 Richard Egelkamp
Richard Egelkamp Dominik Schneider
Dominik Schneider Robert Hertel
Robert Hertel Rolf Daniel
Rolf Daniel- Genomic and Applied Microbiology, Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, University of Goettingen, Göttingen, Germany
Introduction
Nitriles are a diverse group of organic compounds with –C≡N as functional group. Most nitriles are slightly cytotoxic but some cause severe toxic effects. More than 120 naturally occurring nitriles without considering cyanogenic glycosides are present in terrestrial and marine habitats, especially in plant components such as almonds or other fruit pits. The most common group of naturally occurring nitriles are cyanogenic glycosides, which can be found in more than 100 plant families as well as in fungi, bacteria, and animals. This group of molecules can be chemically or enzymatically hydrolyzed, leading to the release of highly toxic hydrogen cyanide and thereby act as natural defense compound (Fleming, 1999). For detoxification, two enzymatic pathways for the degradation of nitriles are known. The first one involves nitrilases (EC 3.5.5.1), a subgroup of the carbon-nitrogen hydrolase superfamily, which degrade nitriles directly to carboxylic acids and ammonia. The second one is a bi-enzymatic pathway using nitrile hydratases (NHases; EC 4.2.1.84) for the degradation of nitriles to amides and amidases (EC 3.5.1.4) for the subsequent degradation to carboxylic acids and ammonia (Gong et al., 2012). The enzymatic hydrolysis of nitriles proceeds under mild reaction conditions, whereas the chemical hydrolysis is dependent on acidic or alkaline conditions and high temperatures. The latter also results in the production of large quantities of byproducts and inorganic waste (Clouthier and Pelletier, 2012; Vergne-Vaxelaire et al., 2013). Consequently, nitrile-converting enzymes are of increasing industrial importance with respect to green chemistry. A constantly increasing number of nitrile-derived amides [e.g., acrylamides or carboxylic acids (e.g., glycolic acid)] are produced with these enzymes (Schmid et al., 2001; Panova et al., 2007). In addition, nitrilases can be used for the treatment of nitrile-polluted wastewater (Li et al., 2016) and other environmentally-friendly bioremediation processes (Gong et al., 2012).
Here, we report data on the taxonomic composition of an enrichment culture with acetonitrile as nitrogen source. In addition, we present eight individual bacterial draft genome sequences of isolates obtained from this enrichment. The genome content of these isolates was analyzed with respect to genes responsible for the nitrile-degrading phenotype. Genome and average nucleotide identity analysis indicated that the isolated bacterial strains are affiliated to the species Rhodococcus erythropolis, Flavobacterium sp., Variovorax boronicumulans, Pseudomonas sp., and Pseudomonas kilonensis.
Materials and Methods
Isolation of the Bacteria
Compost (100 g, pH 7.5) of the Experimental Botanical Garden Göttingen, Germany (51°33′22.6″N 9°57′16.2″E) was suspended in 500 ml H2O and filtered with a 2.7 μm GF/D glass fiber filter (Whatman, Little Chalfont, UK). Enrichment and control cultures were each initiated with 750 μl of the resulting filtrate. Enrichment cultures were grown in 40 ml M9 medium (Atlas, 2010) with ATCC trace mineral supplement (LGC Standards, Teddington, UK), 10 mM glucose as carbon source and 25 mM acetonitrile as sole nitrogen source at 25°C for 4 days. Control cultures were grown in same medium but the medium contained 18.7 mM NH4Cl instead of acetonitrile. The enrichment culture was streaked on solid M9 medium with 25 mM acetonitrile as sole nitrogen source and incubated at 25°C. The obtained 33 colonies showed four distinct colony morphologies, and were picked and purified over five rounds of incubation. Growth in the absence of an added nitrogen source was not recorded.
16S rRNA Gene Sequencing
Genomic DNA of the liquid cultures and the specific isolates was extracted using the MasterPure complete DNA and RNA purification kit (Epicentre, Madison, WI, USA), while DNA isolation of the compost sample was done with the PowerMax soil DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA). The bacterial composition was determined by amplicon-based analysis of the V3-V4 region of the 16S rRNA gene using the bacterial primers S-D-Bact-0341-b-S-17 and S-D-Bact-0785-a-A-21 (Klindworth et al., 2013) with adapters for Illumina MiSeq sequencing (Illumina, San Diego, CA, USA). The PCR reaction mixture (50 μl) contained 10 μl 5-fold Phusion GC buffer, 200 μM of each of the four dNTPs, 2.5 μl DMSO, 0.2 μM of each primer, 200 μM MgCl2, 1 U of Phusion polymerase (Thermo Fisher Scientific, Waltham, MA, USA), and 25 ng of isolated DNA as template. The following cycling scheme was used: initial denaturation at 98°C for 1 min and 25 cycles of denaturation at 98°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 30 s, followed by a final extension at 72°C for 5 min. PCR reactions were performed in triplicate for each sample. The resulting PCR products were pooled in equal amounts and purified using the NucleoMag 96 PCR kit (Macherey-Nagel, Düren, Germany) as recommended by the manufacturer. Quantification of the PCR products was performed using the Quant-iT dsDNA HS assay kit and a Qubit fluorometer as recommended by the manufacturer (Invitrogen, Carlsbad, CA, USA). Indexing of the PCR products was performed with Nextera XT DNA library prep kit as described by the supplier (Illumina). Sequencing was performed with the Illumina MiSeq platform using the dual index paired-end approach (2 × 300 bp) and v3 chemistry.
The 16S rRNA genes of specific isolates were amplified with the primer pair 08f (5′-AGAGTTTGATCCTGGC-3′) and 1504r (5′-TACCTTGTTACGACTT-3′). The previously mentioned cycling scheme was modified to an annealing temperature of 40°C and an extension time of 45 s. Sanger sequencing of the PCR products was done by Seqlab (Göttingen, Germany).
16S rRNA Gene Amplicon Analysis
Demultiplexing and clipping of sequence adapters from raw amplicon sequences were performed by employing CASAVA data analysis software (Illumina). Paired-end sequences were merged using PEAR v0.9.10 with default parameters (Zhang et al., 2014). Subsequently, sequences with an average quality score lower than 20 and containing unresolved bases were removed with the split_libraries_fastq.py script of QIIME 1.9.1 (Caporaso et al., 2010). Non-clipped reverse and forward primer sequences were removed by employing cutadapt 1.12 with default settings (Martin, 2011). USEARCH version 9.2.64 was used following the UNOISE pipeline (Edgar, 2010). In detail, reads shorter than 380 bp were removed, dereplicated, and denoised with the UNOISE2 algorithm of USEARCH resulting in zero-radius operational taxonomic units (zOTUs). Additionally, chimeric sequences were removed using UCHIME2 in reference mode against the SILVA SSU database release 128 (Yilmaz et al., 2014). All quality-filtered sequences were mapped to chimera-free OTUs and a zOTU table was created using USEARCH. Taxonomic classification of the picked reference sequences (zOTUs) was performed with parallel_assign_taxonomy_blast.py against the same SILVA database. Extrinsic domain OTUs, chloroplasts, and unclassified OTUs were removed from the dataset by employing filter_otu_table.py. Sample comparisons were performed at same surveying effort, utilizing the lowest number of sequences by random subsampling (31,000 reads per sample).
Genome Sequencing and Analysis
Genome sequencing was performed using an Illumina MiSeq system with the Nextera XT DNA library prep kit as recommended by the manufacturer (Illumina). Paired-end reads were quality-trimmed with Trimmomatic version 0.36 (Bolger et al., 2014) and verified with FastQC version 0.11.5 (Andrews, 2010). Assembly and first coverage calculation was performed with SPAdes version 3.9.0 (Bankevich et al., 2012). All contigs >500 bp and with a coverage >5 were annotated using Prokka version 1.11 (Seemann, 2014). Final coverage was calculated with Bowtie 2 version 2.2.9 (Langmead and Salzberg, 2012). A BLASTn search (Altschul et al., 1990) was performed against the NCBI non-redundant database to verify assembled contigs. For taxonomic assignment, the full-length 16S rRNA gene sequences of all isolated organisms were searched against the NCBI database. Combination of these data with an analysis of the tetra-nucleotide signatures (Tetra) and the average nucleotide identities (ANI) performed with JSpeciesWS (Richter et al., 2016) as well as in silico DNA-DNA hybridization results calculated with the online tool GGDC 2.1 (Meier-Kolthoff et al., 2013) were employed for taxonomic classification of the isolates. Putative nitrilases, NHases, and amidases were compared with the non-redundant protein database of the NCBI using BLASTp (Altschul et al., 1990). The hit with the highest score is given in Table 1.
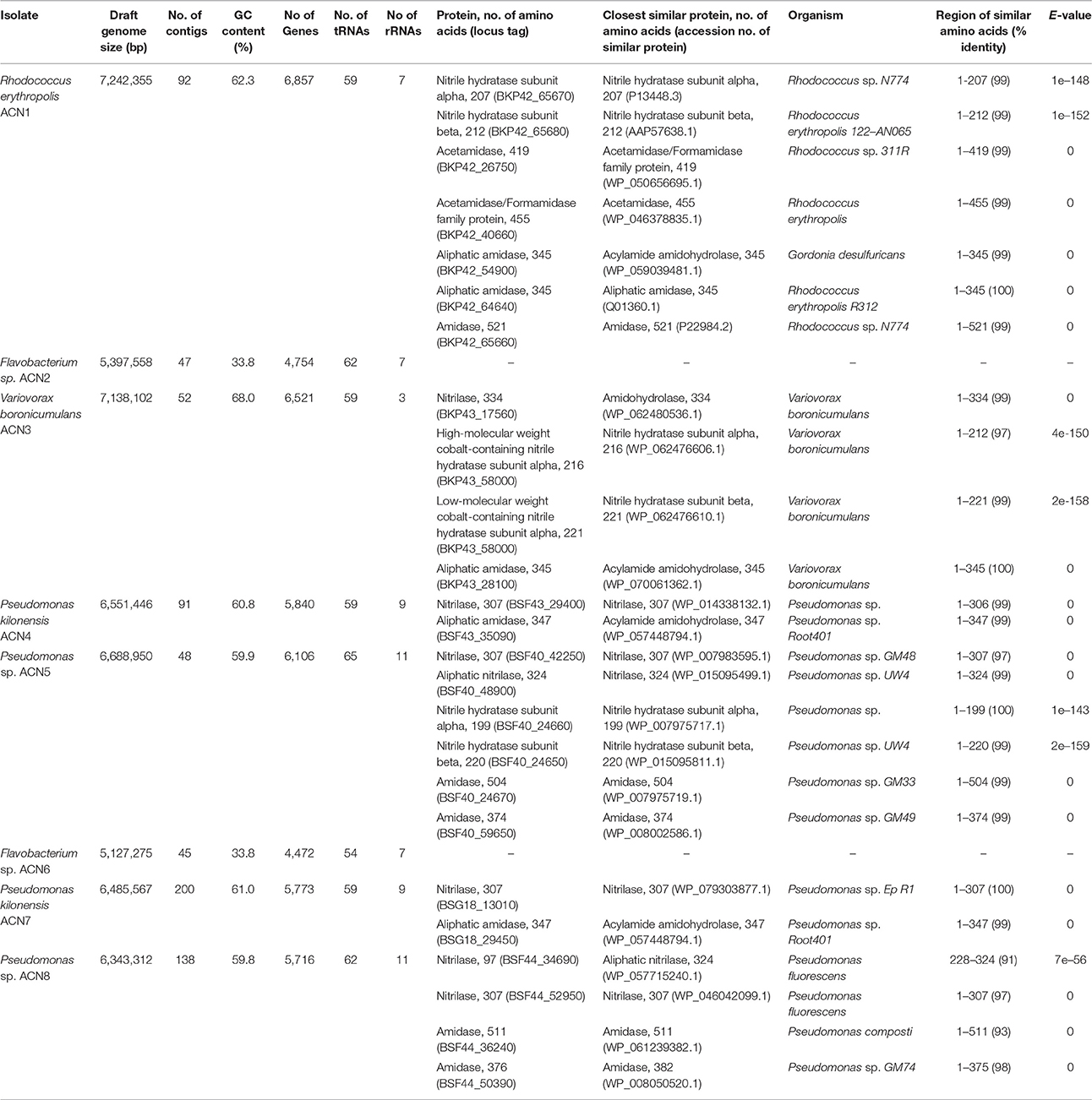
Table 1. Genome data and similarities of putative nitrile-degrading enzymes encoded by the genomes of the isolates.
Results and Discussion
Enrichment of the compost sample with M9 minimal medium containing acetonitrile as sole nitrogen source resulted in microbial growth, indicating the presence of acetonitrile-degrading organisms. The bacterial diversity and composition of the compost sample and the enrichment culture were determined via amplicon-based 16S rRNA gene analysis. The compost sample used as starting material for enrichment comprised 520 zOTUs, which were reduced to 63 during enrichment (Supplementary Table S1). The dominant bacterial genera after enrichment were Aeromonas (33%) and Pseudomonas (55%) with similar abundances in the control sample (Figure 1A). In the enrichment culture the relative abundances of Flavobacterium and Bacillus were 5 and 2%, respectively, and in the control culture 2 and 0.4%, respectively. Chryseobacterium, Paenibacillus, Pedobacter, and Sphingobacterium varied between 1.1 and 1.3% in the control culture, but showed relative abundances below 0.1% in the acetonitrile-containing enrichment. In contrast, Variovorax (3%) was only detected in the acetonitrile enrichment culture in significant amounts. We performed isolation experiments from enrichment cultures on solid media with acetonitrile as sole nitrogen source. A total of 33 isolates (Figure 1B) were recovered of which eight isolates (ACN1 to ACN8, Table 1) showed a different 16S rRNA gene sequence. All isolates were able to grow with acetonitrile as sole nitrogen source in axenic culture. No growth was observed in the absence of an added nitrogen source. The genomes of all eight isolates were sequenced and analyzed with the focus on genes potentially involved in nitrile-degradation (Table 1). Based on average nucleotide identity, tetra-nucleotide signatures and in silico DNA-DNA hybridization, the eight isolates were affiliated to the genera Flavobacterium, Pseudomonas, Rhodococcus and Variovorax. Besides Rhodococcus all isolated strains belonged to genera detected during 16S rRNA gene analysis of the bacterial community in the enrichment culture.
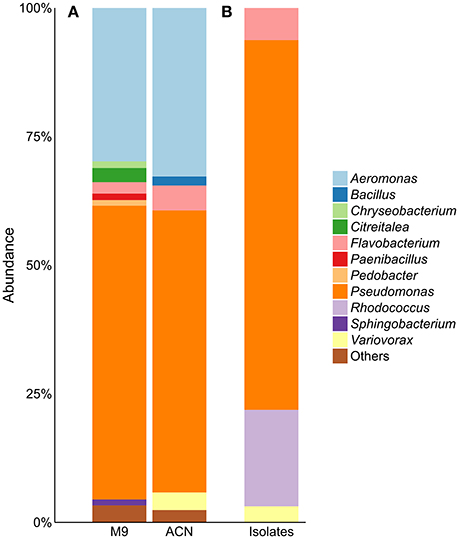
Figure 1. (A) Community composition of acetonitrile-enrichment and control culture and (B) 16S rRNA gene analysis of 33 acetonitrile-degrading isolates; “Others” refers to genera with an abundance of <1%. M9, control; ACN, acetonitril enrichment.
Rhodococcus (ACN1)
In contrast to all other isolates, only slight traces of Rhodococcus could be found during 16S rRNA gene analysis. Nevertheless, this isolate was obtained multiple times from the enrichment, indicating its ability to degrade acetonitrile. The reason for the discrepancy between 16S rRNA gene analysis of the community and the isolation results may be due to non-optimal media conditions for Rhodococcus in the enrichment culture, leading to low abundance. Genome sequencing of R. erythropolis ACN1 and quality-filtering resulted in 2,641,652 paired-end reads resulting in a draft genome of 7.24 Mbp with a 93.9-fold coverage. Similarity searches for putative genes involved in nitrile-degradation revealed two genes (BKP42_65670 and BKP42_65680), coding for one of the two putative NHase subunits. These enzymes are known to be responsible for nitrile degradation in various R. erythropolis strains (Kaufmann et al., 1999; Brandão et al., 2003; Vejvoda et al., 2007; Kamble et al., 2013). The deduced protein sequences showed highest identity to the alpha subunit of Rhodococcus sp. N-774 (Ikehata et al., 1989) and the beta subunit of R. erythropolis deep-sea strain 122-AN065 (Brandão et al., 2003). The deduced acetamidases (BKP42_40660 and BKP42_26750) are most similar to acetamidases of R. erythropolis and Rhodococcus sp. 311R (Ehsani et al., 2015). The two aliphatic amidases of the isolated strain (BKP42_54900 and BKP42_64640) most resemble an acylamide amidohydrolase from Gordonia desulfuricans and an amidase of Rhodococcus sp. R312 (Fournand et al., 1998). An additional amidase (BKP42_65660) is most similar to an amidase of Rhodococcus sp. N-774 (Table 1).
Variovorax (ACN3)
Members of the genus Variovorax were prime candidates for active nitrile degradation, as the genus appeared only in significant abundances in the bacterial community after enrichment with acetonitrile. With the isolation of ACN3 identified as V. boronicumulans a member of this genus was recovered. Sequencing and assembly of 3,471,160 paired-end reads resulted in a draft genome of 7.14 Mbp (131-fold coverage). Although V. boronicumulans is a species described just recently (Miwa et al., 2008), evidence for nitrile degradation by this microorganism was reported before (Zhang et al., 2012; Liu et al., 2013). A search for genes encoding putative nitrile-degrading enzymes resulted in the identification of genes for two possible degradation pathways, the first one via a nitrilase (BKP43_17560) and the second one via an NHase (BKP43_58000 and BKP43_58010) and amidase (BKP43_28100). The most similar enzymes to the nitrilase, alpha and beta subunits of NHase, and the aliphatic amidase are an amidohydrolase, NHase subunits and acylamide amidohydrolase of another V. boronicumulans strain, respectively (Table 1).
Pseudomonas (ACN4, ACN5, ACN7, and ACN8)
With a relative abundance of over 50%, Pseudomonas is the major genus of the bacterial community in the acetonitrile enrichment culture. Several Pseudomonas species such as P. chlorographis and P. fluorescens are able to use nitriles as nitrogen source and harbor genes encoding nitrilases, or NHases and amidases (Nagasawa et al., 1987; Kiziak et al., 2005; Howden et al., 2009). The presence of these genes could be due to the plant habitat of some Pseudomonas species, which is rich in different and unusual nitriles like indole-3-carbonyl nitrile (Rajniak et al., 2015).
Isolates ACN4 and ACN7 were affiliated to P. kilonensis, a species described in 2001 (Sikorski et al., 2001). Sequencing of the ACN4 genome resulted in 2,239,346 paired-end reads, an average coverage of 96.7-fold, and a draft genome of 6.55 Mbp. Genes encoding a putative nitrilase (BSF43_29400) and amidase (BSF43_35090) were detected, which showed highest identity to a Pseudomonas nitrilase and a putative acylamide amidohydrolase of P. sp. Root401 (Bai et al., 2015), respectively.
The genome assembly of isolate ACN7 was based on 2,049,404 paired-end reads and resulted in a draft genome of 6.49 Mbp and a 74.9-fold coverage. The GC-content is 61.0%. The draft genome encodes a nitrilase (BSG18_13010) and an amidase (BSG18_29450) similar to a nitrilase of P. sp. Ep R1 (Chiellini et al., 2014) and an acylamide amidohydrolase of P. sp. Root401, respectively.
The two Pseudomonas isolates ACN5 and ACN8 could not be assigned to a specific Pseudomonas species. The genome of ACN5 (6.69 Mbp, 61.3-fold coverage) was assembled from 1,350,588 paired-end reads. Potential genes for two nitrilases, one NHase and two amidases were predicted in the ACN5 genome. The aliphatic nitrilase (BSF40_48900) showed highest similarity to a nitrilase of Pseudomonas sp. UW4 (Duan et al., 2013), while the other nitrilase (BSF40_42250) most resembles a nitrilase of Pseudomonas sp. GM48 (Brown et al., 2012). The NHase alpha and beta subunits (BSF40_24660 and BSF40_24650) are related to the corresponding ones of Pseudomonas and P. sp. UW4. The predicted amidases (BSF40_24670 and BSF40_59650) are similar to an amidase of P. sp. GM33 and GM49 (Brown et al., 2012). For the assembly of the ACN8 genome, 1,505,826 paired-end reads were used, resulting in a draft genome of 6.34 Mbp and 66.0-fold coverage. Genes for two nitrilases (BSF44_34690 and BSF44_52950) were predicted, which resemble two different nitrilases from P. fluorescens. Furthermore, two genes encoding putative amidases (BSF44_36240 and BSF44_50390) similar to an amidase of P. composti and P. sp. GM74 (Brown et al., 2012) were detected.
Flavobacterium (ACN2 and ACN6)
The two isolates ACN2 and ACN6 from the acetonitrile enrichment culture were affiliated to the Gram-negative genus Flavobacterium. A total of 1,503,824 paired-end reads were used for the assembly of the ACN2 genome, resulting in a draft genome of 5.40 Mbp with an average coverage of 72.7-fold. ACN6 genome assembly of 2,409,950 paired-end reads yielded a draft genome of 5.13 Mbp with average coverage of 120.4-fold. Genes encoding putative nitrile-degrading enzymes were not detected in both draft genomes. As the nitrile degrading capacity of both strains was independently verified multiple times by growth experiments with acetonitrile as sole nitrogen source, the presence of so far unknown genes and pathways for nitrile utilization is indicated. In addition, the ability to overcome the nitrogen limitation via nitrogen fixation from air was experimentally excluded as no growth could be monitored in liquid M9 medium without any added nitrogen source. Furthermore, typical genes for nitrogen fixation such as dinitrogenases and dinitrogenase reductases were not identified in the genome sequences. Thus, two explanations are possible for the discrepancy between the observed phenotype of both strains and the lack of putative genes for nitrile degradation. First, genes responsible for nitrile degradation could not be annotated as they are located in contig gaps, as both genomes are still in the draft state. This is unlikely as most contig gaps are due to repetitive regions longer than the read length of the used sequencing technology (Whiteford et al., 2005; Chaisson et al., 2015). Second, genes for new types of nitrile degradation enzymes are present. The only other available study on nitrile degradation by Flavobacteria reports weak degradation of 3-cyanopyridine by F. aquatile IFO 3772, F. suaveolens IFO 3752 and F. rigense IAM 1238 (Kato et al., 2000), but genes responsible for the observed phenotype were not reported and genomes of the three strains are not available. Thus, further analyses are required to unravel the basis of the nitrile-degrading phenotype of the flavobacterial isolates.
Conclusion
The here presented data revealed the potential of specifically designed enrichment experiments for the isolation of organisms with a desired metabolic activity like nitrile degradation. Our study provided new candidates to cover the industrial demand for new nitrile-degrading biocatalysts as part of a green chemistry. While for the isolates R. erythropolis ACN1, V. boronicumulans ACN2, P. kilonensis ACN4 and ACN7 as well as P. sp. ACN5 and ACN8 nitrilases and/or NHases and amidases could be annotated, the nitrile-degrading pathway of F. sp ACN2 and ACN6 remains to be unraveled.
Accession Numbers
The draft genome sequences of all eight organisms have been deposited at GenBank with the following accession numbers: R. erythropolis ACN1, MRCL00000000; F. sp. ACN2, MRCM00000000; V. boronicumulans ACN3, MRCN00000000; P. kilonensis ACN4, MRCO00000000; P. sp. ACN5, MRCP00000000; F. sp. ACN6, MRCQ00000000; P. kilonensis ACN7, MRCR00000000; P. sp. ACN8, MRCS00000000. Raw sequence data of all genomes are available at the NCBI SRA archive and linked to the respective BioSamples. The sequences of the 16S rRNA gene analysis are linked to the BioSamples SAMN07278921 (inoculum), SAMN0278947 (control) and SAMN07278950 (acetonitrile enrichment). Strains have been submitted to the DSMZ German Collection of Microorganism and Cell Cultures and are available on request.
Author Contributions
RD and RH conceived the study. RE performed the experiments. RE and DS analyzed the data. All authors interpreted the results, wrote the manuscript and reviewed the final version of the manuscript.
Funding
The work of RE was supported by the “Fonds der Chemischen Industrie im Verband der Chemischen Industrie e.V.” The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Melanie Heinemann for technical support. We acknowledge support by the Open Access Publication Funds of the University of Goettingen.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2017.00056/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2010). FastQC: A Quality Control Tool for High throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., and Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brandão, P. F., Clapp, J. P., and Bull, A. T. (2003). Diversity of nitrile hydratase and amidase enzyme genes in Rhodococcus erythropolis recovered from geographically distinct habitats. Appl. Environ. Microbiol. 69, 5754–5766. doi: 10.1128/AEM.69.10.5754-5766.2003
Brown, S. D., Utturkar, S. M., Klingeman, D. M., Johnson, C. M., Martin, S. L., Land, M. L., et al. (2012). Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoids. J. Bacteriol. 194, 5991–5993. doi: 10.1128/JB.01243-12
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chaisson, M. J. P., Wilson, R. K., and Eichler, E. E. (2015). Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 16, 627–640. doi: 10.1038/nrg3933
Chiellini, C., Maida, I., Emiliani, G., Mengoni, A., Mocali, S., Fabiani, A., et al. (2014). Endophytic and rhizospheric bacterial communities isolated from the medicinal plants Echinacea purpurea and Echinacea angustifolia. Int. Microbiol. 17, 165–174. doi: 10.2436/20.1501.01.219
Clouthier, C. M., and Pelletier, J. N. (2012). Expanding the organic toolbox: a guide to integrating biocatalysis in synthesis. Chem. Soc. Rev. 41, 1585–1605. doi: 10.1039/c2cs15286j
Duan, J., Jiang, W., Cheng, Z., Heikkila, J. J., and Glick, B. R. (2013). The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 8:e58640. doi: 10.1371/journal.pone.0058640
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Ehsani, E., Jauregui, R., Geffers, R., Jareck, M., Boon, N., Pieper, D. H., et al. (2015). Draft genome sequence of Rhodococcus sp. strain 311R. Genome Announc. 3:e00378–e00415. doi: 10.1128/genomeA.00378-15
Fleming, F. F. (1999). Nitrile-containing natural products. Nat. Prod. Rep. 16, 567–606. doi: 10.1039/a804370a
Fournand, D., Araud, A., and Galzy, P. (1998). Study of the acyl transfer activity of a recombinant amidase overproduced in an Escherichia coli strain. Application for short-chain hydroxamic acid and acid hydrazide synthesis. J. Mol. Catal. B Enzym. 4, 77–90. doi: 10.1016/S1381-1177(97)00024-6
Gong, J. S., Lu, Z. M., Li, H., Shi, J. S., Zhou, Z. M., and Xu, Z. H. (2012). Nitrilases in nitrile biocatalysis: recent progress and forthcoming research. Microb. Cell Fact. 11:142. doi: 10.1186/1475-2859-11-142
Howden, A. J., Harrison, C. J., and Preston, G. M. (2009). A conserved mechanism for nitrile metabolism in bacteria and plants. Plant J. 57, 243–253. doi: 10.1111/j.1365-313X.2008.03682.x
Ikehata, O., Nishiyama, M., Horinouchi, S., and Beppu, T. (1989). Primary structure of nitrile hydratase deduced from the nucleotide sequence of a Rhodococcus species and its expression in Escherichia coli. Eur. J. Biochem. 181, 563–570. doi: 10.1111/j.1432-1033.1989.tb14761.x
Kamble, A. L., Banoth, L., Meena, V. S., Singh, A., Chisti, Y., and Banerjee, U. C. (2013). Nitrile hydratase of Rhodococcus erythropolis: characterization of the enzyme and the use of whole cells for biotransformation of nitriles. 3 Biotech. 3, 319–330. doi: 10.1007/s13205-012-0104-2
Kato, Y., Ooi, R., and Asano, Y. (2000). Distribution of aldoxime dehydratase in microorganisms. Appl. Environ. Microbiol. 66, 2290–2296. doi: 10.1128/AEM.66.6.2290-2296.2000
Kaufmann, G., Dautzenberg, H., Henkel, H., Müller, G., Schäfer, T., Undeutsch, et al. (1999). Nitrile hydratase from Rhodococcus erythropolis: metabolization of steroidal compounds with a nitrile group. Steroids 64, 535–540. doi: 10.1016/S0039-128X(99)00028-8
Kiziak, C., Conradt, D., Stolz, A., Mattes, R., and Klein, J. (2005). Nitrilase from Pseudomonas fluorescens EBC191: cloning and heterologous expression of the gene and biochemical characterization of the recombinant protein. Microbiology 151, 3639–3648. doi: 10.1099/mic.0.28246-0
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, C., Yue, Z., Feng, F., Xi, C., Zang, H., An, X., et al. (2016). A novel strategy for acetonitrile wastewater treatment by using a recombinant bacterium with biofilm-forming and nitrile-degrading capability. Chemosphere 161, 224–232. doi: 10.1016/j.chemosphere.2016.07.019
Liu, Z.-H., Cao, Y.-M., Zhou, Q.-W., Gui, K., Ge, F., Hou, J.-Y., et al. (2013). Acrylamide biodegradation ability and plant growth-promoting properties of Variovorax boronicumulans CGMCC 4969. Biodegradation 24, 855–864. doi: 10.1007/s10532-013-9633-6
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimination with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Miwa, H., Ahmed, I., Yoon, J., Yokota, A., and Fujiwara, T. (2008). Variovorax boronicumulans sp. nov., a boron-accumulating bacterium isolated from soil. Int. J. Syst. Evol. Microbiol. 58, 286–289. doi: 10.1099/ijs.0.65315-0
Nagasawa, T., Nanba, H., Ryuno, K., Takeuchi, K., and Yamada, H. (1987). Nitrile hydratase of Pseudomonas chlororaphis B23. Purification and characterization. Eur. J. Biochem. 162, 691–698. doi: 10.1111/j.1432-1033.1987.tb10692.x
Panova, A., Mersinger, L. J., Liu, Q., Foo, T., Roe, D. C., Spillan, W. L., et al. (2007). Chemoenzymatic synthesis of glycolic acid. Adv. Synth. Catal. 349, 1462–1474. doi: 10.1002/adsc.200700061
Rajniak, J., Barco, B., Clay, N. K., and Sattely, E. S. (2015). A new cyanogenic metabolite in Arabidopsis required for inducible pathogen defence. Nature 525, 376–379. doi: 10.1038/nature14907
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2016). JSpecies WS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Schmid, A., Dordick, J. S., Hauer, B., Kiener, A., Wubbolts, M., and Witholt, B. (2001). Industrial biocatalysis today and tomorrow. Nature 409, 258–268. doi: 10.1038/35051736
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sikorski, J., Stackebrandt, E., and Wackernagel, W. (2001). Pseudomonas kilonensis sp. nov., a bacterium isolated from agricultural soil. Int. J. Syst. Evol. Microbiol. 51, 1549–1555. doi: 10.1099/00207713-51-4-1549
Vejvoda, V., Šveda, O., Kaplan, O., Přikrylová, V., Elišáková, V., Himl, M., et al. (2007). Biotransformation of heterocyclic dinitriles by Rhodococcus erythropolis and fungal nitrilases. Biotechnol. Lett. 29, 1119–1124. doi: 10.1007/s10529-007-9364-z
Vergne-Vaxelaire, C., Bordier, F., Fossey, A., Besnard-Gonnet, M., Debard, A., Mariage, A., et al. (2013). Nitrilase activity screening on structurally diverse substrates: providing biocatalytic tools for organic synthesis. Adv. Synth. Catal. 355, 1763–1779. doi: 10.1002/adsc.201201098
Whiteford, N., Haslam, N., Weber, G., Prügel-Bennett, A., Essex, J. W., Roach, P. L., et al. (2005). An analysis of the feasibility of short read sequencing. Nucleic Acids Res. 33:e171. doi: 10.1093/nar/gni170
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Zhang, H.-J., Zhou, Q.-W., Zhou, G.-C., Cao, Y.-M., Dai, Y.-J., Ji, W.-W., et al. (2012). Biotransformation of the neonicotinoid insecticide Thiacloprid by the bacterium Variovorax boronicumulans strain J1 and mediation of the major metabolic pathway by nitrile hydratase. J. Agric. Food Chem. 60, 153–159. doi: 10.1021/jf203232u
Keywords: acetonitrile, nitrilase, nitrile hydratase, amidase, Flavobacterium, Pseudomonas, Rhodococcus, Variovorax
Citation: Egelkamp R, Schneider D, Hertel R and Daniel R (2017) Nitrile-Degrading Bacteria Isolated from Compost. Front. Environ. Sci. 5:56. doi: 10.3389/fenvs.2017.00056
Received: 25 July 2017; Accepted: 24 August 2017;
Published: 12 September 2017.
Edited by:
Moisés Canle, University of A Coruña, SpainReviewed by:
Dimitris Tsaltas, Cyprus University of Technology, CyprusFrancesco Degli-Innocenti, Novamont S.p.A., Italy
Copyright © 2017 Egelkamp, Schneider, Hertel and Daniel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rolf Daniel, cmRhbmllbEBnd2RnLmRl