 Kody L. Duhl
Kody L. Duhl Michaela A. TerAvest
Michaela A. TerAvest- Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, United States
Shewanella oneidensis MR-1 is a dissimilatory metal reducing bacterium with a highly branched respiratory electron transport chain. The S. oneidensis MR-1 genome encodes four NADH dehydrogenases, any of which may be used during respiration. We previously determined that a double-knockout of two NADH dehydrogenases, Nuo and Nqr1, eliminated aerobic growth in minimal medium. However, the double-knockout strain was able to grow aerobically in rich medium. Here, we determined that amino acid supplementation rescued growth of the mutant strain in oxic minimal medium. To determine the mechanism of the growth defect, we monitored growth, metabolism, and total NAD(H) pools in S. oneidensis MR-1 and the NADH dehydrogenase knockout strain. We also used a genetically encoded redox sensing system and determined that NADH/NAD+ was higher in the mutant strain than in the wild-type. We observed that the double-knockout strain was able to metabolize D,L-lactate and N-acetylglucosamine when supplemented with tryptone, but excreted high concentrations of pyruvate and acetate. The requirement for amino acid supplementation, combined with an apparent inability of the mutant strain to oxidize pyruvate or acetate suggests that TCA cycle activity was inhibited in the mutant strain by a high NADH/NAD+.
Introduction
Shewanella oneidensis MR-1 is a model organism studied for its ability to respire with a variety of terminal electron acceptors, including oxygen, nitrate, TMAO, metal oxides, and electrodes (Beliaev et al., 2005; Gralnick et al., 2006; Meshulam-Simon et al., 2007; Pinchuk et al., 2011; Coursolle and Gralnick, 2012; TerAvest and Angenent, 2014; Duhl et al., 2018). Underlying the respiratory versatility of S. oneidensis MR-1 is a highly branched electron transport chain, including four NADH dehydrogenases and 3 aerobic terminal oxidases, along with specialized terminal oxidases for a variety of electron acceptors (Heidelberg et al., 2002; Pinchuk et al., 2010; Deutschbauer et al., 2011). Even within use of a single electron acceptor, S. oneidensis MR-1 can remodel its electron transport chain depending on environmental conditions. For example, the cbb3 cytochrome oxidase and the bd quinol oxidase are regulated in response to oxygen concentration, with the bd oxidase being upregulated under microaerobic conditions (Zhou et al., 2013). A third aerobic terminal oxidase, the caa3 cytochrome oxidase, is rarely expressed in S. oneidensis MR-1 and has only been observed under high oxygen, low organic carbon conditions (Le Laz et al., 2016). The variety of respiratory complexes and their differential regulation may allow S. oneidensis MR-1 to optimize metabolic flux and energy conservation under the wide range of redox conditions it experiences in the environment.
Differential expression of respiratory complexes has wide-ranging effects on physiology because the electron transport chain is essential to maintaining redox balance and generating ion-motive forces by translocating H+ or Na+ across the inner cell membrane. The ion-motive forces generated by the electron transport chain power ATP synthesis, transport, and flagellar rotation, making them essential to the growth and maintenance of the cell (Senior, 1988; Paulick et al., 2009, 2015). Differential regulation of electron transport chain complexes will affect cellular energetics because the complexes vary kinetically and in their efficiencies of energy conservation. For example, the NADH dehydrogenases range from pumping 4 H+ per electron pair (Nuo), to 2 Na+ per electron pair (Nqr1 and Nqr2, 2Na+/2e−), to 0 ions per electron pair (Ndh) (Pinchuk et al., 2010). By upregulating Nuo, S. oneidensis can gain the maximum ion-motive force for each NADH oxidized, whereas by upregulating Ndh, it can maintain a homeostatic redox state (NADH/NAD+) even when the demand for ion-motive forces is low.
Evidence of electron transport chain remodeling at the NADH dehydrogenase step has previously been found in Bacillus subtilis. The type II NADH dehydrogenase, Ndh, is directly linked to a regulatory loop driven by NADH/NAD+ within the cell. As NADH levels increase, Ndh is upregulated to ensure large fluctuations in NADH/NAD+ do not occur (Gyan et al., 2006). Escherichia coli has also alters the ratio of its type I and type II NADH dehydrogenases depending on growth conditions (Matsushita et al., 1987; Yagi, 1991; Tran et al., 1997). With four NADH dehydrogenases encoded in the genome, it appears likely that remodeling of the electron transport chain for energy conservation and redox maintenance also occurs in S. oneidensis MR-1. To better understand the S. oneidensis MR-1 electron transport chain, we have begun to characterize the roles of NADH dehydrogenases in physiological processes, such as redox homeostasis and energy conservation.
We previously found that the presence of either Nuo or Nqr1 is required for growth under oxic conditions in minimal medium with D,L-lactate or N-acetylglucosamine (NAG) as the substrate (Duhl et al., 2018). However, we did not determine whether the growth defect was caused by an inability to maintain sufficient ion-motive force across the membrane or lack of capacity to re-oxidize NADH. We hypothesized that the growth defect in a Nuo/Nqr1 double knockout strain was due to an inability to re-oxidize NADH, rather than lack of ion-motive forces, because S. oneidensis is capable of aerobic growth without ATP synthesis via the F0F1-ATP synthase (Hunt et al., 2010). To test this hypothesis, we monitored growth, substrate utilization, product formation, redox state, and NAD(H) pools in the mutant strain. We analyzed redox state in real-time using a genetically encoded redox sensing system based on a transcriptional regulator, Rex, from Bacillus subtilis (Liu et al., 2019). We observed broad metabolic disruption in the double mutant strain. Growth was rescued by amino acid supplementation, suggesting that the growth defect was caused by an inability to synthesize amino acids because of TCA cycle inhibition.
Results
S. oneidensis NADH Dehydrogenase Mutant Requires Amino Acid Supplementation
We previously developed NADH dehydrogenase knockout strains of S. oneidensis MR-1 and found a severe growth defect for a strain lacking Nuo and Nqr1 (ΔnuoNΔnqrF1). The ΔnuoNΔnqrF1 strain was able to grow in lysogeny broth (LB) but not in M5 minimal medium under oxic conditions (Duhl et al., 2018). To determine which components of LB enabled growth, we grew the double-knockout strain in M5 minimal medium supplemented with the major components of LB; 0.5 and 1.0% (w/v) tryptone, or 0.25 and 0.5% (w/v) yeast extract. Both tryptone and yeast extract stimulated growth of ΔnuoNΔnqrF1, but tryptone had a much greater effect than yeast extract. Addition of 1% tryptone enabled growth rates and final densities similar to the double-knockout in LB medium albeit with a prolonged lag phase (Figure 1).
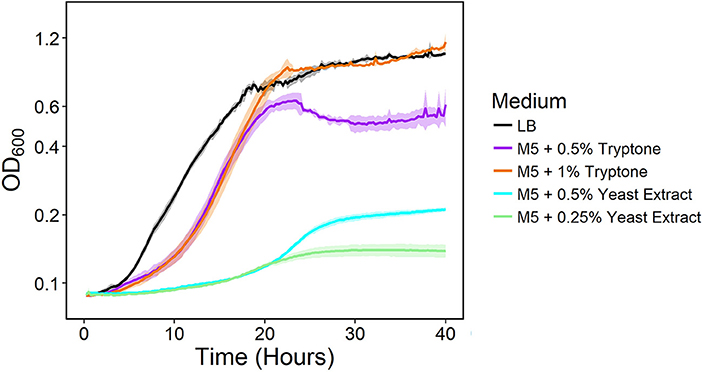
Figure 1. Growth of the ΔnuoNΔnqrF1 strain in LB (black) or M5 medium supplemented with 0.5% (w/v) (purple) and 1.0% (w/v) (orange) tryptone, and 0.5% (w/v) (cyan) and 0.25% (w/v) (light green) yeast extract in 1-mL volumes in 24-well plates at 30°C with shaking.
To investigate why tryptone addition rescued growth of ΔnuoNΔnqrF1, we grew the strain in M5 supplemented with 0.1% tryptone and 20 mM D,L-lactate or 10 mM NAG. To minimize the influence of tryptone as a carbon and energy source, 0.1% (w/v) tryptone was used instead of 0.5 or 1.0%. WT and ΔnuoNΔnqrF1 were grown for 48 h in 24-well plates in the semi-minimal medium with or without an additional carbon source to determine if ΔnuoNΔnqrF1 used either D,L-lactate or NAG in the presence of tryptone. The presence of 20 mM D,L-lactate increased growth of WT and ΔnuoNΔnqrF1 compared with 0.1% tryptone alone, suggesting that the mutant strain was capable of metabolizing D,L-lactate in the presence of tryptone (Figure 2A). HPLC analysis confirmed that after the 48-h incubation period, WT used 100% and ΔnuoNΔnqrF1 used ~75% of the D,L-lactate (Table 1). The same results were observed with NAG as the substrate. Both WT and ΔnuoNΔnqrF1 grew to a higher OD600 with NAG and used 100 and ~70% of the NAG, respectively (Figure 2B and Table 1). The growth rate of ΔnuoNΔnqrF1 was decreased compared to WT with both D,L-lactate and NAG, indicating a growth defect even in the presence of 0.1% tryptone (p ≤ 0.001). To determine whether the effect of the tryptone was due to amino acid supplementation, we also compared growth of the wild-type and mutant supplemented with 0.5% casamino acids or a defined mixture of amino acids and 20 mM D,L-lactate or 10 mM NAG. WT grew with the casamino acids and the defined amino acid mixture, but ΔnuoNΔnqrF1 was unable to grow, suggesting that peptides may be a better amino acid source for S. oneidensis than free amino acids, as observed previously (Serres and Riley, 2006).
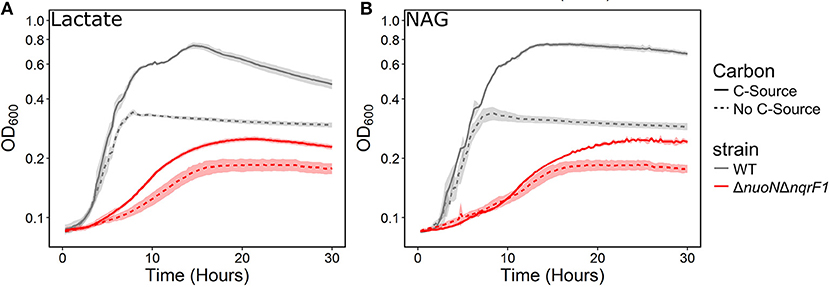
Figure 2. Growth of WT (gray) and ΔnuoNΔnqrF1 (red) in M5 minimal medium supplemented with 0.1% tryptone, with (solid line) or without (dashed line) (A) 20 mM DL-lactate or (B) 10 mM NAG as a carbon source. Growth in 1 mL medium in 24-well plates at 30°C with shaking.
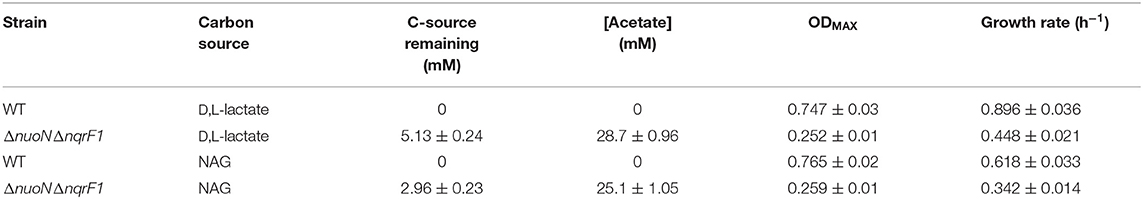
Table 1. Growth and endpoint HLPC analysis of carbon source utilization from 24-well plate experiments after 48-h runtime in M5 supplemented with 0.1% tryptone and 20 mM D,L-lactate or 10 mM NAG from Figure 2.
Analysis of Growth, Metabolism, and NAD(H) in ΔnuoNΔnqrF1
To clarify the effects of knocking out both Nqr1 and Nuo from S. oneidensis MR-1, growth experiments were repeated in 250-mL Erlenmeyer flasks in 50-mL culture volumes. Prior to these experiments, growth was only monitored in 24-well plates in 1-mL culture volumes. Scaling up to 50 mL culture volumes allowed us to sample multiple times throughout growth to monitor metabolic products, and internal redox state (NADH/NAD+). When grown in this culture format in M5 supplemented with 0.1% tryptone and 20 mM D,L-lactate as a carbon source, ΔnuoNΔnqrF1 again exhibited a severe growth defect compared to WT (Figure 3A). The doubling times for WT and ΔnuoNΔnqrF1 were 0.524 ± 0.053 h and 1.300 ± 0.083 h, respectively (p ≤ 0.001). Further, ΔnuoNΔnqrF1 did not fully deplete the D,L-lactate, while WT consumed all available D,L-lactate by 12 h (Figure 3B). WT cultures briefly accumulated small amounts of pyruvate and acetate between hours 6 and 14 of growth, but both were completely consumed by 14 h. In contrast, ΔnuoNΔnqrF1 cultures accumulated large amounts of pyruvate and acetate, i.e., >50% of lactate was converted to these products and excreted (Figures 3C,D). Pyruvate and acetate accumulation in the ΔnuoNΔnqrF1 cultures remained high through the end of the experiment and it appeared that the mutant strain could not utilize these excreted products. To determine if knocking out Nqr1 and Nuo affected redox state in ΔnuoNΔnqrF1, we conducted a colorimetric NADH and NAD+ assay. We investigated NADH/NAD+ and total NAD(H) pools. Overall, we did not find consistent differences in NADH/NAD+ between WT and the mutant, which was unexpected, considering that two NADH dehydrogenases were deleted. However, the total NAD(H) pool of ΔnuoNΔnqrF1 was 1.75-fold higher than that of WT (Table 2).
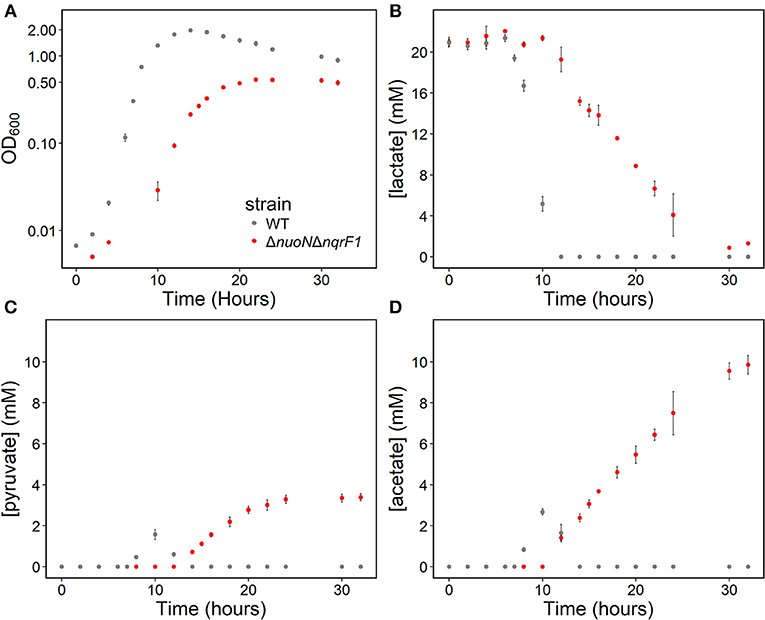
Figure 3. (A) Growth of WT (gray) and ΔnuoNΔnqrF1 (red) in 50-mL culture volumes of M5 minimal medium supplemented with 0.1% tryptone and 20 mM D,L-lactate. (B) D,L-lactate utilization by WT and ΔnuoNΔnqrF1. (C) Pyruvate and (D) acetate concentrations in culture supernatants.
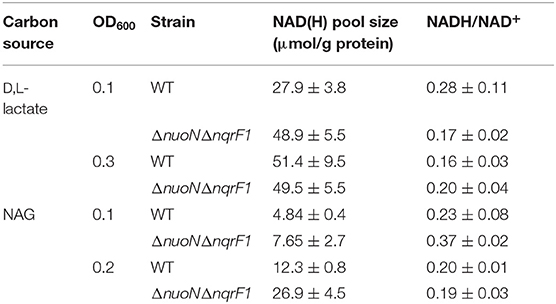
Table 2. Comparison of NAD(H) pools and NADH/NAD+ ratios of WT and ΔnuoNΔnqrF1 when grown in 50 mL cultures of M5 supplemented with 0.1% tryptone and either 20 mM D,L-lactate or 10 mM NAG.
In 50-mL cultures of M5 supplemented with 0.1% (w/v) tryptone and NAG, ΔnuoNΔnqrF1 also exhibited a growth defect compared to WT (Figure 4A). The doubling times for WT and ΔnuoNΔnqrF1 were 0.838 ± 0.065 and 1.823 ± 0.118 h, respectively (p ≤ 0.001). The ability to utilize NAG as a carbon source was also hindered compared to WT (Figure 4B). Similar to what we observed in D,L-lactate, WT cultures accumulated little pyruvate and acetate, while the mutant cultures accumulated high concentrations of these products (>50% of NAG was converted to these products, Figures 4C,D). In this condition, the redox quantification assay data showed inconsistent NADH/NAD+ ratios at the time points sampled. Again, the total NAD(H) pool of ΔnuoNΔnqrF1 was 2-fold larger than that of WT at OD600 = 0.2 (Table 2).
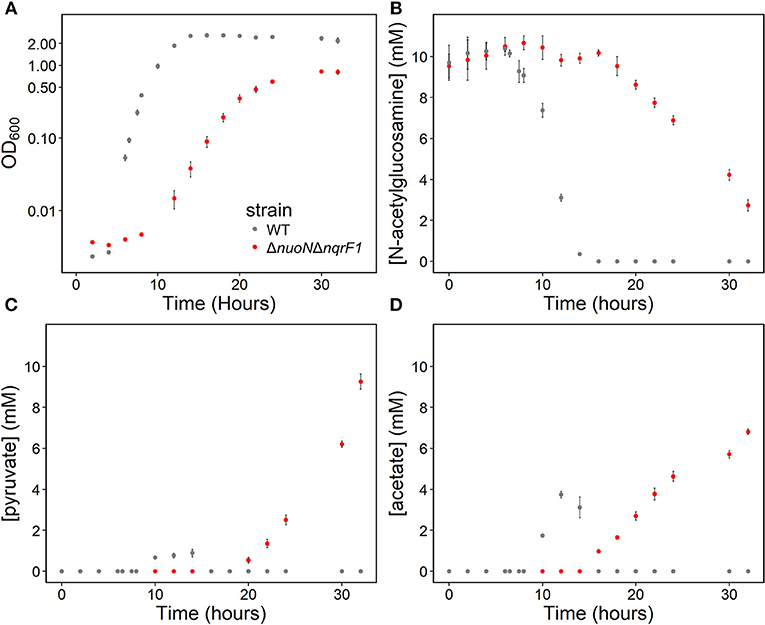
Figure 4. (A) Growth of WT (gray) and ΔnuoNΔnqrF1 (red) in 50-mL culture volumes of M5 minimal medium supplemented with 0.1% tryptone and 10 mM N-acetylglucosamine (NAG). (B) NAG utilization by WT and ΔnuoNΔnqrF1. (C) Pyruvate and (D) acetate concentrations in culture supernatants.
Altogether, our measurements of NADH/NAD+ ratios were not consistently different between the two strains. This was unexpected, because the deletion of NADH dehydrogenases, combined with the accumulation of pyruvate and acetate suggest an accumulation of NADH in the mutant strain. We hypothesized that the sampling procedure for the redox assay may have biased the results because it included a 10-min centrifugation step during which the redox state of the cells could change. To determine whether oxygen levels may have influenced redox state during the centrifugation step, we measured oxygen consumption in centrifuge tubes. We transferred 25 mL of shaking cultures to a 50 mL conical tube and measured dissolved oxygen concentrations over time. We found that WT and ΔnuoNΔnqrF1 consumed oxygen at rates of 0.068 ± 0.004 and 0.058 ± 0.008 mg/L s−1 (normalized to OD600), respectively. At these rates, both strains depleted all dissolved oxygen in the culture samples in 5 min or less (Figure S1).
We also measured the oxygen consumption rates of both strains in a custom device and found that it was 2.273 ± 0.152 μM/s for WT and 0.936 ± 0.134 μM/s for ΔnuoNΔnqrF1 at OD600 = 0.2. During the NADH/NAD+ extraction process, assuming the initial oxygen concentration in the medium is saturated at 8 mg/L (250 μM) and that no additional oxygen dissolves into the liquid during the centrifugation step, all oxygen in the samples would be consumed within the first 2–4 min of the centrifugation. These calculations are complicated by the possibilities that oxygen in the headspace (10 mL) could dissolve into the sample and that as the cells pellet in the centrifuge, they could create a local environment with even less dissolved oxygen. However, these calculations do suggest that oxygen limitation occurs for both strains during the centrifugation step, thereby causing equalization of the NADH/NAD+ ratios of the two strains. This helps to explain why we did not observe consistent differences between redox state and only observed differences in the total NAD(H) pool sizes.
Analyzing NADH Dehydrogenase Knockouts Using a Genetically-Encoded Sensor
Because sampling cells for the enzymatic NADH/NAD+ assay likely biased our results, we monitored redox state in real-time with a transcriptionally regulated redox sensor (Liu et al., 2019). The sensor is based on the transcriptional repressor Rex from B. subtilis (Liu et al., 2019). This redox sensor results in increased green fluorescent protein (GFP) production when NADH/NAD+ increases. We introduced the sensor into S. oneidensis MR-1 and the NADH dehydrogenase knockout strain, ΔnuoNΔnqrF1. To determine whether the redox sensor was functional in S. oneidensis MR-1, we monitored reporter fluorescence during an aerobic to anaerobic transition. Two sets of triplicate 50-mL WT (with redox sensor) cultures were grown aerobically for 6 h. After incubating under oxic conditions, one of the triplicate sets was incubated on the benchtop without shaking for 2 h, then transferred to an anoxic environment and given 50 mM fumarate as a terminal electron acceptor. We expected the reporter output to increase in cultures that were moved to an anaerobic environment because limitation of available terminal electron acceptor will increase intracellular NADH/NAD+. Increased NADH accumulation has previously been observed in E. coli cultures in anaerobic conditions vs. aerobic conditions (de Graef et al., 1999). As predicted, fluorescence output from S. oneidensis with the Rex-based sensor increased in cultures that were transitioned from an aerobic to an anaerobic environment compared to the cultures that remained aerobic (Figure 5). These results suggest that the sensor functions as expected in S. oneidensis MR-1 and responds to increased NADH/NAD+.
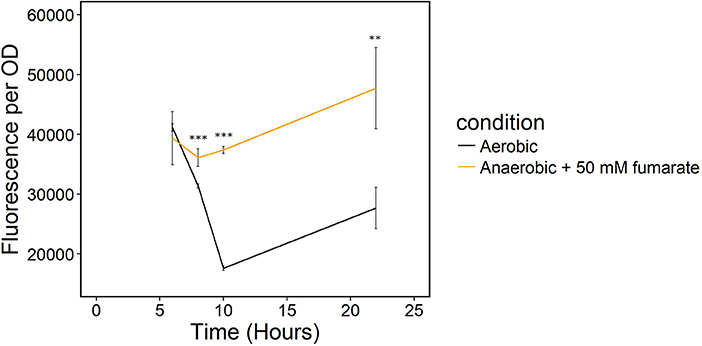
Figure 5. Transition of S. oneidensis MR-1 with the Rex redox sensor growing in triplicate 50-mL cultures of LB from an oxic to an anoxic environment. The aerobic set (black) remained oxic and shaking and the anaerobic set (orange) of triplicates was given 50 mM fumarate as a terminal electron acceptor when moved to an anoxic environment. **p ≤ 0.01 and ***p ≤ 0.001 denote significance of difference from the aerobic culture.
We conducted growth experiments and monitored OD600 and reporter fluorescence in S. oneidensis MR-1 and ΔnuoNΔnqrF1 in 24-well plates. Fluorescence per OD600 was higher in ΔnuoNΔnqrF1 than WT during log-phase and early stationary phase for both carbon sources, suggesting that the mutant strain exhibits increased NADH/NAD+ during growth (Figure 6). However, it is important to note that the redox sensor altered growth of the strains. Therefore, we conducted a side-by-side comparison between strains with and without the sensor. We observed that the presence of the Rex-based redox sensor delayed growth of WT by about 14 h with D,L-lactate and 18 h with NAG (Figure 7). Lag times varied somewhat between experiments (Figures 6, 7), possibly due to small differences in the growth phase of the overnight cultures used for inoculum. However, we consistently observed an extended lag phase in WT containing the Rex sensor. The sensor also affected the growth rates of both strains with either D,L-lactate or NAG as the substrate, although the growth rate of the mutant was still lower than WT in all cases (Table 3). We conducted HPLC analysis of metabolites generated by the strains containing the Rex sensor. Similar to strains without the sensor, WT was able to deplete all initial carbon source, while the mutant was not (Table 4). Also in line with results without the sensor, the mutant strain produced significant amounts of acetate and pyruvate, while WT did not.
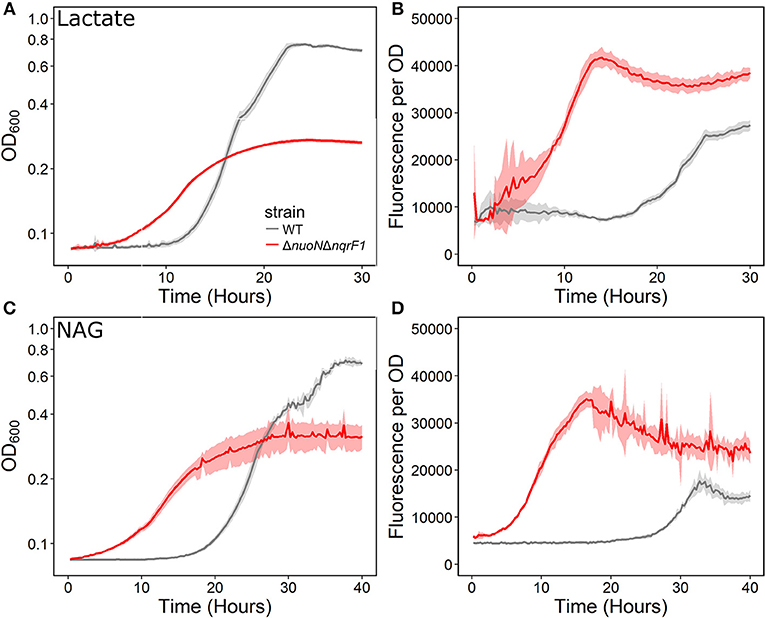
Figure 6. (A) Analysis of growth of the WT and ΔnuoNΔnqrF1 strains that contain the Rex sensor in 1-mL culture volumes of M5 supplemented with 0.1% tryptone and 20 mM D,L-lactate while at 30°C with shaking. (B) Fluorescence output normalized to OD600 with D,L-lactate as the substrate. (C) Analysis of growth of the WT and ΔnuoNΔnqrF1 strains that contain the Rex sensor in 1-mL culture volumes of M5 supplemented with 0.1% tryptone and 10 mM NAG. (D) Fluorescence output normalized to OD600 with NAG as the substrate.
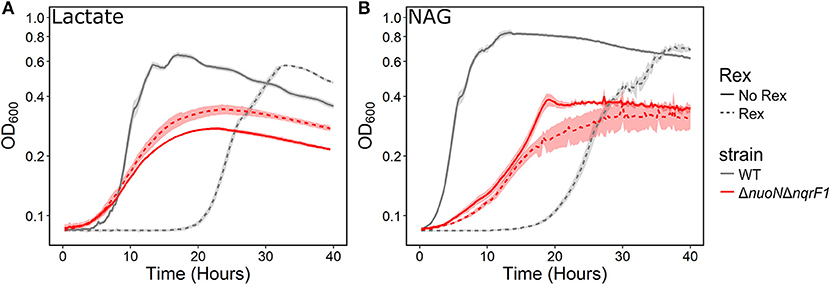
Figure 7. Comparison of growth of WT (gray) and ΔnuoNΔnqrF1 (red) strains with (solid line) and without (dashed line) the Rex redox sensor in M5 supplemented with 0.1% tryptone and (A) 20 mM D,L-lactate or (B) 10 mM NAG. Growth conditions were done in 1-mL culture volumes in 24-well plates at 30°C with shaking.
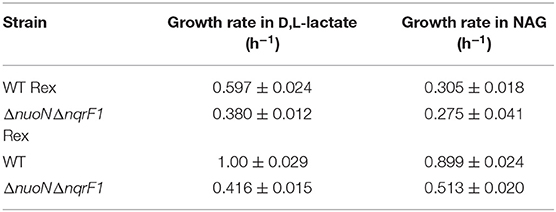
Table 3. Comparison of growth rates of WT and ΔnuoNΔnqrF1 with and without the Rex sensor growing in M5 supplemented with 0.1% tryptone and either 20 mM D,L-lactate or 10 mM NAG from Figure 7.
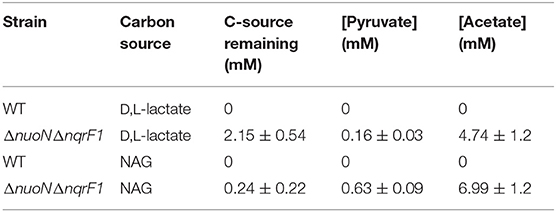
Table 4. Endpoint HLPC analysis of carbon source utilization from 24-well plate experiments with WT and ΔnuoNΔnqrF1 containing the Rex redox sensor after 48-h runtime in M5 supplemented with 0.1% tryptone and 20 mM D,L-lactate or 10 mM NAG.
Discussion
As observed in our previous work, ΔnuoNΔnqrF1 was unable to grow under oxic conditions in M5 minimal medium with either D,L-lactate or NAG as the substrate (Duhl et al., 2018). However, we have now observed that addition of 0.1% (w/v) tryptone to the medium allowed ΔnuoNΔnqrF1 to grow. The major component of tryptone is free amino acids and peptides, suggesting that the mutant strain requires amino acid supplementation and cannot make sufficient amino acids de novo to support growth. Together with accumulation of acetate and pyruvate the requirement for tryptone suggests reduced TCA cycle activity because some TCA reactions are required for de novo amino acid synthesis (Kanehisa and Goto, 2000). However, there are caveats to using tryptone because it is an undefined tryptic digest of casein and could contain other nutrients. We observed that other sources of amino acids, including casamino acids (acid-hydrolyzed casein) or defined amino acids did not rescue growth of the mutant. However, we propose that the rescue was caused by peptides in the tryptone, not by other nutrients. While there are minor differences in carbohydrate and mineral content between tryptone and casamino acids, these differences are small in comparison to the total mineral and carbohydrate content of the overall medium recipe (BD Biosciences, 2006). Further, previous work indicated that S. oneidensis MR-1 is incapable of using individual amino acids as carbon sources but is capable a wide variety of defined dipeptides (Serres and Riley, 2006). This suggests that S. oneidensis MR-1 is much more efficient in peptide uptake than free amino acid uptake, which would explain why tryptone rescues growth of the mutant, while casamino acids do not.
Along with the growth defect observed in the NADH dehydrogenase knockout strain, another finding of this study was that total NAD(H) pool sizes differed significantly between WT and ΔnuoNΔnqrF1. With either D,L-lactate or NAG as carbon sources, we observed roughly 2-fold increases in the total NAD(H) pools. We hypothesize that the increased NAD(H) pool size is caused by NAD+ synthesis to counteract the increase in [NADH] within the cell caused by the limitation of NADH dehydrogenase activity. In S. oneidensis MR-1, NAD+ synthesis is regulated by the repressor NrtR. When [NAD+] decreases, NrtR is released from promotors to allow expression of NAD+ synthesis related genes (Rodionov et al., 2008). Because we observed increased NAD(H) pool sizes in the ΔnuoNΔnqrF1 mutant strain, we propose that excess [NADH] and limited [NAD+] led to overexpression of genes involved in NAD+ synthesis.
The TCA cycle and upstream reactions are also affected by changes in internal redox state and NAD(H) pool size. NADH is an inhibitor of citrate synthase, which converts acetyl-CoA and oxaloacetate to citrate to bring carbon into the TCA cycle (Weitzman and Jones, 1968; Stokell et al., 2003). This suggests that increased NADH/NAD+ may inhibit TCA cycle function by affecting citrate synthase activity. Furthermore, reactions upstream of the TCA cycle maybe affected, because pyruvate dehydrogenase (Pdh) activity is also regulated by NADH/NAD+ and acetyl-CoA concentrations in S. oneidensis MR-1 (Pinchuk et al., 2011; Novichkov et al., 2013). Reduced citrate synthase activity would increase acetyl-CoA concentrations, which may inhibit Pdh function, together with increased NADH/NAD+. It also has been previously shown that Pdh activity can be affected by both NADH/NAD+ and NAD(H) pool size (Shen and Atkinson, 1970), meaning activity is slowed when NADH/NAD+ ratios within the cell increase. Metabolic analysis of the mutant strain supports the hypothesis that pyruvate oxidation and TCA cycle activity were inhibited in ΔnuoNΔnqrF1 and further explains previous data observed in single-knockouts of Nqr1 and Nuo (Duhl et al., 2018). We observed excretion of high levels of pyruvate and acetate by ΔnuoNΔnqrF1, which would be expected if flux through the TCA cycle is blocked (Figure 8). It is also important to note that all four of the NADH dehydrogenases have not been knocked out of S. oneidensis MR-1 in this study. This study sought to understand the roles of the two aerobically expressed NADH dehydrogenases (Pinchuk et al., 2010; Duhl et al., 2018), even though the other NADH dehydrogenases may play a role in redox state regulation in the ΔnuoNΔnqrF1 mutant strain.
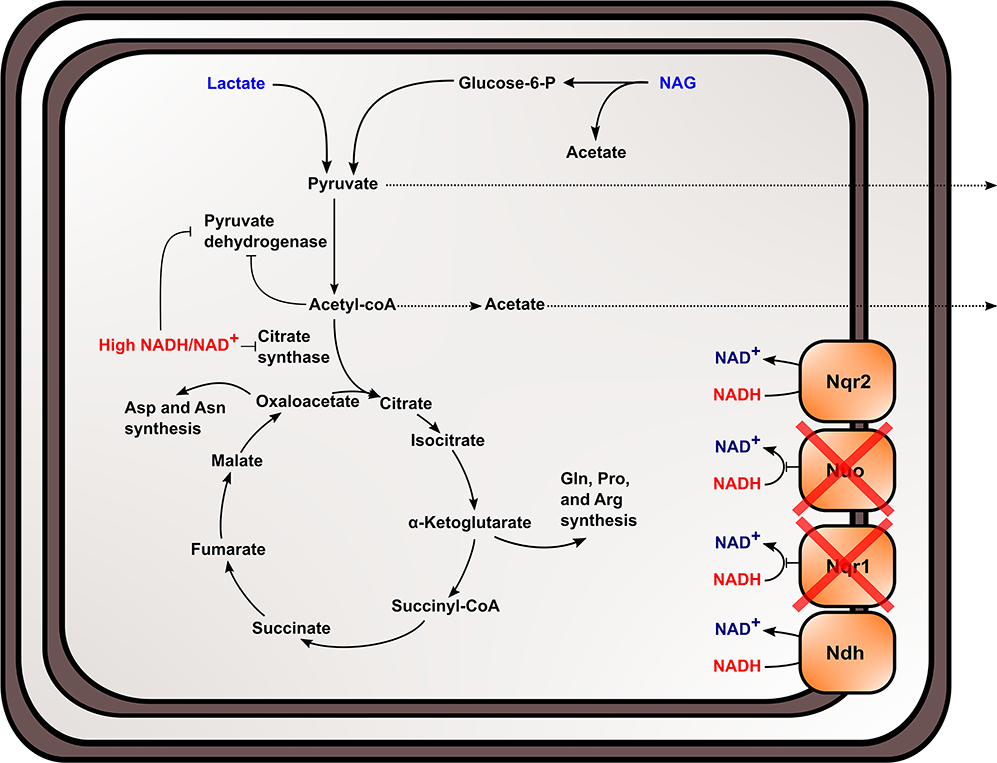
Figure 8. Proposed effect of knocking out NADH dehydrogenases in S. oneidensis MR-1 and the subsequent effects on NADH oxidation, redox state, and TCA cycle inhibition within the cell.
To better understand the effects of NADH dehydrogenase knockouts on the physiological redox state in S. oneidensis MR-1 throughout growth, we used the Rex-based redox sensing system developed by Liu et al. (2019). One of the major limitations to standard NADH and NAD+ quantification assays is the need to remove the bacteria from their growth environment to conduct extractions. We found that the cell harvest and extraction procedure may cause shifts in the cells' redox state. Prior to quenching with acid or base solution in the protocol, shaking of the cultures is ceased and samples are transferred to 15 mL conical tubes (Kern et al., 2014), likely leading to oxygen limitation. We have shown that the cells likely deplete all oxygen within the medium during the centrifugation step, creating an oxygen limited environment that can influence and equalize redox state in both S. oneidensis strains. Conversely, the Rex-based redox sensor directly interacts with intracellular NADH and NAD+ and allows real-time, qualitative measurements of NADH/NAD+ via fluorescent reporter output (Liu et al., 2019). This allowed us to assess NADH/NAD+ without processing the cells in a way that would influence redox state. Our data show that the sensor works as expected in S. oneidensis and that ΔnuoNΔnqrF1 exhibits increased fluorescence output compared to WT.
While the sensor did influence growth of the strains, we were still able to gain qualitative measurements of the internal redox state. It is not clear why the addition of the Rex sensor influenced growth in S. oneidensis MR-1. In E. coli, the Rex sensor did not appear to influence growth (Liu et al., 2019). It is possible that the metabolic burden generated from carrying the redox sensing system caused the changes, and that use of different plasmid backbones or promoters would reduce the effects of the sensor. However, we believe that the output of the sensor is still a valuable source of information for this study for multiple reasons; the sensor generated the expected output for an aerobic to anaerobic transition and the overall phenotypes and differences between WT and the mutant remained similar with the sensor. I.e., the mutant strain still grew to a lower final OD600 and at a slower rate than the WT when the sensor was present (Figure 6 and Table 3). Further, differences in substrate consumption and acetate and pyruvate accumulation remained similar when the sensor was present, with the mutant strain failing to utilize all available carbon source and accumulating acetate and pyruvate. WT with the sensor was able to consume all available substrate and did not accumulate acetate or pyruvate. Although the sensor appeared to affect WT more than the mutant, the general effect on both strains appears similar, and the essential phenotypes remain the same; therefore, we believe that the sensor output reflects real differences in intracellular NADH/NAD+ between the strains.
Altogether, our data indicate that deletion of NADH dehydrogenases affected NAD(H) pool sizes, NADH/NAD+, and upstream metabolic activities, specifically by inhibiting the TCA cycle and blocking amino acid synthesis. Because we have shown that deleting NADH dehydrogenases from S. oneidensis MR-1 led to increased NADH/NAD+ levels, larger NAD(H) pool sizes, and metabolic shifts within the cell, NADH dehydrogenase knockouts may provide an avenue for metabolic engineering. When engineering pathways in bacteria to generate products that are redox cofactor-dependent, it is advantageous to make modifications to that organism to generate higher levels of NADH (Berrios-Rivera et al., 2002). If enzyme concentrations no longer limit the rate of product formation, then the availability of redox cofactors may become limiting (Berrios-Rivera et al., 2002; Balzer et al., 2013). For example, it was necessary to eliminate pathways that compete for NADH in E. coli to improve 1-butanol production (Atsumi et al., 2008). Increasing NADH generation has also been used to enhance electric current production by S. oneidensis MR-1 (Li et al., 2018). These studies show the importance of NADH availability when engineering redox cofactor-dependent pathways. We have shown that knocking out NADH dehydrogenases and eliminating a competing pathway for NADH in S. oneidensis MR-1 increases the availability of NADH, which provides a possible background strain for metabolic engineering in S. oneidensis MR-1. With additional NADH available in these NADH dehydrogenase mutant strains, we could direct NADH into synthetic pathways for product formation, even when oxygen is present, as indicated by the high levels of pyruvate and acetate accumulation by the mutant strain.
Methods
Generating NADH Dehydrogenase Knockout Strains Containing the Rex Redox Sensor
Each single NADH dehydrogenase knockout and the ΔnuoNΔnqrF1 double-knockout strain were generated in a previous study and confirmed by complementation (Duhl et al., 2018). The Rex redox sensor plasmids were received from Dr. Vatsan Raman and Yang Liu at the University of Wisconsin—Madison) and were described in their recent publication (Liu et al., 2019). The two-plasmid system has one plasmid that constitutively expresses Rex and another that contains a fluorescent reporter under Rex regulation. The Rex-containing plasmid was transformed into chemically competent E. coli WM3064 cells. The plasmid was then transferred to S. oneidensis MR-1 and NADH dehydrogenase knockout strains via a conjugation protocol similar to Webster et al. (2014). The plasmid containing the fluorescent reporter was transformed into chemically competent E. coli Mach 1 cells via heat shock. The Mach 1 cells were grown at 37°C while shaking for 18 h in 5 mL cultures. The cultures were used to extract the reporter plasmid via the E.Z.N.A Plasmid DNA Mini Kit I (Omega Bio-tek, D6943-02). Extracted plasmid was then used to transform electrocompetent S. oneidensis MR-1 and NADH dehydrogenase strains containing the Rex plasmid via electroporation (Myers and Myers, 1997). The presence of both plasmids in each strain were confirmed with antibiotic resistance and PCR.
Growth Conditions
Strains were pre-cultured in LB medium for 18 h. Each pre-culture was normalized to OD600 = 1.0 and washed in M5 minimal medium three times by centrifugation and resuspension. Each growth experiment used the following M5 minimal medium recipe: 1.29 mM K2HPO4, 1.65 mM KH2PO4, 7.87 mM NaCl, 1.70 mM NH4SO4, 475 μM MgSO4·7H2O, 10 mM HEPES, 0.01% (w/v) casamino acids, 0.1% tryptone (w/v), 1X Wolfe's mineral solution (aluminum was not included), and 1X Wolfe's vitamin solution (riboflavin was not included), pH adjusted to 7.2 with 1 M NaOH. Carbon donors, either NAG or D,L-lactate, were added to the M5 medium to final concentrations of 10 or 20 mM, respectively. The addition of 0.1% tryptone to the M5 minimal medium recipe was the only adjustment from our previous work (Duhl et al., 2018). To determine if amino acids were the key components required to rescue the growth of the double NADH dehydrogenase knockout, two different modifications to the M5 recipe were made: the addition of 0.5% (w/v) casamino acids, or the addition of a defined amino acid mixture containing 20 mM L-arginine (Alfa Aesar, A15738), 20 mM D,L-aspartic acid (TCI, A0544), 20 mM L-glutamic acid (TCI, G0059), 20 mM L-leucine (VWR, 5811), 20 mM L-lysine monochloride (Acros Organics, 123221000), 20 mM L-proline (TCI P0481), D,L-serine (TCI, S0034), and 20 mM L-valine (V0014). After the amino acids were added to the M5 minimal medium, the pH was readjusted to 7.2 with 5 M NaOH and the medium was filter sterilized.
High-throughput growth experiments containing multiple strains were conducted in clear 24-well culture plates (Greiner Bio-One, 662165) in 1 mL culture volumes of M5 medium, with four replicates per strain. Each well was inoculated with 10 μL of normalized pre-culture (OD600 = 1.0) and monitored in a Synergy H1 plate reader (BioTek Instruments, Winooski, VT) with orbital shaking at 30°C for 48 h. For growth and fluorescence experiments involving the Rex redox sensor, growth was monitored at 600 nm and fluorescence output was monitored at an excitation wavelength of 475 nm, and emission wavelength of 509 nm.
Flask growth experiments were conducted in 50 mL culture volumes in 250-mL Erlenmeyer flasks. Experiments were performed in M5 medium supplemented with 0.1% tryptone and 10 mM NAG or 20 mM D,L-lactate. Flasks were inoculated with 50 μL of standardized pre-cultures and incubated in a floor shaker (New Brunswick Scientific, 12500) at 30°C while shaking at 275 rpm. Cultures were grown in triplicate and sampled by removing 1 mL and reading OD600 on a spectrophotometer (Eppendorf BioPhotometer, D30).
HPLC Analysis
HPLC analysis was conducted as previously described (Duhl et al., 2018).
NADH/NAD+ Quantification Assay
The protocol for the NADH/NAD+ quantification assay was adapted from a previously published method (Kern et al., 2014). Samples (5 mL) for both NADH and NAD+ extractions were taken during early logarithmic growth from 50-mL cultures growing in M5 medium containing 20 mM D,L-lactate or 10 mM NAG. Each 5-mL sample was transferred to a sterile 15-mL conical tube (VWR, 89039-664) and centrifuged at 5,000 × g at 4°C in a Sorvall ST 8R centrifuge (Thermo Scientific) HIGHConic rotor (Thermo Scientific, 75005709) for 10 min. The supernatant was removed from each tube and the pellets were resuspended in 500 μL of 0.1 M HCl containing 500 mM NaCl or 0.1 M NaOH containing 500 mM NaCl for NAD+ or NADH extractions, respectively. Each sample was transferred to a 1.5-mL microcentrifuge tube and incubated for 5 min at 95°C. After incubation, the samples were placed on ice to cool for 10 min and then were centrifuged for 5 min at 5,000 × g at 4°C in the Sorvall ST 8R centrifuge with the microcentrifuge rotor (Thermo Scientific, 75005715). After centrifugation, 300 μL of supernatant were transferred from each sample into new 1.5-mL microcentrifuge tubes and stored at −80°C until the colorimetric assay was conducted.
Each assay was conducted in clear 96-well microplates (Greiner Bio-One, 655101). Standards were prepared from 0.1 mM NAD+ (NAD trihydrate, Amresco, 0455) and 0.1 mM NADH (NADH disodium trihydrate, Amresco, 0384) stocks. Each standard and sample were aliquoted into duplicate sets of wells in 20 μL volumes. To initiate the assay, 180 μL of master mix was added to each well and the plate is placed in the plate reader to orbitally shake and incubate at 30°C. The wells were monitored at 1-min intervals with the Synergy H1 plate reader measuring absorbance at 550 nm. The in-well master mix component concentrations are as follows: 0.1 M bicine (VWR, 0149) buffer pH 8.0, 4 mM EDTA disodium salt (Invitrogen, 15576), 1.66 mM phenazine ethosulfate (Sigma-Aldrich, P4544), 0.42 mM thiazolyl blue tetrazolium bromide (Beantown Chemical, 142015), 10% (v/v) ethanol, and 3.2 units/mL alcohol dehydrogenase (Sigma, A3262). NAD+ and NADH concentrations are determined following the data analysis protocol set out by Kern et al. (2014). Because the extraction efficiencies of 0.1 M HCl and NaOH differed, [NAD+] and [NADH] were normalized to total protein from each extraction determined by conducting the Pierce™ BCA Protein Assay Kit (Thermo Scientific, 23225).
Aerobic to Anaerobic Transition
Pre-cultures of S. oneidensis MR-1 normalized to and OD600 of 1.0 were used to inoculate two sets of triplicate 50-mL cultures of LB to an OD600 of 0.01. Each set was grown for 6 h under oxic conditions at 30°C while shaking at 275 rpm. After the 6-h incubation period each culture was sampled and the one sets that would be transitioned to an anoxic environment was placed at room temperature without shaking for 2 h. After 2 h, each culture was sampled and the anaerobic cultures were moved to an anaerobic chamber and given 50 mM fumarate as a terminal electron acceptor. Each culture set was incubated for another 12 h and samples were taken from each culture 2- and 12-h post-transition.
Oxygen Consumption Measurements
Oxygen consumption of WT and ΔnuoNΔnqrF1 was measured within a microfluidic system designed by Dr. Denis Proshlyakov and Nathan Franz (Michigan State University). Samples were added to the microfluidic system and spectroscopically monitored in a custom device. Each run was conducted in triplicate with cultures normalized to and OD600 of 0.2 that were shaken at 275 rpm for 10 min prior to testing to ensure total oxygen saturation of the medium. Further testing of oxygen consumption was tested in large batch cultures grown in 50 mL of M5 medium supplemented with 0.1% tryptone and 20 mM D,L-lactate. 25 mL of culture that had been continuously shaking at 30°C at 275 rpm were transferred quickly into a 50 mL conical tube. Oxygen consumption was measured by submerging an oxygen probe (Mettler Toledo InLab® OptiOx, 51344621) in the 25 mL of culture and monitoring [O2] over time.
Data Analysis
Analysis of growth, fluorescence, and HPLC data was performed using Rstudio (R Studio Team, 2019) with following packages: ggplot2 (Wickham, 2009), reshape2 (Wickham, 2007), dplyr (Wickham and Francois, 2015), and TTR (Ulrich, 2017). Growth rates were calculated using R package “growthcurver” using default values with background correction set to “min” (Sprouffske and Wagner, 2016). NADH/NAD+ quantification, BCA assay data, and oxygen consumption data were analyzed in Microsoft Excel (2016).
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
KD designed and performed the experiments, analyzed and interpreted the data, and drafted the manuscript. MT designed the study, designed the experiments, analyzed and interpreted the data, and revised the manuscript.
Funding
This research was partially supported by a fellowship from Michigan State University under the Training Program in Plant Biotechnology for Health and Sustainability (T32-GM110523). This work was also supported by the USDA National Institute of Food and Agriculture, Hatch project 1009805.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Cecilia Martinez-Gomez (Michigan State University) for sharing the adapted redox assay protocol, Nicholas Tefft (Michigan State University) for reviewing the manuscript, and Dr. Vatsan Raman and Yang Liu (University of Wisconsin—Madison) for sharing the Rex-based redox sensor and for helpful comments on the manuscript. The authors would also like to thank Dr. Denis Proshlyakov and Nathan Frantz (Michigan State University) for allowing us to use their microfluidic system to measure oxygen consumption.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2019.00116/full#supplementary-material
References
Atsumi, S., Cann, A. F., Connor, M. R., Shen, C. R., Smith, K. M., Brynildsen, M. P., et al. (2008). Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 10, 305–311. doi: 10.1016/j.ymben.2007.08.003
Balzer, G. J., Thakker, C., Bennett, G. N., and San, K.-Y. (2013). Metabolic engineering of Escherichia coli to minimize byproduct formate and improving succinate productivity through increasing NADH availability by heterologous expression of NAD+-dependent formate dehydrogenase. Metab. Eng. 20, 1–8. doi: 10.1016/j.ymben.2013.07.005
BD Biosciences (2006). BD Bionutrients Technical Manual Advanced Bioprocessing. Available online at: https://www.bdbiosciences.com/documents/bionutrients_tech_manual.pdf
Beliaev, A. S., Klingeman, D. M., Klappenbach, J. A., Wu, L., Romine, M. F., Tiedje, J. M., et al. (2005). Global transcriptome analysis of Shewanella oneidensis MR-1 exposed to different terminal electron acceptors. J. Bacteriol. 187, 7138–7145. doi: 10.1128/JB.187.20.7138-7145.2005
Berrios-Rivera, S. J., Bennett, G. N., and San, K.-Y. (2002). Metabolic engineering of Escherichia coli: increase of NADH availability by overexpressing an NAD+-dependent formate dehydrogenase. Metab. Eng. 4, 217–229. doi: 10.1006/mben.2002.0227
Coursolle, D., and Gralnick, J. A. (2012). Reconstruction of extracellular respiratory pathways for iron(III) reduction in Shewanella oneidensis strain MR-1. Front. Microbiol. 3:56. doi: 10.3389/fmicb.2012.00056
de Graef, M. R., Alexeeva, S., Snoep, J. L., and Teixeira de Mattos, M. J. (1999). The steady-state internal redox state (NADH/NAD+) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 181, 2351–2357.
Deutschbauer, A., Price, M. N., Wetmore, K. M., Shao, W., Baumohl, J. K., Xu, Z., et al. (2011). Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet. 7:e1002385. doi: 10.1371/journal.pgen.1002385
Duhl, K. L., Tefft, N. M., and TerAvest, M. A. (2018). Shewanella oneidensis MR-1 utilizes both sodium-and proton-pumping NADH dehydrogenases during aerobic growth. Appl. Environ. Microbiol. 84:e00415-18. doi: 10.1128/AEM.00415-18
Gralnick, J. A., Vali, H., Lies, D. P., and Newman, D. K. (2006). Extracellular respiration of dimethyl sulfoxide by Shewanella oneidensis strain MR-1. Proc. Natl. Acad. Sci. U.S.A. 103, 4669–4674. doi: 10.1073/pnas.0505959103
Gyan, S., Shiohira, Y., Sato, I., Takeuchi, M., and Sato, T. (2006). Regulatory loop between redox sensing of the NADH/NAD+ ratio by rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. J. Bacteriol. 188, 7062–7071. doi: 10.1128/JB.00601-06
Heidelberg, J. F., Paulsen, I. T., Nelson, K. E., Gaidos, E. J., Nelson, W. C., Read, T. D., et al. (2002). Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotech. 20, 1118–1123. doi: 10.1038/nbt749
Hunt, K. A., Flynn, J. M., Naranjo, B., Shikhare, I. D., and Gralnick, J. A. (2010). Substrate-level phosphorylation is the primary source of energy conservation during anaerobic respiration of Shewanella oneidensis strain MR-1. J. Bacteriol. 192, 3345–3351. doi: 10.1128/JB.00090-10
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kern, S. E., Price-Whelan, A., and Newman, D. K. (2014). Extraction and measurement of NAD(P)+ and NAD(P)H. Methods Mol. Biol. 1149, 311–323. doi: 10.1007/978-1-4939-0473-0_26
Le Laz, S., Kpebe, A., Bauzan, M., Lignon, S., Rousset, M., and Brugna, M. (2016). Expression of terminal oxidases under nutrient-starved conditions in Shewanella oneidensis: detection of the A-type cytochrome c oxidase. Sci. Rep. 6:19726. doi: 10.1038/srep19726
Li, F., Li, Y., Sun, L., Chen, X., An, X., Yin, C., et al. (2018). Modular engineering intracellular NADH regeneration boosts extracellular electron transfer of Shewanella oneidensis MR-1. ACS Synth. Biol. 7, 885–895. doi: 10.1021/acssynbio.7b00390
Liu, Y., Landick, R., Raman, S., Landick, B., and Raman, S. (2019). A regulatory NADH/NAD+ redox biosensor for bacteria. ACS Synth. Biol. 8, 264–273. doi: 10.1021/acssynbio.8b00485
Matsushita, K., Ohnishi, T., and Kaback, H. R. (1987). NADH-ubiquinone oxidoreductases of the Escherichia coli aerobic respiratory chain. Biochemistry 26, 7732–7737. doi: 10.1021/bi00398a029
Meshulam-Simon, G., Behrens, S., Choo, A. D., and Spormann, A. M. (2007). Hydrogen metabolism in Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 73, 1153–1165. doi: 10.1128/AEM.01588-06
Myers, C. R., and Myers, J. M. (1997). Replication of plasmids with the p15A origin in Shewanella putrefaciens MR-1. Lett. Appl. Microbiol. 24, 221–225. doi: 10.1046/j.1472-765X.1997.00389.x
Novichkov, P. S., Kazakov, A. E., Ravcheev, D. A., Leyn, S. A., Kovaleva, G. Y., Sutormin, R. A., et al. (2013). RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14:745. doi: 10.1186/1471-2164-14-745
Paulick, A., Delalez, N. J., Brenzinger, S., Steel, B. C., Berry, R. M., Armitage, J. P., et al. (2015). Dual stator dynamics in the Shewanella oneidensis MR-1 flagellar motor. Mol. Microbiol. 96, 993–1001. doi: 10.1111/mmi.12984
Paulick, A., Koerdt, A., Lassak, J., Huntley, S., Wilms, I., Narberhaus, F., et al. (2009). Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol. Microbiol. 71, 836–850. doi: 10.1111/j.1365-2958.2008.06570.x
Pinchuk, G. E., Geydebrekht, O. V., Hill, E. A., Reed, J. L., Konopka, A. E., Beliaev, A. S., et al. (2011). Pyruvate and lactate metabolism by Shewanella oneidensis MR-1 under fermentation, oxygen limitation, and fumarate respiration conditions. Appl. Environ. Microbiol. 77, 8234–8240. doi: 10.1128/AEM.05382-11
Pinchuk, G. E., Hill, E. A., Geydebrekht, O. V., De Ingeniis, J., Zhang, X., Osterman, A., et al. (2010). Constraint-based model of Shewanella oneidensis MR-1 metabolism: a tool for data analysis and hypothesis generation. PLoS Comput. Biol. 6:e1000822. doi: 10.1371/journal.pcbi.1000822
R Studio Team (2019). R Studio: Integrated Development for R. Boston, MA: RStudio, Inc. Available online at: http://www.rstudio.com/
Rodionov, D. A., De Ingeniis, J., Mancini, C., Cimadamore, F., Zhang, H., Osterman, A. L., et al. (2008). Transcriptional regulation of NAD metabolism in bacteria: NrtR family of Nudix-related regulators. Nucleic Acids Res. 36, 2047–2059. doi: 10.1093/nar/gkn047
Senior, A. E. (1988). ATP synthesis by oxidative phosphorylation. Physiol. Rev. 68, 177–231. doi: 10.1152/physrev.1988.68.1.177
Serres, M. H., and Riley, M. (2006). Genomic analysis of carbon source metabolism of Shewanella oneidensis MR-1: predictions versus experiments. J. Bacteriol. 188, 4601–4609. doi: 10.1128/JB.01787-05
Shen, L. C., and Atkinson, D. E. (1970). Regulation of pyruvate dehydrogenase from Escherichia coli interactions of adenylate energy charge and other regulatory parameters. J. Biol. Chem. 245, 5974–5978.
Sprouffske, K., and Wagner, A. (2016). Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17:172. doi: 10.1186/s12859-016-1016-7
Stokell, D. J., Donald, L. J., Maurus, R., Nguyen, N. T., Sadler, G., Choudhary, K., et al. (2003). Probing the roles of key residues in the unique regulatory NADH binding site of type II citrate synthase of Escherichia coli. J. Biol. Chem. 278, 35435–35443. doi: 10.1074/jbc.M302786200
TerAvest, M. A., and Angenent, L. T. (2014). Oxidizing electrode potentials decrease current production and coulombic efficiency through cytochrome c inactivation in Shewanella oneidensis MR-1. ChemElectroChem 1, 2000–2006. doi: 10.1002/celc.201402128
Tran, Q. H., Bongaerts, J., Vlad, D., and Unden, G. (1997). Requirement for the proton-pumping NADH dehydrogenase i of Escherichia coli in respiration of NADH to fumarate and its bioenergetic implications. Eur. J. Biochem. 244, 155–160. doi: 10.1111/j.1432-1033.1997.00155.x
Webster, D. P., TerAvest, M. A., Doud, D. F. R., Chakravorty, A., Holmes, E. C., Radens, C. M., et al. (2014). An arsenic-specific biosensor with genetically engineered Shewanella oneidensis in a bioelectrochemical system. Biosens. Bioelectron. 62, 320–324. doi: 10.1016/j.bios.2014.07.003
Weitzman, P. D. J., and Jones, D. (1968). Regulation of citrate synthase and microbial taxonomy. Nature 219:270. doi: 10.1038/219270a0
Wickham, H. (2007). Reshaping data with the reshape package. J. Stat. Softw. 21, 1–20. doi: 10.18637/jss.v021.i12
Wickham, H., and Francois, R. (2015). dplyr: A Grammar of Data Manipulation. R Package version 0.4.2., 3.
Yagi, T. (1991). Bacterial NADH-quinone oxidoreductases. J. Bioenerg. Biomembr. 23, 211–225. doi: 10.1007/BF00762218
Keywords: Shewanella oneidensis MR-1, NADH dehydrogenase, redox state, TCA cycle, amino acids
Citation: Duhl KL and TerAvest MA (2019) Shewanella oneidensis NADH Dehydrogenase Mutants Exhibit an Amino Acid Synthesis Defect. Front. Energy Res. 7:116. doi: 10.3389/fenrg.2019.00116
Received: 21 June 2019; Accepted: 04 October 2019;
Published: 24 October 2019.
Edited by:
Jeffrey A. Gralnick, University of Minnesota Twin Cities, United StatesReviewed by:
Annette Ruth Rowe, University of Cincinnati, United StatesKai Thormann, University of Giessen, Germany
Copyright © 2019 Duhl and TerAvest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michaela A. TerAvest, dGVyYXZlczJAbXN1LmVkdQ==