 Qi Xu1
Qi Xu1 Markus Alahuhta1Patrick Hewitt2
Markus Alahuhta1Patrick Hewitt2 Nicholas S. Sarai3
Nicholas S. Sarai3 Hui Wei1
Hui Wei1 Neal N. Hengge1
Neal N. Hengge1 Ashutosh Mittal1Michael E. Himmel1
Ashutosh Mittal1Michael E. Himmel1 Yannick J. Bomble1*
Yannick J. Bomble1*- 1BioSciences Center, National Renewable Energy Laboratory, Golden, CO, United States
- 2Boston University, Boston, MA, United States
- 3California Institute of Technology, Pasadena, CA, United States
Cell free biocatalysis is showing promise as a replacement or complement to conventional microbial biocatalysts due to the potential for achieving high yields, titers, and productivities. However, there exist several challenges that need to be addressed before its broader industrial adoption is achieved. New paradigms and innovative solutions are needed to overcome these challenges. In this study we demonstrate high levels of glycerol conversion to 1,3-propanediol using a self-assembling metabolic pathway leveraging the arraying strategy (protein scaffolds) used by thermophilic cellulolytic bacteria to assemble their biomass degrading enzymes. These synthetic metabolons were capable of producing 1,3-PDO at a yield more than 95% at lower glycerol concentration and close to 70% at higher concentrations at a higher productivity rate than the equivalent microbial strain. One of the benefits of our approach is the fact that no enzyme purification is required, and that the assembly of the complex is accomplished in vivo before immobilization, while product formation is conducted in vitro. We also report the recovery of enzymatic activity upon fusion enzymes binding to these protein scaffolds, which could have broader applications when assembling arrayed protein complexes.
Introduction
Although microbial biocatalysts have been successfully used for the production of many high-value chemicals, they often suffer from low product yields due to competing metabolic pathways, low productivities, difficult optimization of metabolic pathways, cellular product toxicity, and expensive isolation of target products from cell cultures (Stephanopoulos, 2007; Chen and Liao, 2016; Chubukov et al., 2016; Chae et al., 2017). Alternatives to production using microbial biocatalysts include cell-free approaches wherein reactions occur in vitro, in isolation, rather than in cells (Kay and Jewett, 2015; Korman et al., 2017; Petroll et al., 2019; Bergquist et al., 2020; Bowie et al., 2020). Cell-free biocatalysis relies either on purified enzyme systems or lysates with both having advantages and disadvantages (Kay and Jewett, 2015; Rollin et al., 2021). Purified enzyme systems are desired as they do not include competing pathways with the complication that enzymes have to be purified individually, unlike in lysate approaches. Additionally, product yields and titers in cell-free biocatalysis can also suffer from diffusion of intermediates and lack of enzyme stability. Some approaches have been used to address these problems using different arraying technologies.
For example, cellulolytic bacteria are known to produce cellulosome complexes that arrays enzymes with different activities together using scaffoldin frameworks (Bayer et al., 2004; Artzi et al., 2017; Bayer, 2017; Smith et al., 2017). Enzymes that bind to the cohesin modules of the scaffoldin have a dockerin module separated by a linker region. The interaction between cohesins and dockerins is species and type-specific and allows controlled binding of several different enzymes next to each other (Carvalho et al., 2003; Cameron et al., 2015). Several synthetic protein scaffolds or designer cellulosomes have been expressed and tested for viability (You et al., 2012; You and Zhang, 2013; Chen et al., 2018; Kahn et al., 2019). Natural and designer cellulosomes can be thermostable (Kahn et al., 2019), allow self-assembly and co-immobilization (You and Zhang, 2013), and show remarkable flexibility in regards to possible activity combinations (Artzi et al., 2017). Designer cellulosomes can also improve the performance of enzymatic pathways by bringing consecutive pathway enzymes in closer proximity to each other to allow for substrate channeling (You et al., 2012).
In this study, we have leveraged the protein arraying approach, used natively by several cellulolytic bacteria to tether their biomass degrading enzymes, to self-organize metabolic pathways (including sensitive enzymes) and simplify their purifications into a single step from a co-expressing enzyme production strain (Figure 1). We applied this approach to the production of 1,3-propanediol (1,3-PDO) from glycerol (Raynaud et al., 2003; Jiang et al., 2016; Lee et al., 2018). Glycerol, a main byproduct of the biofuel industry, represents an ideal conversion intermediate that can be upgraded to a value-added product such as 1,3-PDO (Çelik et al., 2008; Yang et al., 2012; Plácido and Capareda, 2016). Additionally, 1,3-PDO is recognized as a high-valued specialty chemical used as a monomer for manufacturing plastics and cyclic compounds (Saxena et al., 2009; Zeng and Sabra, 2011; Kluge et al., 2019). We first evaluated the residual activity and impact of our co-expressed binding platform in vivo due to the difficulty of handling some of the enzymes in vitro (when expressed as free enzymes) and demonstrated the production of 1,3-PDO in a cell free context enabled by the self-assembling metabolic pathway.
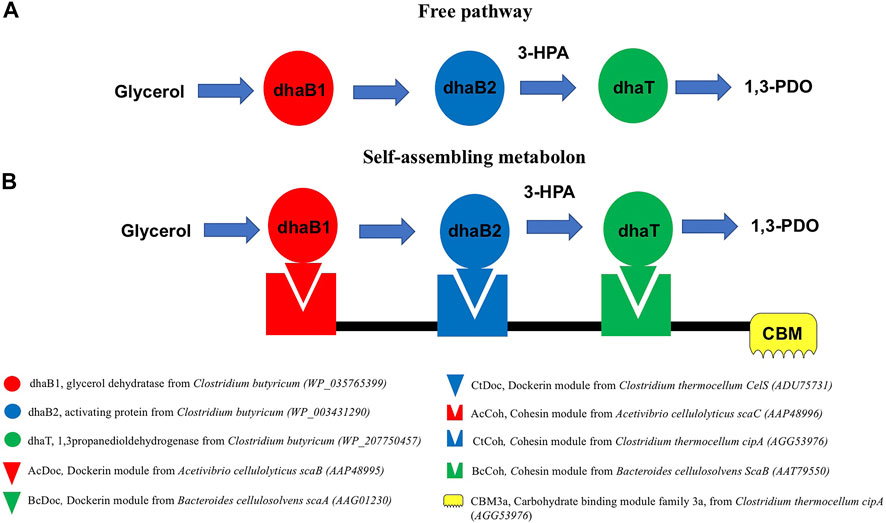
FIGURE 1. Comparison of a traditional cell-free biocatalysis approach using purified enzymes and our self-assembling metabolon using an engineered protein scaffold for the conversion of glycerol to 1,3-PDO. In the self-assembling metabolon, the enzymes are expressed as protein fusions with a dockerin module specifically targeting the corresponding cohesin module on the protein scaffold. The color-coding scheme is used throughout the study to identify the protein modules and enzymes.
Results
Gene Selection and Construction of Self-Assembling Pathway Enzymes for the Conversion of Glycerol to 1,3-PDO
In this study we focused on the production of 1,3-PDO from glycerol using two metabolic steps with 3-hydroxypropionic aldehyde (3-HPA) as an intermediate. The conversion of glycerol to 3-HPA can be achieved using two different types of dehydratases: (1) B12-dependent dehydratases and (2) B12-independent dehydratases (Knietsch et al., 2003; O'Brien et al., 2004; Rieckenberg et al., 2014; Liu et al., 2016). In this study to avoid the mechanism based inactivation of B12-dependent dehydratases that leads to vitamin B12 cleavage (Toraya et al., 1976; Yamanishi et al., 2012), we selected the B12-independent glycerol dehydratase from Clostridium butyricum (Raynaud et al., 2003). However, members of the B12-independent class of dehydratases are extremely oxygen sensitive and very difficult to handle in vitro (Raynaud et al., 2003). In our approach, the conversion of glycerol to 1,3-PDO requires: (1) the dehydration of glycerol to 3-HPA with the B12-independent glycerol dehydratase (dhaB1) and its activating protein (dhaB2) and (2) the hydrogenation of 3-HPA to 1,3 PDO with a 1,3-PDO dehydrogenase (dhaT) (Figure 1) (O'Brien et al., 2004; Lama et al., 2015). All enzymes considered in this study were selected from C. butyricum. These enzymes were expressed individually and as fusion proteins with a dockerin module from either Acetivibrio cellulolyticus, Bacteroides cellulosolvens, or Clostridium thermocellum (Figure 1). These fusion constructs are designed to bind to their cohesin binding partner on a synthetic protein scaffold through their strong natural and specific interactions. The scaffolds are composed of a CBM3a (family 3a carbohydrate binding module) able to specifically bind to cellulose for isolation and purification of the complex, and cohesin modules from different microorganisms that can specifically bind their corresponding dockerins found on the enzymes. All primers and constructs used in this study can be found in Supplementary Tables S1,S2.
In vivo Production of 1,3 PDO and Activity of Fusion Enzymes
Given that one of the key metabolic enzymes, the glycerol dehydratase activating enzyme (dhaB2), involved in the production of 1,3-PDO via 3-HPA is very difficult to express and purify (data not shown), we conducted most of the pathway and enzyme characterization in vivo before demonstrating the promise of our approach in vitro. We first confirmed the production of 1,3-PDO using the glycerol to 1,3-PDO pathway from C. butyricum with the glycerol dehydratase (dhaB1), dhaB1 activating protein (dhaB2), and the NADH-dependent 1,3-propanediol dehydrogenase (dhaT). The resulting E. coli strain encoding this new operon was able to achieve the production of more than 26 mM of 1,3-PDO in 72 h (Figure 2). Note that this strain was not optimized for 1,3-PDO production as this was not the goal of this study but was rather used as a benchmark for subsequent testing. The parent strain of E. coli used in this study was not able to produce 1,3-PDO from glycerol nor was a strain encoding for dhaB1 and dhaB2 even though E. coli possesses an alcohol dehydrogenase, YqhD, known to be promiscuous (Sulzenbacher et al., 2004; Jarboe, 2011) (with a function similar to that of dhaT) (Figure 2). The lack of production of 1,3-PDO in the latter strain indicates that the deletion of YqhD in the parent strains was not necessary to achieve a clean background for 1,3-PDO production.
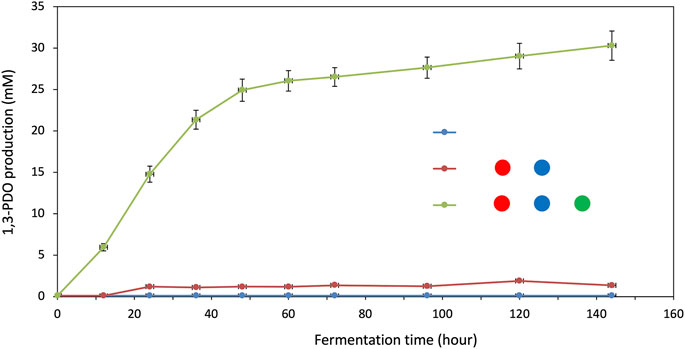
FIGURE 2. Baseline production of 1,3-PDO in E. coli using enzymes from C. butyricum and determination of the contribution of E. coli YqhD to 1,3-PDO production. All symbols are described in Figure 1. Standard deviation is shown as vertical bars for each data point.
We then systematically evaluated the impact of adding dockerin modules to these metabolic enzymes within the pathway. In all cases, the activity of the fusion enzymes that included the dockerin modules was drastically reduced with a corresponding production of 1,3-PDO barely reaching 3.0 mM after 72 h of incubation compared to 26 mM for the strain encoding for the native enzyme operon (Figure 3). There was almost no correlation between the origin of the dockerin module and the impact on activity as all enzymes were equally inhibited by dockerins (Figures 3A–C, and Supplementary Figures S2, S3). Specifically, Figure 3A shows that linking the AcDoc to dhaB1 results in a large activity loss with similar results when AcDoc is linked to dhaB2 (Figure 3B) and dhaT (Figure 3C).
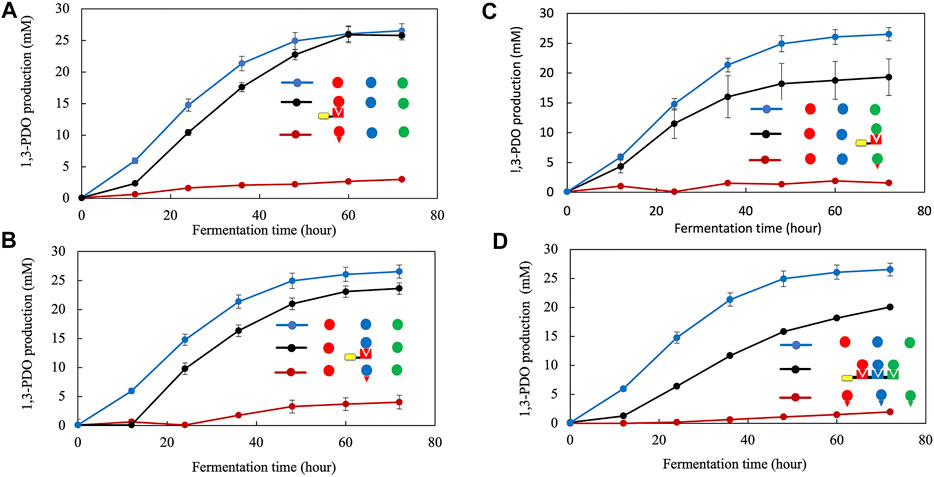
FIGURE 3. Effect of interactions between the cohesin module and the dockerin module on the activity of dhaB1 (A), dhaB2 (B), dhaT (C). Effect of interactions of the full length scaffoldin and the dockerin modules on the activity of the complete dhaB1, dhaB2, dhaT pathway (D). All symbols are described in Figure 1. Standard deviation is shown as vertical bars for each data point.
Surprisingly, the activity of the enzyme and the pathway can be mostly restored when co-expressing a scaffoldin protein (Cohesin-CBM) (Figures 3A–C). Even in the most extreme case with each enzyme being expressed as fusion proteins with a dockerin module, the full length scaffoldin protein restores close to 80% of the 1,3-PDO production, producing 20.1 mM of 1,3-PDO compared to 26.5 mM for the native enzymes after 72 h (Figure 3D). It appears that the binding of the dockerin to its cohesin partner is responsible for this recovery. Another important aspect of the metabolic complex is the presence of a carbohydrate binding module that would allow one step purification of the complex on cellulose for cell-free applications.
Additionally, to evaluate the impact of the CBM module on the production of 1,3-PDO, we considered two different strains both encoding the three fusion enzymes and the protein scaffold but only one containing the CBM. We conducted longer fermentations with all three enzymes as dockerin fusions with the scaffoldin. As shown earlier, in the presence of scaffoldin, 1,3-PDO production was in the vicinity of 80% compared to the free enzyme reference after 72 h of incubation with glycerol but could actually achieve more than 90% recovery after 96 h (Figure 4A). Also, it appears that the impact of the CBM is negligible with achieving a titer of 24.1 mM of 1,3-PDO after 72 h compared to 22.7 mM in the case of the complex lacking the CBM and the same trend continues even after 140 h (Figure 4A).
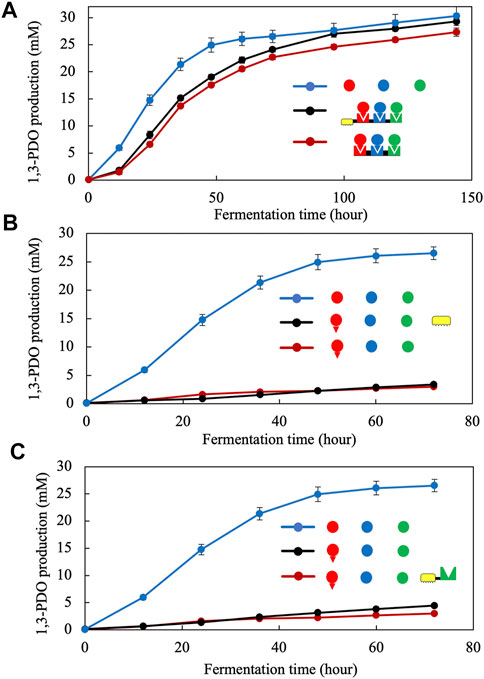
FIGURE 4. Effect of the CBM3a on the activity of the metabolon and comparison at longer fermentation times (A). Effect of potential non-specific interactions between the CBM3a and the dockerin of dhaB1-AcDoc on the activity dhaB1-AcDoc (B). Effect of non-specific interaction between a cohesin and the dockerin of dhaB1-AcDoc on the activity of dhB1-AcDoc (C). All symbols are described in Figure 1. Standard deviation is shown as vertical bars for each data point.
To determine if other mechanisms were at play regarding this interesting activity recovery, we conducted other controls by simply co-expressing a carbohydrate binding module in the presence of a dhaB1-doc fusion enzyme or co-expressing the same dhaB1-doc fusion enzyme with an incompatible cohesin protein. In both cases the production of 1,3-PDO remained negligible with no significant changes after 72 h (Figures 4B,C). These results indicate that only specific binding of the enzyme-dockerin fusion to its cohesin partner allows for restored activity.
Self-Assembly and Isolation of the Metabolon for the Cell-free Conversion of Glycerol to 1,3-PDO
To assemble and isolate the metabolic complex, E. coli cells encoding for the co-expression of the three fusion enzymes and the full length scaffoldin proteins were grown anaerobically and the lysis, immobilization, and assays were also performed under anaerobic conditions (Figure 5). Plans for individual enzyme characterization were abandoned after it became clear that we could not successfully purify dhaB2 contrary to previous reports (O'Brien et al., 2004). Despite attempting anaerobic purification with and without iron-sulfur cluster reconstruction, aerobic purification with metals removed using EDTA followed by iron-sulfur cluster reconstruction, and various expression and purification conditions with or without affinity tags, we were not able to isolate an active dhaB2 (results not shown). Instead, the supernatant of the resulting cell lysate was incubated with cellulose to isolate the complex via interactions with the carbohydrate binding module. The cellulose was then washed extensively to remove all potential substrates, intermediates, and products produced during expression. The resulting immobilized metabolic pathway was incubated with glycerol for 48 h at room temperature (Figure 6). The same procedure was followed for the parent E. coli strain and the E. coli strain encoding for the free metabolic pathway. Three different glycerol concentrations (4/11/40 mM) were selected to determine how yield is affected with increasing glycerol concentration. The purified complex reached 99.5% conversion of glycerol to 1,3-PDO with an initial glycerol concentration of 4.2 mM indicating that the three dockerin-fused metabolic enzymes performed well when self-assembled in the cell on the scaffoldin. The two negative controls, namely the one with empty plasmid and the one with the three wild-type metabolic enzymes did not produce any or very negligible amounts of 1,3-PDO, respectively. The negligible production of 1,3-PDO from the strain encoding wild type enzymes is most likely due to nonspecific binding to the cellulose.
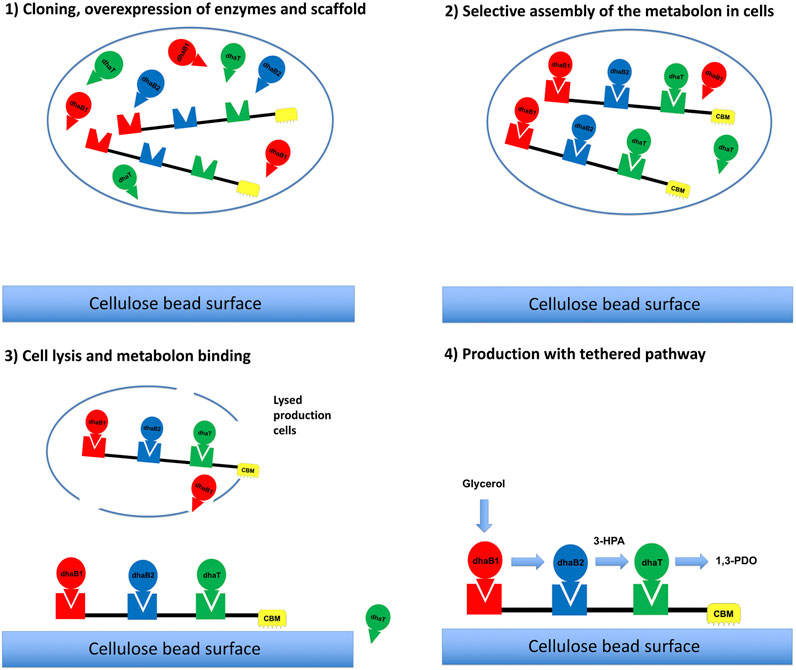
FIGURE 5. Depiction of the process for the in vitro production of 1,3-PDO from glycerol using a self-assembling metabolon including (A) Expression of enzymes and protein scaffolds, (B) Self-assembly of the metabolon in cells, (C) Cell lysis and specific metabolon binding to cellulose, and (D) 1,3-PDO production in vitro. All symbols are described in Figure 1.
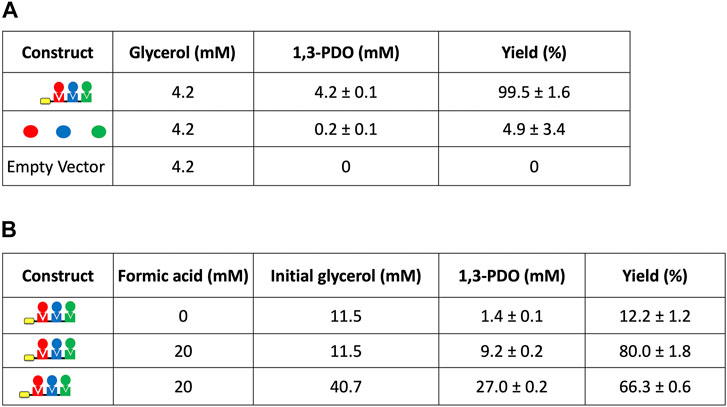
FIGURE 6. Titers and yields of 1,3-PDO from glycerol using the self-assembling metabolon using low levels of glycerol to confirm the specific binding to cellulose before production (5 mM NADH) and using higher levels of glycerol with formate as a sacrificial substrate for cofactor recycling to avoid stoichiometric use of NADH (16 mM NADH in the absence of formate and 1 mM in the presence of formate). All symbols are described in Figure 1.
We also followed the kinetics of the conversion of glycerol to 1,3-PDO using the isolated enzyme complex (Supplementary Figure S1). Glycerol conversion reached 50% after 3.8 h and 70% after 8 h indicating the continuous production by the immobilized complex but also revealing that most of the conversion occurred during the first 8 h of incubation. The conversion ultimately reached 99.5% yield after 48 h (Figure 6A). When considering higher levels of glycerol and stoichiometric levels of the cofactor NADH, this pathway was significantly inhibited (Figure 6B). We hypothesized that providing NADH at stoichiometric levels could lead to inhibition of some of the enzymes in the complex. Therefore, we established a cofactor recycling strategy using a formate dehydrogenase and formate as a sacrificial substrate, to recycle NAD+ to NADH. Using this approach, the immobilized metabolic complex was clearly not limited by the initial amount of NADH (1 mM), owing to cofactor recycling using formate, and was able to produce 9.2 mM of 1,3-PDO with a yield of 80%. The immobilized complex was also able to reach 66% conversion of glycerol with an initial glycerol concentration of about 40 mM which further demonstrates the viability of our approach when using higher glycerol titers. However, follow up studies are needed to fully evaluate the impact of glycerol concentration and potential substrate inhibition on this system.
Discussion
The nascent field of cell-free biocatalysis needs several breakthroughs to demonstrate viability and cost competitiveness for industrial applications. There are several challenges that need to be overcome for cell-free biocatalysis to become a reality especially for the production of commodity fuels and chemicals–high on that list are 1) enzyme cost, (2) ease of enzyme purification, (3) enzyme stability/solubility, and (4) efficient cofactor recycling schemes. Of course, enzyme and production strain engineering can help alleviate some of these problems but purified enzyme-based cell-free processes can still be cumbersome for long metabolic pathways. Additionally, in some cases the stability or solubility of pathway enzymes can become problematic with some enzymatic pathways potentially not being viable in vitro.
In this study we demonstrated that not only complete metabolons can be purified on a cheap cellulosic media directly from the lysate, but we also demonstrated that enzymes or proteins that are very difficult to purify and use in vitro can be expressed and function when tethered to a protein scaffold. This is the case, in this study, for the activating protein dhaB2. Additionally, the fact that all enzymes and necessary proteins can be co-expressed and assembled in vivo before being immobilized for use in vitro is a significant advantage, avoiding cumbersome purification steps, which could lead to a reduced cost for a cell free process. Also, it should be noted that our best 1,3-PDO titer (27 mM), which could be considered unoptimized, compares well to earlier cell-free lysate work conducted by Rieckenberg et. al who used the B12-dependent glycerol dehydratase (Rieckenberg et al., 2014). There are a few examples of other efforts assembling metabolic pathway complexes in vitro but, in these cases, individual proteins had to be purified with traditional approaches, and the purified recombinant proteins were then assembled on protein scaffolds similar to those used in our study (You and Zhang, 2013). Additionally, the enzymes used in previous studies were known to be stable and soluble after purification. In this previous study the authors showed that the activity of the assembled complex was higher compared to the free enzyme pathway, most likely due to channeling of intermediates (You and Zhang, 2013). This is indeed a potential advantage of these tethered enzymatic systems wherein the distance between enzymes can be fine-tuned. Unfortunately, we were not able to accomplish the same comparison as dhaB2 was insoluble. However, in our study we were able to compare to in vivo production and found that the full tethered metabolic pathway was able to produce a similar amount of 1,3-PDO as the strain it originated from but with a higher productivity. These results demonstrate the promise of this approach to assemble pathways with problematic enzymes and to bypass cumbersome purification steps needed for most cell free systems. However, we are also aware that much improvement is needed to increase titers and productivity for these types of systems to be considered for the production of biochemicals at larger scales. Some of these improvements could come from mining for more efficient enzymes and enzyme more suitable for tethering to these protein scaffolds. Additionally, linker length and composition could also impact the activity and stability of these metabolons.
Another very intriguing and fascinating outcome of this study is the finding that binding to the cohesin module found on the scaffolds helps to recover enzymatic activity. This result is scientifically significant as it could have implications for many other applications in enzyme production and engineering. We hypothesize that this phenomenon is due to the dockerin module adversely interacting with the enzymes, more specifically because of the high flexibility of the linker peptides between the enzymes and the dockerin modules (Bomble et al., 2011; Barth et al., 2018). We believe that the flexibility of the overall construct could change upon binding. Indeed, others have observed a similar behavior with glycoside hydrolases constructs natively including a dockerin module. For example, Hammel et. al. showed, using small angle x-ray scattering, that within some cellulosomal biomass degrading enzymes the intrinsic flexibility of the linker between the enzyme and the dockerin module was lost upon binding to a scaffoldin protein leading to a drastic compaction of the overall construct (Hammel et al., 2005). It is possible that a similar phenomenon is occurring within our constructs. It is also likely that this behavior will depend on the type of enzyme, their fold, and their common interactions with other enzymes within cells. It is vital to start understanding these interactions in more details to further enable cell free systems and develop biomimetics means to emulate what can be lost from the cellular environment when using pathways in vitro (Deeds et al., 2007; Barbieri et al., 2015; Cohen and Pielak, 2017; Ziegler et al., 2021). More broadly there is a lot of merit to studying the assembly of individual proteins which conduct independent and discrete biological function through non-covalent interaction. For example, the fatty acid biosynthesis complex is assembled by a highly intertwined, hetero-dodecameric fatty acid synthase with all the fatty acid biosynthesis function. It has been observed that the speed and selectivity of binding interactions between enzymatic complex and substrates can be significantly improved through proximity of individual enzymes leading to higher catalytic efficiency. This is just one example among many others, but it illustrates how these design principles derived from natural systems can help this field as it matures. It also shows that the study of self-assembling complexes could be extended to many other biological processes, in which protein assembly is required, including medical applications.
Materials and Methods
Gene Cloning and Expression in E. coli
Genes used in this study were synthesized by Synbio Technologies (https://www.synbio-tech.com) or cloned from their genomic DNA. Gibson Assembly Cloning Kit (NEB, Ipswich, MA, United States) and other standard gene cloning protocols described previously were used for vector construction (Xu et al., 2017). Primers used in the gene cloning are listed in Supplementary Table S1. 5-alpha E. coli strains (NEB, Ipswich, MA, United States) were used for the gene cloning. pUC57-Kan vector was used for constructing new vectors with various operons related to the 1,3-PDO synthesis pathway. 1,3-PDO synthesis pathway genes were expressed in 5-alpha E. coli strains (NEB, Ipswich, MA, United States) with operons driven by a constitutive promoter from the C. acetobutylicum ATCC 824 thiolase gene. All operons used in this study are listed in Supplementary Table S2. pET vector was used to clone individual genes for their overexpression, and BL21 (DE3) was used for overexpression of individual genes. Cells were grown aerobically in LB supplemented with appropriate antibiotics at 37°C and 225 rpm overnight (seed culture). Twenty milliliters of the seed culture were inoculated into 1 L LB supplemented with appropriate antibiotics, and the cells were grown under the same growth conditions until optical cell density OD600 reached 0.8. The culture was placed on ice for 3 h, then 0.3 mM IPTG was added, and finally grew overnight at 16°C and 225 rpm. The cells were harvested by centrifuge 10,000 xg for 10 min, and were further used for protein extraction and purification.
Protein Purification and Iron-Sulfur Cluster Reconstitution
Frozen cells were lyzed by incubating for 30 min at room temperature followed by BeadBeater lysis with glass beads (Bio Spec Products Inc., Bartlesville, OK, United States; according to manufacturer instructions) in 50 mM Tris, pH 7.0, 100 mM NaCl, and 10 mM imidazole containing EDTA-free protease inhibitor (Thermo Scientific, Rockford, IL, United States; according to manufacturer instructions), 1 U/mL Pierce Universal Nuclease (Thermo Scientific, Rockford, IL, United States) and 5 mg/ml lysozyme (Sigma-Aldrich Corp. St. Louis, MO, United States). The lysate suspension was centrifuged at 15,000 g for 30 min and the resulting supernatant was purified using Ni-NTA chromatography (5 ml HisTrap FF crude column; GE Life Sciences, Piscataway, NJ, United States). Purification was completed with size exclusion chromatography using a HiLoad Superdex 75 (26/60) column (GE Life Sciences, Piscataway, NJ, United States) in 20 mM Tris pH 7.5 and 100 mM NaCl. Strep-tag purification was done according to manufacturer’s instructions for Strep-Tactin XT using a Twin-Strep-tag (IBA Lifesciences GmbH, Göttingen, Germany). When necessary (dhaB2) lysis and purification was done in an anaerobic chamber. Iron-sulfur cluster reconstruction was done several times for different dhaB2 constructs expressed, lyzed, and purified anaerobically or aerobically with and without EDTA removal of the iron-sulfur cluster using two separate protocols by O’Brien et al. (2004) (O'Brien et al., 2004) and Freibert et al. (2018) (Freibert et al., 2018).
Inclusion body preparation: Cells were lyzed as above in 50 mM Tris, pH 7.0, 100 mM NaCl, and 0.02% sodium azide, and the suspension was centrifuged at 15,000 xg for 30 min, its pellet was washed under the same buffer and centrifuge conditions for five times. The pellet suspension was used for SDS-PAGE analysis.
Fermentation for 1,3-PDO Production in E. coli
E. coli cells with operons were grown aerobically in LB medium in the presence of nitrilotriacetic acid (200 mg/L), FeSO4.7H2O (50 mg/L), K2HPO4 (0.5 g/L), Na2SeO4 (30 μg/L), supplemented with 40 g/L glycerol and appropriate antibiotic at 37°C overnight (seed culture), and then the seed culture was transferred to the same medium and continued to be grown anaerobically in an anaerobic chamber (COY, Grass Lake, MI, United States) filled with 5% hydrogen balanced by nitrogen at 37°C and 150 rpm. 2 ml aliquots were withdrawn at designated time points during the fermentation. Concentration of 1,3-PDO produced in the fermentation was measured by HPLC as described below, and the data was used to prepare curves of 1,3-PDO production vs. fermentation time. All fermentations were performed in duplicate, and the reported values are the average of duplicate assays.
Generation and isolation of the self-assembly 1,3-PDO synthesis pathway complex and its conversion of glycerol to 1,3-PDO
E. coli cells transformed with operons were grown anaerobically at 30°C as described above. The protein complex was isolated by amorphous cellulose through procedures described below, under anaerobic conditions. The cells grown from 500 ml culture were lyzed by lysozyme (Hampton Research, Aliso Viejo, CA, United States) or bead-beater (BioSpec Products, Bartlesville, OK, United States) in a buffer containing 100 mM HEPES pH 7.5, 2 mM sodium dithionite, and then supernatant of the cell extract was obtained after centrifugation at 14,000 rpm for 10 min. The supernatant was mixed with 20 mg amorphous cellulose and incubated for 30 min with continuous mixing by inversion at 10 rpm. The suspension was centrifuged at 12,000 rpm for 3 min to recover the pellet, and the pellet was washed by the same buffer as above at least five times. The pellet of cellulose with the bound complex was transferred finally to a 1 ml of reaction solution in the same buffer supplemented with substrate glycerol, SAM (Sigma, St. Louis, MO, United States), NADH (Sigma, St. Louis, MO, United States), and formate when indicated. The reactions were performed at 25°C with continuous mixing by inversion at 10 rpm at designated time points, and finally their supernatants obtained by centrifugation at 12,000 rpm for 3 min. All of the steps described in this section were conducted anaerobically in an anaerobic chamber (5% hydrogen and 95% nitrogen). The supernatants were used to quantify 1,3-PDO generation via HPLC as described below. All assays were performed in triplicate on different cell extracts, and the reported values are the average of the triplicate assays.
Determination of Glycerol, 1,3-PDO and Other Metabolites
Concentration of substrate glycerol, 1,3-PDO and other metabolites produced in fermentation in vivo or by the 1,3-PDO synthesis pathway complex bound to cellulose in vitro, were determined by HPLC. Briefly, samples were filtered through a 0.2-µm filter, and their yield was measured by HPLC using an Aminex HPX87H column with a RID detector maintained at 55°C following standard protocols. The mobile phase used was 0.01 N sulfuric acid at a flow rate of 0.6 ml/min, and a sample injection volume of 20 µL was used. 1,3-PDO standard used to construct calibration curve was purchased from Sigma (St. Louis, MO, United States), whereas glycerol standards were from Fisher Scientific (Denver, CO, United States).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
QX and YB designed the study, QX built the constructs and tested 1,3 PDO production in vivo. MA, PH, and NS conducted protein purification and in vitro assays. HW and NH conducted transformations and protein expression. AM conducted substrate and product analysis. MH helped analyze the results and reviewed the manuscript. YB supervised the research and provided the funding. QX and YB wrote the manuscript and all authors reviewed and approved of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was authored by Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office under Contract DE-AC36-08GO28308 with the National Renewable Energy Laboratory. The publisher, by accepting the article for publication, acknowledges that the U. S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U. S. Government purposes. The views expressed in the article do not necessarily represent the views of the U.S. Department of Energy or the United States Government. We would also like to thank Professor Edward Bayer for sharing the original PCR templates of cohesin and dockerin modules.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2021.680313/full#supplementary-material
References
Artzi, L., Bayer, E. A., and Moraïs, S. (2017). Cellulosomes: Bacterial Nanomachines for Dismantling Plant Polysaccharides. Nat. Rev. Microbiol. 15 (2), 83–95. doi:10.1038/nrmicro.2016.164
Barbieri, L., Luchinat, E., and Banci, L. (2015). Protein Interaction Patterns in Different Cellular Environments Are Revealed by In-Cell NMR. Scientific Rep. 5 (1), 14456. doi:10.1038/srep14456
Barth, A., Hendrix, J., Fried, D., Barak, Y., Bayer, E. A., and Lamb, D. C. (2018). Dynamic Interactions of Type I Cohesin Modules fine-tune the Structure of the Cellulosome of Clostridium Thermocellum. Proc. Natl. Acad. Sci. 115 (48), E11274. doi:10.1073/pnas.1809283115
Bayer, E. A., Belaich, J.-P., Shoham, Y., and Lamed, R. (2004). The Cellulosomes: Multienzyme Machines for Degradation of Plant Cell wall Polysaccharides. Annu. Rev. Microbiol. 58, 521–554. doi:10.1146/annurev.micro.57.030502.091022
Bayer, E. A. (2017). Cellulosomes and Designer Cellulosomes: Why Toy with Nature?. Environ. Microbiol. Rep. 9 (1), 14–15. doi:10.1111/1758-2229.12489
Bergquist, P. L., Siddiqui, S., and Sunna, A. (2020). Cell-Free Biocatalysis for the Production of Platform Chemicals. Front. Energ. Res. 8, 193. doi:10.3389/fenrg.2020.00193
Bomble, Y. J., Beckham, G. T., Matthews, J. F., Nimlos, M. R., Himmel, M. E., and Crowley, M. F. (2011). Modeling the Self-Assembly of the Cellulosome Enzyme Complex*. J. Biol. Chem. 286 (7), 5614–5623. doi:10.1074/jbc.m110.186031
Bowie, J. U., Sherkhanov, S., Korman, T. P., Valliere, M. A., Opgenorth, P. H., and Liu, H. (2020). Synthetic Biochemistry: The Bio-Inspired Cell-free Approach to Commodity Chemical Production. Trends Biotechnol. 38 (7), 766–778. doi:10.1016/j.tibtech.2019.12.024
Cameron, K., Weinstein, J. Y., Zhivin, O., Bule, P., Fleishman, S. J., Alves, V. D., et al. (2015). Combined Crystal Structure of a Type I Cohesin: MUTATION and AFFINITY BINDING STUDIES REVEAL STRUCTURAL DETERMINANTS of COHESIN-DOCKERIN SPECIFICITIES. J. Biol. Chem. 290 (26), 16215–16225. doi:10.1074/jbc.M115.653303
Carvalho, A. L., Dias, F. M. V., Prates, J. A. M., Nagy, T., Gilbert, H. J., Davies, G. J., et al. (2003). Cellulosome Assembly Revealed by the crystal Structure of the Cohesin-Dockerin Complex. Proc. Natl. Acad. Sci. 100 (24), 13809–13814. doi:10.1073/pnas.1936124100
Çelik, E., Ozbay, N., Oktar, N., and Çalık, P. (2008). Use of Biodiesel Byproduct Crude Glycerol as the Carbon Source for Fermentation Processes by RecombinantPichiapastoris. Ind. Eng. Chem. Res. 47 (9), 2985–2990. doi:10.1021/ie071613o
Chae, T. U., Choi, S. Y., Kim, J. W., Ko, Y.-S., and Lee, S. Y. (2017). Recent Advances in Systems Metabolic Engineering Tools and Strategies. Curr. Opin. Biotechnol. 47, 67–82. doi:10.1016/j.copbio.2017.06.007
Chen, C.-T., and Liao, J. C. (2016). Frontiers in Microbial 1-butanol and Isobutanol Production. FEMS Microbiol. Lett. 363 (5), fnw020. doi:10.1093/femsle/fnw020
Chen, H., Huang, R., Kim, E. J., and Zhang, Y. H. P. J. (2018). Building a Thermostable Metabolon for Facilitating Coenzyme Transport and In Vitro Hydrogen Production at Elevated Temperature. ChemSusChem. 11 (18), 3120–3130. doi:10.1002/cssc.201801141
Chubukov, V., Mukhopadhyay, A., Petzold, C. J., Keasling, J. D., and Martín, H. G. (2016). Synthetic and Systems Biology for Microbial Production of Commodity Chemicals. NPJ Syst. Biol. Appl. 2, 16009. doi:10.1038/npjsba.2016.9
Cohen, R. D., and Pielak, G. J. (2017). A Cell Is More Than the Sum of its (Dilute) Parts: A Brief History of Quinary Structure. Protein Sci. 26 (3), 403–413. doi:10.1002/pro.3092
Deeds, E. J., Ashenberg, O., Gerardin, J., and Shakhnovich, E. I. (2007). Robust Protein Protein Interactions in Crowded Cellular Environments. Proc. Natl. Acad. Sci. 104 (38), 14952–14957. doi:10.1073/pnas.0702766104
Freibert, S.-A., Weiler, B. D., Bill, E., Pierik, A. J., Mühlenhoff, U., Lill, R., et al. (2018). “Biochemical Reconstitution and Spectroscopic Analysis of Iron-Sulfur Proteins,” in Methods in Enzymology. Editor S. S. David (Academic Press), 197–226. doi:10.1016/bs.mie.2017.11.034
Hammel, M., Fierobe, H.-P., Czjzek, M., Kurkal, V., Smith, J. C., Bayer, E. A., et al. (2005). Structural Basis of Cellulosome Efficiency Explored by Small Angle X-ray Scattering. J. Biol. Chem. 280 (46), 38562–38568. doi:10.1074/jbc.m503168200
Jarboe, L. R. (2011). YqhD: a Broad-Substrate Range Aldehyde Reductase with Various Applications in Production of Biorenewable Fuels and Chemicals. Appl. Microbiol. Biotechnol. 89 (2), 249–257. doi:10.1007/s00253-010-2912-9
Jiang, W., Wang, S., Wang, Y., and Fang, B. (2016). Key Enzymes Catalyzing Glycerol to 1,3-propanediol. Biotechnol. Biofuels 9 (1), 57. doi:10.1186/s13068-016-0473-6
Kahn, A., Moraïs, S., Galanopoulou, A. P., Chung, D., Sarai, N. S., Hengge, N., et al. (2019). Creation of a Functional Hyperthermostable Designer Cellulosome. Biotechnol. Biofuels 12, 44. doi:10.1186/s13068-019-1386-y
Kay, J. E., and Jewett, M. C. (2015). Lysate of Engineered Escherichia coli Supports High-Level Conversion of Glucose to 2,3-butanediol. Metab. Eng. 32, 133–142. doi:10.1016/j.ymben.2015.09.015
Kluge, M., Pérocheau Arnaud, S., and Robert, T. (2019). 1,3-Propanediol and its Application in Bio-Based Polyesters for Resin Applications. Chem. Africa 2 (2), 215–221. doi:10.1007/s42250-018-0026-4
Knietsch, A., Bowien, S., Whited, G., Gottschalk, G., and Daniel, R. (2003). Identification and Characterization of Coenzyme B 12 -Dependent Glycerol Dehydratase- and Diol Dehydratase-Encoding Genes from Metagenomic DNA Libraries Derived from Enrichment Cultures. Appl. Environ. Microbiol. 69 (6), 3048–3060. doi:10.1128/aem.69.6.3048-3060.2003
Korman, T. P., Opgenorth, P. H., and Bowie, J. U. (2017). A Synthetic Biochemistry Platform for Cell Free Production of Monoterpenes from Glucose. Nat. Commun. 8, 15526. doi:10.1038/ncomms15526
Lama, S., Ro, S. M., Seol, E., Sekar, B. S., Ainala, S. K., Thangappan, J., et al. (2015). Characterization of 1,3-propanediol Oxidoreductase (DhaT) from Klebsiella pneumoniae J2B. Biotechnol. Bioproc. E 20 (6), 971–979. doi:10.1007/s12257-015-0635-6
Lee, J. H., Jung, M.-Y., and Oh, M.-K. (2018). High-yield Production of 1,3-propanediol from Glycerol by Metabolically Engineered Klebsiella pneumoniae. Biotechnol. Biofuels 11 (1), 104. doi:10.1186/s13068-018-1100-5
Liu, J.-z., Xu, W., Chistoserdov, A., and Bajpai, R. K. (2016). Glycerol Dehydratases: Biochemical Structures, Catalytic Mechanisms, and Industrial Applications in 1,3-Propanediol Production by Naturally Occurring and Genetically Engineered Bacterial Strains. Appl. Biochem. Biotechnol. 179 (6), 1073–1100. doi:10.1007/s12010-016-2051-6
O'Brien, J. R., Raynaud, C., Croux, C., Girbal, L., Soucaille, P., and Lanzilotta, W. N. (2004). Insight into the Mechanism of the B12-independent Glycerol Dehydratase fromClostridium Butyricum: Preliminary Biochemical and Structural Characterization‡. Biochemistry 43 (16), 4635–4645. doi:10.1021/bi035930k
Petroll, K., Kopp, D., Care, A., Bergquist, P. L., and Sunna, A. (2019). Tools and Strategies for Constructing Cell-free Enzyme Pathways. Biotechnol. Adv. 37 (1), 91–108. doi:10.1016/j.biotechadv.2018.11.007
Plácido, J., and Capareda, S. (2016). Conversion of Residues and By-Products from the Biodiesel Industry into Value-Added Products. Bioresources and Bioprocessing 3 (1), 23. doi:10.1186/s40643-016-0100-1
Raynaud, C., Sarçabal, P., Meynial-Salles, I., Croux, C., and Soucaille, P. (2003). Molecular Characterization of the 1,3-propanediol (1,3-PD) Operon of Clostridium Butyricum. Proc. Natl. Acad. Sci. 100 (9), 5010. doi:10.1073/pnas.0734105100
Rieckenberg, F., Ardao, I., Rujananon, R., and Zeng, A.-P. (2014). Cell-free Synthesis of 1,3-propanediol from Glycerol with a High Yield. Eng. Life Sci. 14 (4), 380–386. doi:10.1002/elsc.201400034
Rollin, J. A., Bomble, Y. J., St. John, P. C., and Stark, A. K. (2021). Biochemical Production with Purified Cell-free Systems. Biochem. Eng. J. 166, 107002. doi:10.1016/j.bej.2018.07.016
Saxena, R. K., Anand, P., Saran, S., and Isar, J. (2009). Microbial Production of 1,3-propanediol: Recent Developments and Emerging Opportunities. Biotechnol. Adv. 27 (6), 895–913. doi:10.1016/j.biotechadv.2009.07.003
Smith, S. P., Bayer, E. A., and Czjzek, M. (2017). Continually Emerging Mechanistic Complexity of the Multi-Enzyme Cellulosome Complex. Curr. Opin. Struct. Biol. 44, 151–160. doi:10.1016/j.sbi.2017.03.009
Stephanopoulos, G. (2007). Challenges in Engineering Microbes for Biofuels Production. Science 315 (5813), 801–804. doi:10.1126/science.1139612
Sulzenbacher, G., Alvarez, K., van den Heuvel, R. H. H., Versluis, C., Spinelli, S., Campanacci, V., et al. (2004). Crystal Structure of E.Coli Alcohol Dehydrogenase YqhD: Evidence of a Covalently Modified NADP Coenzyme. J. Mol. Biol. 342 (2), 489–502. doi:10.1016/j.jmb.2004.07.034
Toraya, T., Shirakashi, T., Kosuga, T., and Fukui, S. (1976). Substrate Specificity of Coenzyme B12-dependent Diol Dehydrase: Glycerol as Both a Good Substrate and a Potent Inactivator. Biochem. Biophysical Res. Commun. 69 (2), 475–480. doi:10.1016/0006-291x(76)90546-5
Xu, Q., Knoshaug, E. P., Wang, W., Alahuhta, M., Baker, J. O., Yang, S., et al. (2017). Expression and Secretion of Fungal Endoglucanase II and Chimeric Cellobiohydrolase I in the Oleaginous Yeast Lipomyces Starkeyi. Microb. Cel. factories 16 (1), 126. doi:10.1186/s12934-017-0742-5
Yamanishi, M., Kinoshita, K., Fukuoka, M., Saito, T., Tanokuchi, A., Ikeda, Y., et al. (2012). Redesign of Coenzyme B12 Dependent Diol Dehydratase to Be Resistant to the Mechanism-Based Inactivation by Glycerol and Act on Longer Chain 1,2-diols. FEBS J. 279 (5), 793–804. doi:10.1111/j.1742-4658.2012.08470.x
Yang, F., Hanna, M. A., and Sun, R. (2012). Value-added Uses for Crude Glycerol-Aa Byproduct of Biodiesel Production. Biotechnol. Biofuels 5 (1), 13. doi:10.1186/1754-6834-5-13
You, C., Myung, S., and Zhang, Y.-H. P. (2012). Facilitated Substrate Channeling in a Self-Assembled Trifunctional Enzyme Complex. Angew. Chem. Int. Ed. 51 (35), 8787–8790. doi:10.1002/anie.201202441
You, C., and Zhang, Y.-H. P. (2013). Self-assembly of Synthetic Metabolons through Synthetic Protein Scaffolds: One-step Purification, Co-immobilization, and Substrate Channeling. ACS Synth. Biol. 2 (2), 102–110. doi:10.1021/sb300068g
Zeng, A.-P., and Sabra, W. (2011). Microbial Production of Diols as Platform Chemicals: Recent Progresses. Curr. Opin. Biotechnol. 22 (6), 749–757. doi:10.1016/j.copbio.2011.05.005
Keywords: glycerol, biocatalysis, metabolic pathway, biochemical, cellulose, cellulosome assembly
Citation: Xu Q, Alahuhta M, Hewitt P, Sarai NS, Wei H, Hengge NN, Mittal A, Himmel ME and Bomble YJ (2021) Self-Assembling Metabolon Enables the Cell Free Conversion of Glycerol to 1,3-Propanediol. Front. Energy Res. 9:680313. doi: 10.3389/fenrg.2021.680313
Received: 14 March 2021; Accepted: 09 June 2021;
Published: 15 July 2021.
Edited by:
Abdul-Sattar Nizami, Government College University, PakistanReviewed by:
Dehua Liu, Tsinghua University, ChinaShishir P. S. Chundawat, Rutgers The State University of New Jersey, United States
Copyright © 2021 Xu, Alahuhta, Hewitt, Sarai, Wei, Hengge, Mittal, Himmel and Bomble. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yannick J. Bomble, eWFubmljay5ib21ibGVAbnJlbC5nb3Y=