 Ahmed Abdelaziz
Ahmed Abdelaziz Hilde Spahn-Langguth
Hilde Spahn-Langguth Karl-Werner Schramm2,5
Karl-Werner Schramm2,5 Igor V. Tetko
Igor V. Tetko- 1Rosettastein Consulting UG, Freising, Germany
- 2Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt, TUM-Technische Universität München, Freising, Germany
- 3Institute for Medical and Pharmaceutical Proficiency Assessment, Mainz, Germany
- 4Department of Pharmaceutical Sciences, Karl-Franzens-University Graz, Graz, Austria
- 5Molecular EXposomics, German Research Center for Environmental Health, Helmholtz Zentrum München, Neuherberg, Germany
- 6BigChem GmbH, Neuherberg, Germany
- 7Helmholtz Zentrum München - Research Center for Environmental Health (HMGU), Institute of Structural Biology, Neuherberg, Germany
The need for filling information gaps while reducing toxicity testing in animals is becoming more predominant in risk assessment. Recent legislations are accepting in silico approaches for predicting toxicological outcomes. This article describes the results of Quantitative Structure Activity Relationship (QSAR) modeling efforts within Tox21 Data Challenge 20141, which calculated the best balanced accuracy across all molecular pathway endpoints as well as the highest scores for ATAD5 and mitochondrial membrane potential disruption. Automated QSPR workflow systems, OCHEM (http://ochem.eu), the analytics platform, KNIME and the statistics software, CRAN R, were used to conduct the analysis and develop consensus models using 10 different descriptor sets. A detailed analysis of QSAR models for all 12 molecular pathways and the effect of underlying models' accuracy on the quality of the consensus model are provided. The resulting consensus models yielded a balanced accuracy as high as 88.1% ± 0.6 for mitochondrial membrane disruptors. Such high balanced accuracy and use of the applicability domain show a promising potential for in silico modeling to complement design HTS screening experiments. The comprehensive statistics of all models are publicly available online at https://github.com/amaziz/Tox21-Challenge-Publication while the developed consensus models can be accessed at http://ochem.eu/article/98009.
Introduction
High-throughput screening (HTS) allows researchers to conduct millions of chemical, genetic, or pharmacological experiments with minimal intervention. Such procedures may quickly identify potentially active compounds, antibodies, or genes that control particular biochemical pathways. The results of such assays guide the research process. And thus this approach has become a valuable and viable tool for large-scale evaluation of chemicals (Kavlock and Dix, 2010; Judson et al., 2011; Wetmore et al., 2012). The large amounts of data generated by HTS available today may be used to correlate chemical structures to their biological activities. QSARs may support the identification of key characteristics in chemical structures responsible for such activities. This knowledge is then used to provide predictions about the possible activity of test compounds in virtual screening settings for regulatory purposes. The quality of QSAR models based on large chemical libraries from HTS experiments varies. However, the accuracy is usually high enough to support prioritizing chemicals that are worth being subjected to experimental testing. This approach satisfies the imminent need to prioritize chemicals testing, filling information gaps, accelerating the chemical registration process and lowering the overall costs of testing (US EPA, OCSPP).2
Tox21 (Tice et al., 2009; Betts, 2013) represents a multi-agency effort that uses HTS assays for toxicity modeling and prediction in the US. The US Environmental Protection Agency (EPA), The National Institutes of Health (NIH), The National Center for Advancing Translational Sciences (NCATS), The National Institutes of Environmental Health Sciences/National Toxicology Program (NIEHS/NTP) and the Food and Drug Administration (FDA) cooperate in screening chemical substances for some selected potential toxic effects. The data may then be used, with the assistance of in silico techniques, for providing an alternative for expensive, time-consuming, and ethically-questioned animal testing. This implies the potential for providing an economical method for toxicity testing prioritization for thousands of yet untested compounds (Betts, 2013).
Similar efforts to reduce animal testing and utilize computational toxicity modeling are made in Europe. The European Chemical Agency (ECHA) described the role of animals in ensuring the safe use of chemical substances as being the last resort. This is one of the key principles for the REACH (Registration, Evaluation, Authorization, and Restriction of Chemicals) legislations. It encourages the use of so-called “alternative approaches” to reduce animal testing. QSAR modeling is one of the promoted mechanisms for alternative chemicals' risk assessment. Guiding documents exist that explain the best practices and the requirements for accepting QSAR models' predictions (Worth et al., 2005). These guidelines are essential for directing the stakeholders on how to utilize QSAR methodologies in a manner that gets accepted by the regulators. The guidelines warrant evaluating the human and environmental toxicity risks, complying with the regulatory requirements and reducing the need for animal testing at the same time.
The Tox21 Data challenge follows the open-innovation principles (Chesbrough, 2006) to crowdsource scientists' efforts in analyzing HTS data generated through the Tox21 project. It aspires to predict the pathways' interference of chemicals using only their chemical structures. Such predictions can therefore guide regulators and participating governmental agencies in identifying the chemicals (either drugs or industrial) that carry the highest concern for human and environmental risks. The aim of this study is to describe the methodologies used by the winning corresponding author during the challenge (team: AMAZIZ) and to extend the analysis on the chemical libraries beyond what was possible during the limited duration of the challenge. The study investigates a comprehensive approach on consensus modeling and analyzes multiple descriptor packages.
Materials and Methods
Molecular Pathways Screening
In this study, 12 molecular pathway endpoints were investigated, which were selected on the basis of toxicological relevance. The targets were experimentally screened as part of the Tox21 program and the resulting data library made accessible for competitors by the Tox21 Data Challenge organizers (Tox21 Data Challenge 2014—Data).
Estrogen Receptor (ER) (AID 7430773, AID 7430794)
Tox21 compounds library was screened for potentially acting as agonist at the estrogen receptor alpha. Such activators could lead to reproductive dysfunction (Aop:30)5. Two different cell lines were used:
- ER-alpha-UAS-bla GripTiteTM cell line (ER-LBD): This cell line was developed by Invitrogen, Carlsbad, CA, USA. Cells contain a beta-lactamase reporter gene controlled by an Upstream Activator Sequence (UAS) stably integrated into HEK293 cells.
- BG1-Luc-4E2 cell line (ER-full): Dr. Michael Denison from University of California provided the cell line. Cells endogenously express the full-length ER-alpha and are stably transfected with a plasmid containing four estrogen responsive elements (ERE) under the control of an upstream luciferase reporter gene.
Androgen Receptor (AR) (AID 7430406, AID 7430537)
Compounds that agonist the AR may cause reproductive dysfunction (Aop:23)8. The ability of Tox21 compounds to agonist the androgen receptor alpha was measured in two different cell lines.
- GeneBLAzer AR-UAS-bla-GripTite cell line (AR-LBD): This cell line is provided by Invitrogen, Carlsbad, CA, USA. Cells contain a beta-lactamase reporter gene controlled by an upstream activator sequence (UAS) stably integrated into HEK293 cells.
- MDA-kb2 AR-luc cell line (AR-full): This cell line was deposited by Wilson et al. It is a human breast carcinoma cell line that was stably transfected with a luciferase reporter gene under control of the MMTV promoter containing response elements for both androgen receptor (AR) and glucocorticoid receptor (GR).
Aryl Hydrocarbon Receptor (AHR) (AID 743122)9
AHR activation is thought to lead to multiple adverse outcomes including hepatic steatosis (Aop:57)10, uroporphyria (Aop:131)11, developmental abnormalities and embryolethality (in birds) (Aop:22)12, and embryo toxicity in fish (Aop:21)13 inter alia. A cell based HepG2-AhR-luc assay was used to assess the activation of AhR for Tox21 compounds. The HG2L7.5c1 cell line, as developed by Dr. Michael S. Denison (University of California at Davis), was utilized. The human hepatocellular carcinoma (HepG2) cells were stably transfected with an Ah receptor-responsive firefly luciferase reporter gene plasmid carrying 20 dioxin responsive elements and luciferase reporter gene. AhR activation leads to an increase in luciferase activity and therefore ligands can be detected.
Peroxisome Proliferator-Activated Receptor Gamma (PPAR-Gamma) (AID 743140)14
PPAR-gamma activation has been associated with impaired fertility in adult females (Aop:7)15. GeneBLAzer PPAR gamma UAS-bla HEK293H cell line was used in this assay. This cell line is provided by Invitrogen, Carlsbad, CA, USA. Cells contain a beta-lactamase reporter gene controlled by an upstream activator sequence (UAS) stably integrated into HEK293H cells.
Nuclear Factor (erythroid-derived 2)-Like 2/Antioxidant Responsive Element (Nrf2/ARE) (AID 743219)16
The CellSensor ARE-bla Hep-G2 assay was used to assess the activation of the report gene and thus identify chemicals that stimulate oxidative stress. The cells contain a beta-lactamase reporter gene controlled by the Antioxidant Response Element (ARE) stably integrated into HepG2 cells. Fluorescence intensity was measured to assess the activation of the responsive element.
Aromatase Enzyme Inhibitors (AID 743139)17
Aromatase inhibition is associated with reproductive dysfunction among other adverse outcomes (Aop:25)18. The MCF-7 aro ERE cell line (human breast carcinoma), as provided by Dr. Shiuan Chen (Beckman Research Institute of the City of Hope), was used in order to identify aromatase inhibitors. Cells were stably transfected with a promoter plasmid, pGL3-Luc, encompassing three repeats of the estrogen responsive element (ERE).
ATAD5 Receptor (ATAD5) (AID 720516)19
A cell-based assay using embryonic kidney cells (HEK293T) was used to screen the Tox21 compounds library. The assay was developed by Kyungjae Myung (NHGRI, NIH) to detect any enhanced Level of Genome Instability Gene 1 (ELG1; human ATAD5) protein, which increase in response to different kinds of DNA damage. The assay uses a luciferase reporter-gene tagged with ATAD5 to measure the induction of ELG1. Therefore, an increase in luciferase activity marks a chemically induced genetic stress.
Heat Shock Response (HSE) (AID 743228)20
HSE-bla HeLa cell line was utilized in this HTS assay. This cell line is provided by Invitrogen, Carlsbad, CA, USA. Cells contain a beta-lactamase reporter gene controlled by the heat shock response elements.
Disruptors of the Mitochondrial Membrane Potential (MMP) (AID 720637)21
The mitochondrial dysfunction is considered a key event in multiple adverse outcomes (Event:177)22 including neuroinflammation leading to neurodegeneration, excitotoxicity, and learning and memory impairment. A homogenous cell-based assay with a water-soluble mitochondrial membrane potential sensor (m-MPI, Codex Biosolutions, MD) was applied to the Tox21 compounds in order to identify those that can induce mitochondrial toxicity. In healthy cells, the water-soluble dye accumulates in the mitochondria as aggregates, causing red fluorescence. In case of a decrease in MMP, the dye cannot accumulate in the mitochondria and thus remains in the cytoplasm as monomers causing green fluorescence.
Agonists of the p53 Signaling Pathway (P53) (AID 720552)23
p53 gene has been identified as target of AFB1-induced adduction and subsequent mutation which is a key event leading to Hepatocellular Carcinoma (HCC; Aop:46)24. Using CellSensor p53RE-bla HCT-116 cell line, the Tox21 compounds were screened. This cell line is provided by Invitrogen, Carlsbad, CA, USA. Cells contain a stably integrated beta-lactamase (BLA) reporter gene controlled by the p53 response elements. Fluorescence intensity was measured to assess the activation of the responsive element.
Datasets and Data Cleaning
Data were downloaded from the Tox21 challenge website (NIH)25 in both SDF and SMILES formats. The files contained the molecular representation (SDF or SMILES), a molecule name as well as the target response. In addition, SDF files contained few extra tags for the DSSTox compound ID (DSSTox_CID), the chemical formula and the average mass (FW). Both file formats were compared to examine consistency. KNIME (Berthold et al., 2007) was used to compare the structures and responses in both file formats. The data covered 12 pathway endpoints covering the “Nuclear Receptor Signaling Panel” (seven assays) and the “Stress Response Panel” (five assays). All assay endpoints are listed in Table 1.
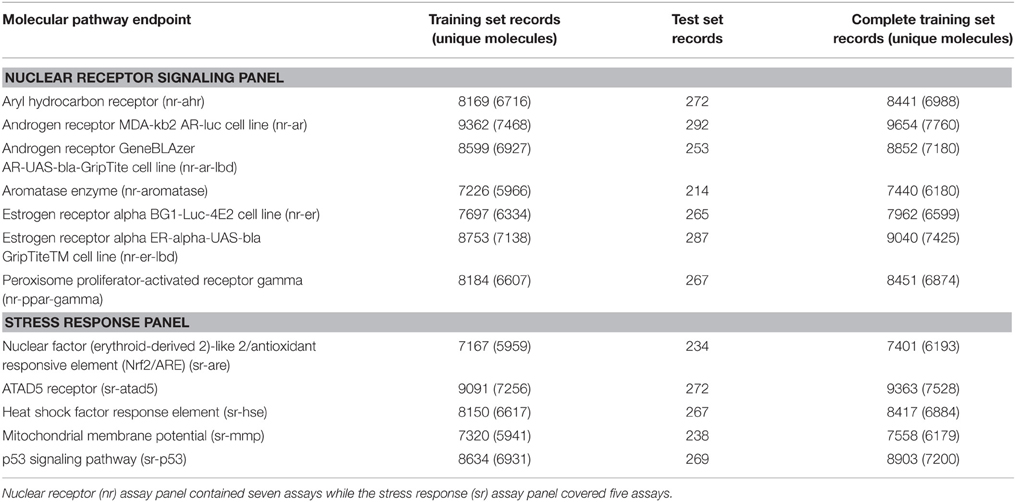
Table 1. Number of records and unique molecules in each dataset.
For each molecular pathway endpoint, both training and leaderboard test sets were combined to form a whole training set. Some molecules were presented multiple times (i.e., exact SMILES representation in spite of different molecule names). The basis for such duplicated records may be the result of intentional repetitive testing for quality control purpose. The Online CHEmical database and Modeling environment platform (OCHEM; Sushko et al., 2011) was used to check records duplication. It calculates the INCHI (James et al., 1995) key structure hash to compare structures. Some records showed different experimental responses despite exhibiting the same molecular structures. Figure 1 shows an example of such duplicates with conflicting experimental measurements. Table 1 shows the number of records per dataset as well as the number of unique molecules.
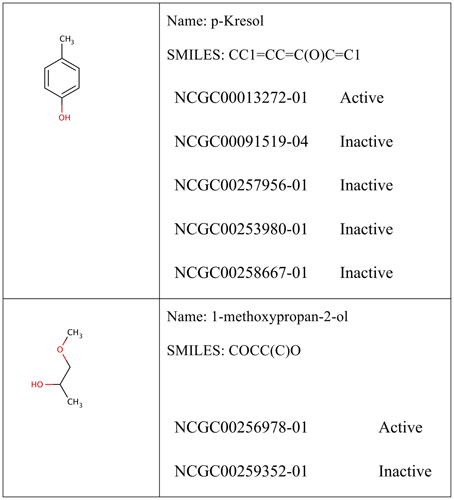
Figure 1. Example of conflicting training data. The examples shown were obtained from the Estrogen Nuclear Receptor dataset. In some cases, it could be reasonable to assume that p-Kresol would be inactive (four records shows inactive against only one active record). In other cases, such as methoxypropan-2-ol, it is not possible tell whether the compound was truly activating the Estrogen nuclear receptor (with one record in every class). Compounds are compared using their calculated INCHI keys generated from the SDF representation. All 12 targets showed similar cases.
Computational Methods
Software Tools
OCHEM (Sushko et al., 2011) offers an interactive web interface (http://www.ochem.eu) that may be used to explore the data, construct QSAR models and run predictions. It also offers the ability to interpret results using prediction-driven matched molecular pairs (Sushko et al., 2014). Handling large datasets and thousands of QSAR models is more convenient using workflow systems such as KNIME (Berthold et al., 2007). For that, OCHEM exposes a number of methods through SOAP web services (Using SOAP web-services—OCHEM user's manual—eADMET docs)26. These methods allow the user to login, upload data, create properties, create or delete QSAR models, download model statistics, and to run predictions on the constructed models. OCHEM implements an xml format that allows users to configure the QSAR modeling tasks with regard to all steps including descriptors calculation, descriptors pre-filtering, and configuring the machine learning algorithms.
Throughout this work, different KNIME (Berthold et al., 2007) workflows were used to explore the data, initialize the QSAR model building process on OCHEM and download the modeling results. All QSAR models were built using OCHEM. CRAN R (R Core Team, 2015) was used to build consensus models and analyze models' performance.
In silico Descriptors Calculation
Ten descriptor packages were selected from OCHEM to be used for constructing QSAR models. These packages were compiled from multiple academic and commercial sources. The selected packages are: GSFrag (Aires-de-Sousa and Gasteiger, 2001), ISIDA fragments (length 2–4; Varnek et al., 2008), Chemaxon descriptors (Introduction to Calculator Plugins—Calculator Plugins—ChemAxon - DOCS)27, Estate indices (Hall et al., 1995; Huuskonen et al., 2000), and AlogPS (Tetko et al., 2001a,b), CDK (using all constitutional, topological, geometrical, electronic, and hybrid descriptors; Steinbeck et al., 2003), Inductive descriptors (Cherkasov et al., 2008), Dragon 6 (Todeschini and Consonni, 2009), Adriana.Code (ADRIANA.Code—Calculation of Molecular Descriptors |Inspiring Chemical Discovery)28, Mera and Mesry (Grishina et al., 2002; Potemkin and Grishina, 2008; Potemkin et al., 2009), QNPR (using SMILES representations—length 1–3 and a threshold of 5; Thormann et al., 2007). Further details on these packages and their integration within OCHEM was reported earlier (Sushko et al., 2011).
The same structure-preprocessing protocol was used prior to the calculation of any descriptor package utilizing Chemaxon Standardizer that is integrated within OCHEM workflow. The standardization workflow consisted of salt counter-ion removal, charge neutralization and the standardizing of certain chemotype representations; such as nitro groups and aromatic rings. For 3D descriptor packages, structural coordinates were optimized using CORINA (Sadowski et al., 1994) starting from a clean SMILES representation. Descriptors calculation failed for some chemicals, the number of failed molecules depends on the nature of the descriptor package. Reasons for calculation failure could be large molecular sizes or undefined chemotypes. The Supplementary Materials (Data Sheet 1) include the count of failed molecules for each constructed model.
Machine Learning
The associative neural networks (ASNN; Tetko, 2002a,b) algorithm was used to construct all models. ASNN is a multilayered perceptron (Rosenblatt, 1957) neural networks algorithm that utilizes ensemble learning. As such, it can be represented by a multilayered graph in which all nodes in a certain layer are linked to the nodes of the preceding one. The resulting class membership is the output of a single neuron in the last layer of the network. ASNN uses a k-Nearest Neighbors (kNN) approach over the space of ensemble predictions to accommodate for a local correction for the ensemble of neural networks. The kNN distance is based on the correlation between the vectors of predicted samples by the networks of the ensemble. All configurations for the algorithm were set to OCHEM defaults [i.e., three neurons in the hidden layer, 1000 iterations, using model ensemble size of 64, the method for neural network training was SuperSAB (Tollenaere, 1990)].
Performance Measures and Validation Protocol
Due to the unbalanced nature of the datasets, balanced accuracy was used throughout the study, as well as during the challenge, as the primary measure for comparing models' performance. It is important to notice that the challenge did not only account for the balanced accuracy but also the Area Under the Receiver Operating Characteristic (AUROC) curve (Hanley and McNeil, 1983).
Bagging (Breiman, 1996) was used to validate the accuracy of the training set. Bagging is a meta-algorithm that involves the aggregation of many models, each of which is based on its own training set (“bag”). Bagging utilizes the random sampling, with repetition, of many subsets of the training set. In each bagging meta-model constructed, an ensemble of 64 models was developed. For each model in the ensemble the training examples were selected randomly from the original training set allowing duplicates (i.e., resampling with replacement). The prediction of each classification was determined by majority voting among the ensemble members. Stratified bagging (Tetko et al., 2013) was used as the validation protocol. It also served to handle the unbalance of the training set (Kotsiantis et al., 2006). In the current implementation, for each of the 64 models in an ensemble, equal numbers of active and inactive compounds were randomly selected. Thus, the size of the training set was always double the size of the minority class.
The calculation of statistical measures was done only using the validation set (out of bag compounds). For molecules with conflicting experimental measurements (see Figure 1), the class with more experimental measurements (majority vote) was selected. Molecules that showed an equal number of active and inactive experimental measurements were excluded.
Consensus Modeling
For each endpoint, consensus models were built using all possible combinations of the underlying 10 models (each built using different in silico descriptor package), i.e., . In total, 12,276 models (1023 × 12 endpoints) were constructed. Simple averaging of the predictions was used for building each of the consensus models.
Two approaches for consensus model selection were investigated in this study. The first approach considers consensus models that show the highest validated balanced accuracy on the training set. The second approach considers consensus models which combine models built with all 10 descriptor packages regardless of the resulting validation balanced accuracy. Both approaches performed comparatively well.
Applicability Domain
In this study, a distance-based method was used to estimate the applicability domain for all models. The distance to model is defined in the property space (rather than the descriptor space; Tetko et al., 2006). This approach uses the standard deviation between the predictions of an ensemble of models (generated through bagging) as a measure of distance.
Results and Discussion
Individual Models
In total 10 descriptor packages were used to model 12 in vitro assay endpoints resulting in 120 QSAR models constructed with 64-bagging-validation. Different endpoints showed varying success. Figure 2 shows the balanced accuracy of all 120 models as grouped by their respective targets with respect to both training and evaluation sets. Other statistical parameters such as specificity, sensitivity, Matthews's correlation coefficient (MCC), and overall accuracy are provided in the Supplementary Materials (Data Sheet 1). All models are published online and may be examined through http://www.ochem.eu/mode/[model-id] replacing [model-id] with the respective model identification number available in the results tables. Users can see a model's summary with performance statistics and applicability domain graphs as well as apply the model to new compounds.
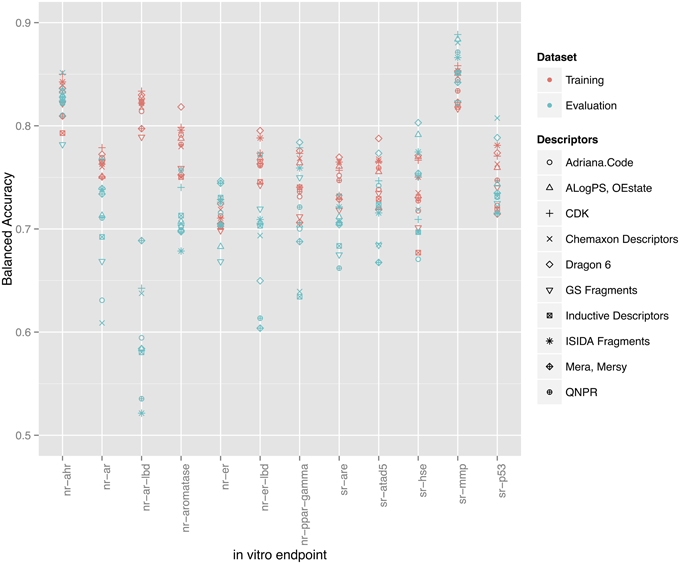
Figure 2. Training set balanced accuracies for all 120 models as grouped by their respective endpoints. Red points represent the validated (through bagging) balanced accuracies calculated on the training set. Blue points represent the balanced accuracy on the evaluation set.
To compare descriptor packages' success, each package was given a score from 1 to 10 according to its rank (a score of 10 was given to the descriptor package contributing to the model with the highest balanced accuracy and a score of 1 for the lowest). The scores were summed for all endpoints. The final rank of descriptors is summarized in Table 2. Dragon and CDK descriptor packages shared the top positions in both training and evaluation sets.
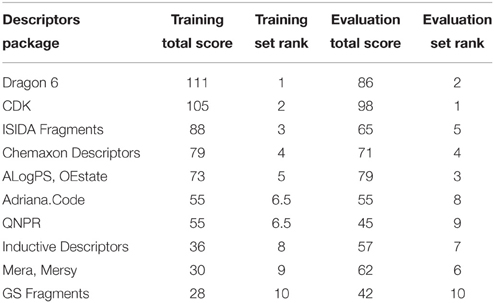
Table 2. Comparison of the performance of different descriptor packages in constructing QSAR models for in vitro pathway disruption prediction.
As shown in Figure 2, a direct correlation exists between the validated training and the evaluation sets' balanced accuracies with the exception of the nr-ar-lbd endpoint. This can also be seen by directly plotting the training set against the evaluation set balanced accuracies as shown in Figure 3.
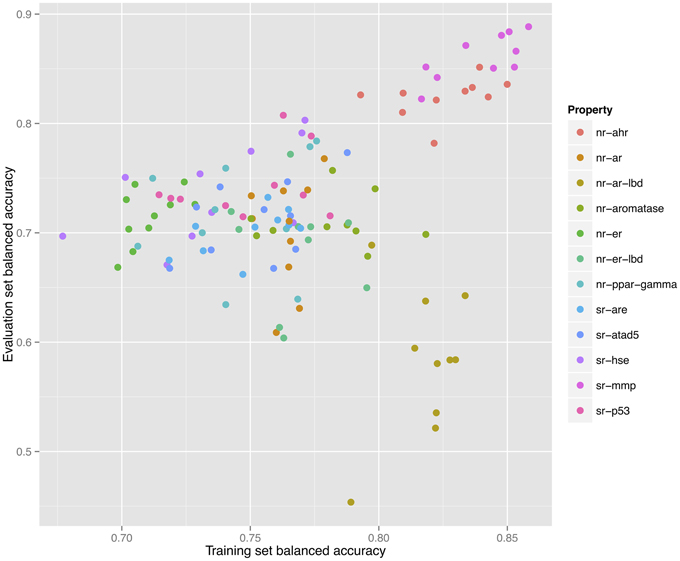
Figure 3. Correlation between training and validation set balanced accuracies for 120 models constructed for 12 endpoints using 10 individual descriptor packages for each endpoint.
Table 3 lists the performance of the single descriptor package models with the highest balanced accuracy for each pathway endpoint together with their corresponding performance on the final evaluation set. The highest balanced accuracy achieved by any of the competing teams (measured on the evaluation set) during the challenge was reported online (Tox21 Data Challenge 2014—Final Leaderboard)29. It is also shown in Table 3 (referred to as “winning balanced accuracy”) for reference.
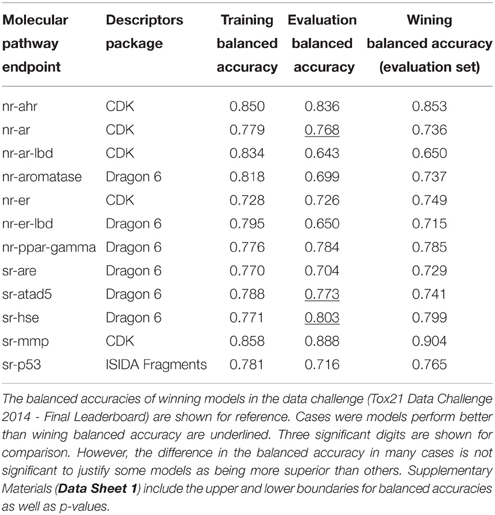
Table 3. Performance of the single-descriptor-package models with the highest training set balanced accuracy for each pathway endpoint.
Consensus Modeling
Table 4 shows the consensus models with highest validated balanced accuracy based on the training set for each endpoint as well as their respective performance on the evaluation set. For all endpoints, consensus modeling was able to improve the performance on the training set. In six endpoints, the consensus models' predictive ability on the evaluation set would also result in a better than winning balanced accuracy.
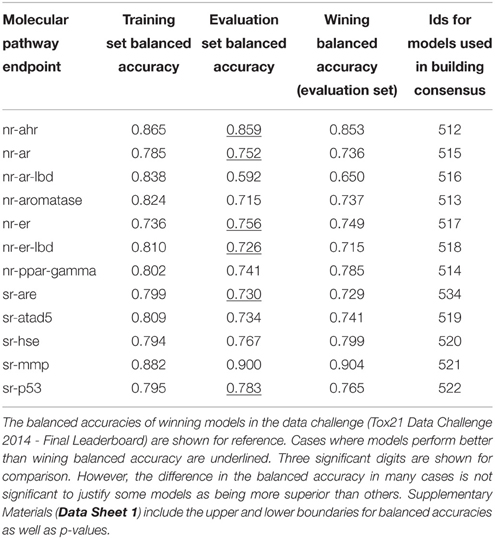
Table 4. Performance of the consensus models with the highest training set balanced accuracy for each pathway endpoint.
For comparison, Table 5 shows the performance of the consensus models involving all 10 underlying descriptor packages for each pathway endpoint. In seven endpoints, the predictive ability of these models on the evaluation set slightly exceeded those of the highest validated balanced accuracy (Table 4).
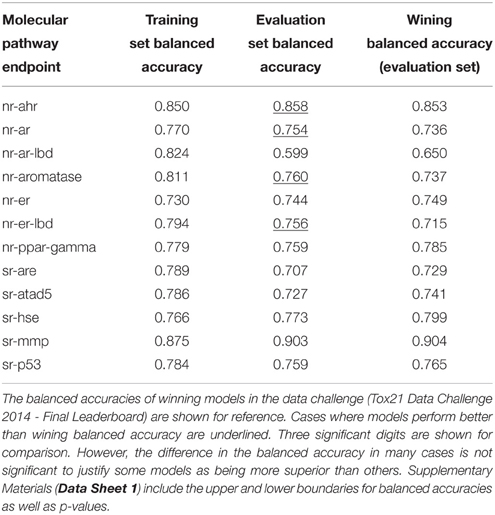
Table 5. Performance of the consensus models involving all 10 descriptor packages for each pathway endpoint.
Descriptor packages differed in their success in representing the chemical structures. Some descriptor packages failed during the calculation phase for some of the molecules (e.g., reporting a chemical structure being too large for calculation). Therefore, models based on them would be deprived from any information gain from those failed molecules (i.e., will have a smaller training set size). A QSAR model built on such descriptors may show good statistics on the smaller training set but fail to perform similarly for an external evaluation set.
The second approach has the advantage of covering the largest number of molecules by compensating for the failure of some packages in descriptors calculation. It can also compensate for some packages bias by offering a wider range of molecular representations. However, it might suffer from the disadvantage of picking noise from descriptor packages with particularly bad performance. It also involves the highest computational expense, as applying such models to new molecules would require calculation of all descriptors from 10 packages. On the other hand, the first approach has the advantage of picking fewer descriptor packages with the highest performance.
Discussion
The combination of the workflow tool (KNIME), the QSAR modeling platform (OCHEM), and the statistical package (CRAN R) allowed the creation and analysis of thousands of models with high efficiency. The use of HTS in vitro assays to construct QSAR models that are able to predict certain molecular pathways' perturbation paves the way toward a better understanding for the mode of chemical toxicity and allows for prioritization of testing efforts. This is in line with the vision of EPA and ECHA for replacing unnecessary animal toxicity testing, rapidly reducing information gaps, and achieving higher outcomes with available efforts and resources.
Due to the time constraint during the challenge, the consensus models selection for team AMAZIZ was based on expert knowledge including the criteria discussed in this study, namely the performance of the model with regard to their balanced accuracy and to a lesser extent the AUROC, preference to descriptor packages, which show more success in representing a larger size of the training set and the simplicity of the underlying descriptor packages (e.g., 2D descriptors are simpler in calculation than 3D descriptors, as they do not need 3D optimization). Table 6 shows the models that were used for the final submission of team AMAZIZ in the challenge. All models can be accessed through their identification numbers for further analysis and to run predictions on new compounds. This study represents a systemic approach to consensus models selection as well as a deeper analysis beyond the challenge.

Table 6. Models used for the final submission by team AMAZIZ during the Tox21 challenge.
The Androgen receptor GeneBLAzer AR-UAS-bla-GripTite cell line endpoint showed exceptional difficulty in modeling. Big discrimination exists between validated performance on the training set and the prediction ability on the evaluation set. Indeed, the endpoint has the lowest success in modeling in the challenge with the winning model being able to achieve a balanced accuracy of only 65% only (the lowest among all endpoints).
Further investigation of the models constructed for this endpoint shows multiple models that would have been able to achieve a higher predictive ability on the evaluation set (0.75–0.80) as shown in Figure 4. However, such models did not show the highest validated balanced accuracy and were thus not selected. The lack of direct correlation between validated balanced accuracy and predictive ability on the evaluation set (Figure 3) can be attributed to the statistical variation in the prediction performance of models for these sets and may also suggest that the split of the whole cluster of chemicals into training and evaluation sets may not have been completely random.
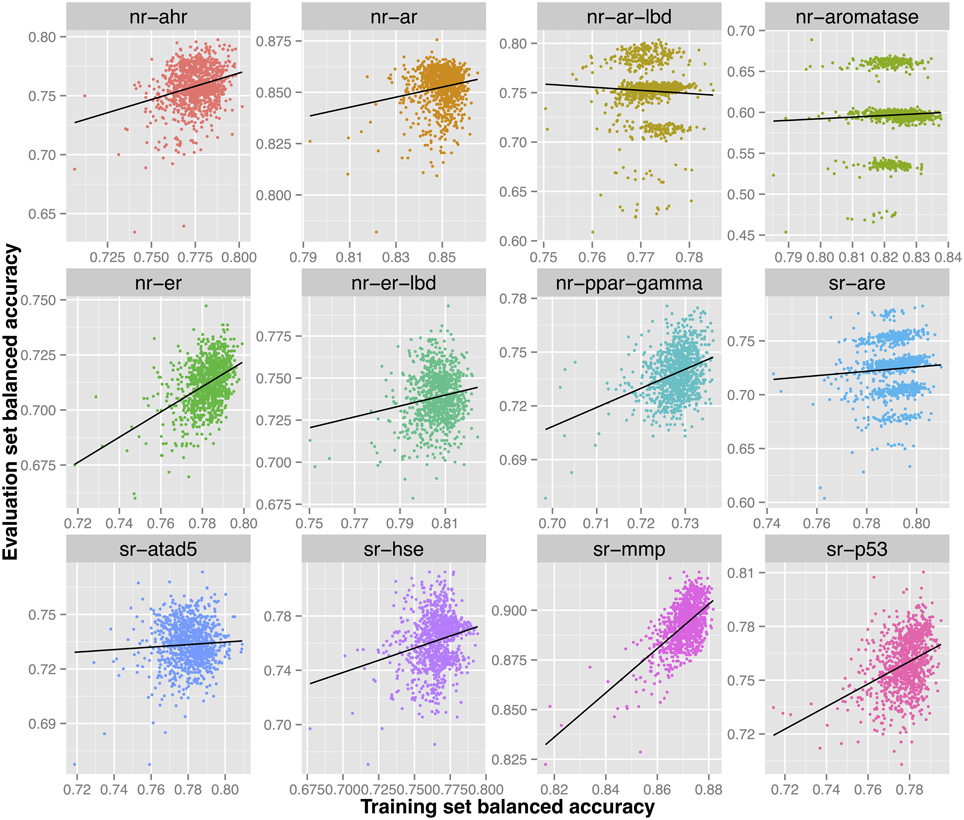
Figure 4. Each sub-figure shows the performance of 1023 consensus models constructed for a single endpoint with x-axis representing the validated balanced accuracy on the training set and y-axis shows the balanced accuracy on the evaluation set. A positive trend line can be noticed with all endpoints except nr-ar-lbd.
Although the alternative approaches for animal testing are highly encouraged, their proper use, and validity must be ensured. For QSAR model building, five OECD principles were established in 2004 (Directorate et al., 2007; OECD Quantitative Structure-Activity Relationships Project [(Q)SARs])30. The OECD principles were taken into consideration during the development of all QSAR models in this study as following:
− The first OECD principle is to have a defined endpoint to ensure the transparency in any physicochemical, biological, or environmental effect that a QSAR model is trying to assess. In this Tox21 challenge, 12 biological targets were well-defined by groups working on the experimental HTS part of the project - for assessment as listed in Table 1.
− The second principle is having an unambiguous algorithm. The “algorithm” refers to the form of relationship between the descriptors of chemical structure and the endpoint in the QSAR model. This can be mathematical/statistical methods or rule-based models defined by experts. Presenting a clear description of the algorithm ensures transparency and allows others to reproduce the model and explain how predictions are generated. In this study, all algorithms used for machine learning, descriptor packages, prefiltering criteria, validation as well as the chemical standardization procedures are described and can be reproduced using the online platform OCHEM. Indeed the process of building high quality QSAR models is tedious and complex. However, by documenting all steps, it is reproducible. Furthermore, by publishing all final models online, the scientific community has continuous access to perform predictions on the constructed QSAR models without a need to reproduce them.
− The third principle, defining domain of applicability, QSAR models are expected to give reliable predictions only for chemicals that are similar to the ones used in the model's training process. In this study, quantitative assessment of the model's confidence in prediction was estimated for all models. This reports the degree of similarity between the compound to be predicted and the model's training set (Sushko, 2011; Sahigara et al., 2012).
− The fourth principle is having appropriate measures of goodness-of-fit, robustness, and predictivity. This principle highlights the need for statistical validation of QSAR models in order to judge models' performance. Such performance validation can be either internal or external. In this study, bootstrap aggregation was used to estimate validation accuracy for the training set. The main statistical parameter applied for comparing all models was balanced accuracy. Performance of all models was also verified against an external test set.
− The fifth and last principle is having a mechanistic interpretation, if possible. The “if possible” phrase shows that the mechanistic interpretation is not mandatory for model acceptance by regulators. Sometimes, the iterative model building process and the involvement of data-mining techniques increase the complexity of the developed QSAR models through multiple training set refinements rendering the mechanistic interpretation hard to directly establish. A different approach for interpretation of complex models using matched molecular pairs was previously suggested (Sushko et al., 2014). All models in this study can be examined using this approach on the OCHEM platform.
The ultimate goal of QSAR models in predictive toxicology, ordinarily, is to forecast an adverse outcome rather than protein binding. In this sense, QSAR prediction of molecular pathways' perturbation is, in itself, an attempt to mechanistically understand toxicological risks. In the context of adverse outcome pathways (AOP), such perturbations are considered as molecular initiating events (MIE), or key events (KE) leading to certain adverse outcome. Such KEs are connected through key event relationships (KERs) to form the network of multiple AOPs. These AOPs form the functional prediction component for real-life circumstances (Villeneuve et al., 2014). In a joint effort between the European Commission—DG Joint Research Centre (JRC) and U.S. EPA, an AOP wiki is being developed. Among its goals is the accommodation of the worldwide efforts for AOP development. The wiki is one of the components of the OECD-sponsored AOP Knowledgebase. The investigated molecular pathways have been suggested to play a role in many adverse outcomes. A comprehensive analysis of the biological impact of the perturbation of these pathways is beyond the scope of this article.
Conclusions
Using QSAR for modeling the outcome of in vitro toxicity assays (representing different molecular pathways) showed promising success with balanced accuracies reaching up to more than 85% for several endpoints as shown in Table 4. The relatively high balanced accuracies among models confirmed the possibility of modeling HTS results from in vitro assays using in silico descriptors as reported in earlier studies (Abdelaziz et al., 2015).
Bagging validation provided a good indication for the models' predictive ability on external validation sets (Figure 3). Stratified bagging addressed the unbalanced nature of the training set and reduced bias toward the majority class. The stratified bagging contributed models, which were optimized toward the balanced accuracy. Moreover, the selection of consensus models also used balanced accuracy as the optimization criteria. This is one of the reasons why models developed in this study calculated the best balanced accuracy across all 12 analyzed targets and did not get the highest AUROC scores, which were used by competition organizers to rank the models. However, despite this, the used strategy allowed to calculate the highest AUROC scores for two targets. It is also important to realize that, due to the model prediction variances, selecting a model with the highest validated accuracy does not guarantee the highest predictive ability for an evaluation set.
Consensus modeling improved the predictive ability of models as signified by both validation and evaluation set accuracies. To a large degree this result was achieved thanks to the diversity of descriptor packages, which captured different aspects of the molecular structures. Use of different descriptors also compensated for failure of some descriptors to represent certain structures and thus covering the entire training set.
In summary, a computational methodology used to develop QSAR models was described. This methodology achieved the highest balanced accuracy for all of the Tox21 Data Challenge organized by the NIH. A similar strategy of consensus modeling was also successful to develop Rank-1 model for another Tox21 Challenge organized by the EPA and TopCoder (Novoratskyi et al., under review). Moreover, the developed models are made publicly available at http://ochem.eu/article/98009 thus allowing other researchers to use them for prospective and retrospective analyses.
Author Contributions
AA designed and executed the study including the R scripts for model building and the KNIME workflows for data handling. He participated as the sole member of team AMAZIZ in the Tox21 data challenge. He is now CEO of Rosettastein Consulting. IT is CEO of BigChem GmbH, who supports and advances the OCHEM platform, which originated in his group in HMGU. He implemented calculation algorithms (ASNN, stratified bagging) and optimized OCHEM API interfaces employed in this study. HSL and KWS provided guidance on the conception and design of the overall study strategy. All authors revised the work, reviewed the manuscript and approved the final version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank ChemAxon (http://www.chemaxon.com), Molecular Networks GmbH (http://www.molecular-networks.com), Talete Srl (http://www.talete.mi.it), and ChemoSophia (http://www.chemosophia.com) for contributing their software tools to OCHEM that were used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2016.00002
Data Sheet 1. Statistical parameters for 8296 models constructed using consensus between 120 models from 10 descriptor packages.
Footnotes
1. ^Tox21 Data Challenge 2014—Data Available at: https://tripod.nih.gov/tox21/challenge/data.jsp
2. ^US EPA, OCSPP, O. Using Predictive Methods to Assess Hazard under TSCA. Available at: http://www2.epa.gov/tsca-screening-tools/using-predictive-methods-assess-hazard-under-tsca#models [Accessed October 15, 2015].
3. ^AID 743077—qHTS assay to identify small molecule agonists of the estrogen receptor alpha (ER-alpha) signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743077 [Accessed July 10, 2015].
4. ^AID 743079—qHTS assay to identify small molecule agonists of the estrogen receptor alpha (ER-alpha) signaling pathway using the BG1 cell line—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743079 [Accessed July 10, 2015].
5. ^Aop:30—Estrogen receptor antagonism leading to reproductive dysfunction-aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:30 [Accessed December 15, 2015].
6. ^AID 743040—qHTS assay to identify small molecule agonists of the androgen receptor (AR) signaling pathway using the MDA cell line—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743040#aDescription [Accessed July 10, 2015].
7. ^AID 743053—qHTS assay to identify small molecule agonists of the androgen receptor (AR) signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743053 [Accessed July 10, 2015].
8. ^Aop:23—Androgen receptor agonism leading to reproductive dysfunction—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:23 [Accessed December 15, 2015].
9. ^AID 743122—qHTS assay to identify small molecule that activate the aryl hydrocarbon receptor (AhR) signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743122 [Accessed July 10, 2015].
10. ^Aop:57—AhR activation leading to hepatic steatosis—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:57 [Accessed December 15, 2015].
11. ^Aop:131—Aryl hydrocarbon receptor activation leading to uroporphyria—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:131 [Accessed December 15, 2015].
12. ^Aop:22—AHR1 activation leading to developmental abnormalities and embryolethality (in birds)—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:22 [Accessed December 15, 2015].
13. ^Aop:21—AhR activation leading to embryo toxicity in fish—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:21 [Accessed December 15, 2015].
14. ^AID 743140—qHTS assay to identify small molecule agonists of the peroxisome proliferator-activated receptor gamma (PPARg) signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743140 [Accessed July 10, 2015].
15. ^Aop:7—PPAR γ activation leading to impaired fertility in adult female- aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:7 [Accessed December 15, 2015].
16. ^AID 743219—qHTS assay for small molecule agonists of the antioxidant response element (ARE) signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743219 [Accessed July 10, 2015].
17. ^AID 743139—qHTS assay to identify aromatase inhibitors: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743139 [Accessed July 10, 2015].
18. ^Aop:25—Aromatase inhibition leading to reproductive dysfunction (in fish)—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:25 [Accessed December 15, 2015].
19. ^AID 720516—qHTS assay for small molecules that induce genotoxicity in human embryonic kidney cells expressing luciferase-tagged ATAD5: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=720516 [Accessed July 10, 2015].
20. ^AID 743228—qHTS assay for small molecule activators of the heat shock response signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=743228 [Accessed July 10, 2015].
21. ^AID 720637—qHTS assay for small molecule disruptors of the mitochondrial membrane potential: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=720637 [Accessed July 10, 2015].
22. ^Event:177—Mitochondrial dysfunction - aopwiki Available at: https://aopkb.org/aopwiki/index.php/Event:177 [Accessed December 15, 2015].
23. ^AID 720552—qHTS assay for small molecule agonists of the p53 signaling pathway: Summary—PubChem BioAssay Summary Available at: https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=720552 [Accessed July 10, 2015].
24. ^Aop:46—Mutagenic Mode-of-Action leading to Hepatocellular Carcinoma (HCC)—aopwiki Available at: https://aopkb.org/aopwiki/index.php/Aop:46 [Accessed December 15, 2015].
25. ^NIH Tox21 Data Challenge 2014. Available at: https://tripod.nih.gov/tox21/challenge/about.jsp
26. ^Using SOAP web-services—OCHEM user's manual—eADMET docs Available at: http://docs.ochem.eu/display/MAN/Using+SOAP+web-services [Accessed January 5, 2015].
27. ^Introduction to Calculator Plugins—Calculator Plugins—ChemAxon—DOCS Available at: https://docs.chemaxon.com/display/CALCPLUGS/Introduction+to+Calculator+Plugins [Accessed January 9, 2015].
28. ^ADRIANA.Code—Calculation of Molecular Descriptors |Inspiring Chemical Discovery Available at: http://www.molecular-networks.com/products/adrianacode [Accessed September 28, 2013].
29. ^Tox21 Data Challenge 2014—Final Leaderboard Available at: https://tripod.nih.gov/tox21/challenge/leaderboard.jsp [Accessed June 18, 2015].
30. ^ECD Quantitative Structure-Activity Relationships Project [(Q)SARs] Available at: http://www.oecd.org/chemicalsafety/testing/oecdquantitativestructure-activityrelationshipsprojectqsars.htm [Accessed June 23, 2015].
References
Abdelaziz, A., Sushko, Y., Novotarskyi, S., Körner, R., Brandmaier, S. V., and Tetko, I. (2015). Using online tool (iPrior) for modeling toxcast™ assays towards prioritization of animal toxicity testing. Comb. Chem. High Throughput Screen. 18, 420–438. doi: 10.2174/1386207318666150305155255
Aires-de-Sousa, J., and Gasteiger, J. (2001). New description of molecular chirality and its application to the prediction of the preferred enantiomer in stereoselective reactions. J. Chem. Inf. Comput. Sci. 41, 369–375. doi: 10.1021/ci000125n
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R., Kötter, T., Meinl, T., et al. (2007). “KNIME: the Konstanz Information Miner,” in Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007), eds C. Preisach, P. D. H. Burkhardt, P. D. D. L. Schmidt-Thieme, and P. D. R. Decker (Freiburg: Springer), 319–326.
Betts, K. S. (2013). Tox21 to date: steps toward modernizing human hazard characterization. Environ. Health Perspect. 121:A228. doi: 10.1289/ehp.121-a228
Cherkasov, A., Ban, F., Santos-Filho, O., Thorsteinson, N., Fallahi, M., and Hammond, G. L. (2008). An updated steroid benchmark set and its application in the discovery of novel nanomolar ligands of sex hormone-binding globulin. J. Med. Chem. 51, 2047–2056. doi: 10.1021/jm7011485
Chesbrough, H. W. (2006). Open Innovation: The New Imperative for Creating and Profiting from Technology. Boston, MA: Harvard Business Press.
Directorate, E., Meeting, J., The, O. F., Committee, C., Working, T. H. E., and On, P. (2007). OECD Environment Health and Safety Publications series on testing and assessment No. 69 GUIDANCE DOCUMENT ON THE VALIDATION OF (QUANTITATIVE) STRUCTURE-ACTIVITY RELATIONSHIP [(Q) SAR] MODELS Environment Directorate.
Grishina, M. A., Bartashevich, E. V., Potemkin, V. A., and Belik, A. V. (2002). Genetic algorithm for predicting structures and properties of molecular aggregates in organic substances. J. Struct. Chem. 43, 1040–1044. doi: 10.1023/A:1023663115138
Hall, L. H., Kier, L. B., and Brown, B. B. (1995). Molecular similarity based on novel atom-type electrotopological state indices. J. Chem. Inf. Comput. Sci. 35, 1074–1080. doi: 10.1021/ci00028a019
Hanley, J. A., and McNeil, B. J. (1983). A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 148, 839–843. doi: 10.1148/radiology.148.3.6878708
Huuskonen, J. J., Livingstone, D. J., and Tetko, I. V. (2000). Neural network modeling for estimation of partition coefficient based on atom-type electrotopological state indices. J. Chem. Inf. Comput. Sci. 40, 947–955. doi: 10.1021/ci9904261
James, C. A., Weininger, D., and Delany, J. (1995). Daylight Theory Manual. Irvine, CA: Daylight Chemical Information Systems. Inc.
Judson, R. S., Kavlock, R. J., Setzer, R. W., Hubal, E. A. C., Martin, M. T., Knudsen, T. B., et al. (2011). Estimating toxicity-related biological pathway altering doses for high-throughput chemical risk assessment. Chem. Res. Toxicol. 24, 451–462. doi: 10.1021/tx100428e
Kavlock, R., and Dix, D. (2010). Computational toxicology as implemented by the U.S. EPA: providing high throughput decision support tools for screening and assessing chemical exposure, hazard and risk. J. Toxicol. Environ. Health. B. Crit. Rev. 13, 197–217. doi: 10.1080/10937404.2010.483935
Kotsiantis, S., Kanellopoulos, D., Pintelas, P., and others (2006). Handling imbalanced datasets: a review. GESTS Int. Trans. Comput. Sci. Eng. 30, 25–36.
Potemkin, V. A., and Grishina, M. A. (2008). A new paradigm for pattern recognition of drugs. J. Comput. Aided. Mol. Des. 22, 489–505. doi: 10.1007/s10822-008-9203-x
Potemkin, V. A., Pogrebnoy, A. A., and Grishina, M. A. (2009). Technique for energy decomposition in the study of “receptor-ligand” complexes. J. Chem. Inf. Model. 49, 1389–1406. doi: 10.1021/ci800405n
R Core Team (2015). R: A Language and Environment for Statistical Computing. Available online at: http://www.r-project.org
Rosenblatt, F. (1957). The Perceptron, A Perceiving and Recognizing Automaton Project Para. New York, NY: Cornell Aeronautical Laboratory.
Sadowski, J., Gasteiger, J., and Klebe, G. (1994). Comparison of automatic three-dimensional model builders using 639 X-ray structures. J. Chem. Inf. Comput. Sci. 34, 1000–1008. doi: 10.1021/ci00020a039
Sahigara, F., Mansouri, K., Ballabio, D., Mauri, A., Consonni, V., and Todeschini, R. (2012). Comparison of different approaches to define the applicability domain of QSAR models. Molecules 17, 4791–4810. doi: 10.3390/molecules17054791
Steinbeck, C., Han, Y., Kuhn, S., Horlacher, O., Luttmann, E., and Willighagen, E. (2003). The Chemistry Development Kit (CDK): an open-source Java library for chemo-and bioinformatics. J. Chem. Inf. Comput. Sci. 43, 493–500. doi: 10.1021/ci025584y
Sushko, I. (2011). Applicability Domain of QSAR Models. Available online at: http://mediatum.ub.tum.de?id=1004002 (Accessed August 31, 2014).
Sushko, I., Novotarskyi, S., Korner, R., Pandey, A. K., Rupp, M., Teetz, W., et al. (2011). Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J. Comput. Aided. Mol. Des. 25, 533–554. doi: 10.1007/s10822-011-9440-2
Sushko, Y., Novotarskyi, S., Körner, R., Vogt, J., Abdelaziz, A., and Tetko, I. V. (2014). Prediction-driven matched molecular pairs to interpret QSARs and aid the molecular optimization process. J. Cheminform. 6, 48. doi: 10.1186/s13321-014-0048-0
Tetko, I. V. (2002a). Associative neural network. Neural Process. Lett. 16, 187–199. doi: 10.1023/A:1019903710291
Tetko, I. V. (2002b). Neural network studies. 4. Introduction to associative neural networks. J. Chem. Inf. Comput. Sci. 42, 717–728. doi: 10.1021/ci010379o
Tetko, I. V., Bruneau, P., Mewes, H.-W., Rohrer, D. C., and Poda, G. I. (2006). Can we estimate the accuracy of ADME-Tox predictions? Drug Discov. Today 11, 700–707. doi: 10.1016/j.drudis.2006.06.013
Tetko, I. V., Novotarskyi, S., Sushko, I., Ivanov, V., Petrenko, A. E., Dieden, R., et al. (2013). Development of dimethyl sulfoxide solubility models using 163,000 molecules: using a domain applicability metric to select more reliable predictions. J. Chem. Inf. Model. 53, 1990–2000. doi: 10.1021/ci400213d
Tetko, I. V., Tanchuk, V. Y., Kasheva, T. N., and Villa, A. E. P. (2001a). Estimation of aqueous solubility of chemical compounds using E-state indices. J. Chem. Inf. Comput. Sci. 41, 1488–1493. doi: 10.1021/ci000392t
Tetko, I. V., Tanchuk, V. Y., and Villa, A. E. P. (2001b). Prediction of n-octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E-state indices. J. Chem. Inf. Comput. Sci. 41, 1407–1421. doi: 10.1021/ci010368v
Thormann, M., Vidal, D., Almstetter, M., and Pons, M. (2007). Nomen est omen: quantitative prediction of molecular properties directly from IUPAC names. Open Appl. Informatics J. 1, 28–32. doi: 10.2174/1874136300701010028
Tice, R., Kavlock, R., and Austin, Christopher (2009). The U.S. “Tox21 Community” and the Future of Toxicology. Available online at: http://www.epa.gov/ncct/bosc_review/2009/posters/1-08_Tice_CompTox_BOSC09.pdf [Accessed January 15, 2014].
Todeschini, R., and Consonni, V. (2009). Molecular Descriptors for Chemoinformatics. 2nd, Rev. Milano: John Wiley & Sons.
Tollenaere, T. (1990). SuperSAB: fast adaptive back propagation with good scaling properties. Neural Netw. 3, 561–573. doi: 10.1016/0893-6080(90)90006-7
Varnek, A., Fourches, D., Horvath, D., Klimchuk, O., Gaudin, C., Vayer, P., et al. (2008). ISIDA-Platform for virtual screening based on fragment and pharmacophoric descriptors. Curr. Comput. Aided. Drug Des. 4, 191–198. doi: 10.2174/157340908785747465
Villeneuve, D. L., Crump, D., Garcia-Reyero, N., Hecker, M., Hutchinson, T. H., LaLone, C. A., et al. (2014). Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol. Sci. 142, 312–320. doi: 10.1093/toxsci/kfu199
Wetmore, B. A., Wambaugh, J. F., Ferguson, S. S., Sochaski, M. A., Rotroff, D. M., Freeman, K., et al. (2012). Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci. An Off. J. Soc. Toxicol. 125, 157–174. doi: 10.1093/toxsci/kfr254
Worth, A. P., Bassan, A., Gallegos, A., Netzeva, T. I., Patlewicz, G., Pavan, M., et al. (2005). The Characterisation of (quantitative) Structure-Activity Relationships: Preliminary Guidance. Institute for Health and Consumer Protection, Toxicology and Chemical Substances Unit, European Chemical Bureau.
Keywords: computational toxicology, alternative testing, Quantitative structure activity relationship, high throughput screening, predictive toxicology, Tox21
Citation: Abdelaziz A, Spahn-Langguth H, Schramm K-W and Tetko IV (2016) Consensus Modeling for HTS Assays Using In silico Descriptors Calculates the Best Balanced Accuracy in Tox21 Challenge. Front. Environ. Sci. 4:2. doi: 10.3389/fenvs.2016.00002
Received: 16 October 2015; Accepted: 12 January 2016;
Published: 04 February 2016.
Edited by:
Ruili Huang, NIH National Center for Advancing Translational Sciences, USACopyright © 2016 Abdelaziz, Spahn-Langguth, Schramm and Tetko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Abdelaziz, Y29udGFjdEBhbWF6aXouY29t