 Alexios Koutsoukas
Alexios Koutsoukas Joseph St. Amand
Joseph St. Amand- Department of Electrical Engineering and Computer Science, Information and Telecommunication Technology Center, University of Kansas, Lawrence, KS, USA
Modern drug discovery and toxicological research are under pressure, as the cost of developing and testing new chemicals for potential toxicological risk is rising. Extensive evaluation of chemical products for potential adverse effects is a challenging task, due to the large number of chemicals and the possible hazardous effects on human health. Safety regulatory agencies around the world are dealing with two major challenges. First, the growth of chemicals introduced every year in household products and medicines that need to be tested, and second the need to protect public welfare. Hence, alternative and more efficient toxicological risk assessment methods are in high demand. The Toxicology in the 21st Century (Tox21) consortium a collaborative effort was formed to develop and investigate alternative assessment methods. A collection of 10,000 compounds composed of environmental chemicals and approved drugs were screened for interference in biochemical pathways and released for crowdsourcing data analysis. The physicochemical space covered by Tox21 library was explored, measured by Molecular Weight (MW) and the octanol/water partition coefficient (cLogP). It was found that on average chemical structures had MW of 272.6 Daltons. In case of cLogP the average value was 2.476. Next relationships between assays were examined based on compounds activity profiles across the assays utilizing the Pearson correlation coefficient r. A cluster was observed between the Androgen and Estrogen Receptors and their ligand bind domains accordingly indicating presence of cross talks among the receptors. The highest correlations observed were between NR.AR and NR.AR_LBD, where it was r = 0.66 and between NR.ER and NR.ER_LBD, where it was r = 0.5. Our approach to model the Tox21 data consisted of utilizing circular molecular fingerprints combined with Random Forest and Support Vector Machine by modeling each assay independently. In all of the 12 sub-challenges our modeling approach achieved performance equal to or higher than 0.7 ROC-AUC showing strong overall performance. Best performance was achieved in sub-challenges NR.AR_LBD, NR.ER_LDB and NR.PPAR_gamma, where ROC-AUC of 0.756, 0.790, and 0.803 was achieved accordingly. These results show that computational methods based on machine learning techniques are well suited to support and play critical role in toxicological research.
Introduction
The average person is exposed to hundreds of chemicals not found naturally in the human organism during his lifespan. Xenobiotic man-made products can be found in wide range of cleaning and healthcare products, as food additives or drugs ingredients among others in various concentrations and mixtures. Advances in modern combinatorial chemistry have led to an unprecedented growth of synthetic chemicals availability on the market. Over the course of the last five decades the number of registered organic and inorganic substances in Chemical Abstract Service (CAS) Registry database grew well over 33 million, when in the 1965 the number was barely exceeding that of 200 thousands (Binetti et al., 2008).
Chemical toxicity may cause life-threating adverse effects on human health, therefore it is necessary to conduct regular risk assessments to ensure and protect public safety (Landrigan and Goldman, 2011). Hazardous toxicological effects on human health that may result due to short or chronic exposure to toxic chemicals include acute toxicity, toxicity to reproduction, mutagenicity and carcinogenicity (Binetti et al., 2008).
The traditional paradigm in toxicity testing consists of in vivo toxicology, where compounds are tested in various and usually high concentrations against tens or even hundreds of rodents or other animals (Merlot, 2010). This paradigm in toxicity testing is not feasible in modern toxicological research due to the large number of chemicals that need to be tested, the high cost of animal models, low throughput readouts, ethical issues, often contradictory findings and poor extrapolability to humans among others and have been extensively discussed in literature (Sun et al., 2012; Calafat et al., 2015).
Safety regulatory agencies are currently dealing with two major challenges. First, the increased number of chemicals that need to be tested for potential harmful effects on human health and second, the time and cost required to evaluate those chemicals (Hartung, 2009). Hence, novel and more efficient assessment methods for evaluation of potential toxicological effects are in high demand. Alternative avenues are currently being explored for chemical risk assessment using in-vivo and in-vitro approaches, such as human cell-based assays and high-throughput screening technologies (HTS; Ekins et al., 2005; Inglese et al., 2006; Shukla et al., 2010). Quantitative high-throughput screening (qHTS) technology has emerged as powerful and efficient way to alleviate limitations of single-point concentration HTS screening and allow to study complex toxicological mechanisms to specific pathways of targeted organs that may lead to disease (Inglese et al., 2006; Lock et al., 2012). qHTS is a titration-based screening approach that utilizes modern screening technologies, such as high-sensitivity detectors, low-volume dispensing and robotic plate handle (Inglese et al., 2006). As opposed to single-point concentration HTS screening, which typically suffers from large number of false positives and false negative readouts, qHTS is capable of identifying and efficiently elucidating structure-activity relationships (SARs) from primary screens. Furthermore, qHTS screening allows thousands compounds (>104 compounds) to be evaluated in different concentrations in cell models in an unprecedented rate (Schmidt, 2009; Attene-Ramos et al., 2013).
Computational approaches for modeling pharmacological and toxicological data combined with powerful data mining algorithms have been steadily gaining popularity by public and private bodies over the last decades (Muster et al., 2008; Kavlock and Dix, 2010). In-silico approaches utilize experimental data generated by in-vivo and in-vitro screening technologies and combined with cutting-edge data mining and cheminformatic techniques are capable of developing powerful predictive models. Such models could be applied to “virtually screen” thousands of chemicals for potential unwanted reactions early on during development cycles or to re-evaluate existing ones. In silico approaches can be applied to generate testable hypothesis for chemicals and direct experimentation toward the most likely unwanted interactions, which can then be validated or invalidated. Hence, in-silico approaches could become the “next big thing” as decision-making tools during the development and risk assessment stages. Therefore, computational approaches could provide more efficient utilization of the limited experimental resources.
The Toxicology in the 21st Century (Tox21) consortium is a major collaborative effort involving several agencies, the National Institutes of Health (NIH), the Environmental Protection Agency (EPA), and the Food and Drug Administration (FDA), was formed to develop and evaluate alternative risk assessment methods (Dix et al., 2007; Judson et al., 2009). A collection of 10,000 compounds composed of environmental chemicals and approved drugs was screened for interference in biochemical pathways of Nuclear and Stress receptor pathways and released for crowdsourcing data analysis.
The datasets released as part of the data challenge were generated by qHTS screening assays and contained compounds activity data against 12 assays, seven of which were part of the Nuclear Receptors (NR) and five of Stress Response (SR) pathways. Nuclear Receptors (NR) are an important family of transcription factors responsible for regulating gene expression and have a wide range of key roles in organisms' cell growth and proliferation, metabolism and homeostasis (Olefsky, 2001). Chemical interference by environmental pollutants or other xenobiotic chemicals can disturb homeostasis and lead to severe toxicities (Janošek et al., 2006). In vivo effects may range from male feminization to reproduction disorders and have been linked with chemical interference of NR (Baker, 2001). Structurally members of NR family present common features, which consist of a DNA binding domain (DBD), which recognize and bind to specific DNA sequences, and a ligand binding domain (LBD), which is located at the C-terminal half, and is responsible for recognizing and interacting with hormone molecules (Wurtz et al., 1996; Moras and Gronemeyer, 1998; Bourguet et al., 2000). NRs included in the challenge were Androgen Receptor (NR.AR), Androgen Receptor Ligand Binding Domain (NR.AR_LBD), Estrogen Receptor (NR.ER) and Estrogen Receptor Ligand Binding Domain (NR.ER_LBD), Aryl hydrocarbon Receptor (NR.AhR) and Peroxisome Proliferator-Activated Receptor gamma (NR.PPAR-γ). Aromatase (NR.Aromatase), member of the Cytochrome P450 protein family, responsible for the biosynthesis of estrogens, was the last included assay part of the NR pathway group (Simpson et al., 1994, 1997).
Cells respond to environmental stress factors, such as elevated and extreme temperature ranges, DNA damages, environmental and chemical toxicants and mechanical damages through a number of mechanisms that belong to Stress Response (SR) pathways (Fulda et al., 2010). Stress Response pathways are responsible for maintaining cell and tissue homeostasis. Five such biochemical assays were included in the challenge namely the ATPase family AAA domain-containing protein 5 (SR.ATAD5), which is involved in DNA damage response (Fox et al., 2012). Heat Shock response Elements (SR.HSE), which are proteins responsible for regulating the expression of heat shock genes (Wu, 1995). Mitochondrial Membrane Potential (SR.MMP) assays are used to evaluate chemically induced mitochondrial toxicity (Varga et al., 2015). Mitochondrial membrane potential changes are commonly measured using fluorescent dyes tools and are linked with cell capacity to generate ATP (Perry et al., 2011). Tumor suppressor protein (SR.p53), typically the p53 pathway is “off” and is activated when cells are under stress or damaged, hence being a good indicator of DNA damage and other cellular stresses (Vogelstein et al., 2000). Tumor suppressor protein p53 is activated by inducing DNA repair, cell cycle arrest and apoptosis (Levine, 1997). The fifth and last SR assay was the antioxidant response element (SR.ARE) signaling pathway. SR.ARE is responsible for regulating the expression of genes in cells exposed to oxidative stress that can change the cellular redox statues (Nguyen et al., 2003).
First the distribution of physicochemical space covered by the Tox21 library by utilizing simple molecular descriptors, the molecular weight (MW) and the octanol/water coefficient (cLogP) were examined. This analysis was performed to obtain an overview of the physicochemical space covered by the Tox21 library and the overlap between the training and testing datasets released during the competition.
It's been shown that chemicals can be active against multiple targets simultaneously, which has been termed as “polypharmacology” (Keiser et al., 2007; Klabunde, 2007). One of the major limitations when analyzing public bioactivity datasets is data incompleteness, which results to sparse bioactivity matrices (Mestres et al., 2008). On the contrary the Tox21 dataset provides a less incomplete bioactivity matrix across the 12 tested assays allowing such analysis to be carried out. The goal here was to investigate relationships between assays in bioactivity space based on the reported chemicals activities across the assays.
Our approach to model the Tox21 data consisted of utilizing circular molecular fingerprints combined with Random Forest and Support Vector Machines by modeling each assay independently. Circular fingerprints were selected for the study as they have been previously shown to perform well in virtual screening applications (Bender, 2010; Hu et al., 2012; Cereto-Massagué et al., 2015). As machine learning techniques two well-established algorithms in the field of cheminformatics were selected and applied, namely the Random Forest (RF) and the Support Vector Machine (SVM). Since their introduction to the field of molecular modeling they have both been successfully applied for a wide range of modeling tasks ranging from virtual screening (Koutsoukas et al., 2011), QSARs/QSPRs (Dudek et al., 2006; Guha, 2008) and to more recent proteocheometric modeling tasks (van Westen et al., 2011). Random Forest (RF), developed by Breiman, is an ensemble of unpruned classification or regression tress formed by applying bootstrap samples of the training data and random features selection in tree induction (Breiman, 2001). On the other hand, the Support Vector Machine (SVM), developed by Cortes and Vapnik, is a non-probabilistic kernel-based supervised learning method that maps input vectors into high-dimensional feature space where the decision hyperplane is constructed (Cortes and Vapnik, 1995). Our main hypothesis was that utilizing circular fingerprints combined with supervised machine learning methods would allow us to develop fast and accurate predictive models well suited for predictive toxicology.
Materials and Methods
In total three datasets were released by the Tox21 data challenge team during the competition: The training set which was designated to serve for model development and hyper-parameters tuning, which from now on will be referred as Tox21_10k, and contained initially 11,764 structures covering activity measurements against 12 assays. The first released test set, which was used to rank teams submissions during the early phase of the competition, which from now on will be referred as Tox21_LDB, and contained 296 structures. The final released dataset was the external validation set, this dataset was used for the final phase of the competition for model evaluation and ranking teams' submissions, which from now on will be referred as Tox21_Ext_Valid, and contained 647 structures. Compounds activities for the external dataset were made publicly available only after the completion of the competition. Final teams submissions were evaluated based on the generated predictions on the external set Tox21_Ext_Valid set.
Data Preprocessing
Prior to modeling steps the datasets were pre-processed and chemical structures standardized with the aim of retaining only suitable structures for the following modeling steps. The importance of data curation prior to modeling steps has been extensively discussed in Fourches et al. (2010). Chemical structures were standardized using the ChemAxon Standardizer software package and stored in SDF (ChemAxon Standardizer, 2014) with the options on: (i) remove salts and solvents, (ii) disconnect metal atoms, (iii) remove fragments (keep largest ones), (iv) add explicit hydrogens, (v) aromatize, (vi) neutralize, (vii) tautomerize, (viii) mesomerize, the protocol utilized is provided in the Supplementary Material named “Stand_Prot.xml.”
The number of unique structures in the Tox21_10k was measured to be 7,502 from the initial 11,764 and 295 for the Tox21_LBD following the standardization process. Following the structure standardization steps compounds activities were normalized by applying the majority rule based on standardized SMILES strings on a per assay basis. In cases where multiple activities were reported against a assay the activity with the most occurrences was retain, else were discarded as ambiguous. Instances where only a single activity was present were retained. Those cases could be attributed to variances in experimental conditions, concentrations, levels of purity and different vendors used, as was also stated by the Tox21 Team during the competition. The number of total instances, the number of active and inactive compounds as also the ratio of inactive/active per assay is shown on Table 1. The number of total instances per assay ranged from 5,747 for NR.Aromatase and up to 6,950 for NR.AR. The ratio of inactive/active instances per assay ranged from 5.5, relatively imbalanced, for SR.ARE and SR.MMP and up to 30.2, highly imbalanced, for NR.AR_LBD. The final datasets that resulted from the above described process are provided in the Supplementary Files “Sup_Tox21_10k” and “Sup_Tox21_LDB.”
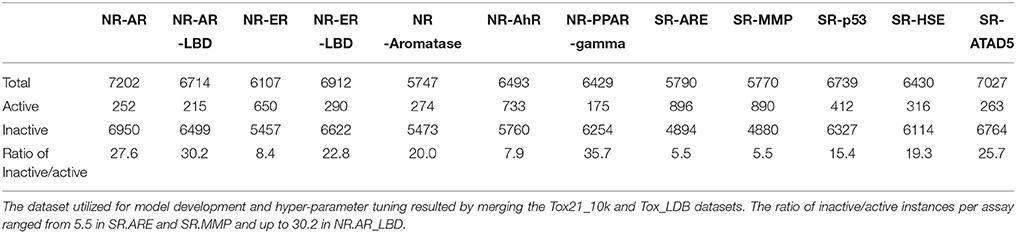
Table 1. Number of data points per assay obtained following the standardization process.
Molecular Descriptors
As molecular descriptors the Morgan Fingerprints (Circular Fingerprints) with radius 3 were utilized, which are equivalent to the extended connectivity fingerprints ECFP_6 (Rogers and Hahn, 2010), with diameter 6. The open source RDKit library (version 2014.09.1) was used to generate the molecular fingerprints from the standardized chemical structures (Landrum, 2015). The descriptors were generated as hashed binary vectors of 1,024 bits length. Morgan fingerprints were the only descriptor utilized during the modeling steps. Molecular Weight (MW) and the octanol/water partition coefficient (cLogP) were calculated using the MOE software package and used to examine and visualize the physicochemical space covered by the Tox21 library (Chemical Computing Group Inc., 2015).
Modeling Approach
Following the data pre-processing the two datasets Tox21_10k and Tox21_LDB were merged to form one larger dataset that was used for model development and hyper-parameters tuning, shown in Figure 3. No external data outside of those provided by the Tox21 Challenge team were utilized in any step of the modeling process. RF and SVM were utilized as implemented in the open-source machine learning library Scikit-learn (Pedregosa et al., 2011).
Each assay/sub-challenge was modeled independently following a single-task approach. 10-fold cross-validation was applied to tune the hyper-parameters for each algorithm. As performance metric the area under the ROC curve (AUC) was used. Receiver Operating Characteristics (ROC) graphs are commonly used in machine learning to compare and visualize the performance of binary classifiers (Fawcett, 2006). The area under the ROC curve (ROC-AUC) of a classifier is a single scalar values represents expected performance, and is equal to the probability that the classifier will rank a random chosen positive instance higher than a negative instance (Bradley, 1997). AUC takes values between (0,1), where values equal to or smaller than 0.5 show that a classifier performs no better or worse than random, instead for values greater than 0.5 a classifier is expected to perform better than random.
In case of SVM the radial basis function “rbf” kernel was considered with values for Cost {103,102,101,1,10−1} and gamma {10−4, 10−3, 10−2, 10−1, 1}. In case of Random Forest the values considered for the number of trees was {50, 100, 300, 500, 1000, 1500} and number of features in each split {log2, sqrt}. The best average AUC and the standard deviation observed over 10-fold cross validation per assay by RF and SVM during the hyper-parameter tuning are shown in Figure 4, named “Best RF 10-CV” and “Best SVM 10-CV” accordingly. The implementations used to tune RF and SVM using ROC-AUC as evaluation metric based on the Scikit-learn are provided in the Supplementary Material “RF_tune.py” and “SVM_tune.py.”
Results
First the chemical space covered by the Tox21 chemical library was examined by calculating and analyzing the distribution of Molecular Weight (MW) and cLogP for the library, shown in Figure 1. As mentioned earlier the total number of structures counted was 7,502 in Tox21_10k, 295 in Tox21_LBD and 647 in Tox21_Ext_Valid following the pre-processing steps. Here it was found that compounds had on average MW of 271.2 and a median of 244.3 Dalton with 50% of compounds having MW between 166 and 337 Dalton. In case of cLogP the average value was 2.41 and median 2.39, with 50% of compounds having values between 0.98 and 3.753. This analysis indicates that a large portion of compounds included in the Tox21 library represent chemicals with drug-like properties, although compounds with MW and cLogP values outside of those typically occupied by drug-like molecules are not rare, e.g., compounds with MW over 1,000 Daltons and cLogP lower than -1 or higher than 6.
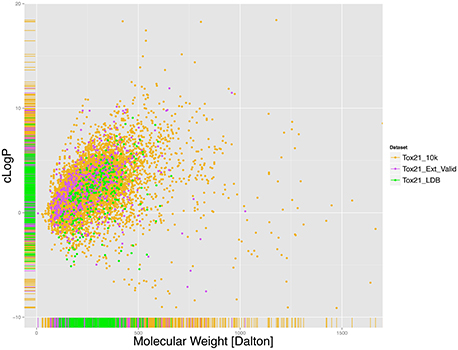
Figure 1. Molecular Weight (MW) and cLogP distribution for the Tox21 chemical library, (gold) Tox21_10k, (green) Tox21_LDB and (purple) Tox21_Ext_Valid. For each color, the rug on x and y-axis represent the areas that are more densely populated. On average the compounds in the Tox21 library present MW of 272.6 and a median of 240.4 Daltons with 50% of compounds having MW between 166 and 339. In case of cLogP the average value was 2.476 and median 2.439, with 50% of compounds being between 1.045 and 3.811.
Next the relationships between assays based on bioactivity profiles of the tested chemicals were examined. Relationships between assays were calculated utilizing the Pearson correlation coefficient r based on compounds activities across tested assays (Todeschini et al., 2012), shown in Figure 2. The analysis was generated using the R programming language (Ihaka and Gentleman, 1996) and the “corrplot” package for visualization, the R script is provided in the Supplementary Material “CorrelationAssaysPlot.R” (Wei, 2013). A cluster was formed between the Androgen and Estrogen Receptors and their ligand bind domains accordingly, which can be seen on the top-left corner of the Figure 2, indicating presence of cross-talks between the two receptors. The correlation between NR.AR and NR.AR_LBD was found to r = 0.66 and between NR.ER and NR.ER_LBD r = 0.5. Furthermore, correlation of r = 0.39 between the NR.AR_LBD and the NR.ER_LBD was observed, indicating that the two ligand binding domains share some structural similarities that can accommodate similar ligands. On the contrary, weak correlations were measured between the NR.AR and the NR.ER_LBD as also between the NR.ER and the NR.AR_LBD, where it was measured to be r = 0.33 and r = 0.22 accordingly. The rest receptors didn't show any correlation between them as the highest observed correlation didn't exceed of r = 0.23 between NR.ER_LBD and SR.p53 and of r = 0.21 between SR.p53 and SR.HSE.
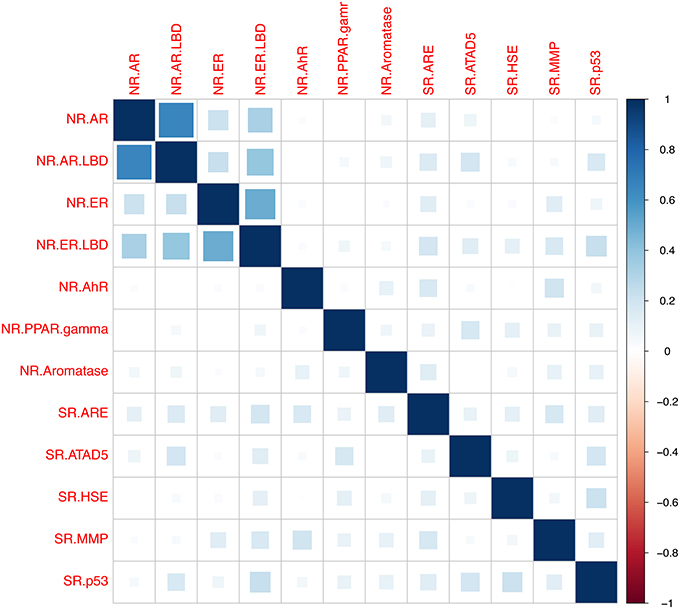
Figure 2. Pearson correlation coefficient r between assays based on compounds activity profiles across the assays. A cluster was formed between the Androgen receptor (NR.AR) and the Androgen receptor ligand-binding domain (NR.AR_LBD), where r was measured 0.66. The Estrogen receptor (NR.ER) and the Estrogen receptor ligand-binding domain (NR.ER.LBD) had r = 0.5. Weaker correlation between the NR.AR_LBD and the NR.ER_LBD was observed (Pearson r = 0.39) showing possible crosstalk between the ligand binding domains. The rest measured correlation were low and did not exceed r = 0.23 between the NR.ER.LBD and SR.p53 and r = 0.21 between SR.HSE and SR.p53.
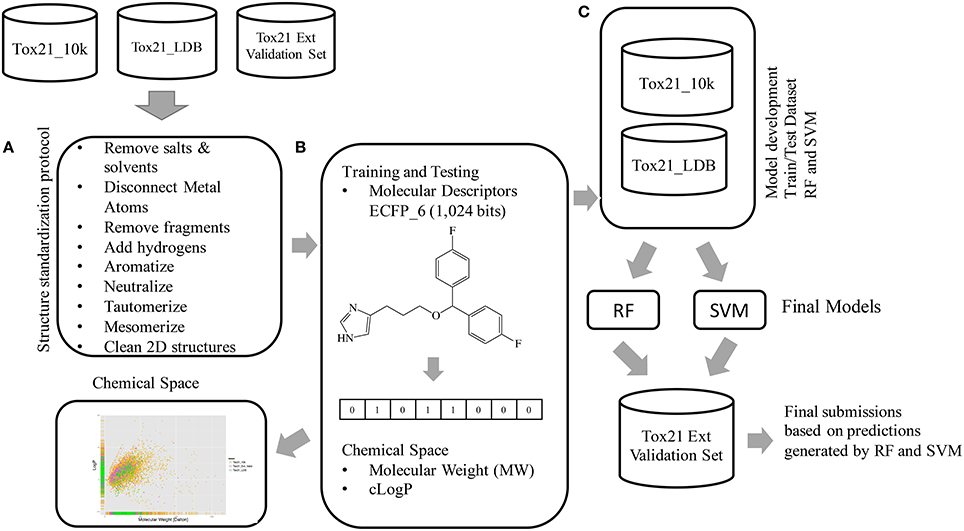
Figure 3. Workflow followed to model the Tox21 dataset (the Tox21_10k dataset containing the initially released data for model development, the Tox21_LDB used for leaderboard ranking and the blind dataset Tox21 External Validation Set for final model evaluation). (A) Structure standardization step, (B) Activities normalization and molecular descriptors calculations. (C) Datasets Tox21_10k and Tox21_LDB were merged into one larger dataset that was subsequently used for model development and hyper-parameters tuning. Final models were evaluated based on the generated predictions on the blind Tox21_Ext_Val Set.
Our group participated in the competition under team aliases frozenarm and ToxFit, where the first submission was based on the results obtained modeling the data using SVM and the latter based on RF independently. In all of the 12 sub-challenges our modeling approaches achieved performance of at least 0.7 ROC-AUC, only for the assay (SR.HSE) the results achieved by SVM were below 0.7 (0.689), showing strong overall performance, shown in Table 2 and Figure 4. As expected the performance achieved by both algorithms during cross validation on the training set and on the external set were different, as shown in Figure 4, with the results achieved on the external dataset being lower. These observed differences could be attributed to several factors, e.g., structural differences between chemical space included in the training and test set, imbalances among inactive/active instances per assay and limitations of utilized molecular descriptors to capture complex chemical features responsible for the bioactivities. When comparing the results achieved by SVM and RF on the Tox21_Ext_Valid, as shown in Table 2, it can be seen that both algorithms achieved comparable results, with RF achieving slightly better ROC-AUC in 7 out of 12 tasks, while SVM in 4 out of 12, and in 1 task (NR.AR) where both algorithms achieved the same ROC-AUC of 0.744.
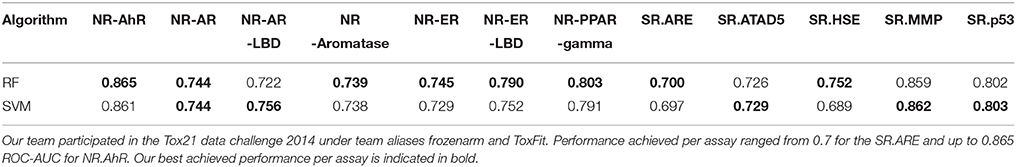
Table 2. Performance achieved by our modeling approach using Random Forest (RF) and Support Vector Machine (SVM) measured by ROC-AUC per sub-challenge on the external To21_Ext_Val.
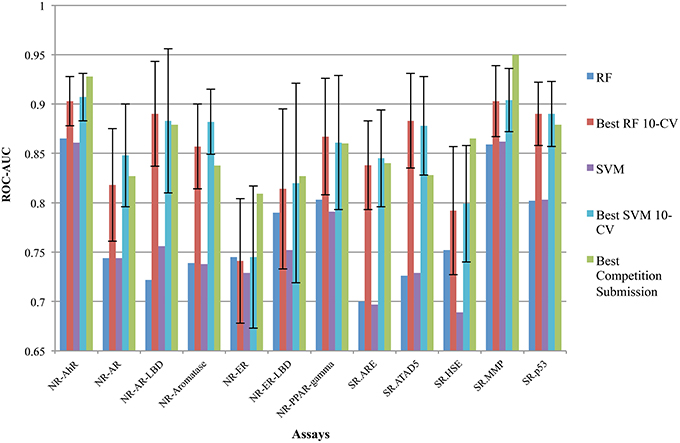
Figure 4. Performance measured by ROC-AUC achieved by RF and SVM accordingly (shown as RF and SVM) per sub-challenge compared to the top-1 reported performance. The best-reported results of the competition per sub-challenge are shown as Best Competition Submission. Best performance achieved during hyper-parameters optimization for RF and SVM over 10-fold cross validation, both the best mean AUC and the standard deviation over the 10-folds are shown as Best RF 10-CV and Best SVM 10-CV, respectively. Our submissions for the To21_Ext_Valid ranged from 0.69 AUC in SR.HSE achieved by SVM, worse performance, and up to 0.865 for SR.AhR by RF, best performance.
It worth noting that in our modeling approach no external data besides of those provided during the competition were utilized and only a single molecular descriptor was used, mainly due to time constrains during the competition. Utilizing external bioactivity data, e.g., from ChEMBL (Gaulton et al., 2012) or PubChem (Wang et al., 2009) databases, and additional molecular descriptors could potentially improve the performance of the models on the external evaluation set.
Discussion
Chemical toxicological risk assessment is a necessary step to ensure public safety and to promote well-being. Potential hazardous side-effects should be detected as early as possible in order to allow informed decisions to be made regarding the future fate of those products. Computational approaches that combine experimental data generated by next generation of high-throughput screening technologies, such as qHTS, and powerful data mining techniques could provide valuable predictive systems for the identification of potential safety alerts for yet untested chemicals, while simultaneously reducing unnecessary animal testing. Furthermore, collaborative research initiatives such as the Toxicology in the 21st Century (Tox21) consortium with the support of the research community could contribute toward the development of novel and powerful approaches for predictive toxicological research. These in-silico approaches could direct experimentation toward the most likely toxic chemicals first, hence providing a far better utilization of the limited experimental resources and ultimately leading to safer chemical products reaching the market or hazardous ones being removed from circulation.
The modeling approach devised by our team to model the Tox21 data challenge 2014 was based on simple circular molecular fingerprints and supervised machine-learning algorithms Random Forest and Support Vector Machine. Here a single task approach was followed, where each assay was modeled independently by RF and SVM. Overall the modeling approach achieved decent performance with results achieving strong performance measured by ROC-AUC equal to or higher than 0.7. The described approach has the advantage of being fast as it is based on simple circular descriptors, which can be generated efficiently for large number of chemical structures and utilized open-source software packages for the main modeling steps. As expected both algorithms selected for the study, RF and SVM, showed good performance and achieved comparable results on the external set.
Author Contributions
The authors AK, JS, and MM designed and ran the experiments, analyzed the data and wrote the manuscript contributing equally. JH is the PI and contributed to experiment design, data analysis and writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by NSF grant 1337899.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2016.00011
References
Attene-Ramos, M. S., Miller, N., Huang, R., Michael, S., Itkin, M., Kavlock, R. J., et al. (2013). The Tox21 robotic platform for the assessment of environmental chemicals–from vision to reality. Drug Discov. Today 18, 716–723. doi: 10.1016/j.drudis.2013.05.015
Baker, V. A. (2001). Endocrine disrupters—testing strategies to assess human hazard. Toxicol. In vitro 15, 413–419. doi: 10.1016/S0887-2333(01)00045-5
Bender, A. (2010). How similar are those molecules after all? Use two descriptors and you will have three different answers. Expert Opin. Drug Discov. 5, 1141–1151. doi: 10.1517/17460441.2010.517832
Binetti, R., Costamagna, F. M., and Marcello, I. (2008). Exponential growth of new chemicals and evolution of information relevant to risk control. Annali dell'Istituto Superiore di Sanita 44, 13–15.
Bourguet, W., Germain, P., and Gronemeyer, H. (2000). Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 21, 381–388. doi: 10.1016/S0165-6147(00)01548-0
Bradley, A. P. (1997). The use of the area under the ROC curve in the evaluation of machine learning algorithms. Patt. Recognit. 30, 1145–1159. doi: 10.1016/S0031-3203(96)00142-2
Calafat, A. M., Valentin-Blasini, L., and Ye, X. (2015). Trends in exposure to chemicals in personal care and consumer products. Curr. Environ. Health Rep. 2, 348–355. doi: 10.1007/s40572-015-0065-9
Cereto-Massagué, A., Ojeda, M. J., Valls, C., Mulero, M., Garcia-Vallvé, S., and Pujadas, G. (2015). Molecular fingerprint similarity search in virtual screening. Methods 71, 58–63. doi: 10.1016/j.ymeth.2014.08.005
Chemical Computing Group Inc. (2015). Molecular Operating Environment (MOE), 2013.08, 1010, Montreal, QC.
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Dix, D. J., Houck, K. A., Martin, M. T., Richard, A. M., Setzer, R. W., and Kavlock, R. J. (2007). The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. Off. J. Soc. Toxicol. 95, 5–12. doi: 10.1093/toxsci/kfl103
Dudek, A., Arodz, T., and Galvez, J. (2006). Computational methods in developing quantitative structure-activity relationships (QSAR): a review. Combin. Chem. High Through. Screen. 9, 213–228. doi: 10.2174/138620706776055539
Ekins, S., Nikolsky, Y., and Nikolskaya, T. (2005). Techniques: application of systems biology to absorption, distribution, metabolism, excretion and toxicity. Trends Pharmacol. Sci. 26, 202–209. doi: 10.1016/j.tips.2005.02.006
Fawcett, T. (2006). An introduction to ROC analysis. Pattern Recognit. Lett. 27, 861–874. doi: 10.1016/j.patrec.2005.10.010
Fourches, D., Muratov, E., and Tropsha, A. (2010). Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 50, 1189–1204. doi: 10.1021/ci100176x
Fox, J. T., Sakamuru, S., Huang, R., Teneva, N., Simmons, S. O., Xia, M., et al. (2012). High-throughput genotoxicity assay identifies antioxidants as inducers of DNA damage response and cell death. Proc. Natl. Acad. Sci. U.S.A. 109, 5423–5428. doi: 10.1073/pnas.1114278109
Fulda, S., Gorman, A. M., Hori, O., and Samali, A. (2010). Cellular stress responses: cell survival and cell death. Int. J. Cell Biol. 2010, 1–23. doi: 10.1155/2010/214074
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., et al. (2012). ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107. doi: 10.1093/nar/gkr777
Guha, R. (2008). On the interpretation and interpretability of quantitative structure–activity relationship models. J. Comput. Aided Mol. Des. 22, 857–871. doi: 10.1007/s10822-008-9240-5
Hartung, T. (2009). Toxicology for the twenty-first century. Nature 460, 208–212. doi: 10.1038/460208a
Hu, G., Kuang, G., Xiao, W., Li, W., Liu, G., and Tang, Y. (2012). Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 52, 1103–1113. doi: 10.1021/ci300030u
Ihaka, R., and Gentleman, R. (1996). R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5, 299–314.
Inglese, J., Auld, D. S., Jadhav, A., Johnson, R. L., Simeonov, A., Yasgar, A., et al. (2006). Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 103, 11473–11478. doi: 10.1073/pnas.0604348103
Janošek, J., Hilscherová, K., Bláha, L., and Holoubek, I. (2006). Environmental xenobiotics and nuclear receptors—Interactions, effects and in vitro assessment. Toxicol. In vitro 20, 18–37. doi: 10.1016/j.tiv.2005.06.001
Judson, R. S., Houck, K. A., Kavlock, R. J., Knudsen, T. B., Martin, M. T., Mortensen, H. M., et al. (2009). In vitro screening of environmental chemicals for targeted testing prioritization: the toxcast project. Environ. Health Pers. 118, 485–492. doi: 10.1289/ehp.0901392
Kavlock, R., and Dix, D. (2010). Computational toxicology as implemented by the U.S. EPA: providing high throughput decision support tools for screening and assessing chemical exposure, hazard and risk. J. Toxicol. Environ. Health B 13, 197–217. doi: 10.1080/10937404.2010.483935
Keiser, M. J., Roth, B. L., Armbruster, B. N., Ernsberger, P., Irwin, J. J., and Shoichet, B. K. (2007). Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206. doi: 10.1038/nbt1284
Klabunde, T. (2007). Chemogenomic approaches to drug discovery: similar receptors bind similar ligands. Br. J. Pharmacol. 152, 5–7. doi: 10.1038/sj.bjp.0707308
Koutsoukas, A., Simms, B., Kirchmair, J., Bond, P. J., Whitmore, A. V., Zimmer, S., et al. (2011). From in silico target prediction to multi-target drug design: current databases, methods and applications. J. Proteomics 74, 2554–2574. doi: 10.1016/j.jprot.2011.05.011
Landrigan, P. J., and Goldman, L. R. (2011). Children's vulnerability to toxic chemicals: a challenge and opportunity to strengthen health and environmental policy. Health Aff. 30, 842–850. doi: 10.1377/hlthaff.2011.0151
Landrum, G. (2015). RDKit: Open-Source Cheminformatics. Available online at: http://www.rdkit.org
Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. doi: 10.1016/S0092-8674(00)81871-1
Lock, E. F., Abdo, N., Huang, R., Xia, M., Kosyk, O., O'shea, S. H., et al. (2012). Quantitative high-throughput screening for chemical toxicity in a population-based in vitro model. Toxicol. Sci. Off. J. Soc. Toxicol. 126, 578–588. doi: 10.1093/toxsci/kfs023
Merlot, C. (2010). Computational toxicology–a tool for early safety evaluation. Drug Discov. Today 15, 16–22. doi: 10.1016/j.drudis.2009.09.010
Mestres, J., Gregori-Puigjané, E., Valverde, S., and Solé, R. V. (2008). Data completeness—the Achilles heel of drug-target networks. Nat. Biotechnol. 26, 983–984. doi: 10.1038/nbt0908-983
Moras, D., and Gronemeyer, H. (1998). The nuclear receptor ligand-binding domain: structure and function. Curr. Opin. Cell Biol. 10, 384–391. doi: 10.1016/S0955-0674(98)80015-X
Muster, W., Breidenbach, A., Fischer, H., Kirchner, S., Muller, L., and Pahler, A. (2008). Computational toxicology in drug development. Drug Discov. Today 13, 303–310. doi: 10.1016/j.drudis.2007.12.007
Nguyen, T., Sherratt, P. J., and Pickett, C. B. (2003). Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229
Olefsky, J. M. (2001). Nuclear receptor minireview series. J. Biol. Chem. 276, 36863–36864. doi: 10.1074/jbc.R100047200
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830.
Perry, S., Norman, J., Barbieri, J., Brown, E., and Gelbard, H. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98–115. doi: 10.2144/000113610
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754. doi: 10.1021/ci100050t
Schmidt, C. W. (2009). TOX21 new dimensions of toxicity testing. Environ. Health Pers. 117, A348–A353. doi: 10.1289/ehp.117-a348
Shukla, S. J., Huang, R., Austin, C. P., and Xia, M. (2010). The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today 15, 997–1007. doi: 10.1016/j.drudis.2010.07.007
Simpson, E. R., Mahendroo, M. S., Means, G. D., Kilgore, M. W., Hinshelwood, M. M., Graham-Lorence, S., et al. (1994). Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr. Rev. 15, 342–355.
Simpson, E. R., Zhao, Y., Agarwal, V. R., Michael, M. D., Bulun, S. E., Hinshelwood, M. M., et al. (1997). Aromatase expression in health and disease. Recent Prog. Horm. Res. 52, 185–213. discussion: 213–214.
Sun, H., Xia, M., Austin, C. P., and Huang, R. (2012). Paradigm shift in toxicity testing and modeling. AAPS J. 14, 473–480. doi: 10.1208/s12248-012-9358-1
Todeschini, R., Consonni, V., Xiang, H., Holliday, J., Buscema, M., and Willett, P. (2012). Similarity coefficients for binary chemoinformatics data: overview and extended comparison using simulated and real data sets. J. Chem. Inf. Model. 52, 2884–2901. doi: 10.1021/ci300261r
van Westen, G. J. P., Wegner, J. K., Ijzerman, A. P., van Vlijmen, H. W. T., and Bender, A. (2011). Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. Med. Chem. Commun. 2, 16–30. doi: 10.1039/C0MD00165A
Varga, Z. V., Ferdinandy, P., Liaudet, L., and Pacher, P. (2015). Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circul. Physiol. 309, H1453–H1467. doi: 10.1152/ajpheart.00554.2015
Vogelstein, B., Lane, D., and Levine, A. J. (2000). Surfing the p53 network. Nature 408, 307–310. doi: 10.1038/35042675
Wang, Y., Xiao, J., Suzek, T. O., Zhang, J., Wang, J., and Bryant, S. H. (2009). PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37, W623–W633. doi: 10.1093/nar/gkp456
Wei, T. (2013). corrplot: Visualization of a Correlation Matrix. R package version 0.73. Available online at: http://CRAN.R-project.org/package=corrplot
Wu, C. (1995). Heat shock transcription factors: structure and regulation. Annu. Rev. Cell Dev. Biol. 11, 441–469. doi: 10.1146/annurev.cb.11.110195.002301
Keywords: in silico, toxicology, tox21 data challenge 2014, machine learning, data-mining, cheminformatics, predictive toxicology
Citation: Koutsoukas A, St. Amand J, Mishra M and Huan J (2016) Predictive Toxicology: Modeling Chemical Induced Toxicological Response Combining Circular Fingerprints with Random Forest and Support Vector Machine. Front. Environ. Sci. 4:11. doi: 10.3389/fenvs.2016.00011
Received: 01 October 2015; Accepted: 05 February 2016;
Published: 02 March 2016.
Edited by:
Ruili Huang, NIH National Center for Advancing Translational Sciences, USAReviewed by:
Xiaoqing Chang, Integrated Laboratory System, USAJui-Hua Hsieh, Kelly Government Solutions, USA
Copyright © 2016 Koutsoukas, St. Amand, Mishra and Huan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Huan, amh1YW5AaXR0Yy5rdS5lZHU=