 Wenguang Xiong
Wenguang Xiong Yongxue Sun
Yongxue Sun Xueyao Ding
Xueyao Ding Yiming Zhang
Yiming Zhang Zhenling Zeng
Zhenling Zeng- National Laboratory of Safety Evaluation (Environmental Assessment) of Veterinary Drugs, College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
Antibiotic resistance genes (ARGs) in the environment have paid great concern due to their health risk. We investigated antibiotics concentrations (tetracyclines, sulfonamides, and fluoroquinolones), ARGs abundances (tetracycline, sulfonamide, and plasmid-mediated quinolone resistance (PMQR) genes), and bacterial community composition in sediment and water samples in the Liuxi River, China. Antibiotics concentrations were determined by ultra-performance liquid chromatography-electrospray tandem mass spectrometry. ARGs abundances were quantified by a culture-independent method. Bacterial community composition was analyzed by metagenomic approach based on Ion Torrent Personal Genome Machine platform. Antibiotics concentrations were at the levels of 1.19–622 ug kg−1 in sediment samples and below the limit of detection to 127 ng L−1 in water samples. Relative abundances (ARGs copies/16S rRNA gene copies) of detected ARGs were at the range of 10−5 to 10−2. The dominant phyla were Proteobacteria, Bacteroidetes, and Verrucomicrobia in sediment samples, and were Proteobacteria, Actinobacteria, and Bacteroidetes in water samples. The results indicated that the river environment was contaminated by antibiotics and may be as a reservoir of ARGs. This study provided quantitative data on antibiotics, ARGs and bacterial community composition in the Liuxi River, a geographical location different from the reported studies.
Introduction
Antibiotic resistance genes (ARGs) in the environment have drawn great concern due to their health risk. The environment may be as reservoirs of ARGs, which could be acquired by clinical pathogens through horizontal gene transfer (Forsberg et al., 2012; Yang et al., 2013). Aquatic system serves as a key milieu for the environmental release, mixing, persistence and spread of ARGs, and antibiotic resistance bacteria (Allen et al., 2010; Taylor et al., 2011). Evidences suggested that waste discharges from anthropogenic compartments, such as animal feedlots and wastewater treatment plants significantly contributed to the pollution of ARGs in downstream rivers (Pruden et al., 2012; Hsu et al., 2014). The presence of ARGs in river environment may pose a potential risk to public health, as river water is often used for irrigation, recreation and also used as a source of drinking water. Riverine ARGs and antibiotic resistance bacteria could spread into drinking water and threaten public health via food chain (Walsh et al., 2011; Jiang et al., 2013). However, only a few studies investigated the abundance of ARGs including tetracycline (tet) resistance genes and sulfonamide (sul) resistance genes in the global rivers, such as the Almendares River in Cuba (Graham et al., 2010) and South Platte River in USA (Pruden et al., 2012) by culture independent methods; additionally, only one study have investigated the abundance of plasmid-mediated quinolone resistance (PMQR) genes by using a culture independent method in river environment in India (Rutgersson et al., 2014), which need complementary local studies at different geographical locations. What is more, bacterial community composition in river environment has not been comprehensive investigated. More studies have identified the bacteria in river environment by culture-based methods (Mitch et al., 2010; Marti et al., 2013b). To date, however, not enough studies have identified the bacterial community composition in river environment by metagenomic approach.
The Liuxi River is one of the most important rivers in Guangzhou, China. It flows through an agriculture area and a resident urban area, before discharges into South China Sea. In the past decades, there have been hundreds of chemical plants and thousands of animal feedlots adjacent to the river's bank. The liquid effluents from chemical plants and animal feedlots are commonly discharged into the Liuxi River. Therefore, ARGs in the Liuxi River may be significantly promoted by anthropogenic activities. Furthermore, ARGs in the Liuxi River may be accessible by humans, since the water from the river is usually used for irrigation, recreation and as a source of drinking water for metropolitan Guangzhou city, with a population of more than 15 million people.
The objective of this study was to investigate ARGs occurrence and bacterial community composition in the Liuxi River. A comprehensive report was addressed, by (1) detecting the concentrations of antibiotics (tetracyclines, sulfonamides and fluoroquinolones) (2) quantifying the abundances of ARGs (tet resistance genes, sul resistance genes and PMQR genes) by using a culture independent method (3) analyzing the bacterial community composition by metagenomic approach. This study provided quantitative data on antibiotics, ARGs and bacterial community composition in the Liuxi River, a geographical location different from the reported studies.
Materials and Methods
Study Site
The Liuxi River initially flows Conghua city (site A, upstream 1), and then loops Zhongluotan town (site B, upstream 2) and Renhe town (site C, downstream 1), finally passes through Nangang town (site D, downstream 2). The four sample sites cover the whole river. Sampling locations and potential pollution sources are marked in Figure 1. In Figure 1, animal unit (AU), equivalent to 454 kg animal body weight is a standardized method for quantification of animal feedlots (Kellogg et al., 2000); chemical oxygen demand (COD) is an important mark for wastewater pollution. The values of COD in Figure 1 were referenced to He et al. (2012).
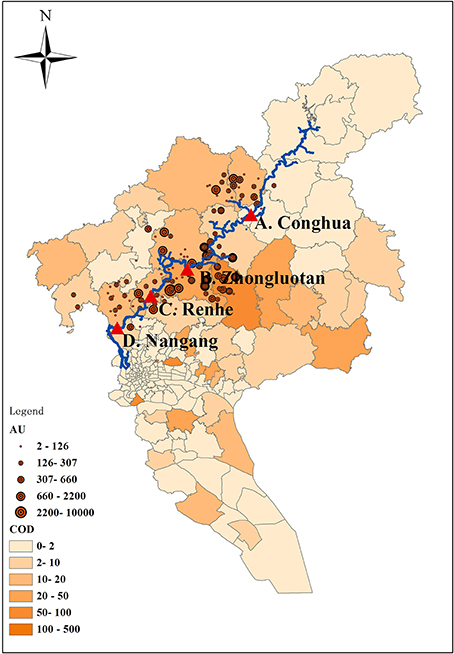
Figure 1. Sampling locations and potential pollution sources. AU, animal unit, equivalent to 454 kg animal body weight. COD, Chemical Oxygen Demand.
Sample Collection
Surface water (3 L) from the top 0.5 m of the water surface was collected using sterile polystyrene bottles. Sediment (~500 g) was collected from the top 5 cm layer using a gravity-corer. Sample sites are defined as A, B, C, and D. For example, A-w and A-s are paired water and sediment samples from A site; this is also the same for the samples from the other sample sites. For one sample site, three samples collected from different locations along the river cross section were combined to analyze. Samples were stored as sterile as possible in a cooler box, until immediate returned to the laboratory for processing (<12 h). After homogeneous mixing, each water or sediment sample was divided into two aliquots under aseptic condition. One aliquot was used for the molecular analyses; the other one was used for the quantification of antibiotics. Each aliquot was further divided into three subsamples for repeated analysis.
Antibiotic Analysis
Antibiotics including tetracyclines (oxytetracycline, chlorotetracycline and doxycycline), sulfonamides (sulfametoxydiazine, sulfamethazine and sulfamethoxazole) and fluoroquinolones (ciprofloxacin, norfloxacin and enrofloxacin) were chosen to analyze due to their important role in human and animal medicine. Antibiotic analysis was determined by ultra-performance liquid chromatography-electrospray tandem mass spectrometry according to the method of Luo et al. (2010). Antibiotic analysis was performed on an Agilent 1200 liquid chromatograph with a Waters Quattro Micro triple quadrupole mass spectrometer. The recoveries for antibiotics based on matrix-matched calibration were at the range of 71–93% in water samples and 65–88% in sediment samples, and the quantification limits were at the range of 5.3–12 ng L−1 for water and 2.1–5.6 μg kg−1 for sediment.
DNA Extraction
Particulates in water samples were removed by layered filtration with Whatman paper (20–25, 6 and 1 μm, respectively). Approximately 500 mL of prefiltered water subsample was immediately concentrated in duplicate by using a sterile filter (0.2 μm). Subsequent extraction steps were conducted according to the manufacture's protocol from Power Water DNA Kit (Mo Bio Laboratories, Inc., CA, USA). Sediment samples were centrifuged at 10,000×g for 30 s at room temperature to remove liquid. Weighted 0.25 g of sediment sample was extracted in duplicate by using a commercial Power Soil DNA Kit (Mo Bio Laboratories, Inc., CA, USA) according to the manufacturer's instructions. The concentration and quality of the extracted DNA were determined by spectrophotometer analysis and agarose gel electrophoresis.
PCR Assay
Qualitative PCR assays were used to determine the presence of various ARGs, including tet resistance genes [tet(M), tet(O), tet(W), tet(S), tet(Q), tet(X) and tet(B/P)], sul resistance genes [sul(1), sul(2) and sul(3)], PMQR genes [qep(A), oqx(A), oqx(B), aac(6′)-Ib and qnr(S)], and bacterial 16S rRNA gene. These ARGs are usually observed in human affecting compartments. All PCR assays were performed in a 25 μL volume reaction by using TaKaRa Ex Taq PCR Kit according to the manufacturer's instructions. The details regarding PCR assay (primers sequences, amplicon size, and annealing temperature) are described in Supplementary Material Sections Table S1. PCR products were analyzed by gel electrophoresis using 1% (w/v) agarose in 1× TAE buffer.
qPCR Assay
The PCR product of each gene was purified, and ligated into pMD20-T Vector (TaKaRa, Dalian, China) before being cloned into Escherichia coli DH5à. Clones containing targeted gene inserts were selected and confirmed by PCR. Plasmids carrying targeted genes were extracted, purified and sequenced. Ten-fold serially diluted plasmids over six orders of magnitude were performed to generate standard curves for quantification of each gene in qPCR. qPCR amplifications were performed on a Bio-Rad IQ5 instrument (Bio-Rad Company, USA) by using SYBR Premix Ex Taq II (TaKaRa, Dalian, China) according to the manufacturer's instructions. The positive (using plasmid DNA as template DNA) and negative (using nuclease-free water instead of template DNA) controls were included in each run. Duplicate real-time assays were performed for all subsamples and controls. The details regarding qPCR assay (primers, amplicon size and annealing temperature) are described in Supplementary Material Sections Table S1. The specificity was verified by the melting curves and gel electrophoresis. qPCR efficiency of each gene (93–105%) was examined with R2 values more than 99.1% for all calibration curves. To minimize the variance caused by overall extraction efficiencies, total bacterial community and possible sample degradation, the relative abundances of targeted genes were obtained by normalizing their copies to 16S rRNA gene copies.
Metagenomic Sequencing
Most bacterial species could be distinguished by partial V6 region in 16S rRNA gene (Chakravorty et al., 2007). This region was targeted by the universal bacterial primers (F: 5′-ACTCAAATGAATTGACGGGG-3′) and (R: 5′-GCTCGTTGCGGGACTTAA-3′). Each primer was labeled with an adaptor sequence and unique multiplex identifier code. PCRs were performed in 50 uL reaction volume containing 0.5 μL Plantium Taq (5 U μL−1), 0.5 μL of each primer (50 μM), 10 ng genomic DNA, 0.5 μL dNTP (10 mM each), and added up to 50 μL by molecular biology-grade water. PCR cycle consisted of 30 s initial denaturation at 94°C, 5 cycles of 20 s at 94°C, 20 s at 45°C, and 60 s at 65°C, followed by 20 cycles of denaturation at 94°C for 20 s and annealing at 60°C for 20 s, then 72°C for 20 s, and a final elongation step of 5 min at 72°C. PCR amplicons were gel purified and quantified. The emulsion PCR was carried out by applying the Ion Torrent Personal Genome Machine (PGM) Template 200 Kit as described in the appropriate user Guide provide by the manufacturer. Amplicon libraries were sequenced on the Ion Torrent PGM system with Ion 316™ chip by using Ion Sequencing 200 kit. The reason why we chosen Ion Torrent PGM system was because microbial community profiles produced by Ion Torrent PGM are highly comparable to those produced by 454 sequencing (Yergeau et al., 2012), and Ion Torrent PGM provides a low cost, scalable and high throughput solution for metagenomic analyses (Rothberg et al., 2011).
Identifier codes were trimmed from each sequence. Raw sequences were filtered using a moving average Q20 cutoff. Raw sequences were submitted to Sequence Read Archive database in NCBI (accession No. PRJNA230277).
Results
Antibiotics Concentrations
As summarized in Figure 2 and Supplementary Material Sections Table S2, three classes of antibiotics including nine antibiotics (oxytetracycline, chlorotetracycline, doxycycline, sulfametoxydiazine sulfamethazine, sulfamethoxazole, ciprofloxacin, norfloxacin, and enrofloxacin) were detected with concentrations ranging from 1.19 to 622 μg kg−1 in sediment samples and below the limit of detection to 127 ng L−1 in water samples. Of the tetracyclines analyzed in this study, oxytetracycline had the highest concentrations in sediment samples, ranging from 3.77 to 147 μg kg−1, and chlorotetracycline had the highest concentrations in water samples, ranging from from 25.4 to 127 ng L−1. Of the sulfonamides analyzed in this study, sulfamethoxazole had the highest concentrations in sediment samples, ranging from 1.49 to 2.74 μg kg−1, and it is also had the highest concentrations in water samples, ranging from 12.6 to 45.0 ng L−1. Of the fluoroquinolones analyzed in this study, enrofloxacin had the highest concentrations in sediment samples, ranging from 371 to 622 μg kg−1, and norfloxacin had the highest concentrations in water samples, ranging from 27.6 to 118 ng L−1.
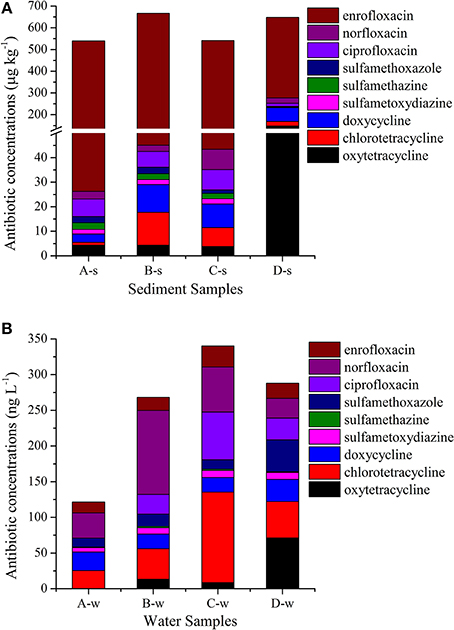
Figure 2. Antibiotic concentrations in sediment samples (A) and in water samples (B).
Occurrence and Levels of Resistance Genes
Absolute abundance (copies per mL or per g) and relative abundance (ARGs copies/16S rRNA gene copies) of ARGs are shown in Supplementary Material Sections Table S3 and Table S4, respectively. Among 15 ARGs investigated, most of them were detected in all sediment and water samples; tet(M) in A-w, tetS in A-w, B-w, and C-w samples were not detected; and tet(B/P) and qep(A) were not observed at all (Figure 3 and Supplementary Material Sections Table S4). ARGs [tet(M), tet(O), tet(W), tet(S), tet(Q), tet(X), sul(1), sul(2), sul(3), oqx(A), oqx(B), aac(6′)-Ib and qnr(S)] were detected with relative abundance ranging from 1.59 × 10−5 to 3.49 × 10−2. oqx(B) had the highest abundance among all detected genes for all samples, ranging from 2.29 × 10−3 to 3.49 × 10−2. Of tet resistance genes analyzed in this study, tet(W) had the highest abundance, ranging from 1.22 × 10−3 to 1.06 × 10−2. Of sul resistance genes analyzed in this study, sul(1) had the highest abundance, ranging from 4.19 × 10−3 to 1.72 × 10−2.
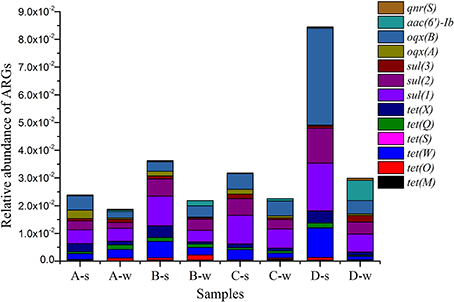
Figure 3. Relative abundance of antibiotic resistance genes (ARGs) in all samples.
Bacterial Community Composition
A total of 226, 795 reads with average 28, 349 high quality sequences per sample were obtained. RDP Classifier tool was used to identify the phylogenetic classification of sequences (Cole et al., 2009). Most of the sequences were assigned to twenty one phyla in the samples (Figure 4). Generally, the dominant phyla were Proteobacteria, Bacteroidetes, and Verrucomicrobia in sediment samples as high as 48.81, 4.82, and 4.28%, respectively, and the dominant phyla were Proteobacteria, Actinobacteria, and Bacteroidetes in water samples as high as 86.39, 11.22, and 4.75%, respectively. Some phyla, such as Fusobacteria, Chlamydiae, Synergistetes were at low abundance of <0.1%. Some bacterial classes might specifically associated with antibiotic containing environment were also observed in this study, such as Deltaproteobacteria (1.06–4.28%), Epsilonproteobacteria (0.01–0.2%), Clostridia (1.11–1.84%) and Bacilli (0.16–0.54%) in sediment samples; Deltaproteobacteria (0.03–0.16%), Epsilonproteobacteria (0.01–0.68%), Clostridia (0.04–0.35%) and Bacilli (0.03–1.26%) in water samples.
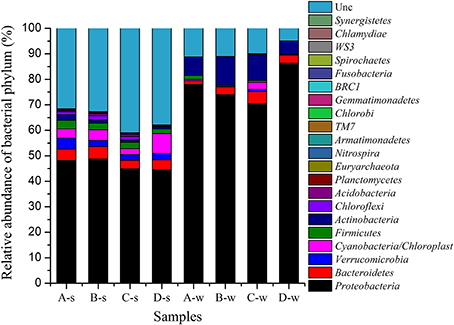
Figure 4. Phylum for bacterial community composition in all samples.
Discussion
Several studies have reported antibiotic pollution in river environments in China, such as the Huangpu River (Jiang et al., 2011), the Yellow River, Hai River, and Liao River (Zhou et al., 2011). In those studies, the concentrations of most antibiotics detected in the river environments were elevated above background concentrations. In sediment samples collected from the Yellow River, Hai River, Liao River, oxytetracycline had the concentrations as high as 653 μg kg−1, sulfamethoxazole had not detected at all, and norfloxacin had the highest concentrations of up 5770 μg kg−1 among the fluoroquinolones (ciprofloxacin, norfloxacin, and enrofloxacin) analyzed. Those results were different from the results from the present study, supporting that the distribution of antibiotics in the environment are dependent on the antibiotic consumption, use patterns, antibiotic partition characteristics and chemical stability (Hari et al., 2005). The waste effluents containing antibiotics from animal feedlots and chemical plants (Figure 1) were probably responsible for the antibiotic pollution in the Liuxi River.
Since ARGs were defined as emerging contaminants (Pruden et al., 2006), several studies have investigated the occurrence and abundance of ARGs in aquatic environment worldwide. In aquaculture environment in China, the relative abundances of sul(1 and 2) and tet(W, O and M) were at levels of 10−5 to 10−3 and 10−5 to 10−2, respectively in sediment samples (Gao et al., 2012). In river environment in USA, tet(W) was ubiquitous in sediment and water samples, with the relative abundance of 10−4 to 10−2 (LaPara et al., 2011). And also in river environment in Pakistan, the relative abundance of sul(1) was at the level of 10−4 to 10−2 in water samples (Khan et al., 2013). The results of the relative abundance of sul and tet resistance genes in those studies were comparable to the results from the present study. To date, only a few studies have investigated the occurrence of PMQR genes such as oqx(A), oqx(B), aac(6′)-Ib and qnr(S) in river environments using a culture independent method. In the present study, relative abundances of these PMQR genes were at a range of 10−5 to 10−2 in sediment and water samples. If as Graham et al. (2010) suggested that relative abundance of 10−6 to10−8 is typical of pristine areas as background, whereas highly contaminated sites often have the relative abundance of >10−4, the Liuxi River were highly contaminated by tet resistance genes, sul resistance genes and PMQR genes. Riverine ARGs were correlated with human activities (Pruden et al., 2012). Evidences suggested that human affecting compartments, such as wastewater treatment plants, animal feedlots and pharmaceutical factories were as significant point sources of ARGs into the receiving water bodies (Graham et al., 2010; LaPara et al., 2011; Li et al., 2012), which may also explain the reason why high contaminations of ARGs were observed in the Liuxi River. Besides antibiotics, metals also co-select for ARGs (Knapp et al., 2011). Furthermore, another important mechanism horizontal gene transfer plays a critical role in the spread and persistence of ARGs in the environment (Andam et al., 2011). Aquatic environments provide ideal settings for the horizontal exchange (O′Brien, 2002; Baquero et al., 2008). Mobile genetic elements, often observed in river environments and other environmental compartments may drive the transfer of resistance determinants between the bacteria of anthropogenic origin and environmental indigenous bacteria (Kristiansson et al., 2011; Zhang et al., 2011; Hsu et al., 2014). The Liuxi River is usually used for irrigation, recreation and as a source of drinking water for metropolitan people, which probably accelerated the exchange of resistance genes between the water bacteria and human microbe.
The dominant phyla were Proteobacteria, Bacteroidetes, and Verrucomicrobia in sediment samples and Proteobacteria, Actinobacteria, and Bacteroidetes in water samples in the Liuxi River. The dominant bacterial groups in water samples in this study were as the same as those in water samples collected from water supply reservoirs in Spain (Huerta et al., 2013), but it was different from the result from a river influenced by a wastewater treatment plant in Spain (Marti et al., 2013a), with the dominant phyla of Proteobacteria (or Firmicutes) and Cyanobactera (or Actinobacteria). The dominant phyla in sediment samples were also variable in different studies. In the above water supply reservoirs in Spain, Actinobacteria, Firmicutes, and Proteobacteria were observed as dominant phyla in sediment samples. In an antibiotic-contaminated river influenced by a wastewater treatment plant in India (Kristiansson et al., 2011), Proteobacteria, Bacteroidetes, and Firmicutes were dominated in sediment samples. Those results from the rivers with or without receiving wastewater compared together indicated that dominant bacterial phyla in river environments were influenced by antibiotic contaminations. Furthermore, the ubiquitous presence of Deltaproteobacteria, Epsilonproteobacteria, Clostridia, and Bacilli in sediment and water samples also indicated that the Liuxi River was contaminated by antibiotics, since Li suggested that Deltaproteobacteria, Epsilonproteobacteria, Clostridia, and Bacilli might be specifically associated with antibiotic containing environment (Li et al., 2011). In the present study, some phyla such as Synergistetes and TM7 were recently recognized, and were reported for the first time in river environments by using metagenome analysis. Some species/strains belonging to the phyla reported in the present study may have implication in human diseases. Fusobacteria have been shown to play important role in intra-amniotic infections, premature labor, and tropical ulcers (Sadek et al., 1998); some species/strains of Chlamydiae are obligate intracellular pathogens (Byrne and Ojcius, 2004); and Synergistetes have been suggested to be involved in periodontal disease, soft tissue infections and gastrointestinal infections (Horz et al., 2006; Vartoukian et al., 2007).
There were several limitations to our study. First, we just collected samples at four sites in the main stream and did not analyze the contaminations in details. In future studies, we would analyze the correlation of antibiotic concentration and ARG abundance between sediment and pared water samples, and also analyze the correlation between the pollutant levels and emission source typology in more samples collected from the main stream and tributaries of this river. Second, we had chosen tet resistance genes only belonging to Gram-positive bacteria. A more extensive determination of tet resistance genes harbored by Gram-positive and negative bacteria would permit a more detailed assessment of the distribution of tet resistance genes in the environment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grant 2013CB 127203 from the National Key Basic Research Program of China and grant 31172368 from the National Natural Science Foundation of China.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fenvs.2014.00061/abstract
References
Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., and Handelsman, J. (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259. doi: 10.1038/nrmicro2312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andam, C. P., Fournier, G. P., and Gogarten, J. P. (2011). Multilevel populations and the evolution of antibiotic resistance through horizontal gene transfer. FEMS Microbiol. Rev. 35, 756–767. doi: 10.1111/j.1574-6976.2011.00274.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baquero, F., Martínez, J.-L., and Cantón, R. (2008). Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotech. 19, 260–265. doi: 10.1016/j.copbio.2008.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Byrne, G. I., and Ojcius, D. M. (2004). Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat. Rev. Microbiol. 2, 802–808. doi: 10.1038/nrmicro1007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chakravorty, S., Helb, D., Burday, M., Connell, N., and Alland, D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Meth. 69, 330–339. doi: 10.1016/j.mimet.2007.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. doi: 10.1093/nar/gkn879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forsberg, K. J., Reyes, A., Wang, B., Selleck, E. M., Sommer, M. O., and Dantas, G. (2012). The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–1111. doi: 10.1126/science.1220761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, P., Mao, D., Luo, Y., Wang, L., Xu, B., and Xu, L. (2012). Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res. 46, 2355–2364. doi: 10.1016/j.watres.2012.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graham, D. W., Olivares-Rieumont, S., Knapp, C. W., Lima, L., Werner, D., and Bowen, E. (2010). Antibiotic resistance gene abundances associated with waste discharges to the Almendares River near Havana, Cuba. Environ. Sci. Technol. 45, 418–424. doi: 10.1021/es102473z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hari, A. C., Paruchuri, R. A., Sabatini, D. A., and Kibbey, T. C. (2005). Effects of pH and cationic and nonionic surfactants on the adsorption of pharmaceuticals to a natural aquifer material. Environ. Sci. Technol. 39, 2592–2598. doi: 10.1021/es048992m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, L., Yu, L. S., Jiang, D. P., and Li, Z. Q. (2012). Research on spatial distribution of livestock and poultry pollutant in Guangzhou City. Ecol. Sci. 31, 6. doi: 10.3969/j.issn
Horz, H.-P., Citron, D. M., Warren, Y. A., Goldstein, E. J., and Conrads, G. (2006). Synergistes group organisms of human origin. J. Clin. Microbiol. 44, 2914–2920. doi: 10.1128/JCM.00568-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, J.-T., Chen, C.-Y., Young, C.-W., Chao, W.-L., Li, M.-H., Liu, Y.-H., et al. (2014). Prevalence of sulfonamide-resistant bacteria, resistance genes and integron-associated horizontal gene transfer in natural water bodies and soils adjacent to a swine feedlot in northern Taiwan. J. Hazard. Mater. 277, 34–43. doi: 10.1016/j.jhazmat.2014.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huerta, B., Marti, E., Gros, M., López, P., Pompêo, M., Armengol, J., et al. (2013). Exploring the links between antibiotic occurrence, antibiotic resistance, and bacterial communities in water supply reservoirs. Sci. Total Environ. 456, 161–170. doi: 10.1016/j.scitotenv.2013.03.071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, L., Hu, X., Xu, T., Zhang, H., Sheng, D., and Yin, D. (2013). Prevalence of antibiotic resistance genes and their relationship with antibiotics in the Huangpu River and the drinking water sources, Shanghai, China. Sci. Total Environ. 458, 267–272. doi: 10.1016/j.scitotenv.2013.04.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, L., Hu, X., Yin, D., Zhang, H., and Yu, Z. (2011). Occurrence, distribution and seasonal variation of antibiotics in the Huangpu River, Shanghai, China. Chemosphere 82, 822–828. doi: 10.1016/j.chemosphere.2010.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kellogg, R. L., Lander, C. H., Moffitt, D. C., and Gollehon, N. (2000). Manure nutrients relative to the capacity of cropland and pastureland to assimilate nutrients: Spatial and temporal trends for the United States. P. Water Environ. Federation 2000, 18–157. doi: 10.2175/193864700784994812
Khan, G. A., Berglund, B., Khan, K. M., Lindgren, P.-E., and Fick, J. (2013). Occurrence and abundance of antibiotics and resistance genes in rivers, canal and near drug formulation facilities-a study in Pakistan. PLoS ONE 8:e62712. doi: 10.1371/journal.pone.0062712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knapp, C. W., McCluskey, S. M., Singh, B. K., Campbell, C. D., Hudson, G., and Graham, D. W. (2011). Antibiotic resistance gene abundances correlate with metal and geochemical conditions in archived Scottish soils. PLoS ONE 6:e27300. doi: 10.1371/journal.pone.0027300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kristiansson, E., Fick, J., Janzon, A., Grabic, R., Rutgersson, C., Weijdegård, B., et al. (2011). Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS ONE 6:e17038. doi: 10.1371/journal.pone.0017038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
LaPara, T. M., Burch, T. R., McNamara, P. J., Tan, D. T., Yan, M., and Eichmiller, J. J. (2011). Tertiary-treated municipal wastewater is a significant point source of antibiotic resistance genes into Duluth-Superior Harbor. Environ. Sci. Technol. 45, 9543–9549. doi: 10.1021/es202775r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, D., Qi, R., Yang, M., Zhang, Y., and Yu, T. (2011). Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 45, 6063–6073. doi: 10.1016/j.watres.2011.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J., Wang, T., Shao, B., Shen, J., Wang, S., and Wu, Y. (2012). Plasmid-mediated quinolone resistance genes and antibiotic residues in wastewater and soil adjacent to swine feedlots: potential transfer to agricultural lands. Environ. Health Persp. 120, 1144–1149. doi: 10.1289/ehp.1104776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, Y., Mao, D., Rysz, M., Zhou, Q., Zhang, H., Xu, L., et al. (2010). Trends in antibiotic resistance genes occurrence in the Haihe River, China. Environ. Sci. Technol. 44, 7220–7225. doi: 10.1021/es100233w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marti, E., Jofre, J., and Balcazar, J. L. (2013a). Prevalence of antibiotic resistance genes and bacterial community composition in a river influenced by a wastewater treatment plant. PLoS ONE 8:e78906. doi: 10.1371/journal.pone.0078906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marti, R., Gannon, V. P., Jokinen, C., Lanthier, M., Lapen, D. R., Neumann, N. F., et al. (2013b). Quantitative multi-year elucidation of fecal sources of waterborne pathogen contamination in the South Nation River basin using Bacteroidales microbial source tracking markers. Water Res. 47, 2315–2324. doi: 10.1016/j.watres.2013.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mitch, A. A., Gasner, K. C., and Mitch, W. A. (2010). Fecal coliform accumulation within a river subject to seasonally-disinfected wastewater discharges. Water Res. 44, 4776–4782. doi: 10.1016/j.watres.2010.05.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Brien, T. F. (2002). Emergence, spread, and environmental effect of antimicrobial resistance: how use of an antimicrobial anywhere can increase resistance to any antimicrobial anywhere else. Clin. Infect. Dis. 34 (Suppl. 3), S78–S84. doi: 10.1086/340244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pruden, A., Arabi, M., and Storteboom, H. N. (2012). Correlation between upstream human activities and riverine antibiotic resistance genes. Environ. Sci. Technol. 46, 11541–11549. doi: 10.1021/es302657r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pruden, A., Pei, R., Storteboom, H., and Carlson, K. H. (2006). Antibiotic resistance genes as emerging contaminants: studies in northern Colorado. Environ. Sci. Technol. 40, 7445–7450. doi: 10.1021/es060413l
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rothberg, J. M., Hinz, W., Rearick, T. M., Schultz, J., Mileski, W., Davey, M., et al. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352. doi: 10.1038/nature10242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rutgersson, C., Fick, J., Marathe, N., Kristiansson, E., Janzon, A., Angelin, M., et al. (2014). Fluoroquinolones and qnr genes in sediment, water, soil, and human fecal flora in an environment polluted by manufacturing discharges. Environ. Sci. Technol. 48, 7825–7832. doi: 10.1021/es501452a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sadek, F., Drucker, D., Boote, V., Bennett, K., and Eley, A. (1998). Phospholipids of Fusobacterium spp. J. Appl. Microbiol. 85, 302–308. doi: 10.1046/j.1365-2672.1998.00506.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, N. G., Verner-Jeffreys, D. W., and Baker-Austin, C. (2011). Aquatic systems: maintaining, mixing and mobilising antimicrobial resistance? Trends Ecol. Evol. 26, 278–284. doi: 10.1016/j.tree.2011.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vartoukian, S. R., Palmer, R. M., and Wade, W. G. (2007). The division “Synergistes.” Anaerobe 13, 99–106. doi: 10.1016/j.anaerobe.2007.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, T. R., Weeks, J., Livermore, D. M., and Toleman, M. A. (2011). Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect. Dis. 11, 355–362. doi: 10.1016/S1473-3099(11)70059-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, J., Wang, C., Shu, C., Liu, L., Geng, J., Hu, S., et al. (2013). Marine sediment bacteria harbor antibiotic resistance genes highly similar to those found in human pathogens. Microb. Ecol. 65, 975–981. doi: 10.1007/s00248-013-0187-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yergeau, E., Lawrence, J. R., Sanschagrin, S., Waiser, M. J., Korber, D. R., and Greer, C. W. (2012). Next-generation sequencing of microbial communities in the Athabasca River and its tributaries in relation to oil sands mining activities. Appl. Environ. Microb. 78, 7626–7637. doi: 10.1128/AEM.02036-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, T., Zhang, X.-X., and Ye, L. (2011). Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS ONE 6:e26041. doi: 10.1371/journal.pone.0026041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, L.-J., Ying, G.-G., Zhao, J.-L., Yang, J.-F., Wang, L., Yang, B., et al. (2011). Trends in the occurrence of human and veterinary antibiotics in the sediments of the Yellow River, Hai River and Liao River in northern China. Environ. Pollut. 159, 1877–1885. doi: 10.1016/j.envpol.2011.03.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: antibiotics, antibiotic resistance genes, bacterial community composition, metagenomic approach, river
Citation: Xiong W, Sun Y, Ding X, Zhang Y and Zeng Z (2014) Antibiotic resistance genes occurrence and bacterial community composition in the Liuxi River. Front. Environ. Sci. 2:61. doi: 10.3389/fenvs.2014.00061
Received: 13 August 2014; Accepted: 30 November 2014;
Published online: 17 December 2014.
Edited by:
Aziz Ullah, Kohat University of Science and Technology, PakistanReviewed by:
Ulisses Miranda Azeiteiro, Universidade Aberta, PortugalFrancesco Di Natale, Università di Napoli Federico II, Italy
Copyright © 2014 Xiong, Sun, Ding, Zhang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenling Zeng, National Laboratory of Safety Evaluation (Environmental Assessment) of Veterinary Drugs, College of Veterinary Medicine, South China Agricultural University, 483 Wushan Road, Guangzhou 510642, China e-mail:emx6ZW5nQHNjYXUuZWR1LmNu
†These authors have contributed equally to this work.