 Stephen J. Capuzzi
Stephen J. Capuzzi Regina Politi
Regina Politi Olexandr Isayev
Olexandr Isayev Sherif Farag
Sherif Farag- Laboratory for Molecular Modeling, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
The ability to determine which environmental chemicals pose the greatest potential threats to human health remains one of the major concerns in regulatory toxicology. Computational methods that can accurately predict a chemical's toxic potential in silico are increasingly sought-after to replace in vitro high-throughput screening (HTS) as well as controversial and costly in vivo animal studies. To this end, we have built Quantitative Structure-Activity Relationship (QSAR) models of 12 stress response and nuclear receptor signaling pathways toxicity assays as part of the 2014 Tox21 Challenge. Our models were built using the Random Forest, Deep Neural Networks and various combinations of descriptors and balancing protocols. All of our models were statistically significant for each of the 12 assays with the balanced accuracy in the range between 0.58 and 0.82. Our results also show that models built with Deep Neural Networks had higher accuracy than those developed with simple machine learning algorithms and that dataset balancing led to a significant accuracy decrease.
Introduction
The ability to determine which environmental chemicals pose the greatest potential threats to human health remains one of the major concerns in regulatory toxicology. In addition, the inability to recognize potentially toxic substances during the initial steps of drug development contributes to the failure of promising pharmaceutical leads in more than 30% of human clinical trials (Kola and Landis, 2004). Historically, the estimated human health impact of these chemicals has been assessed through in vivo animal studies. Animal studies, however, are costly, laborious, impractical for evaluating large numbers of chemicals, and are being progressively eliminated due to their controversial nature (Anastas et al., 2010). However, over the past several years, the focus has switched to high-throughput in vitro screening (HTS) in order to identify chemical hazards and prioritize chemicals for additional in vivo testing (O'Brien et al., 2006). The ToxCast project and the Tox21 consortium have used high-throughput screening to characterize the in vitro biological activity of chemicals across multiple cellular pathways and biochemical targets (Dix et al., 2007). HTS campaigns, however, can also be costly and time-consuming because every new series of chemicals must be screened against multiple toxicity endpoints and at various concentrations. Therefore, in silico methods that can accurately predict toxicity toward the prioritization of chemicals for experimental testing are in-demand. To this end, the 2014 Tox21 Challenge sought to “crowdsource” predictive models from various researchers across the globe to assess how well their models can predict the toxic potential of a compound in several biological pathways screened against the Tox21 10,000 compound library (https://tripod.nih.gov/tox21/challenge/about.jsp).
Quantitative Structure-Activity Relationship (QSAR) models provide such a computational method toward the in silico prediction of chemical toxicity. QSAR models utilize complex machine learning algorithms to establish a relationship between chemical structure and the modeled endpoint (toxicity). Robust and rigorously-validated QSAR models are then used to provide in silico predictions of the endpoint-of-interest for yet-untested chemicals (Tropsha, 2010). Thus, the Tox21 program aimed to identify new methods for assessing chemical toxicity in the form of QSAR models in order to improve the identification of chemicals that may affect the functions of seven nuclear receptors (AR, AR-LBD, ER, ER-LBD, AhR, Aromatase, PPAR-gamma) and five stress response pathways (ARE, ATAD5, HSE, MMP, p53) in the human body.
Several of these pathways of interest regulate normal endocrine function. Endocrine disrupting chemicals (EDCs) interfere with the endocrine system through interactions with nuclear receptors (Diamanti-Kandarakis et al., 2009). EDCs engender myriad adverse developmental, reproductive, neurological, and immunological effects in both humans and wildlife. Unfortunately, both humans and wildlife are ubiquitously exposed to EDCs, as EDCs have widespread industrial applications, resulting in endocrine toxicity (Casals-Casas and Desvergne, 2011). For instance, bisphenol-A and its analogs—EDCs which are used heavily in the manufacturing of polycarbonate plastics and epoxy resins (Bae et al., 2002)—have been shown to bind to the estrogen receptor (ER), androgen receptor (AR), and peroxisome proliferator-activated receptor (PPAR) gamma (Han et al., 2003). Moreover, there is ample evidence that EDCs also interact with stress response pathways, such as mitochondrial membrane potential (MMP) and tumor suppressor p53 (Min et al., 2003; Chandra, 2013). For these reasons, the identification of endocrine disrupting chemicals (EDCs) is of particular interest to the Tox21 program and environmental chemical hazard screening in general.
The overall goal of the Tox21 Challenge was to predict compound activity (toxic or non-toxic) in pathway assays provided by the Challenge organizers using only chemical structure data. The data provided was generated from seven nuclear receptor and five stress response pathway assays run against the Tox21 compound library. We performed various permutations of curation and balancing protocols to generate Random Forest (RF) and deep neural net (DNN) models employing either Dragon or SiRMS descriptors.
Methods
Datasets
All datasets (training and test sets) of compound toxicity in 12 different pathway assays were downloaded from the Tox21 Challenge website (https://tripod.nih.gov/tox21/challenge/index.jsp). The training set included 11,764 compounds with activities 0 (non-toxic) and 1 (toxic) in each of the 12 assays. Test set 1 comprised 296 compounds with various activities in each of the 12 assays. This test set, initially used to evaluate model performance, was subsequently merged into the training set. Test set 2 included 647 compounds with various activities in each of the 12 assays. This set was used to evaluate model performance and to rank model submissions of various participants. For all datasets in each assay, a compound was active (1), inactive (0), or untested.
Dataset Curation
Each dataset was curated according to our well-established protocol (Fourches et al., 2010). Structural standardization, the cleaning of salts, and the removal of mixtures, inorganics, and organometallics was performed using Instant JChem software (version 6.2, ChemAxon).
In the case of replicate compounds, InChI Keys were generated using Instant JChem software. For replicates with the same activities in a given assay, a single representative compound was selected for inclusion into the training set. For replicates with the different activities in a given assay, all compounds were excluded.
After curation, the sizes of the training set, test set 1, and test set 2 were reduced to 9323 compounds, 291 compounds, and 641 compounds, respectively.
Dataset Balancing
For each pathway assay, only compounds that were explicitly tested (active or inactive) were used. Inactive (non-toxic) compounds were the predominant majority (ratio 10:1 or higher) as compared to active (toxic) compounds in the training sets for each of the 12 assays. Inactive compounds were down-sampled such as to make the remaining number of inactives similar to the respective number of active compounds in each of the individual assays either (a) randomly or (b) according to highest Tanimoto similarity to compounds in test set 2. In a separate study (c), the training set was left unbalanced (see Supplemental for individual assay counts).
Molecular Descriptors
Dragon Descriptors
An ensemble of 2489 molecular descriptors was computed with the Dragon software (version 5.4) for all compounds (with explicit hydrogen atoms) in every dataset.
SiRMs
2D Simplex Representation of Molecular Structure (SiRMS) descriptors (Muratov et al., 2010) were generated by the HiT QSAR software (Kuz'min et al., 2008). At the 2D level, the connectivity of atoms in a simplex, atom type, and bond nature (single, double, triple, or aromatic) have been considered. SiRMS descriptors account not only for the atom type, but also for other atomic characteristics that may impact biological activity of molecules, e.g., partial charge, lipophilicity, refraction, and atom ability for being a donor/acceptor in hydrogen-bond formation (H-bond). Detailed description of HiT QSAR and SiRMS can be found elsewhere (Kuz'min et al., 2008; Muratov et al., 2010).
Model Building and Evaluation
Random Forest (RF)
QSAR models were built using an in-house implementation on Chembench (http://chembench.mml.unc.edu) of the original RF algorithm (Breiman, 2001).
External 5-fold Cross Validation
All RF models were evaluated using external 5-fold cross validation (Tropsha et al., 2003). Every training set for each of the 12 assays was randomly partitioned into five equal parts with the same active (toxic)/inactive (non-toxic) ratio before modeling. In turn, each of the five parts was “left out” to form an external set used to validate the model developed on the remaining four parts that collectively amounted to the modeling set.
Score Threshold
The ensemble of selected RF models outputs a continuous consensus score (RF score) ranging from 0 (non-toxic) to 1 (chemical predicted to be toxic by all models). When there is a disagreement between those individual RF models, the consensus RF score can thus take any value between 0 and 1. When computed for a set of chemicals, RF scores can be used to rank those chemicals based on their increasing RF-evaluated likelihood of being toxic. For all assays, a RF score threshold was arbitrarily set to 0.5, with scores ≥ 0.5 being active (toxic) and scores <0.5 being inactive (non-toxic).
Y-Randomization
Models were further validated through Y-randomization, wherein activities (i.e., the response variable Y) observed for the original training set are randomly assigned to the training set compounds multiple times and the models are built for all datasets generated by these multiple permutations of the response variable. This procedure ensures that the models built for the original datasets do not reflect a chance correlation between multiple independent variables (i.e., chemical descriptors) and the dependent variable.
Deep Learning Models
We trained deep neural net (DNN) (Schmidhuber, 2015) models with the rectified linear units (ReLU) activation function (Nair and Hinton, 2010) instead of typical sigmoidal units. The rectified linear unit computes the function f(x) = max(0,x). In other words, the activation is simply thresholded at zero when x < 0 and then linear with a of slope 1 when x > 0.
Neural networks can have many hyperparameters. Therefore, in order to choose the best network architecture, we performed a grid search over the parameters based on the 10% randomly selected validation set from the training data. The parameter space include number of hidden layers {2, 3}, number of neurons {100, 200, 400, 800, 1600}, amount of dropout {0, 0.25, 0.5}, and L2 regularization.
All networks were trained using mini-batched stochastic gradient descent (SGD) and AdaGrad (Duchi et al., 2011). AdaGrad is an Adaptive Gradient Method that utilizes different adaptive learning rates for every feature. It was shown to significantly accelerate convergence and slightly improve performance of DNNs (Dean et al., 2012). The output layer is a standard softmax classifier and cross entropy objective function. For every endpoint, DNN models were trained independently.
In addition, we also investigated the performance of a multitask network (one model for all 12 tasks trained jointly) using the identical training approach. Learning several tasks at the same time is performed with the aim of mutual benefits between different tasks. The similarity (and dissimilarity) between the tasks is exploited to enrich a model (Caruana, 1997).
All models were trained using in-house software based on Theano framework (Bastien et al., 2012). We also used normalized DRAGONH descriptors as our input vectors.
Data Visualization
We use a multidimensional scaling (MDS) approach (Borg and Groenen, 1997), implemented in Python, to seek a low-dimensional representation of the data that conserves the distances in the original high-dimensional space. ECFP6 fingerprints are used to calculate the similarity matrix between the chemicals. MDS applied on this similarity matrix attempts to model the similarity or dissimilarity of data as distances in geometric space. In this way, higher similarity between the chemicals results in shorter distances between the chemicals in the projection.
Results
Overview
We have developed several Random Forest models using different descriptors and balancing approaches as described in Methods; these models are summarized in Table 1. Models 1, 2, and 3 were submitted for final evaluation and ranking; whereas, Model 4 was built after the Tox21 Challenge had closed (Table 1).

Table 1. Description of models implemented using Random Forest.
Evaluation and Ranking
The performance of all submitted models was evaluated by AUC-ROC resulted from predictions made for test set 2. Results for all of our models in comparison with the winning model for each assay are summarized in Figure 1. None of our submitted models (Model 1, Model 2, and Model 3) were ranked in the top 10. Additionally, differences in balancing protocol and descriptor type in our submitted models had little effect on the overall performance. Model 4 was built using unbalanced data. It was not submitted for evaluation by the organizers, and therefore was ineligible for ranking. Nevertheless, Model 4 showed a greater AUC value for 10 of the 12 assays over our three submitted models. Model 4 also showed comparable predictive performance to the winning models: seven of the 12 AUCs (AhR, Aromatase, ATAD5, ER, ER-LBD, MMP, p53) differ from the winner by 0.05. Interestingly, when comparing the external balanced accuracy, defined as (Sensitivity + Specificity)/2, of our models to those of winning models (based on AUC), a different trend emerges (Figure 2). For nine out of the 12 assays, the external balanced accuracy of at least one of our models is higher than that of the winning model. Indeed, for all submitted models in the Challenge, our Model 2 had the highest external balanced accuracy for AR (0.74); Model 4 had an even higher external balanced accuracy (0.82).
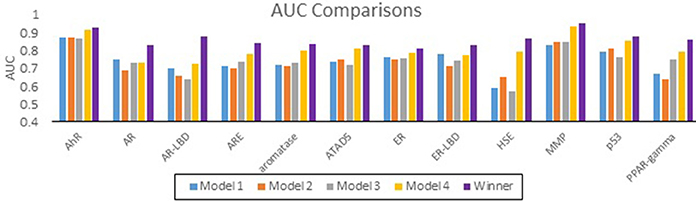
Figure 1. Comparison of AUC values for the Tox21 assays. AUC values of our models (blue, orange, gray, and yellow) as well as the AUC values of the winning model (purple).
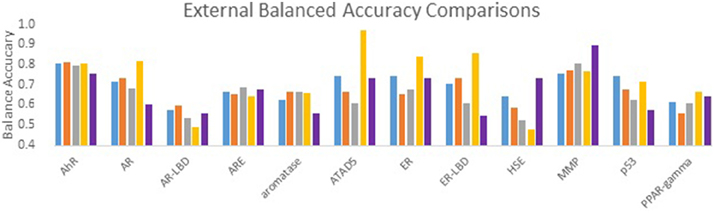
Figure 2. Comparison of external balanced accuracy (BA) values for the Tox21 assays. BA values of our models (blue, orange, gray, and yellow) as well as the BA values of the winning model (purple).
Figure 3 visualizes the distribution of active and inactive compounds from the training dataset and test set 2 based on fingerprint similarity (see Section Model Building and Evaluation for details). Figures 3A,B show the distribution of compounds in the training sets of Models 2 and 4, respectively, as well as in test set 2 for one of our most accurately predicted endpoints, AhR. Figures 3C,D show the same type of distribution for one of the least accurately predicted endpoints, HSE, that has largest increase in AUC as a result of using unbalanced dataset for modeling (Model 4). This analysis reveals that balanced training dataset used in Model 2 for AhR (Figure 3A) has tight clustering of active compounds in addition to broad coverage for the compounds to be predicted in test set 2. Thus, an increase in the number of compounds in the training dataset when unbalanced dataset is used does not result in a significant gain in AUC. However, as opposed to AhR, no distinct clusters are observed in the balanced dataset for HSE. Active and inactive compounds are widely dispersed, which calls into question the assay quality of this endpoint. This dispersion results in the misclassification of inactive compounds in test set 2. Figure 3D, however, shows that using unbalanced data increases the chemical diversity, which provides better coverage of test set 2, and enhances representation of inactives found in the test set 2. This expansion reflected in an increase of AUC (see Figure 1).
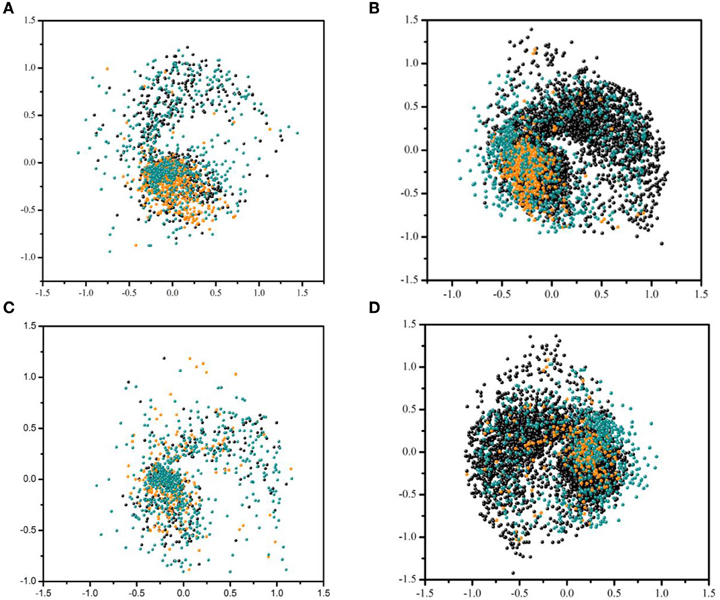
Figure 3. Distribution of active and inactive compounds from the training dataset and test set 2 based on fingerprint similarity. (A) Balacned training set used for endpoint AhR in Model 2 and test set 2. (B) Unbalanced training set used for AhR in Model 4 and test set 2. (C) Unbalanced training set used for endpoint HSE in Model 2 and test set 2. (D) Unbalanced training set used for HSE in Model 4 and test set 2. Each point represent either compounds from the test set 2 (cyan) and training set inactives (black) and actives (orange).
After the results of the challenge were announced, we also decided to evaluate the limit of model performance even further. We used Model 4 as our base line (see Table 2). We combined all three datasets and retrained Model 4 with the same RF parameters using 5-fold external cross validation (Model 4/5CV column in Table 2). Unexpectedly, we obtained significant performance boost. AUCs for three endpoints, AR, AR-LBD, and ER-LBD are significantly higher as compared to AUC values achieved by Model 4. Accuracy for the other nine assays were approximately on par with the balanced models. It is not clear why such discrepancy is observed, most likely it is due to the small size of the test set and very small number of active compounds in each of them.
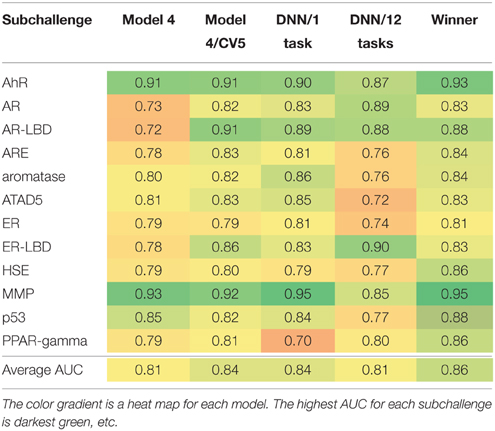
Table 2. Post-challenge assessment of the accuracy (AUC) of different models and their comparison with the wining solution.
Given that the overall challenge winner used DNN (Mayr et al., 2015), we decided to investigate the utility of DNN after the completion of the challenge. Due to very limited technical information released by the winning team, however, we were not able to independently verify their models. Instead, we trained DNN according to our own protocol (See Section Model Building and Evaluation). Table 2 also reports performance of DNN models in single task and multitask regimes. On average, both approaches were not able to match the winning model, AUCs 0.84 and 0.81 vs. 0.86. However, the difference between DNN/1task (our best overall model) and winning team is small with a notable exception of PPAR-gamma, ΔAUC = 0.16. The single task DNN model was also significantly better than Model 4 for AR and AR-LBD. Very recently, models of drug-induced liver injury (DILI) with DNN were also found to provide better performance than previously described “shallow” prediction models (Xu et al., 2015). Therefore, DNN architectures seems to be beneficial for toxicity prediction. In strike contrast, performance of the multitask model was poor for five assays (ARE, Aromatase, ATAD5, ER, and p53). Due to the limited dataset size, we were not able to reliably train all DNN models. In order to take full advantage of deep learning methods at least an order of magnitude larger number of training examples is necessary.
Discussion
The results of our submitted models (Model 1, Model 2, and Model 3) indicate that for these data no combination of descriptors or balancing protocol outperforms any other combination. Intriguingly, our unbalanced (and un-submitted) Model 4 outperformed our submitted models and had AUC values comparable to the winning models. This observation demonstrates that for these assays balancing actually decreases model performance. This may be because balancing restricts the chemical space covered in toto by inactives. Since the number of actives in test set 2 is much smaller than the number of inactives (between 1 and 14% of test set 2 compounds are actives for a given assay), reducing the chemical space of inactives through balancing may have resulted in the misclassification of inactives in test set 2. In general, when training set compounds are highly imbalanced toward the inactive class, QSAR classification will favor the majority (inactive) class, resulting in low sensitivity for the minority (active) class (Chen et al., 2005). For this reason, datasets are usually balanced as to maximize the sensitivity and specificity of the training set. In the current challenge, however, models were evaluated on an external dataset that was highly populated with the inactive class. Therefore, for future challenges and/or modeling efforts regarding these assay endpoints, using unbalanced data may be preferable.
Conflicting performance trends obtained in Table 2 also emphasizes the following community needs:
1. Judging model performance using a very small test set can be suboptimal.
2. Deep Learning can provide some accuracy improvement compared to regular machine learning methods. However, model reproducibility is very hard to achieve, especially for this rapidly emerging field.
3. Further methodological developments are required to investigate applicability and methods of training multitask-DNN method. There is a significant room for model improvement and exploiting information about assay relations as well as target features and other biological information.
Conclusion
In this work, we investigated the use of different QSAR approaches for toxicity assays prediction in the 2014 Tox21 challenge. We carefully curated all datasets according the well-established protocol. We used Random Forest and Deep Neural Nets to train models. In addition we also explored several balancing strategies. The model performance was evaluated by the area under the receiver operating characteristic curve (AUC-ROC) and by the balanced accuracy (BA). The values for AUC-ROC were in the range of 0.55–0.87 and those for BA were in the range of 0.58–0.82; the highest predictive power was achieved for the AR pathway assay. No significant difference in respective model performance was found when using different curation protocols or different descriptors. Marginal increase in AUC-ROC as well as in BA was observed for some of the pathways when the dataset was balanced based on the similarity to the external test set (test set 2). Moreover, a significant increase in the balanced accuracy of prediction for external datasets was found once the unbalanced datasets were used to build the model. Our results show that overall neural networks achieved improvement over simple machine learning algorithms and that balancing lead to a significant accuracy decrease.
The Tox21 Challenge was evaluated using the AUC metric. Interestingly we noticed, when evaluated using BA, at least one of our models outperformed the winning model in 10/12 assays. Furthermore, our Model 2 had the highest balanced accuracy for AR (0.74) against all submissions. Our models, therefore, can be used for future screening of compounds for toxicity in these pathways. Our models have the additional advantage of being freely and publicly available through our Chembench platform (https://chembench.mml.unc.edu/; Walker et al., 2010).
The goal of the 2014 Tox21 Challenge was to predict toxicity in the various biological pathways using chemical structure data only. The availability of these chemical structures and their associated biological activity in the pathways of interest affords the opportunity to build pathway-based hybrid QSAR models. Hybrid QSAR models utilize in vitro bioactivity as biological descriptors in conjunction with chemical descriptors in order to improve the predictivity of QSAR models (Liu et al., 2015). These hybrid QSAR models could be employed toward the prediction of in vivo toxic effects, which is a considerable challenge for predictive toxicology.
In sum, the 2014 Tox21 Challenge successfully enabled academic groups, industrial teams, and fans of machine-learning from around the world to compare and contrast various in silico methodologies toward the prediction of toxicity in several different assays. These modeling efforts and their associated findings will be of great use to the scientific community and will enhance the quality of toxicity prediction going forward.
Author Contributions
SC, RP, and OI contributed equally to this work, collaborating on all sections of the manuscript. SF provided information related to pathways. AT provided final edits.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors kindly thank the following funding source: National Institutes of Health (1R01GM096967-01A1). The authors would like to acknowledge ChemAxon for providing an academic license and the organizers of the 2014 Tox21 Challenge. OI gratefully acknowledge the support and hardware donation of NVIDIA Corporation and personally Mark Berger. OI also acknowledges partial support from 2015 CSA Trust grant for Deep Learning work.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2016.00003
Abbreviations
AR, androgen receptor; AR-LBD, androgen receptor—ligand binding domain; AhR, aryl hydrocarbon receptor; ER, estrogen receptor alpha—full; ER-LBD, estrogen receptor alpha—ligand binding domain; PPAR-gamma, peroxisome proliferator-activated receptor gamma; ARE, nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element; HSE, heat shock factor response element; MMP, mitochondrial membrane potential; p53, tumor suppressor p53; QSAR, Quantitative Structure-Activity Relationship; HTS, High-Throughput Screening; AUC, Area under the curve; BA, Balanced accuracy; DNN, Deep Neural Network.
References
Anastas, P., Teichman, K., and Hubal, E. C. (2010). Ensuring the safety of chemicals. J. Expo. Sci. Environ. Epidemiol. 20, 395–396. doi: 10.1038/jes.2010.28
Bae, B., Jeong, J. H., and Lee, S. J. (2002). The quantification and characterization of endocrine disruptor bisphenol-a leaching from epoxy resin. Wat. Sci. Technol. 46, 381–387. Available online at: http://wst.iwaponline.com/content/46/11-12/381
Bastien, F., Lamblin, P., Pascanu, R., Bergstra, J., Goodfellow, I., Bergeron, A., et al. (2012). Theano: New Features and Speed Improvements. Symbolic Computation; Learning. Available online at: http://arxiv.org/abs/1211.5590
Borg, I., and Groenen, P. (1997). Modern Multidimensional Scarles. Springer Series in Statistics. New York, NY: Springer-Verlag.
Casals-Casas, C., and Desvergne, B. (2011). Endocrine disruptors: from endocrine to metabolic disruption. Annu. Rev. Physiol. 73, 135–162. doi: 10.1146/annurev-physiol-012110-142200
Chandra, D. (ed.). (2013). Mitochondria as Targets for Phytochemicals in Cancer Prevention and Therapy. New York, NY: Springer.
Chen, J. J., Tsai, C. A., Young, J. F., and Kodell, R. L. (2005). Classification ensembles for unbalanced class sizes in predictive toxicology. SAR QSAR Environ. Res. 16, 517–529. doi: 10.1080/10659360500468468
Dean, J., Corrado, G., Monga, R., Chen, K., Devin, M., Mao, M., et al. (2012). “Large scale distributed deep networks,” in Advances in Neural Information Processing Systems, 1223–1231. Available online at: http://papers.nips.cc/paper/4687-large-scale-distributed-deep-networks
Diamanti-Kandarakis, E., Bourguignon, J.-P., Giudice, L. C., Hauser, R., Prins, G. S., Soto, A. M. R., et al. (2009). Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr. Rev. 30, 293–342. doi: 10.1210/er.2009-0002
Dix, D. J., Houck, K. A., Martin, M. T., Richard, A. M., Woodrow Setzer, R., Kavlock, R. J., et al. (2007). The toxcast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 95, 5–12. doi: 10.1093/toxsci/kfl103
Duchi, J., Hazan, E., and Singer, Y. (2011). Adaptive subgradient methods for online learning and stochastic optimization. J. Mach. Learn. Res. 12, 2121–2159. Available online at: http://dl.acm.org/citation.cfm?id=1953048.2021068
Fourches, D., Muratov, E., and Tropsha, A. (2010). Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 50, 1189–1204. doi: 10.1021/ci100176x
Han, W.-D., Mu, Y.-M., Lu, X.-C., Xu, Z.-M., Li, X.-J., Yu, L., et al. (2003). Up-regulation of LRP16 mRNA by 17beta-estradiol through activation of estrogen receptor alpha (ERalpha), but not ERbeta, and promotion of human breast cancer MCF-7 cell proliferation: a preliminary report. Endocr. Relat. Cancer 10, 217–224. doi: 10.1677/erc.0.0100217
Kola, I., and Landis, J. (2004). Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715. doi: 10.1038/nrd1470
Kuz'min, V. E., Artemenko, A. G., and Muratov, E. N. (2008). Hierarchical QSAR technology based on the simplex representation of molecular structure. J. Comput. Aided Mol. Des. 22, 403–421. doi: 10.1007/s10822-008-9179-6
Liu, J., Mansouri, K., Judson, R. S., Martin, M. T., Hong, H., Chen, M., et al. (2015). Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 28, 738–751. doi: 10.1021/tx500501h
Mayr, A., Klambauer, G., Unterthiner, T., and Hochreiter, S. (2015). DeepTox: toxicity prediction using deep learning. Front. Environ. Sci. 3:80. doi: 10.3389/fenvs.2015.00080
Min, J., Lee, S.-K., and Bock Gu, M. (2003). Effects of endocrine disrupting chemicals on distinct expression patterns of estrogen receptor, cytochrome P450 aromatase and p53 genes in oryzias latipes liver. J. Biochem. Mol. Toxicol. 17, 272–227. doi: 10.1002/jbt.10089
Muratov, E. N., Artemenko, A. G., Varlamova, E. V., Polischuk, P. G., Lozitsky, V. P., Fedchuk, A. S., et al. (2010). Per Aspera Ad Astra: application of simplex QSAR approach in antiviral research. Fut. Med. Chem. 2, 1205–1226. doi: 10.4155/fmc.10.194
Nair, V., and Hinton, G. E. (2010). “Rectified linear units improve restricted boltzmann machines,” in Proceedings of the 27th International Conference on Machine Learning (ICML-10). 807–814. Available online at: http://citeseer.ist.psu.edu/viewdoc/summary?
O'Brien, P. J., Irwin, W., Diaz, D., Howard-Cofield, E., Krejsa, C. M., Slaughter, M. R., et al. (2006). High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 80, 580–604. doi: 10.1007/s00204-006-0091-3
Schmidhuber, J. (2015). Deep learning in neural networks: an overview. Neural Networks 61, 85–117. doi: 10.1016/j.neunet.2014.09.003
Tropsha, A. (2010). Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 29, 476–488. doi: 10.1002/minf.201000061
Tropsha, A., Gramatica, P., and Gombar, V. K. (2003). The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 22, 69–77. doi: 10.1002/qsar.200390007
Walker, T., Grulke, C. M., Pozefsky, D., and Tropsha, A. (2010). Chembench: a cheminformatics workbench. Bioinformatics 26, 3000–3001. doi: 10.1093/bioinformatics/btq556
Keywords: Tox21, machine-learning, stress response signaling pathways, nuclear receptor signaling pathways, endocrine disrupting chemicals, QSAR, deep learning
Citation: Capuzzi SJ, Politi R, Isayev O, Farag S and Tropsha A (2016) QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci. 4:3. doi: 10.3389/fenvs.2016.00003
Received: 30 September 2015; Accepted: 12 January 2016;
Published: 04 February 2016.
Edited by:
Ruili Huang, NIH National Center for Advancing Translational Sciences, USAReviewed by:
Patrick D. McMullen, The Hamner Institutes for Health Sciences, USAZhichao Liu, National Center for Toxicological Research, Food and Drug Administration, USA
Copyright © 2016 Capuzzi, Politi, Isayev, Farag and Tropsha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Tropsha, YWxleF90cm9wc2hhQGVtYWlsLnVuYy5lZHU=
†These authors have contributed equally to this work.