) Slight peak shifts to the left can be observed with the bimetallic catalyst. (B–D) show the individual peaks for the (111), (200), and (220) facets, respectively. The broken line indicates the peak positions for Pt/C (from the literature) and the solid line shows the corresponding peak positions for E. coli-Pd (20%). The peaks for the bimetallic catalyst (atom ratio ~2:1 of Pd:Pt) fall between these for all three peaks.
Previous work using Pd (Courtney et al., 2016) showed that apparently large NPs of Bio-Pd comprised agglomerations of smaller NPs within them, presumably maintained as separate small entities by the supporting biomatrix. The Pt-agglomerations seen in Figure 4A were expected to have a similar structure however, E. coli-Pt (10%) XRD powder patterns, taken previously, showed no discernible peaks and were indistinct (Williams, 2015). By use of the metal mixture, a well-defined XRD pattern was obtained (Figure 5A). No peaks could be seen that corresponded to Pt(0) and alloying was assumed. This was checked by comparing E. coli-Pt/Pd (10%:10%) XRD peaks with Pt FCC nanoparticle XRD peaks (Jain et al., 2013) and E. coli-Pd (20%) XRD peaks. Figures 5B–D show the E. coli-Pt/Pd peaks for the 111, 200, and 220 planes, respectively (broken line indicates peak positions for Pt; solid line for E. coli-Pd (20%) peaks). The peaks lie between those for Pd and Pt, implying alloying of the metals, which was seen previously with a chemically-made Pt-Pd alloy, where TEM images showed octahedral nanoparticles with complete mixing of Pt and Pd and XRD peaks similar to those obtained here (Lee et al., 2011). The Scherrer equation was used to calculate the mean particle size of the alloyed NPs as 5.2 nm as described in Materials and Methods.
Using Vergard's equation as described in Materials and Methods the elemental compositions of Pd and Pt were calculated as ~40 and 60% which was further confirmed by ICP-MS (Supplementary Information, S2). The data indicate that the presence of a pre-deposit of Pd “protects” the Pt against formation of agglomerated Pt-deposits by intercepting and patterning incoming Pt onto the Pd-NPs laid down in the first step. It is assumed, from previous work on formation of Pd/Au NPs (Deplanche et al., 2012), that the Pd(0) galvanically reduces incoming Pt(IV) to Pt(0) that then forms a co-crystal with Pd(0).
The bioreduction of Pd(II) (Mikheenko et al., 2008; Deplanche et al., 2010) and Pt(IV) (Riddin et al., 2009) involves hydrogenases. In the latter case the initial Pt(IV) to Pt(II) reduction was via an oxygen-sensitive cytoplasmic hydrogenase and the subsequent Pt(II) to Pt(0) reduction involved an oxygen-tolerant periplasmic (i.e., exterior to the cell membrane) hydrogenase. Not only does this imply an uptake mechanism for Pt(IV) by the bacterial cells [in this case a mixed sulfate-reducing consortium (Riddin et al., 2009)] followed by efflux of Pt(II) but a localization of bio-reduction of Pt(II), presumably near the point of the Pt(II) efflux into the periplasm. In the case of Pd(II), loss of Pd(0) NP-patterning and of the small periplasmic Pd-NPs, with formation of larger Pd(0) deposits at the inner membrane, was noted in Desulfovibrio fructosovorans following removal of the periplasmic NiFe hydrogenase (Mikheenko et al., 2008). Govender et al. (2009) hypothesized that the incoming octahedral PtCl complex [Pt(IV)] is too large to fit into the active region of the primary hydrogenase, which would be functionally equivalent to deleting it, giving the same result as using the smaller Pd(II) [equivalent to Pt(II)]. The actual mechanisms by which PGM are reduced by more than one hydrogenase are outside the scope of this study but the additional complexity of reduction of Pt(IV), if taken up by the cells and effluxed as partially reduced Pt(II) (above) implies that Pt(IV) “interception” and patterning by extant Pd(0)-NPs followed by galvanic reduction and alloying, may give a more dispersed product than by using Pt(IV) and bioreduction alone.
Electrochemical Characterization and Performance of the Bio-Metallic Catalysts
The bimetallic catalyst (Pd/Pt; 10%/10%) was evaluated electrochemically against E. coli-Pt (20%) and E.coli-Pd (20%) after 50 cleaning scans at a voltage scan rate of 100 mV s−1 as detailed above. A CV was taken at this scan rate and used to evaluate the electrochemical surface areas (ECSAs) with activity related to mass of metal via the known electrode loading which was estimated using ICP-MS of solubilized metal recovered from the electrode (above).
As shown in Figures 6A,B, the CVs acquired for E. coli-Pt/Pd (10%:10%) had more clearly defined peaks for hydrogen adsorption and desorption than either E. coli-Pd (20%) or E. coli-Pd (20%) indicating better ECSAs for the bimetallic catalyst, as indicated by the dispersion of small NPs at the cell surface shown in Figure 4C (inset). The ECSA was calculated for each material as described in Materials and Methods. E. coli-Pt/Pd (10%:10%) had a higher ECSA with a value of 11.27 m2/gMetal as compared to 6.74 and 8.77 m2/gMetal for E. coli-Pt(20%) and E. coli-Pd(20%) respectively (Figure 7).
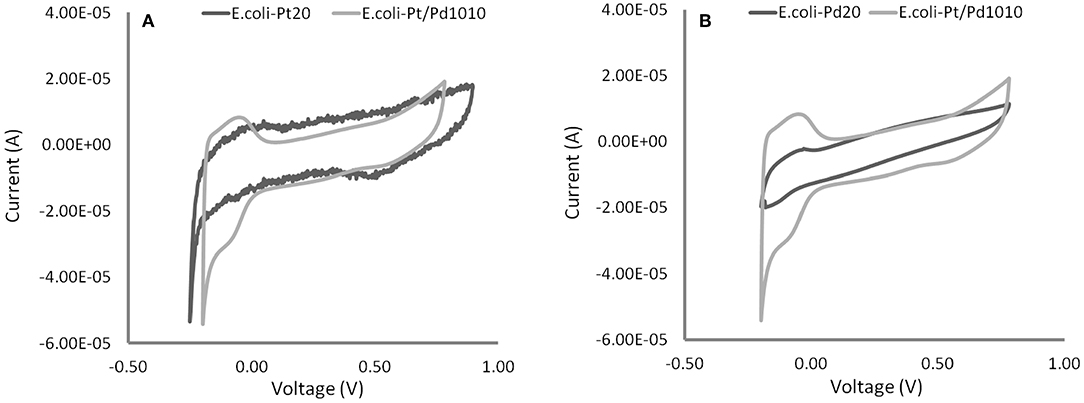
Figure 6. Electrochemical tests (cyclic voltammograms) of the bimetallic catalyst compared against 20 wt% Bio-Pd in (A) and 20 wt% Bio-Pt in (B) in 0.1 M HNO3 at 25°C and in N2-saturated solutions with a scan rate of 100 mV/s. Distinct peaks are apparent for E. coli-Pt/Pd (10%:10%) relative to both E. coli-Pd(20%) and E. coli-Pt (20%).
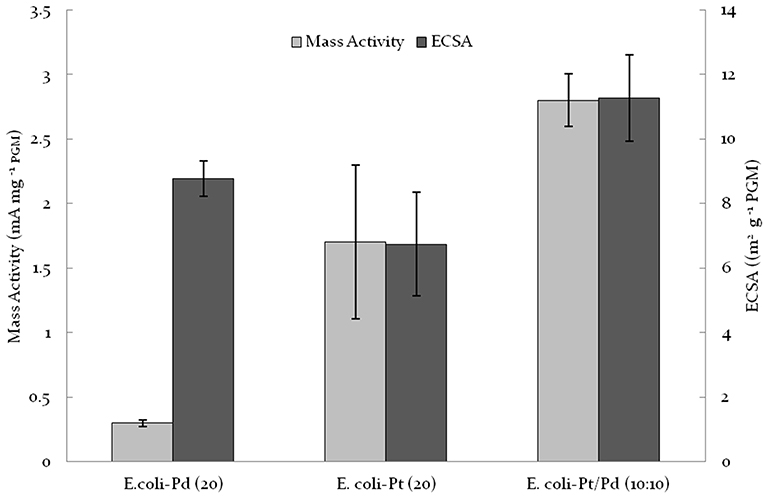
Figure 7. Comparison of E. coli-Pt/Pd (10%:10%) mass activities and ECSA against E. coli-Pd (20%) and E. coli-Pt (20%).
To evaluate the ORR kinetic performance of the catalysts, linear sweep voltammetries (LSVs) were acquired at varying rotating speeds under O2-saturated conditions. From this the mass activities of the catalysts were evaluated as detailed in Materials and Methods. The results are shown in Figure 7 where E. coli-Pt/Pd (10%:10%) shows better mass activity (2.8 mA/mgMetal) relative to both E. coli-Pd (20%: 0.3 mA/ mgMetal) and E. coli-Pt (20%: 1.7 mA/mgMetal) i.e., nearly 10 times higher activity for the alloy than by using bio-Pd and 1.6-fold higher than by using bio-Pt. It was concluded that the alloying of the two metals enhanced the electrochemical activity of the catalyst, attributed to a synergy of the higher catalytic activity of Pt coupled with the better dispersion afforded by the Pd “host” at a mol fraction of Pd:Pt of approx. 2:1.
In order to evaluate whether the metallic “shell” around the cell (Figure 4C) has had an effect on alleviating the inherent resistance of bacteria, impedance tests were done as described in Materials and Methods. Figure 8 shows a marked decrease in impedance (i.e., increase in conductivity) between E. coli-Pt (20%) and E. coli-Pd (10%) and E. coli-Pd (20%) thus giving credence to the role of the palladium shell in increasing conductivities. E. coli-Pt/Pd (10%:10%) further shows similar impedance to E. coli-Pd (10%) and increased impedance relative to E. coli-Pd (20%) which further validates the role of palladium in conduction. However, it is important to note that the impedances seen for all samples are in the kΩ range which is significantly high relative to the commercial catalyst TKK which comprises Pt nanoparticles on carbon at a metal loading of 45.9%, i.e., more than twice the metal as the biomaterial. Attempts to increase the conductivity of the latter by incorporating nanocarbon into the bio-preparations to mimic the role of TKK-carbon were unsuccessful (Stephen, unpublished work).
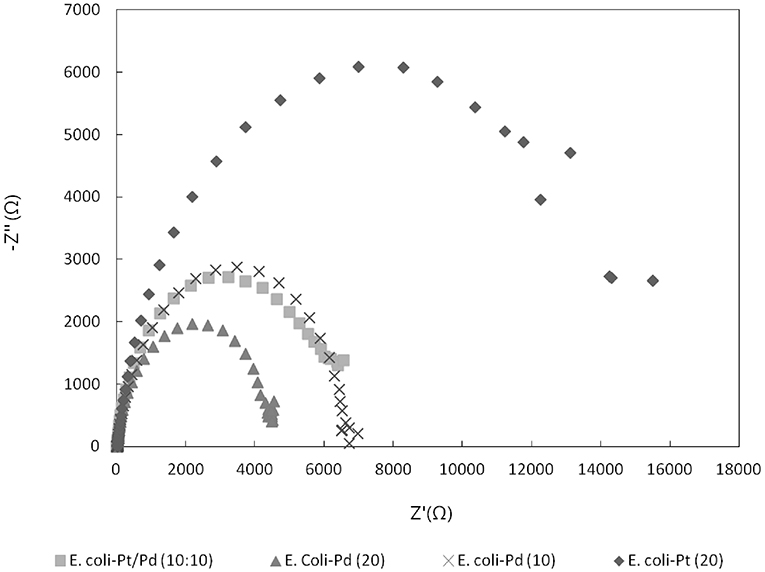
Figure 8. Nyquist plot comparing E.coli-Pt/Pd impedances against E. coli-Pd (20%), E. coli-Pd (10%), and E. coli-Pt (20%).
Previous groups have used highly destructive temperature-dependent methods to clean and access bio-nanoparticles. Yong et al. (2010) synthesized E-coli–Pd and Desulfovibrio desulfuricans-Pd via carbonizing the catalyst at 700°C under N2 following bioreduction of the metal. A similar strategy was utilized by Xiong et al. (2015) where Shewanella oneidensis–Pd was carbonized at 420°C with KOH. The catalyst obtained using both strategies showed low power outputs and while Xiong et al. (2015) had a more durable catalyst than 20%Pt/C; it had a poor mass activity relative to current commercial catalysts such as TKK (0.016 vs. 0.2 mA/μg). Following co-synthesis of a material by Priestley et al. (2015) which comprised Pd(0), reduced graphene oxide and contributory E. coli bacteria (but was not tested for catalytic activity). Liu et al. (2016) developed a similar concept where S. oneidensis–Pd/Au NPs were synthesized using graphene oxide (GO) sheets for the anodic reaction. Post NP synthesis, the catalyst was treated with GO sheets and then hydrothermally reacted; this simultaneously reduced GO to rGO, alloyed PdAu and carbonized the bacteria thus making the material conductive. While the addition of GO prevented extreme agglomeration and increased conductance, the high temperature required for carbonization would increase the overall CO2 and energy cost, negating the low-cost advantages potentially delivered by using a purely bio-manufactured catalyst. As such there is a need for a more engineered approach to biosynthesizing cheap NPs as FC catalysts and E. coli-Pt/Pd (10%:10%) serves as a starting point. Figure 9 compares the biogenic catalyst against commercial TKK catalyst; clearly the potential advantages of the biosynthetic approach are currently outweighed by a significant shortfall in performance. Williams (2015) produced bio-Pt NPs which performed comparably to TKK catalyst but this required removal of much of the biomaterial using several changes of concentrated NaOH over several weeks; this is clearly unsustainable but shows that biogenesis has the potential to make a competitive catalyst.
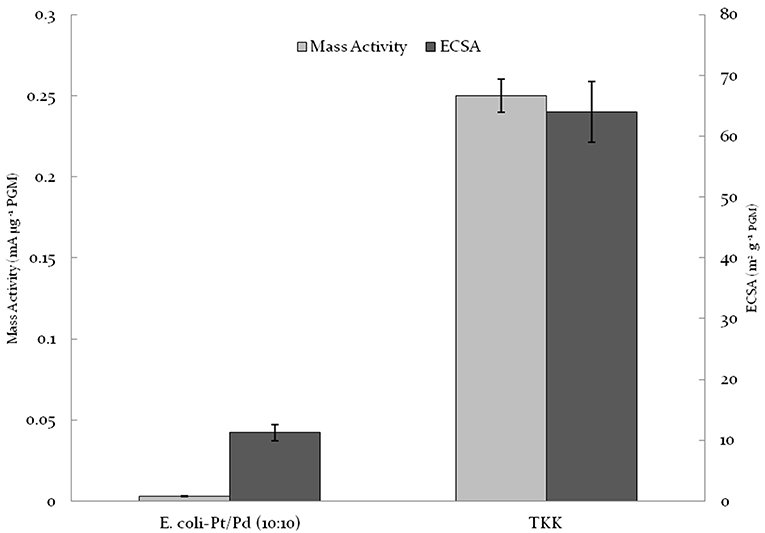
Figure 9. Comparison of E.coli-Pt/Pd (10%/10%) mass activities and ECSA against TKK. Note that the loading of Pt on TKK is nearly 50% whereas that on biomass is 20 wt% in total. The specific activity (mA/g metal) is shown but the conductivity between metallic NPs separated by carbon or biomass is not factored-in.
Conclusions and Future Perspectives
E. coli-Pt/Pd (10%:10%) produced an alloyed catalyst that had better mass activities and ECSAs relative to single metal versions of this catalyst (Figure 7). The increased performance is attributable to the localization of nanoparticles on/in the cell. As seen in Figure 4, alongside a nanoparticle “shell” around the cells, the periplasm also hosted agglomerations that protruded outside the cell. It could be suggested that these would be the TPB sites where the ORR would take place and the shell would act to conduct electrons away to the circuit. E.coli-Pt per se has these agglomerations where the ORR could occur without a conductive support, whereas E. coli-Pd supplies the conductive “shell” but makes less effective ORR catalyst. Hence, this combination of the two would result in the marked increase in mass activities and ECSAs. However, as shown in Figure 9, these values are low compared to commercial catalyst TKK (Pt-NPs on carbon at a metal loading of 45.9%). The relatively low performance of the biocatalyst can be attributed to three factors (1) the inaccessible intracellular nanoparticles, (2) agglomerated NPs and finally (3) the high impedance relative to TKK. As such the strategy to improve performance would address these issues.
The dual requirements for development are targeted and controlled bio-NP synthesis and to decrease the inherent resistance of the bacterial scaffold. The presence of intracellular NPs indicates a metal trafficking mechanism [presumably assimilating Pd(II) in lieu of an essential metal such as Ni(II) (discussed by Omajali et al., 2015)]. As continued cell viability is not required, once the appropriate metal uptake genes are identified, the pathway could be disabled following bacterial growth. Identification of key genes is underway with some early success (Torgeman, 2017), while, on the other hand, over expression of metal efflux genes [known to regulate the intracellular content of essential metals like Ni(II)] would also assist in increasing the proportion of the cell surface NPs and hence increasing the ECSA. It is known that hydrogenase enzymes are involved in periplasmic Pd-NP synthesis with an active NiFe hydrogenase being required for distribution of Pd-NPs throughout the periplasm (Mikheenko et al., 2008) (see earlier). Furthermore, for production of the TBP the NPs must preferably breach the barrier of the outer membrane. Bio-Pd NPs made in a liposome structure containing entrapped hydrogenase had little catalytic activity (Sammons et al., 2013) presumably due to occlusion of catalyst surface by the lipid materials which would also restrict conductivity. The use of appropriate solvents (e.g., acetone) may be appropriate for membrane dissolution or, as an alternative, ethanol which is produced industrially via fermentation processes. Dimitriadis et al. (2007) showed that waste yeast cells from the ethanolic fermentation could function as a fuel cell (FC) anode when palladized. However, for membrane-dissolution, the ethanol would require concentration by distillation. Simply disrupting the cells reduced the catalytic activity of Bio-Pd (Mikheenko, unpublished) An alternative approach could explore the use of Gram positive bacteria as these produce Pd-NPs similarly to Gram negative types (Omajali et al., 2015) but lack the outer membrane. However studies using Gram positive Micrococcus luteus (Courtney, 2017) and Bacillus benzeovorans (Omajali et al., 2015) showed internalization of Pd-NPs, in the latter case to a greater extent than the Gram negative sulfate-reducing D. desulfuricans (Omajali et al., 2015) the bio-Pd of which was shown to outperform bio-Pd of E. coli in the PEMFC anodic reaction (Yong et al., 2010).
D. desulfuricans, which produces H2S (normally considered as a catalyst poison), is not available in large quantities commercially but a continuous process has been developed for remediation of mine-wastes using biogenic H2S as a metal precipitant. The waste sulfidogenic biomass from this process, when palladized, was shown to be active in chemical catalysis as well as in a green chemistry reaction as Pd/Ru bimetallic (Mikheenko et al., 2019). It has been reported that monodisperse palladium sulfide is an efficient electrocatalyst for the ORR (Du et al., 2018), attributed to optimized oxygen absorption onto a Pd4S surface, which is counter-intuitive as sulfide is generally regarded as a catalyst poison. Despite washing the biomass, the presence of palladium sulfide was confirmed in both D. desulfuricans and the waste sulfidogenic culture using X-ray photoelectron spectroscopy (Mikheenko et al., 2019). Hence, the waste biomass is under current evaluation as a potential fuel cell ORR catalyst using the methods described in this study.
However, for this approach to work effectively there would need to be a simultaneous decrease in the inherent bacterial resistance. Various strategies to achieve this have already been employed for microbial fuel cells (MFC's) (Saratale et al., 2017). These are fuel cells that employ live bacteria on the anodic side and the inherent resistance of live cells is a major limiting factor in their commercialization. Hence, various strategies including the use of cheap conductive polymers have been developed. Song et al. (2017) polymerized polypyrrole on the surface of various bacterial strains to decrease their inherent resistances in a one pot synthesis; an effective strategy would be to employ bacteria to localize nanoparticle synthesis on their surface and increase conductivity by using inexpensive conductive materials.
With respect to sustainable economic synthesis, this study shows that a bimetallic of Pd/Pt is advantageous over single metal versions of Bio-NP catalyst. This opens the way to bio-fabrication of mixed metallic NPs from wastes as these usually contain more than one metal and proof of principle was indicated by the use of a metal mixture made from an industrial processing waste (Yong et al., 2010). A major cost is biomass growth. However waste yeast cells have been used to make FC catalyst (Dimitriadis et al., 2007). Other studies have shown good activity of palladized waste E. coli from a biohydrogen process (Zhu et al., 2016) in both chemical catalysis and as a FC catalyst (Orozco et al., 2010) (notably, the bio-H2 is poison free and is sufficiently pure to be used directly in a PEMFC). The potential availability of large quantities of E. coli from industrial processes (e.g., pharmaceuticals production) makes this a potentially attractive “option” also forestalling the environmental impact and cost of waste disposal; indeed pre-production strains of E. coli that exported a primary protein of interest had a catalytic activity comparable to E. coli MC4100 once palladized for use in “second life” (Zajac et al., unpublished).
Finally it should be remembered that in a PEMFC the ORR is rate-limiting. Hence, the use of a commercial catalyst in the anodic reaction risks some catalyst redundancy and a bio-derived substitute may find immediate application here, pending biotechnological development of a competitive ORR catalyst.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
AS carried out all the experimental work and authored the paper with author contributions from NR and LM. IM provided training in making Bio-NPs and provided specialist assistance in their characterization.
Funding
AS acknowledges with thanks a studentship from the EPSRC Doctoral Training Centre Fuel Cells and their Fuels. This work was supported in part by NERC (grant NE/L014076/1) to LM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge, with thanks, useful discussions with Dr. S. Sharma and Mr. J. Gomez-Bolivar for the use of Figures 3A,B. The authors also acknowledge the Centre for Electron Microscopy at University of Birmingham for the help with TEM imaging and Ms. Jie Chen for her invaluable assistance with XRD.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2019.00066/full#supplementary-material
Footnotes
1. ^Anon. https://www.kitco.com/commentaries/2018-01-17/The-48-Year-Record-of-Pt-Pd-Ratios.html. accessed 2/12/12018.
References
Attard, G., Casadesús, M., Macaskie, L. E., and Deplanche, K. (2012). Biosynthesis of platinum nanoparticles by Escherichia coli MC4100: can such nanoparticles exhibit intrinsic surface enantioselectivity? Langmuir 28, 5267–5274. doi: 10.1021/la204495z
Baeshen, M. N., Al-Hejin, A. M., Bora, R. S., Ahmed, M. M. M., Ramadan, H. A. I., Saini, K. S., et al. (2015). Production of biopharmaceuticals in E. coli: current scenario and future perspectives. J. Microbiol. Biotechnol. 25, 953–962. doi: 10.4014/jmb.1412.12079
Banham, D., and Ye, S. (2017). Current status and future development of catalyst materials and catalyst layers for proton exchange membrane fuel cells: an industrial perspective. ACS Energy Lett. 2, 629–638. doi: 10.1021/acsenergylett.6b00644
Butera, R. A., and Waldeck, D. H. (1997). X-ray diffraction investigation of alloys. J. Chem. Educ. 74:115. doi: 10.1021/ed074p115
Cooper, J., and Beecham, J. (2013). A study of platinum group metals in three-way autocatalysts Plat. Met. Rev. 57, 281–288. doi: 10.1595/147106713X671457
Courtney, J. (2017). Alternative chemical methods for the catalytic processes within hydrogen fuelled proton exchange membrane fuel cells (PhD thesis). University of Birmingham, Birmingham, United Kingdom.
Courtney, J., Deplanche, K., Rees, N. V., and Macaskie, L. E. (2016). Biomanufacture of nano-Pd(0) by Escherichia coli and electrochemical activity of Bio-Pd(0) made at the expense of H2 and formate as electron donors. Biotechnol. Lett. 38, 1903–1910. doi: 10.1007/s10529-016-2183-3
Deplanche, K., Caldelari, I., Mikheenko, I. P., Sargent, F., and Macaskie, L. E. (2010). Involvement of hydrogenases in the formation of highly catalytic Pd(0) nanoparticles by bioreduction of Pd(II) using Escherichia coli mutant strains. Microbiology 156, 2630–2640. doi: 10.1099/mic.0.036681-0
Deplanche, K., Merroun, M. L., Casadesus, M., Tran, D. T., Mikheenko, I. P., Bennett, J. A., et al. (2012). Microbial synthesis of core/shell gold/palladium nanoparticles for applications in green chemistry. J. R. Soc. Interface 9, 1705–1712. doi: 10.1098/rsif.2012.0003
Dimitriadis, S., Nomikou, N., and McHale, A. P. (2007). Pt-based electro-catalytic materials derived from biosorption processes and their exploitation in fuel cell technology. Biotechnol. Lett. 29, 545–551. doi: 10.1007/s10529-006-9289-y
Dincer, I., and Acar, C. (2015). Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 40, 11094–11111. doi: 10.1016/j.ijhydene.2014.12.035
Du, C., Li, P., Yang, F., Cheng, G., Chen, S., and Luo, W. (2018). Monodisperse palladium sulfide as efficient electrocatalyst for oxygen reduction reaction. ACS Appl. Mater. Interfaces 10, 753–761. doi: 10.1021/acsami.7b16359
Dutta, S. (2014). A review on production, storage of hydrogen and its utilization as an energy resource. J. Ind. Eng. Chem. 20, 1148–1156. doi: 10.1016/j.jiec.2013.07.037
Garsany, Y., Baturina, O. A., Swider-Lyons, K. E., and Kocha, S. S. (2010). Experimental methods for quantifying the activity of platinum electrocatalysts for the oxygen reduction reaction. Anal. Chem. 82, 6321–6328. doi: 10.1021/ac100306c
Govender, Y., Riddin, T., Gericke, M., and Whiteley, C. G. (2009). Bioreduction of platinum salts into nanoparticles: a mechanistic perspective. Biotechnol. Lett. 31, 95–100. doi: 10.1007/s10529-008-9825-z
Holton, O. T., and Stevenson, J. W. (2013). The role of platinum in proton exchange membrane fuel cells. Plat. Met. Rev. 57, 259–271. doi: 10.1595/147106713X671222
Jain, A., Ong, S. P., Hautier, G., Chen, W., Richards, W. D., Dacek, S., et al. (2013). Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1:11002. doi: 10.1063/1.4812323
Lee, Y.-W., Ko, A.-R., Han, S.-B., Kim, H.-S., and Park, K.-W. (2011). Synthesis of octahedral Pt-Pd alloy nanoparticles for improved catalytic activity and stability in methanol electrooxidation. Phys. Chem. Chem. Phys. 13, 5569–5572. doi: 10.1039/c0cp02167a
Liu, J., Zheng, Y., Hong, Z., Cai, K., Zhao, F., and Han, H. (2016). Microbial synthesis of highly dispersed PdAu alloy for enhanced electrocatalysis. Sci. Adv. 2:e1600858. doi: 10.1126/sciadv.1600858
Mabbett, A. N., Sanyahumbi, D., Yong, P., and Macaskie, L. E. (2006). Biorecovered precious metals from industrial wastes: single single-step conversion of a mixed metal liquid waste to a bioinorganic catalyst with environmental application. Environ. Sci. Technol. 40, 1015–1021. doi: 10.1021/es0509836
Macaskie, L. E., Mikheenko, I. P., Omajai, J. B., Stephen, A. J., and Wood, J. (2017). Metallic bionanocatalysts: potential applications as green catalysts and energy materials. Microb. Biotechnol. 10, 1171–1180. doi: 10.1111/1751-7915.12801
Mikheenko, I. P., Gomez-Bolivar, J., Merroun, M. L., Macaskie, L. E., Sharma, S., Walker, M., et al. (2019). Upconversion of cellulosic waste into a potential “drop in fuel” via novel catalyst generated using Desulfovibrio desulfuricans and a consortium of acidophilic sulfidogens. Front. Microbiol. 10:970. doi: 10.3389/fmicb.2019.00970
Mikheenko, I. P., Rousset, M., Dementin, S., and Macaskie, L. E. (2008). Bioaccumulation of palladium by Desulfovibrio fructosovorans wild-type and hydrogenase-deficient strains. Appl Environ Microbiol. 74, 6144–6146. doi: 10.1128/AEM.02538-07
Murray, A. J., Zhu, J., Wood, J., and Macaskie, L. E. (2017). A novel biorefinery: biorecovery of precious metals from spent automotivecatalyst leachates into new catalysts effective in metal reduction and in the hydrogenation of 2-pentyne. Mins. Eng. 113, 102–108. doi: 10.1016/j.mineng.2017.08.011
Murray, A. J., Zhu, J., Wood, J., and Macaskie, L. E. (2018). Biorefining of platinum group metals from model waste solutions into catalytically active bimetallic nanoparticles. Microb. Biotechnol. 11, 359–368. doi: 10.1111/1751-7915.13030
Nørskov, J. K., Rossmeisl, J., Logadottir, A., Lindqvist, L., Kitchin, J. R., Bligaard, T., et al. (2004). Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 106, 17886–17892. doi: 10.1021/jp047349j
Omajali, J. B., Mikheenko, I. P., Merroun, M. L., Wood, J., and Macaskie, L. E. (2015). Characterization of intracellular palladium nanoparticles synthesized by Desulfovibrio desulfuricans and Bacillus benzeovorans. J. Nanopart. Res. 17:264. doi: 10.1007/s11051-015-3067-5
Orozco, R. L., Redwood, M. D., Yong, P., Caldelari, I., Sargent, F., and Macaskie, L. E. (2010). Towards an integrated system for bio-energy: hydrogen production by Escherichia coli and use of palladium-coated waste cells for electricity generation in a fuel cell. Biotechnol. Lett. 32, 1837–1845. doi: 10.1007/s10529-010-0383-9
Priestley, R. E., Mansfield, A., Bye, J., Deplanche, K., Jorge, A. B., Brett, D., et al. (2015). Pd nanoparticles supported on reduced graphene-E. coli hybrid with enhanced crystallinity in bacterial biomass. RSC Adv. 5, 84093–84103. doi: 10.1039/C5RA12552A
Rice, C. A., Urchaga, P., Pistono, A. O., McFerrin, B. W., McComb, B. T., and Hu, J. (2015). Platinum dissolution in fuel cell electrodes: enhanced degradation from surface area assessment in automotive accelerated stress tests. J. Electrochem Soc. 162, F1175–F1180. doi: 10.1149/2.0371510jes
Riddin, T. L., Govender, M., Gericke, M., and Whiteley, C. G. (2009). Two different hydrogenase enzymes from sulphate-reducing bacteria are responsible for the bioreductive mechanism of platinum into nanoparticles. Enz. Microb. Technol. 45, 267–273. doi: 10.1016/j.enzmictec.2009.06.006
Sammons, R. L., Wang, A., Mikheenko, I. P., Handley-Sidhu, S., and Macaskie, L. E. (2013). Bacterially derived nanomaterials and enzyme-driven lipid-associated metallic particle catalyst formation. Adv. Planar Lipid Bilayers Liposomes. 18, 237–261. doi: 10.1016/B978-0-12-411515-6.00009-6
Saratale, G. D., Saratale, R. G., Shahid, M. K., Zhen, G., Kumar, G., Shin, H. S., et al. (2017). A Comprehensive overview on electro-active biofilms, role of exo-electrogens and their microbial niches in microbial fuel cells (MFCs). Chemosphere 178, 534–547. doi: 10.1016/j.chemosphere.2017.03.066
Schröfel, A., Kratošová, G., Šafarík, I., Šafaríková, M., Raška, I., and Shor, L.M. (2014). Applications of biosynthesized metallic nanoparticles - a review. Acta Biomater. 10, 4023–4042. doi: 10.1016/j.actbio.2014.05.022
Simonsen, S. B., Chorkendorff, I., Dahl, S., Skoglundh, M., Meinander, K., Jensen, T. N., et al. (2012). Effect of particle morphology on the ripening of supported Pt nanoparticles. J. Phys. Chem. C. 116, 5646–5653. doi: 10.1021/jp2098262
Singh, S., Vidyarthi, A. S., and Dev, A. (2015). “Microbial synthesis of nanoparticles,” in Bio-Nanoparticles: Biosynthesis and Sustainable Biotechnological Implications, ed O. V. Singh (Hoboken, NJ: John Wiley and Sons, Inc, 155–186.
Song, R. B., Wu, Y. C., Lin, Z. Q., Xie, J., Tan, C. H., Say, J., et al. (2017). Living and conducting: coating individual bacterial cells with in situ formed polypyrrole. Ang. Chem. Int. Ed. 56, 10516–10520. doi: 10.1002/anie.201704729
Staffell, I., Brett, D. J. L., Brandon, N. J., and Hawkes, A. D. (2015). Domestic microgeneration: renewable and distributed energy technologies. Hous. Stud. 31, 749–750. doi: 10.4324/9781315697109
Stephen, A. J., Archer, S. A., Orozco, R. L., and Macaskie, L. E. (2017). Advances and bottlenecks in microbial hydrogen production. Microb. Biotechnol. 10, 1120–1127. doi: 10.1111/1751-7915.12790
Torgeman, E. (2017). Biosynthesis of gold and palladium nanoparticles via bacteria (M.Sc thesis). University of Oslo, Olso, Norway.
Williams, A. (2015). BioGenic precious metal-based magnetic nanocatalyst for enhanced oxygen reduction (PhD thesis). University of Birmingham, Birmingham, United Kingdom.
Xiong, L., Chen, J. J., Huang, Y. X., Li, W. W., Xie, J. F., and Yu, H. Q. (2015). An oxygen reduction catalyst derived from a robust Pd-reducing bacterium. Nano Energy 12, 33–42. doi: 10.1016/j.nanoen.2014.11.065
Yong, P., Mikheenko, I. P., Deplanche, K., Redwood, M. D., and Macaskie, L. E. (2010). Biorefining of precious metals from wastes: an answer to manufacturing of cheap nanocatalysts for fuel cells and power generation via an integrated biorefinery? Biotechnol. Lett. 32, 1821–1828. doi: 10.1007/s10529-010-0378-6
Yong, P., Paterson-Beedle, M., Mikheenko, I. P., and Macaskie, L. E. (2007). From bio-mineralisation to fuel cells: biomanufacture of Pt and Pd nanocrystals for fuel cell electrode catalyst. Biotechnol. Lett. 29, 539–544. doi: 10.1007/s10529-006-9283-4
Yousfi-Steiner, N., Moçotéguy, P., Candusso, D., and Hissel, D. (2009). A review on polymer electrolyte membrane fuel cell catalyst degradation and starvation issues: causes, consequences and diagnostic for mitigation. J. Power Sources 194, 130–145. doi: 10.1016/j.jpowsour.2009.03.060
Zhang, J., Zhang, H., Wu, J., and Zhang, J. (2013a). “Chapter 1 - PEM fuel cell fundamentals,” in PEM Fuel Cell Testing and Diagnosis (Amsterdam: Elsevier B.V.), 1–42.
Zhang, J., Zhang, H., Wu, J., and Zhang, J. (2013b). “Chapter 4 - the effects of temperature on PEM fuel cell kinetics and performance,” in PEM Fuel Cell Testing and Diagnosis (Amsterdam: Elsevier B.V.), 121–141.
Zhang, J., Zhang, H., Wu, J., and Zhang, J. (2013c). “Chapter 12 - electrochemical half-cells for evaluating PEM fuel cell catalysts and catalyst layers,” in PEM Fuel Cell Testing and Diagnosis (Amsterdam: Elsevier B.V.), 337–361.
Zhang, S., Ding, Y., Liu, B., and Chang, C. C. (2017). Supply and demand of some critical metals and present status of their recycling in WEEE. Waste Manag. 65, 113–127. doi: 10.1016/j.wasman.2017.04.003
Zhu, F., Kim, J., Tsao, K. C., Zhang, J., and Yang, H. (2015). Recent development in the preparation of nanoparticles as fuel cell catalysts. Curr. Opin. Chem. Eng. 8, 89–97. doi: 10.1016/j.coche.2015.03.005
Keywords: biosynthesized nanoparticles, nanoparticles, PEMFC, fuel cells, palladium platinum bimetallic
Citation: Stephen AJ, Rees NV, Mikheenko I and Macaskie LE (2019) Platinum and Palladium Bio-Synthesized Nanoparticles as Sustainable Fuel Cell Catalysts. Front. Energy Res. 7:66. doi: 10.3389/fenrg.2019.00066
Received: 05 April 2019; Accepted: 04 July 2019;
Published: 24 July 2019.
Edited by:
Kirk T. Semple, Lancaster University, United KingdomReviewed by:
Eric D. van Hullebusch, UMR7154 Institut de physique du globe de Paris (IPGP), FranceIoan Stamatin, University of Bucharest, Romania
Copyright © 2019 Stephen, Rees, Mikheenko and Macaskie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lynne E. Macaskie, bC5lLm1hY2Fza2llQGJoYW0uYWMudWs=