 Amanda R. Lawter
Amanda R. Lawter Nikolla P. Qafoku
Nikolla P. Qafoku Hongbo Shao2
Hongbo Shao2- 1Pacific Northwest National Laboratory, Richland, WA, USA
- 2Illinois State Geological Survey, University of Illinois at Urbana-Champaign, Champaign, IL, USA
The sequestration of carbon dioxide (CO2) in deep underground reservoirs has been identified as an important strategy to decrease atmospheric CO2 levels and mitigate global warming, but potential risks on overlying aquifers currently lack a complete evaluation. In addition to CO2, other gasses such as methane (CH4) may be present in storage reservoirs. This paper explores for the first time the combined effect of leaking CO2 and CH4 gasses on the fate of major, minor and trace elements in an aquifer overlying a potential sequestration site. Emphasis is placed on the fate of arsenic (As) and cadmium (Cd) released from the sediments or present as soluble constituents in the leaking brine. Results from macroscopic batch and column experiments show that the presence of CH4(at a concentration of 1% in the mixture CO2/CH4) does not have a significant effect on solution pH or the concentrations of most major elements (such as Ca, Ba, and Mg). However, the concentrations of Mn, Mo, Si and Na are inconsistently affected by the presence of CH4 (i.e., in at least one sediment tested in this study). Cd is not released from the sediments and spiked Cd is mostly removed from the aqueous phase most likely via adsorption. The fate of sediment associated As [mainly sorbed arsenite or As(III) in minerals] and spiked As [i.e., As5+] is complex. Possible mechanisms that control the As behavior in this system are discussed in this paper. Results are significant for CO2 sequestration risk evaluation and site selection and demonstrate the importance of evaluating reservoir brine and gas stream composition during site selection to ensure the safest site is being chosen.
Introduction
The capture and subsequent sequestration of CO2 has become an attractive strategy for addressing global climate change. Proximity from CO2 producing industries to suitable sequestration sites, as well as the size of available reservoirs, makes geologic carbon sequestration a viable option for mitigating the increasing trend of CO2 emissions (Bachu, 2000; Bruant et al., 2002; IPCC, 2005; Benson and Cole, 2008). However, many concerns must be addressed before large scale implementation of CO2 storage reservoirs can begin. One of the major concerns is possible leakage of CO2 out of the storage reservoir, and how this might affect environmental quality.
Such leakage can happen as a quick, large release of CO2, such as a well failure, or a slow, gradual leak through an undetected fault (Harvey et al., 2013). Previous studies have been conducted with various conclusions over the past few years. Results range from indication that potential CO2 leakage poses a serious risk to overlying groundwater resources (Wang and Jaffe, 2004; Zheng et al., 2009; Kharaka et al., 2010; Little and Jackson, 2010; Vong et al., 2011; Wei et al., 2011; Cahill et al., 2013), to a low level of risk (Smyth et al., 2009; Keating et al., 2010; Frye et al., 2012; Mickler et al., 2013; Kirsch et al., 2014).
In addition to CO2, CH4 may be present in the deep subsurface reservoirs through co-injection with CO2 (Blanco et al., 2012; Mohd Amin et al., 2014). Blanco et al. (2012) reported CH4 levels as high as 6% in industrial emissions in Spain (generated by waste management); however, a study conducted by de Visser et al. (2008), in agreement with the European Enhanced Capture (ENCAP) project, suggested a limit of 4% CH4 in gas injection streams. CH4 can also be present as a native gas prior to injection (Taggart, 2010; Oldenburg and Doughty, 2011; Hosseini et al., 2012). The production of methane by methane-producing bacteria is also a source of CH4 within storage reservoirs (Leu et al., 2011). Experimental data and modeling have shown that CH4 causes an increase in gas plume size and is more buoyant than CO2 (Blanco et al., 2012; Hosseini et al., 2012) and, according to modeling done by Taggart (2010), will form a leading CH4 plume. It is therefore likely that the aquifer sediments would be exposed to a leaking stream of mixed gasses that in addition to CO2 may also contain CH4.
Exposure of the aquifer sediment to a stream of mixed gasses becomes even more likely if one considers that methanogenesis is promoted within the aquifer following a CO2 leakage event (Harvey et al., submitted). The addition of CH4 in the aquifer can create a unique and not well studied subsurface geochemical system with many associated uncertainties, especially in terms of the mobility of major, minor, and trace elements. We are not aware of studies that have investigated the effects of CH4 gas and/or the combined effect of CO2/CH4 gas leakage on groundwater quality.
In addition to CO2and CH4 gasses, the leakage may contain contaminants such as As and Cd that may already be present in reservoir brines, or could be released into the brine from the reservoir rocks during CO2-rich brine: rock interactions (Karamalidis et al., 2013). If these contaminants were transported to the groundwater aquifer by leaking brine and CO2, water quality could be deteriorated. Previous modeling studies have shown As and Cd, present in deep saline reservoir brine, can be brought up by leaking CO2 into an overlying groundwater aquifer (Bacon, 2013; Carroll et al., 2014), leading to a realistic possibility of groundwater quality reduction through leakage of sequestered CO2. Although brine and the associated contaminants are not always transported along with gas leakage (Keating et al., 2013), the possibility for this transport has been shown through modeling (Bacon, 2013; Carroll et al., 2014) and field experiments (Keating et al., 2013). The Environmental Protection Agency (EPA) has developed drinking water standards using maximum contamination levels (MCLs). The MCL for As is 10 μg L−1 and the MCL for Cd is 5 μg L−1 (Environmental Protection Agency, 2003). While the risk of exceeding EPA MCLs could be low based on previous results from our research team (Qafoku et al., 2014; Lawter et al., 2015; Shao et al., 2015; Wang et al., 2015) and others (Smyth et al., 2009; Mickler et al., 2013; Kirsch et al., 2014), an increase in contaminant levels above the current levels within the aquifer can still represent a deleterious impact to groundwater quality (Bacon et al., in press; Carroll et al., 2014). In addition, the combined effect of the CO2/CH4 gasses on the fate of leaked As and Cd has not been previously investigated.
This paper explores the effect of a mixture of gasses (i.e., CO2 and CH4) on the pH and the fate of major, minor and trace elements that may be released from sediments exposed to CO2/CH4 gas streams. The paper also investigates the effects of possible contaminants (i.e., As and Cd), which may be transported with reservoir brine, on groundwater quality of the overlying aquifer. A series of batch and column experiments were conducted as part of this investigation combined with solid phase inspections [scanning electron microscopy (SEM) combined with energy-dispersive spectroscopy (EDS)] and interrogations [laser ablation and inductively coupled plasma mass spectrometry (ICP-MS)]. The ultimate goal was to study trends and determine experimentally-derived parameters, which may then be used by modelers to evaluate potential risks to groundwater sources related to deep subsurface CO2 sequestration.
Materials and Methods
High Plains Sediments
High Plains aquifer sediments were selected because the High Plains aquifer overlies several potential CO2 sequestration sites, and is representative of largely unconsolidated aquifers elsewhere that are likely to overlie potential reservoirs. The sediments used in this study were obtained from the same well, named “CNG,” from two different depths (59–61′ and 109–111′). The sediments are referred to as CNG 60 and CNG 110, respectively. The samples collected by the Kansas Geological Survey were received as loose sediments, and were mostly <2 mm upon arrival. Characterization of these sediments has been conducted and included in other reports and papers written by the members of our group, including acid extractions, quantitative XRD (QXRD) and SEM combined with EDS (Qafoku et al., 2013; Lawter et al., 2015; Shao et al., 2015). Briefly, the QXRD results indicate that quartz is the major component for both sediments, but varying amounts of feldspar and mica are also present (Table S2). Small amounts of carbonate minerals were detected by QXRD in sediment CNG 110 but not CNG 60 (Table S2) (Qafoku et al., 2013; Shao et al., 2015). Acid extractions revealed the sediments contained elements of concern for groundwater quality, such as As, Sr, Cu, and Pb (Lawter et al., 2015; Shao et al., 2015).
Sediment Characterization
SEM/EDS
SEM combined with EDS was used to characterize morphological features of sediment particles and determine the identity of possible adsorbents within the sediments based on particle chemical compositions. Individual clasts of the <53 μm fraction were carbon coated to make them electrically conductive and then examined using a FEI Quanta 3D FEG SEM. Images were collected using a backscattered electron detector. As discussed in Lawter et al. (2015), EDS results show a small amount of detectable Ca in CNG 60. The rough surfaces seen in the SEM images are indicative of high reactive surfaces able to adsorb aqueous species of different elements (Figure 1).

Figure 1. SEM image and EDS spectra for CNG 60. “Spectrum” tag marks where EDS analysis was conducted.
Laser Ablation
Laser ablation was used to analyze sediments for solid phase associated As and/or As bearing minerals. A J100 Series Applied Spectra laser ablation unit was connected to a Thermo Scientific X-Series 2 ICP-MS allowing immediate readings of As levels at various locations within the sample in counts per second (cps). Cps is a measure of intensity that allows for a relative (semi-quantitative) comparison between similar samples. Ablation was first conducted using 50% power and a 75 μm spot size for As analysis to confirm the presence of As in the pre-treatment sediments. Multiple grains of different minerals from both CNG sediments used in this study were then visually selected and ablated to obtain data from distinct minerals. The spot size used was 35 μm with 30% power and approximately 70 shots per run. The ICP-MS tracked cps for As, as well as Si, Ca, Al, and Fe to assist in determining the identity of the minerals where As was found.
Synthetic Groundwater
SGW was used in this work for both batch and column experiments. The SGW recipe in Table S1 was developed using USGS data for the composition of central High Plain aquifer groundwater (Becker et al., 2002). To make the SGW, CaCO3, and MgCO3 were dissolved in 2% HNO3 to ensure complete dissolution before being added to the groundwater. The final pH of the SGW was adjusted to 7.50 with 1 M KOH and/or 2% HNO3. An As and Cd spike was added to the SGW at concentrations of 114 and 40 μg L−1, respectively, for one set of columns and for the batch experiments. These concentrations were chosen based on previous geochemical modeling results, representing the highest predicted concentration to reach the aquifer when As and Cd contaminated brine migrates along with the CO2 (Carroll et al., 2014).
Batch Experiments
Batch studies were conducted on the benchtop under atmospheric conditions. Room temperature ranged from approximately 19°C to 22°C during the course of the experiments.
Batch experiments were conducted using 60 mL poly bottles as batch reactors, with an inlet and outlet in the lid of each (Figure S1). A mixture of 1% CH4 and 99% CO2 gasses was injected into each batch reactor for a pre-determined amount of time. Calculations obtained using STOMP-WNE modeling showed this mixture of gas gives an expected aqueous concentration of 1700 mg L−1 CO2 and 0.24 mg L−1 CH4. For each pre-determined experimental time, a replicate and a blank reactor with no gas injection were also sampled. Blank reactors contained the same sediment and solution as the experiment reactors, including the As/Cd spike, but were not injected with any gas. Sediments CNG 60 and CNG 110 were used for these experiments. Each reactor received 5 g of sediment and 15 mL of spiked SGW, and was left to equilibrate overnight before beginning gas injection. The gas rate (70 ± 10 mL/min) was monitored several times during the experiment. Experimental times included 4 h, 12 h, 1, 4, and 8 days based on previous data which showed that the SGW saturated with CO2 gas reacted fast with the sediments (hours to a few days) (Lawter et al., 2015; Shao et al., 2015; Wang et al., 2015). At each of the predetermined times, the batch reactors were weighed to determine liquid loss and the pH was measured soon thereafter in an aliquot of the liquid phase (about 1.5 mL). The weight change in the samples was negligible (less than 5%); loss of liquid was minimized by directing the gas through a gas washing bottle before entering the batch reactors. The remaining liquid was then centrifuged, filtered through a 0.22 μm syringe filter and subsampled, then acidified with concentrated HNO3 for ICP-MS and inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis.
ICP analyses followed strict analytical quality assurance methods as specified in the Hanford Analytical Services Quality Assurance Requirements Documents (HASQARD, www.hanford.gov/files.cfm/AS_rl-96-68_vol-1.pdf).
A MetalSoft Center [Pscataway, NJ; (Meng et al., 2001)] As speciation cartridge was used to determine aqueous As speciation in the liquid samples collected at the end of the 8 day batch experiment. Immediately following the 8 day sampling, a filtered (0.22 μm) 5 mL subsample was passed through an As speciation cartridge. The first 2–3 mL were collected in a separate container and then discarded, and the final 2–3 mL were used for ICP-MS As analysis. The speciation cartridge selectively removes As5+ from the liquid, leaving only As3+ in the liquid sample (subsequently measured with ICP-MS). Total As was also obtained from ICP-MS, allowing for As5+ calculations from simple subtraction of the As3+ results from the total As concentrations.
Column Experiments
Column studies were conducted under the same temperature and pressure conditions as the batch study. The same Kansas sediments (CNG 110 and CNG 60, <2 mm) from the High Plains aquifer were used in the column studies. For each sediment, columns were leached with 1% CH4 and 99% CO2 saturated SGW spiked with As and Cd. A control column with CO2/CH4 saturated SGW with no As/Cd spike was also conducted. A picture of the apparatus used in the column experiment is shown in the ESI (Figure S2). Polyvinyl chloride (PVC) columns with an inner diameter of 2.4 cm and length of 5.7 cm were packed using the method described in Qafoku et al. (2010). Briefly, sediments were added to the columns in small increments (i.e., 10 g at a time), then lightly pushed down with a dowel. The surface was scratched before adding the next increment, to avoid forming layers inside the column. Filters were placed at the top and bottom of the column to promote uniform distribution of the influent solution and to prevent sediment from clogging tubing.
The columns were initially leached with N2 purged SGW for several hours to fully saturate before being leached with CO2/CH4gas-saturated SGW or spiked SGW. The SGW was continuously bubbled with the gas at a rate of 0.5 mL/min, and the influent was then injected through the bottom inlet of the column at a flow rate of 0.03 mL/min. The pH and Eh was continuously measured on line with an Accumet Benchtop XL15 meter. Measurements were recorded once per hour during the experiment, with increased measurements recorded following each stop flow event. Stop flow events were applied to increase fluid residence time to observe the effect of time dependent (or pH-dependent) reactions and processes on the release of elements from the sediments. Approximately one sample per pore volume (PV) was collected during the experiment, with an increased collection rate immediately following stop flow events. At the conclusion of the experiment, select samples were analyzed by ICP-OES and ICP-MS for elemental concentration determination.
Results and Discussion
Effect of CH4
A major objective of our study was to determine the effect of CH4 on aquifer chemistry if it were to leak with CO2 from a sequestration site into an overlying aquifer.
pH and Eh
During the CO2/CH4 batch experiments, the solution pH of the CNG 60 reactors decreased from approximately 7.5 to 5 immediately following gas injection (Figure 2). The solution pH of CNG 110 reactors decreased as well, but remained steady around pH 6, presumably due to pH buffering by calcite. These pH trends matched those observed in similar batch experiments conducted without the addition of CH4(Shao et al., 2015).
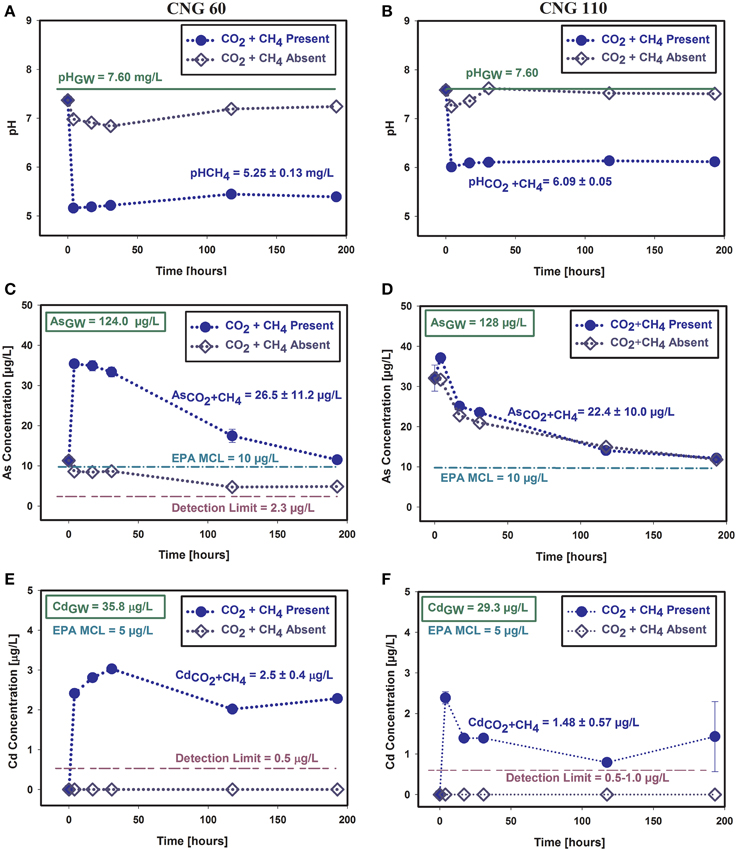
Figure 2. Batch study results: pH changes (A,B), and concentrations changes of As (C,D) and Cd (E,F). Experiments represented here had As/Cd spiked SGW and were injected with the CO2/CH4 gas mixture. Experimental results are represented by closed circles; blank replicates (no gas injection) are represented by open diamonds. Left: CNG 60; Right: CNG 110.
In the column experiments, trends of pH changes between the experiments conducted with and without CH4 were also similar (Figure 3, Figure S5). Effluent pH values in the columns conducted without CH4 remained slightly higher (i.e., up to 1.5 pH units) than those conducted with CH4, although differences were observed prior to the gas injection, meaning the effect may not be attributed only to the addition of CH4. The pH could have been affected by the addition of CH4, due to the additional H+ and carbonic acid from CH4dissolution in water (Wolery and Jarek, 2003):
However, calculations using PHREEQC (http://wwwbrr.cr.usgs.gov/projects/GWC_coupled/phreeqc/) showed no effect on pH, which is in agreement with experimental results. Modeling results (STOMP-WNE) showed an aqueous concentration of 0.24 mg L−1 CH4 when using 1% CH4 and 99% CO2 gas (calculated using 1 atm, 2.5 psi and 20°C, representative of the batch experiment conditions), but calculated aqueous CH4 concentrations were significantly higher (e.g., 1.2 and 2.4 mg L−1) when the concentration of CH4 was increased to 5 and 10%, respectively. Future studies should investigate the effect of these greater concentrations of CH4 in the gas mixture on pH. While 10% may be an overestimate of CH4 present in CO2 sequestration projects, Blanco et al. (2012) stated that industrial emissions can reach CH4 concentrations of >5%.
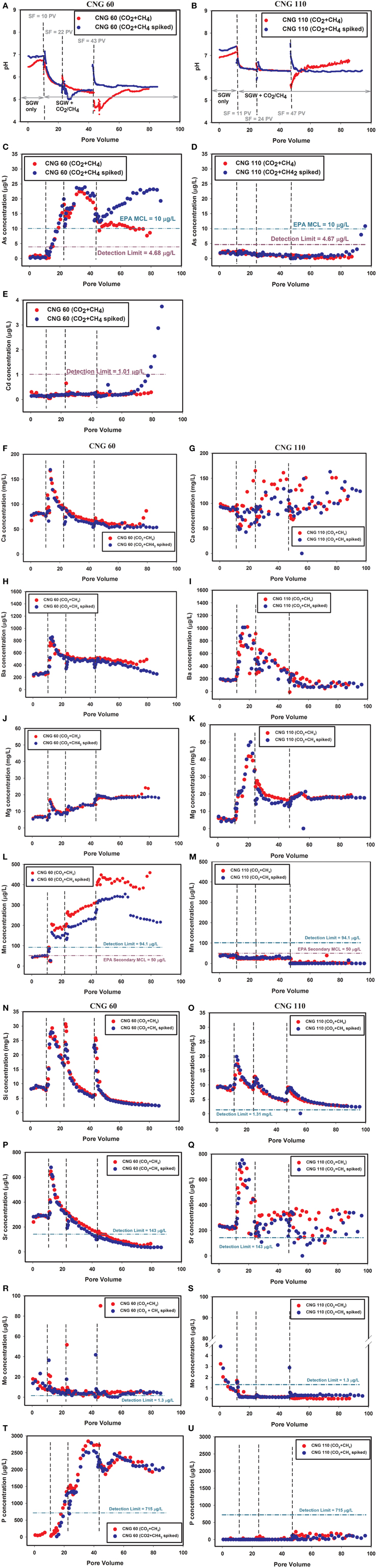
Figure 3. Column study results: pH (A,B) and concentration of As (C,D) and Cd (E). Red represents columns with the control SGW injected with CO2/CH4; blue represents the As/Cd spiked SGW injected with CO2/CH4. CNG 60 columns are on the left and CNG 110 columns are on the right. A Cd graph is not included for CNG 110 because no Cd was detected by ICP analysis. Ca (F, G), Ba (H,I), Mg (J,K), Mn (L,M). Si (N,O), Sr (P,Q), Mo (R,S), and P (T,U).
The Eh measured in the columns with and without CH4 were similar at the beginning of the experiments. However, there was a slight, temporary increase in Eh values thereafter in the experiment conducted with sediment CNG 60 in the absence of CH4, while the Eh increased to approximately 550 mV in the experiment conducted in the presence of CH4, and stayed at this relatively elevated Eh for the duration of the experiment (Figure S6). In the experiments conducted with sediment CNG 110, the Eh remained steady until the end of the experiments, and then increased in both columns to approximately 500 mV. As with CNG 60, however, the increase in Eh was temporary in the experiment conducted with the CO2 gas alone, but remained relatively elevated in the CO2/CH4 column (Shao et al., 2015). The elevated redox potential in the columns conducted with CH4 was unexpected, as a reduction in dissolved O2 was predicted, as shown in the reaction above. Clearly, the addition of CH4 in the gas mixture at the concentration of 1% did not cause a significant change in Eh values.
Chemical Element Release and Fate
While there were slight variations in the concentrations of major elements in batch experiments conducted with and without CH4, the majority of these elements followed the same trends in both sets of experiments (Figure 2, Figure S4). However, in the CNG 60 batch experiment, the concentration of Na was 7–20% lower in the CO2/CH4 experiment than in the CO2 experiment. For CNG 110, Ba and S were lower in the batch test conducted with CH4 than the test without it (approximately 25 and 11.5% lower, respectively). Minor elements, such as Sr and P, showed mostly similar trends in the CO2 and CO2/CH4 batch experiments (Figure S4).
The results from the column experiments showed that major element release trends were similar for Ca, Ba and Mg, but Si trends for sediment CNG 60 were different (Figure S5). The Si concentrations in the study conducted with CO2/CH4 reached a consistent maximum of 25–30 mg L−1 after each stop flow, whereas in the study conducted with only CO2, a decreasing peak with each subsequent stop flow was observed (Shao et al., 2015). This indicates that either a time- or pH-dependent dissolution reaction of one or more Si bearing minerals was controlling Si aqueous concentrations in the presence of CH4, or Si followed different pathways after release in the CO2 vs. CO2/CH4 systems (Shao et al., 2015).
Similar trends were observed in the CO2/CH4 columns for Sr in both sediments compared to the results in Shao et al. (2015). Concentrations of P were measured in the experiment conducted with sediment CNG 60 only and followed similar trends of release regardless of the presence or absence of CH4. The Mn concentrations were similar in the experiments with and without CH4 conducted with sediment CNG 60. In the experiments conducted with sediment CNG 110, Mn levels were below the detection limit (94 μg L−1) in columns with and without CH4 until the end of the experiment, when the CO2 only column effluent Mn concentration increased to approximately 150 μg L−1 (Figure S5 and Shao et al., 2015).
As and Cd Release and Fate
Importantly, the As concentrations in the batch experiments followed decreasing trends for both sediments, from a spiked concentration of approximately 114 μg L−1 to a concentration of 35–40 μg L−1 after the initial overnight equilibration period, and decreasing to less than 15 μg L−1 by the end of the experiment (Figure 2). Similar trends were observed in the CO2 batch experiments conducted without CH4 (reported in the paper by Shao et al., 2015, see Figure S4). Cadmium concentrations of less than 3.5 μg L−1 were measured within the first day of the batch experiments (from an initial spike of approximately 40 μg L−1). As in the previous CO2 experiments conducted without CH4, the results of the CO2/CH4 experiments showed a continuous decrease in Cd in CNG 110 samples, ending the experiment below 1 μg L−1. The trend was slightly different in the experiment conducted with sediment CNG 60, where a steady concentration of around 2 μg L−1 was measured during the experiment (Figure 2). Both As and Cd concentrations were below the U.S. EPA MCL during and by the end of these experiments.
The difference in As concentration between the CNG 60 gas injected reactors and blank (no gas) reactors is likely due to the concentration of aqueous P, a source of competition for As adsorption. Phosphorous was not released in detectable amounts in the blank reactor, but was detected in the gas injected reactors (Figure S4), creating a source of competition for the As in these reactors, resulting in less As adsorption compared to the blank reactors.
In the column experiment conducted with sediment CNG 110, the effluent As concentration remained below detection limits until the last few data points, when As increased above the detection limit of 4.7 μg L−1. This pattern was observed in previous studies conducted with CO2 only, although the increase in As concentration occurred sooner than in the CO2/CH4 experiments. Cadmium was only detected in the CNG 60 column, and only at the end of the experiment. Cadmium was not detected in the CNG 110 columns conducted with CO2/CH4, or either column conducted by Shao et al. (2015) without CH4.
Although CNG 110 released a greater amount of Ca, Mg, Sr and Ba, which we attributed to the presence of calcite in this sediment, CNG 60 liquid samples contained slightly higher concentrations of As and Cd, as expected (Figure 2, Figure S4). Only the spiked SGW CNG 110 column reached above detectable amounts of As, and only near the end of the experiment. Previous acid extractions have shown variability in the As content of CNG 60 and CNG 110, with concentrations ranging from 0.9 to 1.6 μg L−1 for CNG 60 and 0.9 to 4.1 μg L−1 for CNG 110 (Lawter et al., 2015; Shao et al., 2015). The lower pH measured in the effluents of the batch experiments conducted with the CNG 60 sediment indicated likeliness for this sediment to release a greater concentration of metals than CNG 110 most likely due to increased mineral dissolution (Harvey et al., 2013). The pH of CNG 60 is consistently around 1 pH unit lower than CNG 110, likely due to lower buffering ability in CNG 60, resulting from lower calcite content in the sediment.
QXRD results indicated the presence of micas in the High Plains aquifer sediments, which may explain the high As and Cd adsorption capacity of these sediments. Micas contain planar surfaces which have a permanent negative charge, and therefore are good sorbent of aqueous cations, such as Cd. On the other hand, anions, such as arsenate and arsenite, may be sorbed onto micas as micas have variable charge reactive edge groups that are positively charged under acidic conditions (Yang et al., 2010). The adsorption study conducted by Yang et al. (2010) found Cd and As are both adsorbed by natural muscovite, with Cd adsorption increasing at pH 8.7 or higher, and maximum As adsorption at pH 5.6, with As(V) adsorbed more readily than As(III) (adsorption capacity of 0.791 and 0.330 mg g−1, respectively). Studies by Di Benedetto et al. (2006) and Yokoyama et al. (2012) found negatively charged arsenate can also adsorb to calcite due to the positive surface charge of carbonates at low pH, but arsenite is neutral below pH 9.3 and therefore not adsorbed onto carbonates.
Effect of As/Cd Spike on pH, Eh, and Element Release and Fate
Column experiments were conducted with and without an As/Cd spike in the solutions, which were saturated with a mixture of 99% CO2 and 1% CH4 gasses. Few differences were observed until after the third stop flow in the experiments conducted with sediment CNG 60 using the control or spiked SGW (Figure 3, Figures S5, S6). The pH of the spiked SGW and control SGW columns varied slightly throughout the experiment (varying up to 1 pH unit) but reached a similar pH (approximately 5.5) by the end of the experiment (Figure 3). The pH in the CNG 110 columns similarly varied, but unlike the CNG 60 columns, the control SGW column pH increased to a final pH higher than the spiked SGW column (Figure 3). The Eh for all the columns started around 350 mV, and increased during the experiment. The Eh for the control SGW columns was variable (possibly due to an inconsistently functioning probe) while the spiked SGW columns were steadier; all fell within the range of 350–600 mV (Figure S6).
The concentrations of Ca, Mg, and Ba in the CNG 60 control SGW column experiment were similar to the CNG 60 column experiment conducted with spiked SGW prior to an increase that occurred at the very end of the experiment in only the control SGW column (Figure S5). Concentrations of Ba, Mg, and Ca were very similar in the spiked and control SGW CNG 110 columns (Figure 3, Figure S5).
Manganese concentrations in the column experiment conducted with sediment CNG 60 were consistently higher in the control SGW column than in the spiked SGW column. This difference may be related to interactions between As and Mn (i.e., oxidization of sorbed As3+ by MnO2) or due to the increased precipitation of Mn from increased Eh in the spiked SGW column (Young, 2003). Manganese was not detected in the CNG 110 columns. While most other elements in the CNG 110 columns fell below the detection limits and therefore are not reported here (i.e., P, Sn, Cr, Pb, Zn, and Cu), Mo, Sr, and Si levels are nearly identical between the control and spiked SGW columns (Figure S6). Tin, Cr, Zn, and Cu were below detection limits for the CNG 60 columns as well. Lead, Mo, Sr, Si, and P were above detection limits, but similar trends were observed between the spiked and control SGW CNG 60 columns (Figure S5; Pb and Mo not reported).
The As levels were nearly identical between the two CNG 60 columns until the 3rd stop flow, indicating a similar As source. After the 3rd stop flow event, the As concentration in the control experiment (not spiked SGW) decreased and that of the experiment conducted with the spiked SGW increased (Figure 3). Cadmium concentrations during these experiments were mostly non-detectable in all columns; however, an increase in the effluent Cd concentration was observed at the end of the experiment conducted with the spiked SGW and CNG 60 sediment (Figure 3).
Concentrations of As were below the detection limit for both of the CNG 110 columns, until the end of the experiment when the effluent As concentration increased in the column experiment conducted with the spiked SGW (Figure 3). Cadmium did not rise above the detection limit of 1.01 μg L−1 during the experiment.
The low concentration of Cd in CNG 110 and CNG 60 effluents is important, as the SGW was spiked with approximately 40 μg L−1 Cd, yet the concentration of Cd increased above the 1.01 μg L−1 detection limit only at the very end in the CNG 60 spiked SGW column experiment. Arsenic immobilization is also important, as the initial SGW spike concentration was 114 μg L−1 but the effluent As concentration started below the 4.68 μg L−1 detection limit in all column experiments. The As concentration in the experiment conducted with sediment CNG 60 increased above this detection limit after the influent solution was switched from SGW to the gas saturated SGW and continued to increase above the 10 μg L−1 EPA MCL, following identical trends between the control SGW and spiked SGW columns, again most likely indicating the mechanism of release was similar in both of these columns. After a decrease in concentration following the third stop flow event, As concentrations increased in the experiment conducted with the spiked SGW while remaining steady in the control experiments conducted with SGW.
Possible Mechanisms That Control As Fate
While Cd behavior in these systems appears to be less complex (Cd likely undergoes adsorption to minerals that are present in the sediments), As behavior, on the other hand, is multifaceted. Arsenic is a redox sensitive contaminant and commonly occurs in the soil aqueous phase as As3+ (usually in the form of arsenite) and As5+ (usually in the form of arsenate). Arsenite and arsenate have different sorption properties; arsenite adsorbs strongly only on Fe oxides whereas arsenate adsorbs on almost all adsorbents present in soils (So et al., 2008; Yokoyama et al., 2012; Martin et al., 2014).
In the batch experiments, aqueous As speciation was determined on the 8th day of the experiment and the results showed a greater reduction of As5+ than As3+ compared to the concentrations of each speciation in the spiked SGW (Table S3). At least 90% of the total As was removed in all of the batch experiments, including the blank reactors. Despite having low concentrations of As3+ in the SGW, some As3+ was still present in the 8 day samples. One CNG 60 replicate contained more than 40% of the As3+ found in the SGW, but less than 10% of the As5+ remained in solution. The spiked blank samples contained 1.7–7.8% of the As5+ concentration found in the SGW, but 33–39% of the As3+ concentration. This suggests that there was As5+/As3+ (as arsenate and arsenite) competition for adsorption sites on mineral surfaces that were available for these aqueous species at circumnetrual pH. As pH decreases to more acidic values, variable charge minerals (e.g., Fe oxides) and edge groups on phyllosilicates (e.g., micas, kaolinite) develop additional positive surface charge, increasing the anion exchange capacity and the adsorption extent of arsenate and arsenite; this may help explain why the As3+ concentrations were below detection limits in some of the CO2 injected sample, as the pH was lower in these samples than in the blanks.
It is difficult to identify the source of As in the sediments due to low As detection limits on solid phase characterization techniques, such as SEM/EDS. In addition, the samples are heterogeneous, and it is therefore difficult to conclusively identify the source of As within the samples. Laser ablation measurements conducted on the sediments were used in an attempt to find As on the sediment surface. A grain with high As concentration (determined by preliminary laser ablation) was ablated in the same place with the same pattern several times to determine if As concentrations changed with depth. The As intensity decreased with each subsequent ablation, as did the Fe intensity, while Si intensity increased (Figure 4). The grain, which was most likely quartz, had a reddish coating that was visible under an optic microscope. These measurements and observations suggest that the As may be associated with an Fe oxide coating on the surface of the sediment grains. Additional solid phase characterization of these grains is necessary to determine the As sources within the sediment. Additional information on the laser ablation analysis can be found in the ESI.
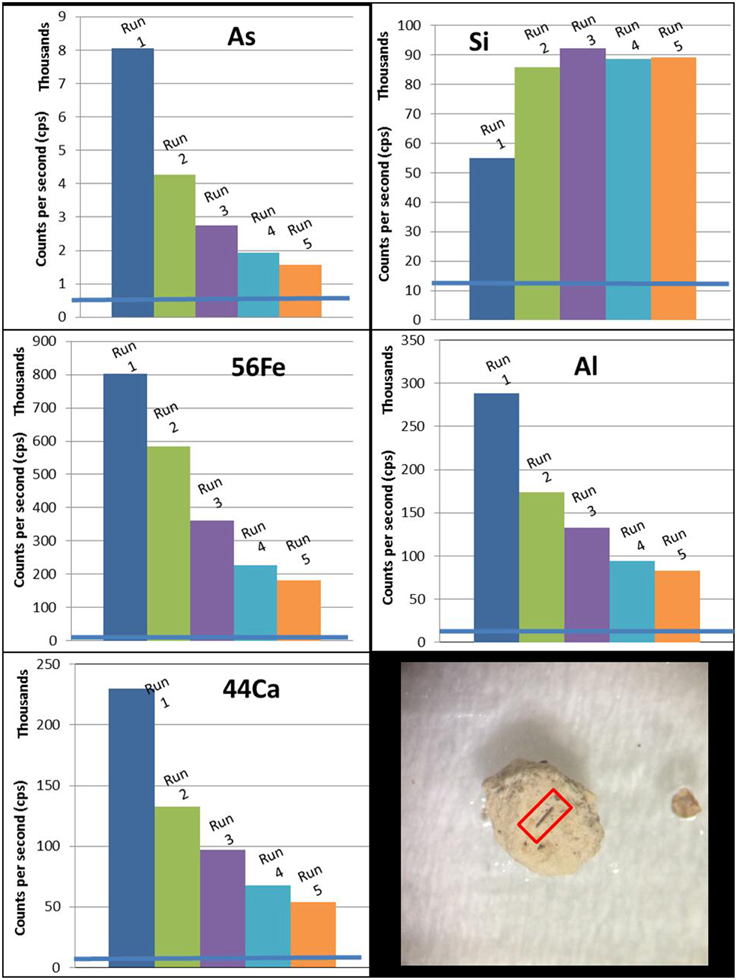
Figure 4. LA-MS count per second (cps) graphs for the “coated” CNG 110 grain for As, Fe, Al, Ca, and Si. Also included is a picture of the grain, with a red box around the ablation site. The ablation site was approximately 0.5 mm in length.
In the column experiments, a CO2/CH4 gas saturated SGW which was either not spiked (control) or spiked with As5+ was injected through the columns. The identical As trends in the control SGW and spiked SGW CNG 60 columns (Figure 3) suggest that the As in the effluent was not the As5+ from the spiked SGW, but As released from another source within the sediment. Data presented in a previous study from our research group (Shao et al., 2015) showed that high concentrations of As3+ were present in the column effluent. The authors concluded that As3+ was released from a source within the sediment.
In addition to sorbed As3+, two other sources of trivalent As are possible. First, spiked As5+ may undergo reduction in isolated locations within the sediment. This is unlikely to be a significant source of As3+, however, because the redox conditions remained oxidizing during these experiments (Figure S6) and this would not explain the As3+ present in the control (not spiked) SGW column. Second, another possible source for trivalent As could be an As(III)-bearing phase which may be unstable under acidic conditions. However, we were not able to identify such minerals in the sediment matrix. Laser ablation results indicated the As present in the pre-treatment samples was present in coatings, that most likely are Fe oxides (see the below and in ESI for additional laser ablation results). The release of As is therefore more likely to be caused by arsenite desorption from these coatings, and not during dissolution of As(III)-bearing minerals. The relatively high As3+ content of the blank batch samples supports this conclusion as well; the blank samples have a higher pH than the CO2/CH4 injected solutions and the sediments were therefore less likely to undergo dissolution, so the high aqueous As3+ content was more likely related to competition with As5+ from the SGW spike than from dissolution of an As(III)-bearing phase.
Implications and Future Needs
CO2 sequestration has the potential to lessen the increasing atmospheric CO2 trend on a global scale if substantial sequestration efforts are undertaken. This is important as part of the solution for slowing and possibly stabilizing the current global warming trend. While the benefits of CO2 sequestration may be intuitive, there are many potential risks related to this strategy that must be carefully identified and researched to prevent unintentional degradation to groundwater quality. This research focuses on the technical risks which will be used to further evaluate the strategy and aid in policy development.
Sequestered CO2 leakages pose a risk to overlying groundwater aquifers not only through the reduction in pH within the aquifer, but also through the possible transport or production of other gasses, such as CH4, and transport of contaminants found in reservoir brine, such as As and Cd. As an important natural resource, it is imperative to thoroughly evaluate the effect of these gasses and contaminants on overlying groundwater aquifer quality.
The release of metals from aquifer sediment when exposed to CO2/CH4 is similar to the metal release seen with only CO2 exposure. This indicates that the addition of CH4, whether present in the initial gas stream, as a native gas, or produced within the reservoir or aquifer, may not have a negative effect on the overlying unconsolidated aquifer when it occurs at low concentrations (i.e., 1% of the total gas mixture mass). A low potential risk related to methane presence can lead to reduced sequestration costs through reduced gas stream purification requirements, increased reservoir possibilities due to inclusion of reservoirs containing native CH4, and reduced groundwater quality concerns. However, the effects of CH4 in this study could be limited by the low concentration of CH4 in the aqueous phase (i.e., 0.24 mg L−1). The aqueous concentration increases significantly with the increasing percentage of the CH4 gas, as indicated by modeling results. This increased aqueous concentration may result in much greater effects in the geochemistry of the aquifer and requires further research.
Despite relatively high concentrations of As and Cd spiked in the SGW for these experiments, both elements from the spike were quickly removed or significantly reduced in the solution phase. This indicates a large removal capacity for contaminants in similar unconsolidated aquifers. The release of As from CNG 60 sediments, however, indicates the importance of site specific evaluations prior to sequestration. Sites where overlying aquifer sediments contain releasable As, for example, may not be chosen after evaluation due to potential groundwater quality concerns. Alternatively, sites with overlying aquifer sediments containing calcite may be favored due to the expected pH buffering capabilities of the calcite during a CO2 leakage event. Developing site selection protocols and identifying evaluation requirements can support policy development to make CO2 sequestration a viable option for the reduction of CO2 emission levels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding for this research was provided by the National Risk Assessment Partnership (NRAP) in the U.S. DOE Office of Fossil Energy under DOE contract number DE AC05 76RL01830. XRD and SEM analysis were performed in the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at PNNL. PNNL is operated by Battelle for the U.S. DOE under Contract DE-AC06-76RLO 1830. ICP analysis was provided by Ian Leavy, Keith Geiszler and Steven Baum, PNNL. Sediment samples were obtained from the Kansas Geological Survey at Kansas University. The authors would like to thank Drs. Matt Shinderman, Julie Pett-Ridge, and Susan Capalbo of Oregon State University for their reviews of this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2015.00049
References
Bachu, S. (2000). Sequestration of CO2 in geological media: criteria and approach for site selection in response to climate change. Energy Convers. Manage. 41, 953–970. doi: 10.1016/S0196-8904(99)00149-1
Bacon, D. (2013). Reduced Order Model for the Geochemical Impacts of Carbon Dioxide, Brine, and Trace Metal Leakage into an Unconfined, Oxidizing Carbonate Aquifer, Version 2.1. Technical Report, Richland, WA: Pacific Northwest National Laboratory.
Bacon, D., Qafoku, N. P., Dai, Z., Keating, E. H., and Brown, C. F. in press. Modeling the impact of carbon dioxide leakage into an unconfined, oxidizing carbonate aquifer. Int. J. Greenhouse Gas Control. doi: 10.1016/j.ijggc.2015.04.008
Becker, M. F., Bruce, B. W., Pope, L. M., and Andrews, W. J. (2002). Ground-Water Quality in the Central High Plains Aquifer, Colorado, Kansas, New Mexico, Oklahoma, and Texas, 1999. Oklahoma, OK: U.S. Geological Survey.
Benson, S. M., and Cole, D. R. (2008). CO2 sequestration in deep sedimentary formations. Elements 4, 325–331. doi: 10.2113/gselements.4.5.325
Blanco, S. T., Rivas, C., Fernandez, J., Artal, M., and Velasco, I. (2012). Influence of methane in CO2 transport and storage for CCS technology. Environ. Sci. Technol. 46, 13016–13023. doi: 10.1021/es3037737
Bruant, R. G., Guswa, A. J., Celia, M. A., and Peters, C. A. (2002). Safe storage of CO2 in deep saline aquifers. Environ. Sci. Technol. 36, 240A–245A. doi: 10.1021/es0223325
Cahill, A. G., Jakobsen, R., Mathiesen, T. B., and Jensen, C. K. (2013). Risks attributable to water quality changes in shallow potable aquifers from geological carbon sequestration leakage into sediments of variable carbonate content. Int. J. Greenhouse Gas Control 19, 117–125. doi: 10.1016/j.ijggc.2013.08.018
Carroll, S. A., Keating, E., Mansoor, K., Dai, Z., Sun, Y., Trainor-Guitton, W., et al. (2014). Key factors for determining groundwater impacts due to leakage from geologic carbon sequestration reservoirs. Int. J. Greenhouse Gas Control 29, 153–168. doi: 10.1016/j.ijggc.2014.07.007
de Visser, E., Hendriks, C., Barrio, M., Molnvik, M. J., de Koeijer, G., Liljemark, S., et al. (2008). Dynamis CO2 quality recommendations. Int. J. Greenhouse Gas Control 2, 478–484. doi: 10.1016/j.ijggc.2008.04.006
Di Benedetto, F., Costagliola, P., Benvenuti, M., Lattanzi, P., Romanelli, M., and Tanelli, G. (2006). Arsenic incorporation in natural calcite lattice: evidence from electron spin echo spectroscopy. Earth Planet. Sci. Lett. 246, 458–465. doi: 10.1016/j.epsl.2006.03.047
Frye, E., Bao, C., Li, L., and Blumsack, S. (2012). Environmental controls of cadmium desorption during CO2 Leakage. Environ. Sci. Technol. 46, 4388–4395. doi: 10.1021/es3005199
Harvey, O. R., Qafoku, N. P., Cantrell, K. J., Lee, G., Amonette, G. E., and Brown, C. F. (2013). Geochemical implications of gas leakage associated with geologic CO2 storage-A qualitative review. Environ. Sci. Technol. 47, 23–26. doi: 10.1021/es3029457
Hosseini, S. A., Mathias, S. A., and Javadpour, F. (2012). Analytical model for CO2 injection into brine aquifers-containing residual CH4. Transp. Porous Media 94, 795–815. doi: 10.1007/s11242-012-0025-x
IPCC. (2005). Special Report on Carbon Dioxide Capture and Storage Working Group III of the Intergovernmental Panel on Climate Change. Cambridge; New York, NY: Cambridge University Press.
Karamalidis, A. K., Torres, S. G., Hakala, J. A., Shao, H., Cantrell, K. J., and Carroll, S. (2013). Trace metal source terms in carbon sequestration environments. Environ. Sci. Technol. 47, 322–329. doi: 10.1021/es304832m
Keating, E. H., Fessenden, J., Kanjorski, N., Koning, D. J., and Pawar, R. (2010). The impact of CO2 on shallow groundwater chemistry: observations at a natural analog site and implications for carbon sequestration. Environ. Earth Sci. 60, 521–536. doi: 10.1007/s12665-009-0192-4
Keating, E. H., Newell, D. L., Viswanathan, H., Carey, J. W., Zyvoloski, G., and Pawar, R. (2013). CO2/brine transport into shallow aquifers along fault zones. Environ. Sci. Technol. 47, 290–297. doi: 10.1021/es301495x
Kharaka, Y. K., Thordsen, J. J., Kakouros, E., Ambats, G., Herkelrath, W. N., Beers, S. R., et al. (2010). Changes in the chemistry of shallow groundwater related to the 2008 injection of CO2 at the ZERT field site, Bozeman, Montana. Environ. Earth Sci. 60, 273–284. doi: 10.1007/s12665-009-0401-1
Kirsch, K., Navarre-Sitchler, A. K., Wunsch, A., and McCray, J. E. (2014). Metal release from sandstones under experimentally and numerically simulated CO2 leakage conditions. Environ. Sci. Technol. 48, 1436–1442. doi: 10.1021/es403077b
Lawter, A., Qafoku, N. P., Wang, G., Shao, H., and Brown, C. F. (2015). Evaluating impacts of CO2 intrusion into an unconsolidated aquifer: 1. Experimental data. Int. J. Greenhouse Gas Control.
Leu, J. Y., Lin, Y. H., and Chang, F. L. (2011). Conversion of CO2 into CH4 by methane-producing bacterium FJ10 under a pressurized condition. Chem. Eng. Res. Design 89, 1879–1890. doi: 10.1016/j.cherd.2011.02.033
Little, M. G., and Jackson, R. B. (2010). Potential impacts of leakage from deep CO2 geosequestration on overlying freshwater aquifers. Environ. Sci. Technol. 44, 9225–9232. doi: 10.1021/es102235w
Martin, M., Violante, A., Ajmone-Marsan, F., and Barberis, E. (2014). Surface interactions of arsenite and arsenate on soil colloids. Soil Sci. Soc. Am. J. 78, 157–170. doi: 10.2136/sssaj2013.04.0133
Meng, X., Korfiatis, P. G., Jing, X., and Christodoulatos, C. (2001). Redox transformations of arsenic and iron in water treatment sludge during aging and TCLP extraction. Environ. Sci. Technol. 35, 3476–3481. doi: 10.1021/es010645e
Mickler, P. J., Yang, C. B., Scanlon, B. R., Reedy, R., and Lu, J. M. (2013). Potential impacts of CO2 leakage on groundwater chemistry from laboratory batch experiments and field push-pull tests. Environ. Sci. Technol. 47, 10694–10702. doi: 10.1021/es401455j
Mohd Amin, S., Weiss, D. J., and Blunt, M. J. (2014). Reactive transport modelling of geologic CO2 sequestration in saline aquifers: the influence of pure CO2 and of mixtures of CO2 with CH4 on the sealing capacity of cap rock at 37°C and 100 bar. Chem. Geol. 367, 39–50. doi: 10.1016/j.chemgeo.2014.01.002
Oldenburg, C. M., and Doughty, C. (2011). Injection, flow, and mixing of CO2 in porous media with residual gas. Transp. Porous Media 90, 201–218. doi: 10.1007/s11242-010-9645-1
Qafoku, N., Bown, C., Wang, G., Sullivan, E., Lawter, A., Harvey, O., et al. (2013). Geochemical Impacts of Leaking CO2 from Subsurface Storage Reservoirs to Unconfined and Confined Aquifers. Rep. No. PNNL-22420. Richland, WA.
Qafoku, N. P., Dresel, E. P., Ilton, E., McKinley, J. P., and Resch, C. T. (2010). Chromium(VI) transport in an acidic waste contaminated subsurface medium: the role of reduction. Chemosphere 81, 1492–1500. doi: 10.1016/j.chemosphere.2010.08.043
Qafoku, N. P., Lawter, A. R., Shao, H., Wang, G., and Brown, C. F. (2014). Evaluating impacts of CO2 gas intrusion into a confined sandstone aquifer: experimental Results. Energy Proced. 63, 3275–3284. doi: 10.1016/j.egypro.2014.11.355
Shao, H., Qafoku, N., Lawter, A., Bowden, M., and Brown, C. (2015). Coupled geochemical impacts of leaking CO2 from subsurface storage reservoirs on the mobilization of metals from sediments. Environ. Sci. Technol. doi: 10.1021/acs.est.5b01004
Smyth, R. C., Hovorka, S. D., Lu, J., Romanak, K. D., Partin, J. W., Wong, C., et al. (2009). Assessing risk to fresh water resources from long term CO2 injection-laboratory and field studies. Energy Proced. 1, 1957–1964. doi: 10.1016/j.egypro.2009.01.255
So, H. U., Postma, D., Jakobsen, R., and Larsen, F. (2008). Sorption and desorption of arsenate and arsenite on calcite. Geochim. Cosmochim. Acta 72, 5871–5884. doi: 10.1016/j.gca.2008.09.023
Taggart, I. (2010). Extraction of dissolved methane in brines by CO2 injection: implication for CO2 sequestration. Spe Reservoir Eval. Eng. 13, 791–804. doi: 10.2118/124630-PA
Vong, C. Q., Jacquemet, N., Picot-Colbeaux, G., Lions, J., Rohmer, J., and Bouc, O. (2011). Reactive transport modeling for impact assessment of a CO2 intrusion on trace elements mobility within fresh groundwater and its natural attenuation for potential remediation. Int. Conf. Greenhouse Gas Control Technol. 4, 3171–3178. doi: 10.1016/j.egypro.2011.02.232
Wang, G., Qafoku, N., Lawter, A., Bowden, M., Harvey, O., Sullivan, C., et al. (2015). Geochemical impacts of leaking CO2 from substorage reservoirs to an unconfined aquifer. Int. J. Greenhouse Gas Control.
Wang, S., and Jaffe, P. R. (2004). Dissolution of a mineral phase in potable aquifers due to CO2 releases from deep formations; effect of dissolution kinetics. Energy Convers. Manage. 45, 2833–2848. doi: 10.1016/j.enconman.2004.01.002
Wei, Y., Maroto-Valer, M., and Steven, M. D. (2011). Environmental consequences of potential leaks of CO2 in soil. Energy Proced. 4, 3224-3230. doi: 10.1016/j.egypro.2011.02.239
Wolery, T. J., and Jarek, R. L. (2003). Software User's Manual EQ3/6 (Version 8.0). New Mexico, NM: Sandia National Laboratories, Albuquerque.
Yang, J.-S., Lee, J. Y., Park, Y.-T., Baek, K., and Choi, J. (2010). Adsorption of As(III), As(V), Cd(II), Cu(II), and Pb(II) from Aqueous Solutions by Natural Muscovite. Sep. Sci. Technol. 45, 814–823.
Yokoyama, Y., Tanaka, K., and Takahashi, Y. (2012). Differences in the immobilization of arsenite and arsenate by calcite. Geochim. Cosmochim. Acta 91, 202–219. doi: 10.1016/j.gca.2012.05.022
Young, S. (2003). Soil mineralogy with environmental applications, SSSA Book Series 7, ed. J. B. DIXON & D. G. SCHULZE. xxix+866 pp. Madison, Wisconsin: Soil Science Society of America (2002). US $90 (hardback). ISBN 0 89118 839 8. J. Agric. Sci. 140, 125–127. doi: 10.1017/S0021859603213101
Keywords: CO2 sequestration, water quality, risk assessment, fate of As and Cd, reservoir brine
Citation: Lawter AR, Qafoku NP, Shao H, Bacon DH and Brown CF (2015) Evaluating impacts of CO2 and CH4 gas intrusion into an unconsolidated aquifer: fate of As and Cd Front. Environ. Sci. 3:49. doi: 10.3389/fenvs.2015.00049
Received: 07 May 2015; Accepted: 29 June 2015;
Published: 10 July 2015.
Edited by:
Tangfu Xiao, Chinese Academy of Sciences, ChinaReviewed by:
Quan Yuan, Chinese Academy of Sciences, ChinaCarlos Paulo, University of Toronto, Canada
Copyright © 2015 Lawter, Qafoku, Shao, Bacon and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda R. Lawter, Geosciences Group, Earth Systems Science Division, Pacific Northwest National Laboratory, 902 Battelle Boulevard, PO Box 999, MSIN: P7-54, Richland, WA 99352, USA,YW1hbmRhLmxhd3RlckBwbm5sLmdvdg==