Abstract
Recent studies are unraveling the impact of microorganisms from the roots and rhizosphere on interactions between plants and herbivorous insects and are gradually changing our perception of the microorganisms' capacity to affect plant defenses, but the reverse effect has seldom been investigated. Our study aimed at determining how plant herbivory influences the dynamics of root and rhizosphere microbial community assemblages and whether potential changes in root metabolites and chemical elements produced during herbivory can be related to microbial community diversity. We conducted our study on oilseed rape (Brassica napus) and its major belowground herbivore, the cabbage root fly (Delia radicum). We further assessed the influence of initial soil microbial diversity on these interactions. Different microbial diversities based on a common soil matrix were obtained through a removal-recolonization method. Root and rhizosphere sampling targeted different stages of the herbivore development corresponding to different perturbation intensities. Root bacterial communities were more affected by herbivory than some rhizosphere bacterial phyla and fungal communities, which seemed more resistant to this perturbation. Root herbivory enhanced the phylum of γ-Proteobacteria in the roots and rhizosphere, as well as the phylum of Firmicutes in the rhizosphere. Herbivory tended to decrease most root amino acids and sugars, and it increased trehalose, indolyl glucosinolates, and sulfur. Higher abundances of four bacterial genera (Bacillus, Paenibacillus, Pseudomonas, and Stenotrophomonas) were associated following herbivory to the increase of trehalose and some sulfur-containing compounds. Further research would help to identify the biological functions of the microbial genera impacted by plant infestation and their potential implications in plant defense.
Introduction
Interactions between plants and herbivorous insects are known to influence the evolution of plant defense mechanisms such as defensive and toxic metabolites or volatiles attracting natural enemies of the pest (Fürstenberg-Hägg et al., 2013; Nishida, 2014). Plants constitute an important crossroad in biological interactions since they are also involved in interactions with microorganisms that can improve plant growth, nutrient acquisition, and protection from different plant bioagressors (Richardson et al., 2009; Schnitzer et al., 2011). Recent studies are unraveling the impact of microorganisms from the rhizosphere on plant-insect interactions and are gradually changing our perception of the microorganisms' capacity to affect plant defenses (Pineda et al., 2017). A single strain or a whole microbial community has been shown to increase plant defenses, which in turn can negatively impact insect fitness (Hol et al., 2010; Pangesti et al., 2015a,b; Lachaise et al., 2017). However, the reverse effect has seldom been investigated while herbivore attacks on plants could represent important perturbations for belowground microbial communities.
Perturbations are seen as major drivers of ecosystems stability (Milchunas et al., 1988). Since the 90's, the positive relationship between an ecosystem's stability and its diversity was addressed in different systems and particularly in grasslands facing different abiotic stresses (Ives and Carpenter, 2007). It seemed that the recovery of an ecosystem following a stress could be conditioned by the ecosystem's initial diversity prior to this stress. For example, an ecosystem with high plant diversity that suffers a drought will better recover than an ecosystem with a low plant diversity (Tilman and Downing, 1994; van Ruijven and Berendse, 2010). Effects of such stresses were investigated on resistance (the ability to sustain a perturbation or stress) and resilience (the capacity to come back to a stable state) of different soil processes such as nitrogen and carbon cycles, organic matter decomposition, or respiration (Griffiths and Philippot, 2013). Resistance and resilience of grassland communities toward biotic stresses such as herbivory has also been investigated (see review by Cingolani et al., 2005). However, these studies concerned either grazing mammals or insect herbivory in grasslands, with the latter having variable effects on mycorrhiza (Johnson and Rasmann, 2015). To our knowledge however, this question has never been addressed for microbial communities of agroecosystem plants confronted to insect herbivory.
Insect herbivory can modify plant physiology (Bolton, 2009) and alter metabolite concentrations in plant tissues (Griffiths et al., 1994; Hopkins et al., 1995; Ponzio et al., 2017) as well as nutrient uptake (Katayama et al., 2014; Aziz et al., 2016). It can also modify the release of organic matter by plant roots (rhizodeposition), a major driver of soil microbial communities (Singh et al., 2004; Paterson et al., 2007). Some studies showed that root herbivory can influence plant carbon sources and rhizosphere chemistry, which in turn modify the abundance of bacteria and fungi, and community physiological profiles in the rhizosphere (Grayston et al., 2001; Dawson et al., 2004; Treonis et al., 2005). Katayama et al. (2014) showed that herbivory could also alter chemical element uptakes by the roots as well as nitrogen concentration of microbial origin. So far, when the effect of herbivory on plant-associated microorganisms was covered, the experiments either neglected to take into account the plant metabolites and chemical elements or did not integrate the notion of perturbation dynamics, or cultivable methods and fluorimetry were used to study microbial communities.
Our study aimed at determining the impact of insect herbivory on the plant-microorganism interactions. We hypothesized that (i) the dynamics of root and rhizosphere microbial communities would be influenced by belowground herbivory, (ii) these dynamics would depend on root metabolites and chemical elements induced by herbivory, (iii) the initial soil microbial diversity would influence plant chemistry and hence the dynamics of microbial communities. We conducted our experiment on oilseed rape (“OSR,” Brassica napus) and its belowground pest, the cabbage root fly (“CRF,” Delia radicum) (Ahuja et al., 2010). This fly is a specialist of brassicaceous plants. Females lay their eggs aboveground, at the base of plant stems and, upon hatching, which occurs a few days later, larvae feed by tunneling into the roots for 2–3 weeks, before pupating in the nearby soil and emerging. Herbivory of the CRF on brassicaceous plants is known to change the concentration of primary (e.g., sugars such as glucose and sucrose) and secondary metabolites (e.g., indolyl glucosinolates) in the roots (Hopkins et al., 1999; van Dam and Raaijmakers, 2006; Pierre et al., 2012; van Geem et al., 2015). However, nothing is known about how CRF herbivory and resulting biochemical changes in the roots influence the belowground microbial communities which interact with the plant.
Materials and methods
Soil preparation and inoculation
A batch of soil was collected in November 2014 from the layer −10 to −30 cm deep of a field in Brittany (La Gruche, Le Rheu, France, 48°08′44″N, 01°47′97″ W) where wheat was cultivated for 20 years, and it was stored in containers at ambient temperature in the dark. After a year of storage in containers, the soil was homogenized, ground, sieved at 4 mm to remove the macrofauna and mixed with 1/3 silica. As described in Lachaise et al. (2017), this mixture was sterilized at 35 kGy and left 2 months to stabilize before inoculation while the remaining unsterilized soil with no silica was ground and sieved at 2 mm before being suspended in water. Following the detailed protocol of Lachaise et al. (2017), this suspension was then undiluted (100) or diluted at 106 before inoculating the sterilized soil, hence creating the two levels of soil microbial diversity used in our experiment: respectively “high” and “low” initial soil microbial diversities, also referred to as soil microbial modalities (Figure 1). This dilution-inoculation method was performed three times in order to obtain three soil biological replicates per soil microbial modality. After inoculation, the soil was incubated in the dark for seven weeks at 18°C and 50% humidity. During this period, the bags containing the soil were opened weekly under sterile conditions using a laminar flow cabinet to homogenize the soil and facilitate microbial respiration and recolonization. This allowed the soil to reach optimal bacterial and fungal densities and similar abundances of Colony Forming Units between the two soil microbial modalities at the end of the recolonization period. A part of this soil was collected before sowing to evaluate initial soil microbial diversities (N = 18, 9 samples per soil microbial modality).
Figure 1
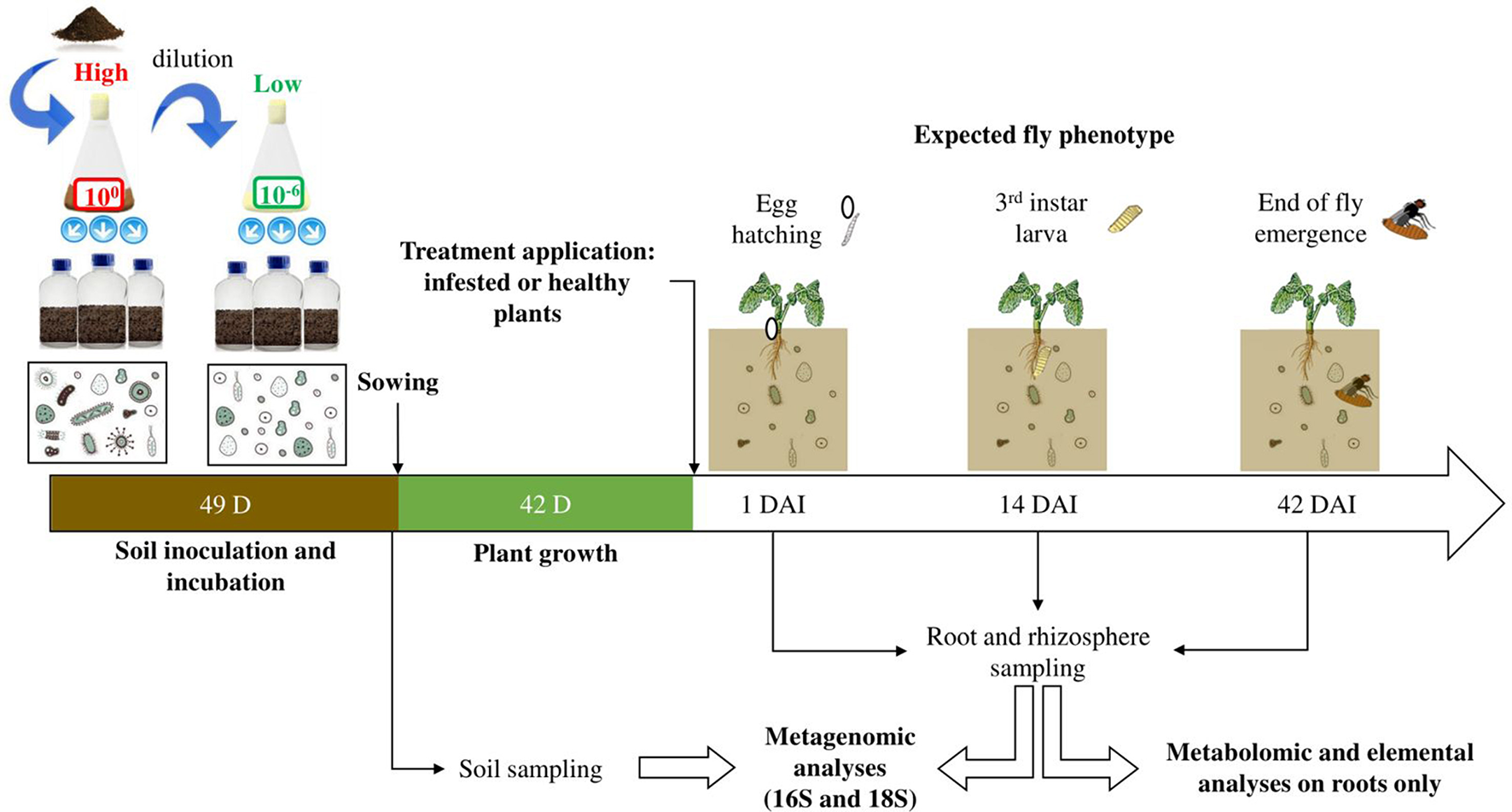
Experimental design. Collected soil was suspended in water and diluted or not, to obtain two solutions of respectively low and high microbial diversities. Sterilized soil was inoculated with one of these two solutions and incubated for 49 days. After sowing, plants were cultivated for 42 days and then two treatments were applied, obtaining healthy and infested plants. The experiment started after the infestation (i.e., D. radicum eggs deposited on the plant crown) and sampling was carried out according to the fly biological cycle and important development stages: egg hatching, 3rd instar larva, end of adult emergence, occurring around 1 day after infestation (DAI), 14 and 42 DAI respectively. These sampling times also corresponded to the beginning, the peak and the end of herbivory. Metagenomics, metabolomics and elemental analyses were performed on the obtained samples.
Insect rearing
The population of cabbage root fly (“CRF,” Delia radicum) used in our experiment was collected in the field in 2015 (Le Rheu, Bretagne, France). In the laboratory, flies were fed on sugar, milk powder and yeast (ratio 1:1:1) and they were reared on rutabaga roots (Brassica napus subsp. rapifera) in a climatic room (16:8 LD, 21 ± 2°C; 60% ± 10% RH) as described in Neveu Bernard-Griffiths (1998). Adult flies were left to oviposit on rutabaga roots for 24h and black-headed eggs (i.e., ready to hatch) were collected 3 days later for our experiment.
Plant growth, infestation, and sampling
Seeds of Brassica napus L. (subsp. oleifera cv. Tenor) were sown in individual pots using a layer of pozzolan at the bottom and the soil previously obtained (characterized either with a “high” or “low” initial soil microbial diversity). Plants were watered twice a week by sub-irrigation with a nutritive solution based on Hoagland and Arnon (1950) during the whole experiment. This solution was obtained by blending three separate solutions: a macronutrient solution (3 mM of KNO3, 0.5 mM of KH2PO4, 1 mM of MgSO4 7H2O, 2.5 mM of Ca(NO3)2 4H2O), a micronutrient solution (10 μM of MnSO4 H2O, 1 μM of ZnSO4 7H2O, 0.5 μM of CuSO4, 30 μM of H3BO3, 1 μM of Na2MoO4 2H2O, 0.5 μM of Co(NO3)2 6H2O), and a Fe EDTA solution (27 μM of Fe EDTA Na). Plants were cultivated during 6 weeks in a greenhouse under natural late 2016 winter photoperiod (mean temp. 15.3°C, min/max 5.5/25.6°C). Two treatments were applied on the plants: half of the plants were infested by depositing eight black-headed eggs on the crown per plant, the other half remained untreated. These two batches of plants were then referred to as “infested plants” and “healthy plants” respectively. All plants were then moved to a climatic chamber (photoperiod 16:8 LD and thermoperiod 20:18°C LD) for the rest of the experiment. Afterwards, healthy and infested plants were sampled at 1, 14, and 42 days after infestation (“DAI”), which respectively represented (i) the hatching stage which corresponds to the initiation of herbivory, (ii) the third larval instar which corresponds to the peak of herbivory, and (iii) the end of the fly emergence which corresponds to the end of herbivory (Figure 1). A total of 108 plants were harvested, with 9 plants per condition (2 soil microbial modalities, 2 treatments being healthy and infested, and 3 sampling times). Roots and rhizosphere were sampled as follows: the root fraction was collected, corresponding to the area from the crown to the tip roots, and was washed twice in 20 mL of sterile permuted water before being transferred to a clean Falcon tube; the whole root bath (i.e., 40 mL in a Falcon tube) corresponded to the rhizosphere fraction; both tubes were immersed in liquid nitrogen and stored at −80°C before being freeze-dried; only roots were ground using glass beads. In order to have sufficient material for molecular, metabolomic and elemental analyses, roots from three different plants of the same treatment, cultivated on the same soil microbial diversity and biological replicates, were pooled to make one sample, hence obtaining a total of 36 root samples (N = 3 samples per condition). The rhizosphere fraction (N = 36 rhizosphere samples) was treated similarly.
Molecular and bioinformatic analysis
Initial bulk soil (i.e., before sowing), as well as root and rhizosphere samples collected during the experiment, were analyzed.
According to the protocol developed by the GenoSol platform (Dijon, France) and described in Plassart et al. (2012), soil DNA was extracted from 2 g of wet bulk soil (i.e., initial soil before sowing) or 1 g of freeze-dried rhizosphere respectively in 8 or 5 mL of lysis buffer containing 100 mM of Tris-HCl (pH 8), 100 mM of EDTA (pH 8), 100 mM of NaCl, 2% SDS, and ultrapure water. The following modifications were performed: tubes were vortexed at mid and at the end of incubation (i.e., bath at 70°C), then centrifuged at 3,500 rpm at 15°C for 10 min; during deproteinization, samples were centrifuged at 14,000 g at 4°C during 10 min; during DNA precipitation, tubes were placed at −20°C for 30 min. To obtain DNA pellets, tubes were centrifuged at 13,000 rpm at 4°C for 30 min and supernatant was discarded. DNA pellets were washed as follows: 400 μL of 70% ice-cold ethanol were added to samples, which were centrifuged at 13,000 rpm at 4°C for 5 min and supernatants were removed. Remaining traces of ethanol were removed by heating open tubes at 60°C for at least 15 min or more if needed. Pellets of DNA were resuspended with 100 μL of ultrapure water and the duplicated samples were finally pooled. Bulk soil and rhizosphere samples were purified twice. The first purification required Microbiospin (Biorad, Hercules, California, USA) columns of PVPP (PolyVinyl PolyPirrolidone, Sigma-Aldrich). To prepare the columns, their tips were removed and columns were placed in 2 mL Eppendorf tubes. Then, columns were washed with 400 μL of ultrapure water and centrifuged at 1,000 g and at 10°C during 2 min. After emptying the tubes, the previous step was carried out a second time, before centrifuging empty tubes at 1,000 g at 10°C for 4 min. Following Plassart et al. (2012), 100 μL of DNA were injected into the columns, previously transferred to a clean tube, however our samples were incubated on ice for 5 min, before a 4 min centrifugation at 1,000 g at 4°C. The obtained DNA (~95 μL) was used for the second purification, performed using the Geneclean® Turbo kit (MP Biomedicals) with the following modifications: samples were centrifuged at 10,000 g for 10 s; after adding the GTE (GeneClean Turbo Elution Solution), samples were incubated on ice for 5 min and centrifuged at 10,000 g for 1 min; the GTE, incubation and centrifugation steps were repeated a second time, to finally obtain ~60 μL of clean DNA.
Root DNA was extracted using the NucleoSpin® Plant II kit and protocol (Macherey-Nagel, Düren, Germany) with the following modifications: 30 g of freeze-dried root powder were used; (i) cell lysis was done with buffer PL1; (ii) incubation after adding RNase A lasted 30 min; (iii) the crude lysate was centrifuged before its filtration.
DNA quantification was performed using a Quantus fluorometer (Promega, Madison, Wisconsin, USA) and the Quantifluor kit (dsDNA: E2670).
PCR amplification and sequencing were performed at the GenoScreen platform (Lille, France) using the Illumina Miseq platform to a 2 × 250 bases paired-end version. We used PCR primer pairs 799F (5′-AACMGGATTAGATACCCKG-3′) and 1223R (5′-CCATTGTAGTACGTGTGTA-3′), and NS22B (5′-AATTAAGCAGACAAATCACT-3′) and SSU0817 (5′-TTAGCATGGAATAATRRAATAGGA-3′) (Lê Van et al., 2017) to amplify 16S and 18S rDNA genes, respectively.
To manage mismatch between reads 1 and 2 in the overlap region, we used bases trimming at Q30 with PRINSeq, trimming of specific primers with Cutadapt, assembly with FLASH starting with trimmed reads 1 and 2, a minimum of 30 bases overlapping was required with a 97% homology between reads. Regarding 18S rDNA, only read 1 was analyzed because i) amplicon size did not allow a sufficient overlapping area between read 1 and read 2 and ii) read 1 was of better quality. The GnS-PIPE bioinformatical pipeline was used for the bioinformatical analyses of 16S rDNA and 18S rDNA (Terrat et al., 2012). Raw data sets were deposited on the European Nucleotide Archive database system under the project number (PRJEB25217). Bulk soil samples accession numbers range from ERS2281263 to ERS2281298, those of root and rhizosphere samples range from ERS2255945 to ERS2256016 for 16S rDNA and from ERS2256770 to ERS2256841 for 18S rDNA.
Metabolites and chemical elements analysis
Primary metabolites: amino acids, carbohydrates, polyols, and organic acids
Quantification of free amino acids (AAs), non-structural carbohydrates, polyols, and organic acids (CPOs) was based on the method described by Gravot et al. (2010) and performed using 9 to 12 mg of freeze-dried root powder, with the same methanol-chloroform-water-based extraction. Minor adjustments were made to this protocol: after adding 100% chloroform, tubes were rapidly vortexed and then agitated for 10 min at room temperature; after adding water, samples were vortexed for 20 s and centrifuged for 5 min at 12,000 g and 15°C.
For AA derivatization and profiling, 50 μL of methanol-water extract were vacuum-dried. The dry residue was resuspended in 50 μL of ultrapure water and the tubes were rapidly vortexed, put in an ultrasonic bath for 5 min and centrifuged for 5 s at room temperature. Derivatization of AAs was performed using the AccQTag Ultra Derivatization kit (Waters) with the following modified volumes: 5 μL of the resuspended sample, 35 μL of AccQTag Ultra Borate Buffer and 10 μL of AccQTag Reagent were placed in a new tube, which was vortexed and placed in a water bath for 10 min at 55°C. The whole volume was transferred in a vial and derivatizated AAs were analyzed using liquid chromatography (Acquity UPLC-DAD system, Waters, Milford, MA, USA) according to Jubault et al. (2008). However, the column used for analyses was heated at 53°C and AAs were detected at 265 nm using a photodiode array detector. Identification of AAs was realized by comparison with a standard solution and quantification was made thanks to the internal standard BABA (3-aminobutyric acid).
Analysis of CPOs was performed by gas chromatography coupled to a flame ionization detector (GC-FID, Thermo Fisher Scientific, Waltham, MA, USA) and based on the protocol described by Lugan et al. (2009), which however required several modifications. The online derivatization for CPOs was performed with a Trace 1300 GC-FID (Thermo Scientific) equipped with a Tri Plus RSH (Thermo Scientific). Fifty microliters of the methanol-water extract were sampled in injection vials and vacuum-dried. This online derivatization was performed as follows: the dried extract was resuspended in 50 μL of pyridine containing 20 mg.mL−1 of methoxyaminehydrochloride, under orbital shaking at 40°C for 90 min. Fifty microliters of MSTFA (N-methyl-trimethylsilyltrifluoroacetamide) were added before incubation at 40°C for 30 min. One microliter of the mixture was injected into the GC-FID with a split/splitless injector (split mode set to 1:20) at 260°C, on a TG-5MS column (30 m × 0.32 × 0.25 mm, Agilent Technologies) connected to a flame ionization detector at 300°C. The temperature gradient of the GC oven was: 4 min at 100°C followed by an increase of 10°C.min−1 up to 198°C and maintained at this temperature for 2 min; an increase of 1°C.min−1 up to 202°C; then an increase of 15°C.min−1 ramp up to 268°C and held for 3 min followed by an increase of 1°C.min−1 up to 272°C and raised to 210°C at 10°C.min−1 maintained for 7 min. Identification of CPOs was realized by comparison with a standard solution, while quantification was achieved with adonitol as the internal standard.
Secondary metabolites: glucosinolates
Extraction and analysis of glucosinolates (GSLs) were performed based on the protocol from Hervé et al. (2014) with the following modifications: GSLs were extracted from 12 to 15 mg of freeze-dried root powder and tubes were centrifuged for sedimentation at 12,000 g and 15°C for 5 min. Analysis of GSLs was performed using liquid chromatography coupled with mass spectrometry (Acquity UPLC-TQD, Waters) with electrospray ionization in a negative mode. Chromatographic conditions, A and B-eluents and the gradient used are described in Hervé et al. (2014). Quantification of GSLs was realized using an external calibration with a standard solution containing glucoerucin, gluconasturtiin, and glucobrassicin in the range of 4 to 80 μmol.L−1. These compounds were respectively used to quantify aliphatic, aromatic and indolyl GSLs.
Chemical elements
Analysis of root chemical elements composition was performed according to Maillard et al. (2015). About 4 mg of freeze-dried root powder were used to measure total N and S contents, using a continuous flow isotope mass spectrometer (Nu Instrument, Wrexham, United Kingdom) linked to a C/N/S analyzer (EA3000, Euro Vector, Milan, Italy). For other elements such as K, Ca, P, Mg, Fe, Mn, Zn, Cu, Mo, Ni, and B, roots samples were submitted to microwave acid sample digestion (Multiwave ECO, Anton Paar, les Ulis, France) using 800 μL of concentrated HNO3, 200 μL of H2O2 and 1 mL of Milli-Q water for 40 mg of root powder. All samples were previously spiked with two internal-standard solutions of Gallium and Rhodium, respectively, for a final concentration of 10 and 2 μg.L−1. Mineralized samples were then diluted to 50 mL with Milli-Q water to obtain solutions containing 2.0% (v/v) of nitric acid, then filtered at 0.45 μm using a teflon filtration system (Filtermate, Courtage Analyses Services, Mont-Saint-Aignan, France). Samples were then analyzed by High Resolution Inductively Coupled Plasma Mass Spectrometry (HR ICP-MS, Thermo Scientific, Element 2TM) and quantification of each element was performed using external standard calibration curves. Additional information about mineralization and the utilization of HR ICP-MS can be found in the annexes of Maillard (2016).
Statistical analyses
Analyses were performed using the R software (R Core Team, 2016) and a 5% threshold for statistical significance.
Microbial communities
When analyses were performed on microbial data, some samples had to be removed from the dataset for the following reasons: one root sample was lost before sequencing (from the low diversity-healthy plants-14 DAI condition), one rhizosphere bacterial sample (from the high diversity-healthy plants-1 DAI condition) showed an abnormal smaller total read count after normalization while one root fungal sample (from the high diversity-healthy plants-1 DAI condition) had abnormal phyla abundances compared to other samples.
Bacterial and fungal richnesses and diversities, represented by the number of observed Operational Taxonomic Units (OTUs) and the Shannon index (obtained with the diversity function in the “vegan” package) respectively, were obtained from non-rarefied OTU data and analyzed in roots and rhizosphere separately using a linear model. Models took into account the following factors: sampling time (1, 14, and 42 DAI), plant treatment (healthy vs. infested plants), soil microbial diversity (high vs. low diversity), and soil replicate, as well as paired interactions between the three first factors. Linear models were adjusted depending on the significance of interactions: interactions were all removed if none was significant while they were kept if at least one was significant or close, which was respectively the case of analyses on the rhizosphere and root compartments. A type II analysis of variance table was performed on the models, followed by comparisons based on least-squares means when possible.
In order to analyze the bacterial and fungal community structure, distance-based redundancy discriminant analysis (dbRDA, dbrda function in the “vegan” package) was performed on a Bray-Curtis dissimilarity matrix, obtained from OTU data, which were filtered using a 1%0 threshold and log2-transformed. A type II permutation test for constrained multivariate analyses was performed on the dbRDA using the “RVAideMemoire” package (Hervé, 2016a,b) to evaluate the contribution of each factor (i.e., compartment, time, treatment, soil microbial diversity, and soil biological replicates) to microbial community structure. Quantified contributions expressed as r-squared, were obtained from the varpart function (“vegan” package).
Phyla analyses were performed according to the scripts provided by Bulgarelli et al. (2015) but P-values were corrected using the False Discovery Rate (FDR) method. Plotting was done with the “ggplot2” package.
Genewise negative binomial generalized linear models were conducted using the “edgeR” package (Anders et al., 2013) on filtered data to recover genera differences between plant treatments within sampling time. The obtained P-values were corrected with the FDR method. Using “ggplot2” package, genus count differences between infested and healthy (Xi – Xh) were plotted when the treatment was significant on one hand and when these differences exceeded 250 or −250 counts.
Metabolites and chemical elements
For each sampling time, two different analyses were performed on metabolites and chemical elements, taking into account the plant treatment (healthy vs. infested plants), the soil microbial diversity (high vs. low diversity), and the interaction between both factors, as well as soil biological replicates. Both analyses required the data to be transformed using the fourth root. First, a redundancy analysis (RDA), associated to a permutation significance test based on cross-validation, was performed to determine the influence of the previously mentioned factors on the root metabolomic and elemental profiles. Second, linear models were used to assess precisely which metabolites and elements were affected by the above factors. Finally, a type II analysis of variance was performed on the linear models and the obtained P-values were corrected with the FDR method. The content differences between infested and healthy (Xi – Xh) was plotted.
Relationships between root microbial communities and root chemistry
To assess the relationship between root microbial communities and root chemistry under herbivory, we applied the DIABLO (Data Integration Analysis for Biomarker discovery using Latent cOmponents) method on our data with the plant treatment (i.e., infested vs. healthy plants) defined as the grouping factor, using the scripts provided by Hervé et al. (2018) and two R packages (“mixOmics and “RVAideMemoire”). Only metabolites or chemical elements significantly impacted by the treatment at a given time were kept for this analysis at this same given time. Bacterial and fungal genera used in this analysis were also significantly impacted by the plant treatment and bacterial count differences respected the 250/−250 threshold mentioned above. Hence, a three block-DIABLO was performed at 14 DAI on one hand, using three datasets (i.e., absolute abundance (expressed in counts of OTUs) of microbial genera, metabolites, and chemical elements) while a two block-DIABLO was performed at 42 DAI on the other hand with two datasets: microbial genera and combined metabolites and elements. In the latter case, an analysis on three blocks was not possible using a single column dataset for chemical elements, which was why it was included in the metabolite dataset. Discrimination of the grouping factor was assessed with a permutation significance test based on cross-validation.
Results
Our experiment was based on two soils of different bacterial and fungal alpha-diversities (Table S1) and phyla abundances (Figure S1).
Diversity and structure of microbial communities
After 6 weeks of OSR culture in soils of different microbial diversities and 1 DAI, the bacterial and fungal alpha-diversities were similar in both root and rhizosphere compartments.
Bacteria
Root bacterial richness and diversity were significantly impacted by sampling time, initial soil microbial diversity and the interaction between time and treatment (Table 1). Diversity was also influenced by the interaction between time and initial soil microbial diversity.
Table 1
| Factors | Roots | Rhizosphere | ||||||
|---|---|---|---|---|---|---|---|---|
| F-value | df | P-value | F-value | df | P-value | |||
| Bacteria | Richness (observed OTUs) | Sampling time | 4.61 | 2 | 0.020 | 11.40 | 2 | <0.001 |
| Treatment | 0.80 | 1 | 0.370 | 0.16 | 1 | 0.680 | ||
| Soil microbial diversity | 32.90 | 1 | <0.001 | 95.80 | 1 | <0.001 | ||
| Soil biological replicates | 1.58 | 2 | 0.220 | 1.57 | 2 | 0.220 | ||
| Sampling time:Treatment | 19.10 | 2 | <0.001 | _ | _ | _ | ||
| Sampling time:Soil microbial diversity | 4.07 | 2 | 0.030 | _ | _ | _ | ||
| Treatment:Soil microbial diversity | 0.35 | 1 | 0.550 | _ | _ | _ | ||
| Diversity (Shannon index) | Sampling time | 9.34 | 2 | 0.001 | 4.94 | 2 | 0.014 | |
| Treatment | 0.006 | 1 | 0.930 | 4.06 | 1 | 0.053 | ||
| Soil microbial diversity | 20.80 | 1 | <0.001 | 39.80 | 1 | <0.001 | ||
| Soil biological replicates | 2.04 | 2 | 0.150 | 1.28 | 2 | 0.290 | ||
| Sampling time:Treatment | 22.10 | 2 | <0.001 | _ | _ | _ | ||
| Sampling time:Soil microbial diversity | 1.23 | 2 | 0.300 | _ | _ | _ | ||
| Treatment:Soil microbial diversity | 0.03 | 1 | 0.850 | _ | _ | _ | ||
| Fungi | Richness (observed OTUs) | Sampling time | 9.19 | 2 | 0.001 | 1.50 | 2 | 0.230 |
| Treatment | 0.07 | 1 | 0.780 | 0.56 | 1 | 0.450 | ||
| Soil microbial diversity | 4.27 | 1 | 0.050 | 27.70 | 1 | <0.001 | ||
| Soil biological replicates | 0.91 | 2 | 0.410 | 3.58 | 2 | 0.040 | ||
| Sampling time:Treatment | 3.28 | 2 | 0.056 | _ | _ | _ | ||
| Sampling time:Soil microbial diversity | 0.09 | 2 | 0.900 | _ | _ | _ | ||
| Treatment:Soil microbial diversity | 1.10 | 1 | 0.300 | _ | _ | _ | ||
| Diversity (Shannon index) | Sampling time | 9.25 | 2 | 0.001 | 4.20 | 2 | 0.024 | |
| Treatment | 0.70 | 1 | 0.410 | 2.34 | 1 | 0.130 | ||
| Soil microbial diversity | 4.06 | 1 | 0.056 | 6.38 | 1 | 0.017 | ||
| Soil biological replicates | 1.24 | 2 | 0.300 | 1.68 | 2 | 0.200 | ||
| Sampling time:Treatment | 3.71 | 2 | 0.040 | _ | _ | _ | ||
| Sampling time:Soil microbial diversity | 0.05 | 2 | 0.940 | _ | _ | _ | ||
| Treatment:Soil microbial diversity | 0.88 | 1 | 0.350 | _ | _ | _ | ||
Statistical output of bacterial and fungal alpha-diversity indices in the root and rhizosphere compartments, associated to Figure 2.
The above table presents the outcome of variance analyses (F test) performed on linear models, based on richness (i.e., observed OTUs) or diversity (Shannon index) of each compartment, and taking into account sampling time (1, 14, and 42 days after infestation “DAI”), treatment (healthy vs. infested plants), soil microbial diversity (high vs. low soil diversity) and 2 × 2 interactions between these factors, as well as soil replicates. F-value, degrees of freedom (df), and P-value (in bold when significant) are given for each tested factor or interaction. Dashes indicate that all interactions in the rhizosphere were non-significant and have thus been removed from the models.
At high initial soil microbial diversity, the bacterial richness (observed OTUs) of healthy plants increased with time, reaching a peak of 900 observed OTUs at 14 DAI, and then decreased at 42 DAI (Figure 2A). Herbivory reversed this trend and richness was significantly increased at 42 DAI. Richness was significantly different between healthy and infested plants both at 14 and 42 DAI. Root bacterial diversity (Shannon index) had a similar profile at high and low initial diversities whether or not plants were infested. The Shannon index was significantly lower in infested plants than in healthy plants at 14 DAI but it was higher at 42 DAI (Figure 2A).
Figure 2
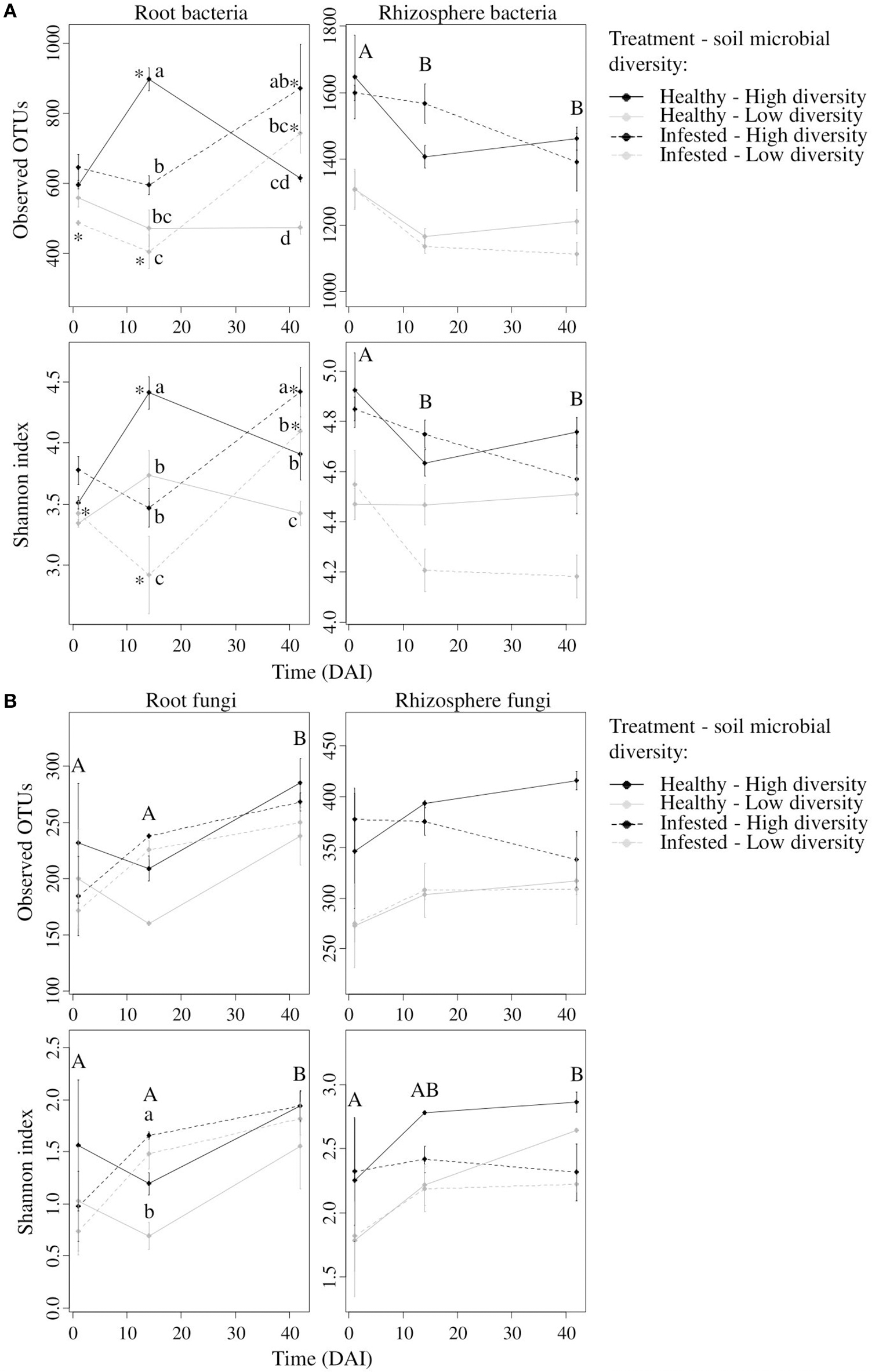
Bacterial and fungal alpha-diversity indices in the root and rhizosphere compartments. Mean value (± se) of bacterial (A) and fungal (B) richness (i.e., Observed OTUs) and diversity (i.e., Shannon index) for the root (left column) and rhizosphere (right column) compartments at 1, 14, and 42 days after infestation (DAI). In the root compartment, lowercase letters represent significant differences between modalities at one given time while asterisks represent significant differences between times for one given modality. Regarding the rhizosphere compartment, capital letters represent significant differences between times for all modalities.
At low initial soil microbial diversity, the bacterial richness (observed OTUs) of healthy plants did not change with time but increased significantly after herbivory (i.e., 42 DAI) (Figure 2A). At this low initial soil diversity, root bacterial diversity (Shannon index) increased in healthy plants at 14 DAI and remained stable at 42 DAI while infested plants displayed an opposite profile (Shannon index decreased at 14 DAI and increased at 42 DAI).
Rhizosphere bacterial richness and diversity were both influenced by sampling time and initial soil microbial diversity (Table 1, Figure 2A). Richness and Shannon index decreased over time in both healthy and infested plants, the latter being similar between 14 and 42 DAI. These indices showed higher values in plants grown on high soil microbial diversity than on low soil diversity.
A dbRDA confirmed that bacterial community structure was driven by compartment, time, treatment and initial soil microbial diversity (Figure 3A). Differences in bacterial community structures between healthy and infested plants were highly significant.
Figure 3
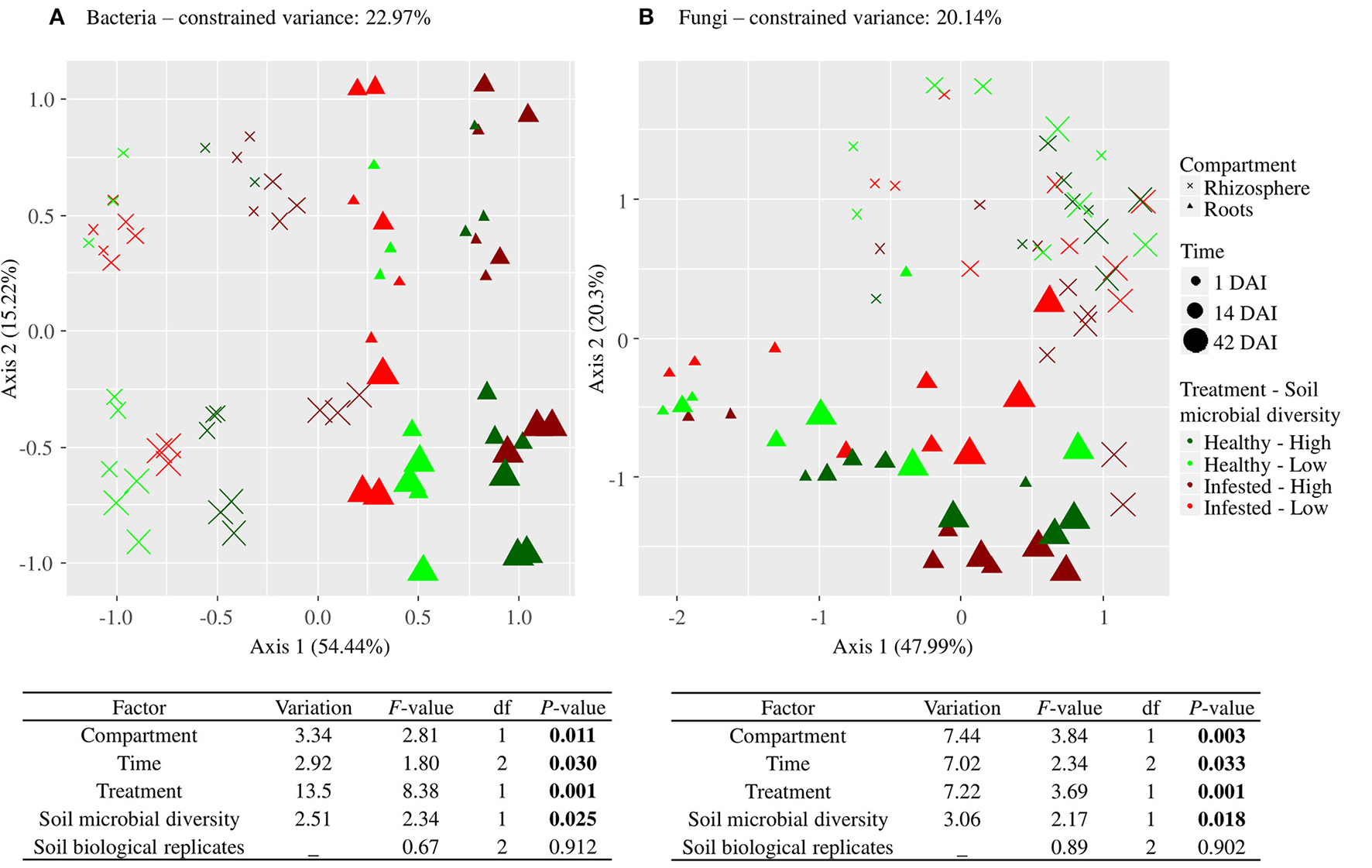
Driving factors of bacterial (A) and fungal (B) community structure. Presented for each microbial kingdom is an ordination plot and the output of the type II permutation test performed on the dbRDA, using the Bray-Curtis dissimilarity index and OTU data set. It includes the variation (adjusted r-squared) explained by each factor, the F-value, the degrees of freedom (df), and the P-value (in bold when significant). Compartment refers to roots and rhizosphere, time refers to 1, 14, and 42 days after infestation (DAI), treatment refers to healthy and infested plants and soil microbial diversity refers to high and low soil microbial diversities.
Fungi
Root fungal richness was four times lower than root bacterial richness and ranged from 150 to 300 observed OTUs. It was only influenced by sampling time (Table 1). The richness of healthy and infested plants at both soil diversities similarly increased over time and reached 250 OTUs at 42 DAI (Figure 2B).
Root fungal diversity (Shannon index) was impacted by time and by the interaction between time and treatment but not by soil microbial diversity (Table 1). Both richness of healthy and infested plants increased over time but the Shannon index was higher in infested plants at 14 DAI, hence during the peak of herbivory (Figure 2B).
Rhizosphere fungal richness was similar to root richness, with 250 to 350 observed OTUs, and it did not vary as much as bacterial richness. Rhizosphere fungal richness was impacted by initial soil microbial diversity and by the soil biological replicates (Table 1). Richnesses of healthy and infested plants remained similarly stable over time (Figure 2B). Richness was greater in plants grown on high soil microbial diversity.
Rhizosphere fungal diversity was influenced by both sampling time and initial soil microbial diversity (Table 1). The Shannon index of healthy and infested plants increased similarly over time and diversity was greater in plants grown on high soil microbial diversity (Figure 2B).
Fungal community structure drivers were compartment, time, and treatment, which were highly significant, as well as initial soil microbial diversity (dbRDA, Figure 3B).
Identification of microbial phyla and genera associated with herbivory and initial soil microbial diversity
Relative abundance of microbial phyla
The roots (Figure 4A) and rhizosphere (Figure 4B) contained four major bacterial phyla, of which Bacteroidetes, Firmicutes, and Proteobacteria were the most abundant. The root bacterial phyla were mainly influenced by infestation but not at 1 DAI. Significant differences occurred during the peak of herbivory at 14 DAI: the phylum of γ-Proteobacteria increased under herbivory, while the phyla of α-, δ-Proteobacteria, Actinobacteria, Bacteroidetes, and “Others” decreased. At 42 DAI, infestation still influenced the phyla of γ-Proteobacteria, Actinobacteria and “Others.” Initial soil microbial diversity impacted β-Proteobacteria at 14 and 42 DAI with a larger abundance in high diversity-infested plants while on the contrary Actinobacteria was more abundant in low diversity-infested plants at 42 DAI.
Figure 4
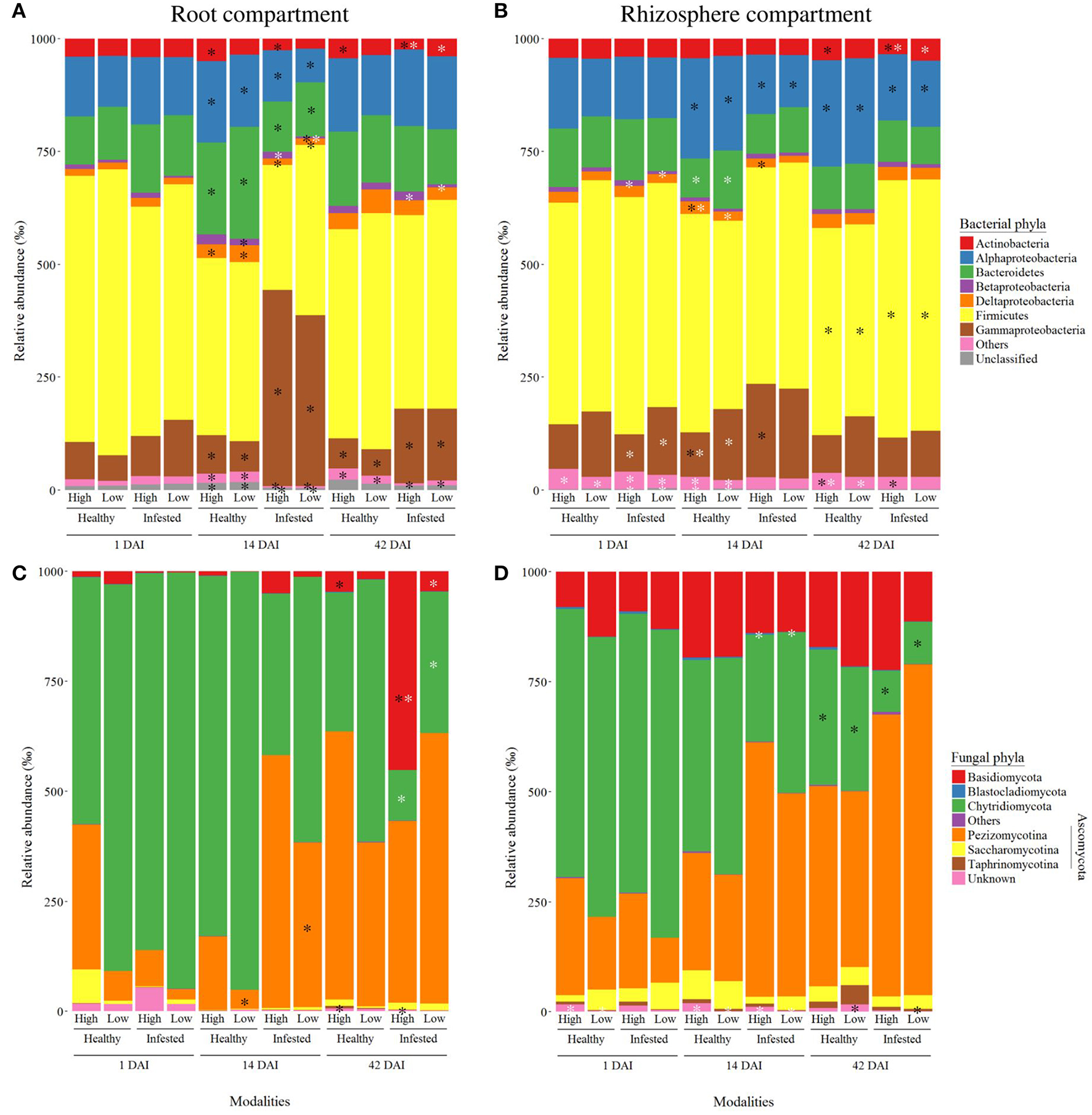
Dominant bacterial and fungal phyla detected in the roots and rhizosphere. Mean relative abundance are presented for bacterial (A,B) and fungal (C,D) phyla from the root (A,C), and rhizosphere (B,D) compartments. All modalities are represented, taking into account the sampling time (1, 14, and 42 days after infestation), the treatment (healthy and infested plants), and initial soil microbial diversity (high and low levels). Black asterisks indicate significant differences between healthy and infested plants at a given time and soil microbial diversity, while white asterisks indicate significant differences between soil microbial diversities at a given time and for a given treatment.
The bacterial phyla of the rhizosphere (Figure 4B) were also influenced by infestation as well as the initial soil microbial diversity and their abundances were in the same range as in the roots. Overall, the phyla of Actinobacteria, β-, δ-, γ-Proteobacteria, and “Others” differed between high and low microbial diversities at the different times. Infestation started to impact phyla at 14 DAI, with more γ-Proteobacteria and less α-Proteobacteria in infested plants. At 42 DAI, the phyla of α-Proteobacteria and Firmicutes were still affected by infestation and more abundant in infested plants.
In the roots and the rhizosphere, there were four major fungal phyla among which Ascomycota (divided into three sub-phyla), Basidiomycota and Chytridiomycota were the most abundant. Relative abundances of root fungal phyla (Figure 4C) were very variable. At 1 DAI, there was no difference between modalities. Pezizomycotina and Basidiomycota abundances increased in infested plants at 14 and 42 DAI respectively while Taphrinomycotina abundance decreased at 42 DAI. The phyla of Basidiomycota and Chytridiomycota were influenced by the soil microbial diversity at 42 DAI in infested plants.
Fungal phyla (Figure 4D) seemed to vary less in the rhizosphere than in the roots but proportions of Chytridiomycota seemed to decrease in favor of Basidiomycota, Pezizomycotina, and Saccharomycotina. There was no effect of herbivory on fungal phyla at 1 and 14 DAI, but Chytridiomycota abundance decreased in infested plants at 42 DAI. The phyla of Blastocladiomycota and “Others” were lightly influenced by the initial soil microbial diversity, with larger abundances in plants of high diversity.
Absolute abundance of microbial genera
A total of 2,031 bacterial genera were detected in the roots and in the rhizosphere.
The root compartment presented 74 bacterial genera from nine different phyla, which varied significantly following herbivory. Most of these genera showed a different abundance between healthy and infested plants at 14 and/or 42 DAI but not at 1 DAI (Table S2). Out of these 74 genera, Bacillus, Clostridium, Paenibacillus, Pseudomonas, and Stenotrophomonas were the most abundant genera influenced by herbivory (Figure 5A). Bacillus decreased in infested plants while the other four genera increased and none of them returned to an abundance similar to the one detected in healthy plants at 42 DAI. Abundance varied with the initial soil microbial diversity. At low soil diversity, the increase of Paenibacillus abundance following herbivory was greater than at high soil diversity while that of the other genera were similar between both diversities.
Figure 5
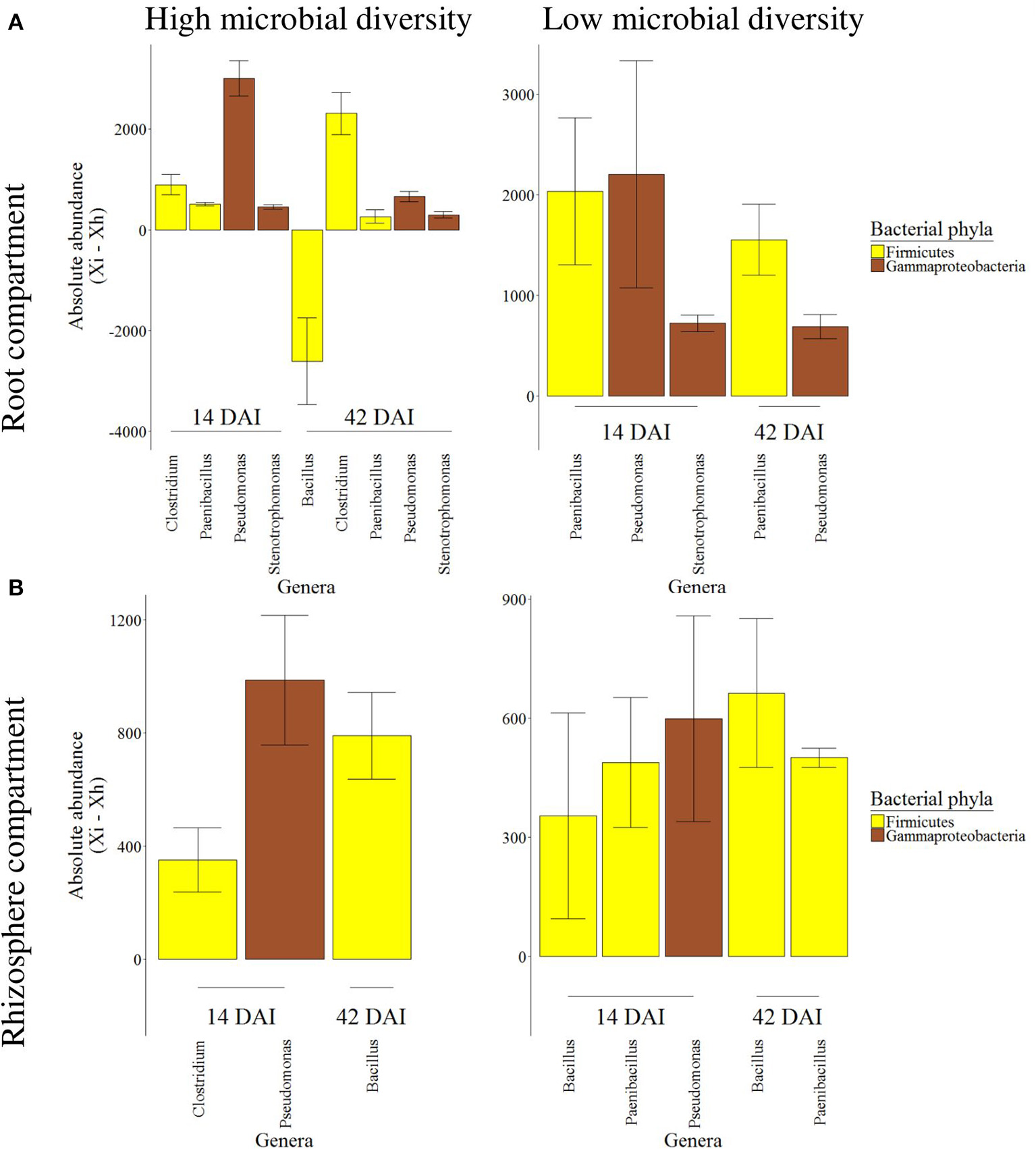
Absolute abundance variation of root and rhizosphere bacterial genera between healthy and infested plants. Mean differential absolute abundance (± se) are presented for bacterial genera from the root (A) and rhizosphere (B) compartment, associated to either high or low soil microbial diversity, at 14 and 42 days after infestation (DAI). Genera represented in the figure were significantly impacted by the plant treatment (i.e., healthy vs. infested) at a given time and exceeded the 250/–250 threshold. Genus abundance is expressed as the difference between absolute abundance of infested plants (Xi) and absolute abundance of healthy plants (Xh).
In the rhizosphere compartment, fewer bacterial genera (46 genera from 9 phyla) were impacted by the infestation at 14 and/or 42 DAI (Table S3). Compared to the roots, the range of bacterial absolute abundance in the rhizosphere was lower. This time, only Bacillus, Clostridium, Paenibacillus, and Pseudomonas were the most abundant genera influenced and increased by herbivory (Figure 5B). As previously shown in the roots, the rhizosphere was also characterized by a change of range in the differential abundance associated to the initial soil microbial diversity, where the range was lower at low diversity.
A total of 2,593 fungal genera were detected in the roots and in the rhizosphere. The fungal genera varied as much as their phyla (Table S4). In the roots, four out of the five herbivory-influenced genera were more abundant in infested plants, including Ajellomyces and Filobasidiella from the subphylum of Pezizomycotina and the phylum of Basidiomycota respectively. No herbivory-influenced genus was significantly impacted at low microbial diversity in the roots. In the rhizosphere, eight genera were influenced by infestation, Torulaspora and Tuber being more abundant in infested plants at 1 and 42 DAI respectively (Table S4).
Root compounds modulated by herbivory
Primary and secondary metabolites
Metabolomic profile of OSR roots was significantly influenced by the treatment at 1, 14, and 42 DAI (Table 2) where infested and healthy plants showed two different profiles, associated to metabolite variations (Figure S2). Their profiles were also impacted by the initial soil microbial diversity but only at 14 DAI. The constrained variance (i.e., part of the variance explained by our variables) had the highest value (73%) at 14 DAI.
Table 2
| Compounds | Time | Total variance (%) | Constrained variance (%) | Factor | RDA test |
|---|---|---|---|---|---|
| Metabolites | 1 DAI | Constrained = 67.28 Unconstrained = 32.72 | Axis 1 = 67.96 Axis 2 = 16.73 | Treatment | F = 8.11; df = 1; P = 0.002 |
| Soil microbial diversity | F = 0.97; df = 1; P = 0.375 | ||||
| Soil biological replicates | F = 1.07; df = 2; P = 0.354 | ||||
| Interaction Treatment-Soil microbial diversity | F = 1.09; df = 1; P = 0.313 | ||||
| 14 DAI | Constrained = 73.49 Unconstrained = 26.51 | Axis 1 = 66.70 Axis 2 = 14.45 | Treatment | F = 9.95; df = 1; P = 0.001 | |
| Soil microbial diversity | F = 3.22; df = 1; P = 0.039 | ||||
| Soil biological replicates | F = 1.28; df = 2; P = 0.247 | ||||
| Interaction Treatment-Soil microbial diversity | F = 0.87; df = 1; P = 0.463 | ||||
| 42 DAI | Constrained = 61.18 Unconstrained = 38.82 | Axis 1 = 52.32 Axis 2 = 23.88 | Treatment | F = 4.48; df = 1; P = 0.002 | |
| Soil microbial diversity | F = 1.29; df = 1; P = 0.271 | ||||
| Soil biological replicates | F = 1.45; df = 2; P = 0.184 | ||||
| Interaction Treatment-Soil microbial diversity | F = 0.77; df = 1; P = 0.602 | ||||
| Elements | 1 DAI | Constrained = 54.38 Unconstrained = 45.62 | Axis 1 = 46.08 Axis 2 = 39.89 | Treatment | F = 2.66, Df = 1, P = 0.055 |
| Soil microbial diversity | F = 2.00, Df = 1, P = 0.122 | ||||
| Soil biological replicates | F = 0.80, Df = 2, P = 0.610 | ||||
| Interaction Treatment-Soil microbial diversity | F = 0.87, Df = 1, P = 0.488 | ||||
| 14 DAI | Constrained = 61.26 Unconstrained = 38.74 | Axis 1 = 61.20 Axis 2 = 23.68 | Treatment | F = 5.20, Df = 1, P = 0.006 | |
| Soil microbial diversity | F = 2.02, Df = 1, P = 0.105 | ||||
| Soil biological replicates | F = 0.50, Df = 2, P = 0.897 | ||||
| Interaction Treatment-Soil microbial diversity | F = 1.24, Df = 1, P = 0.301 | ||||
| 42 DAI | Constrained = 55.99 Unconstrained = 44.01 | Axis 1 = 55.54 Axis 2 = 27.07 | Treatment | F = 2.68, Df = 1, P = 0.045 | |
| Soil microbial diversity | F = 1.16, Df = 1, P = 0.298 | ||||
| Soil biological replicates | F = 0.73, Df = 2, P = 0.650 | ||||
| Interaction Treatment-Soil microbial diversity | F = 2.31, Df = 1, P = 0.079 |
Statistical output of multivariate analysis performed on root metabolites and elements.
The above table presents the outcome of permutation test performed on RDA, based on root metabolite and element contents, and taking into account the treatment (healthy vs. infested plants), soil microbial diversity (high vs. low soil diversity), soil biological replicates and the interaction between treatment and diversity. Total and constrained variances, F-value, degrees of freedom (df), and P-value (in bold when significant) are given for the tested factors and interaction, at each sampling time (1, 14, and 42 days after infestation “DAI”).
Overall, 33 metabolites (17 AAs, 9 CPOs, and 7 GSLs presented in Figures 6A–C respectively) were significantly affected by infestation at one or several moments of the plant-insect interaction and 2 metabolites were impacted by the soil microbial diversity (Table S5). Many of them differed at 1 and/or 14 DAI and most of these 33 metabolites were less abundant in infested plants except 2 AAs (β-alanine, SMCSO), 2 CPOs (trehalose, glycerate), and 2 GSLs (glucobrassicin, neoglucobrassicin), which production increased. Twenty-three of these metabolites reached back a stable state, close to the profile of healthy plants at 42 DAI. All GSLs varied due to infestation but six were still influenced at 42 DAI.
Figure 6
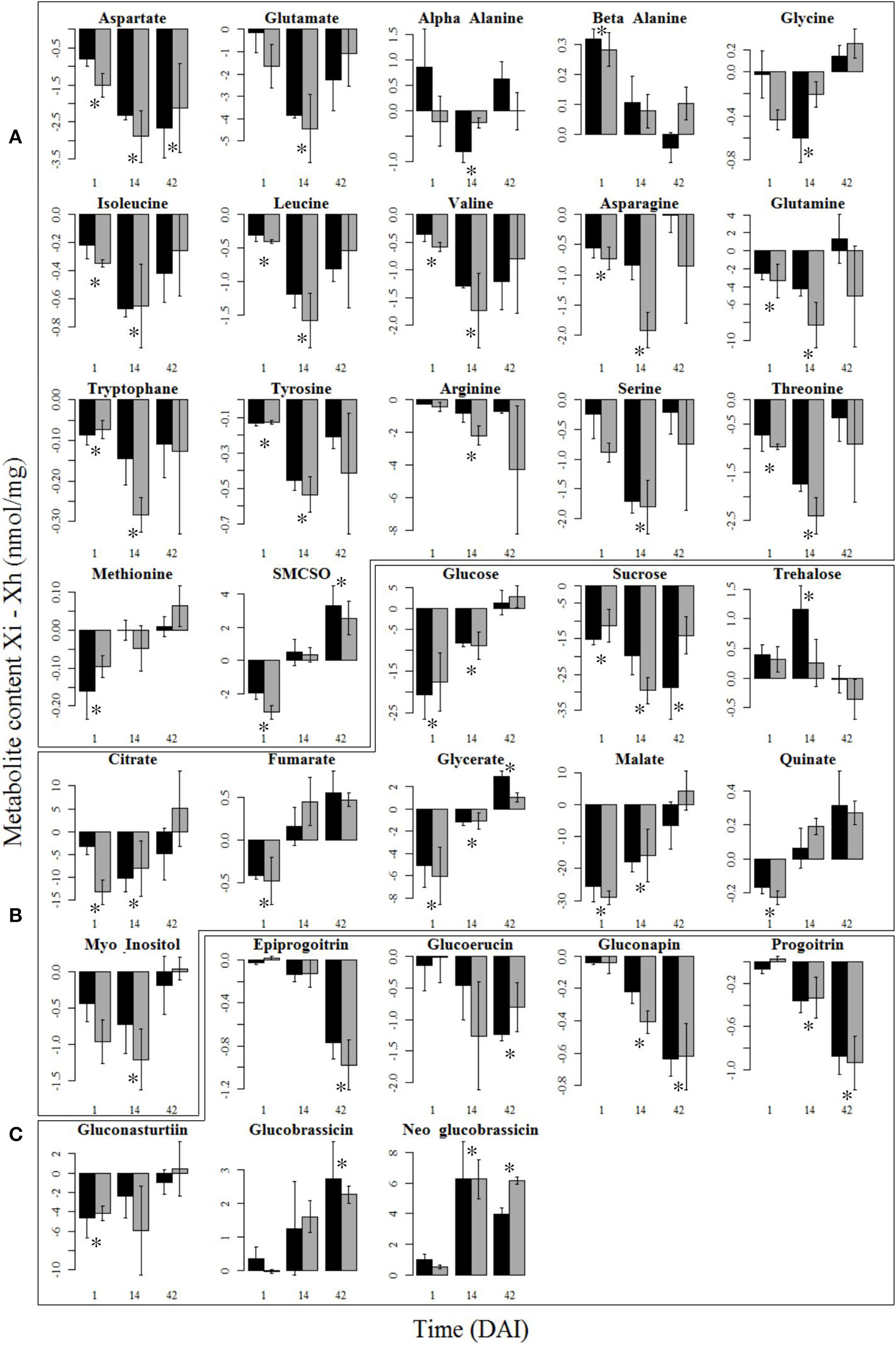
Variation of root metabolite contents between healthy and infested plants. Mean differential content (± se) of the root amino acids (A), carbohydrates, polyols and organic acids (B), and glucosinolates (C) at 1, 14, and 42 days after infestation (DAI) that were significantly impacted by the plant treatment (i.e., healthy vs. infested), are represented in this figure. Metabolite content is expressed as the difference between content of infested plants (Xi) and content of healthy plants (Xh). Asterisks show significant differences (P < 0.05) between healthy and infested plants at a given time. High and low initial soil microbial diversities are represented in black and gray bars respectively. Metabolites are sorted out in the same order as in Table S5, with only the significant metabolites remaining.
Chemical elements
Elemental profiles of OSR roots were only impacted by the treatment at 14 and 42 DAI (Table 2, Figure S3). At 14 DAI, the profile of infested plants was highly modified compared to healthy plants. As for metabolites, the constrained variance was the highest at 14 DAI (61%).
Only 8 chemical elements (4 macroelements, 3 microelements, and 1 heavy metal) were significantly impacted by the infestation, while soil microbial diversity had no effect on chemical element content (Figure 7, Table S6). Six elements (Mg, K, Fe, Na, V, Cd) decreased in infested plants at 14 DAI while N content increased. All of these elements reached a state similar to healthy plants at 42 DAI. Sulfur also increased in infested plants, but only at 42 DAI.
Figure 7
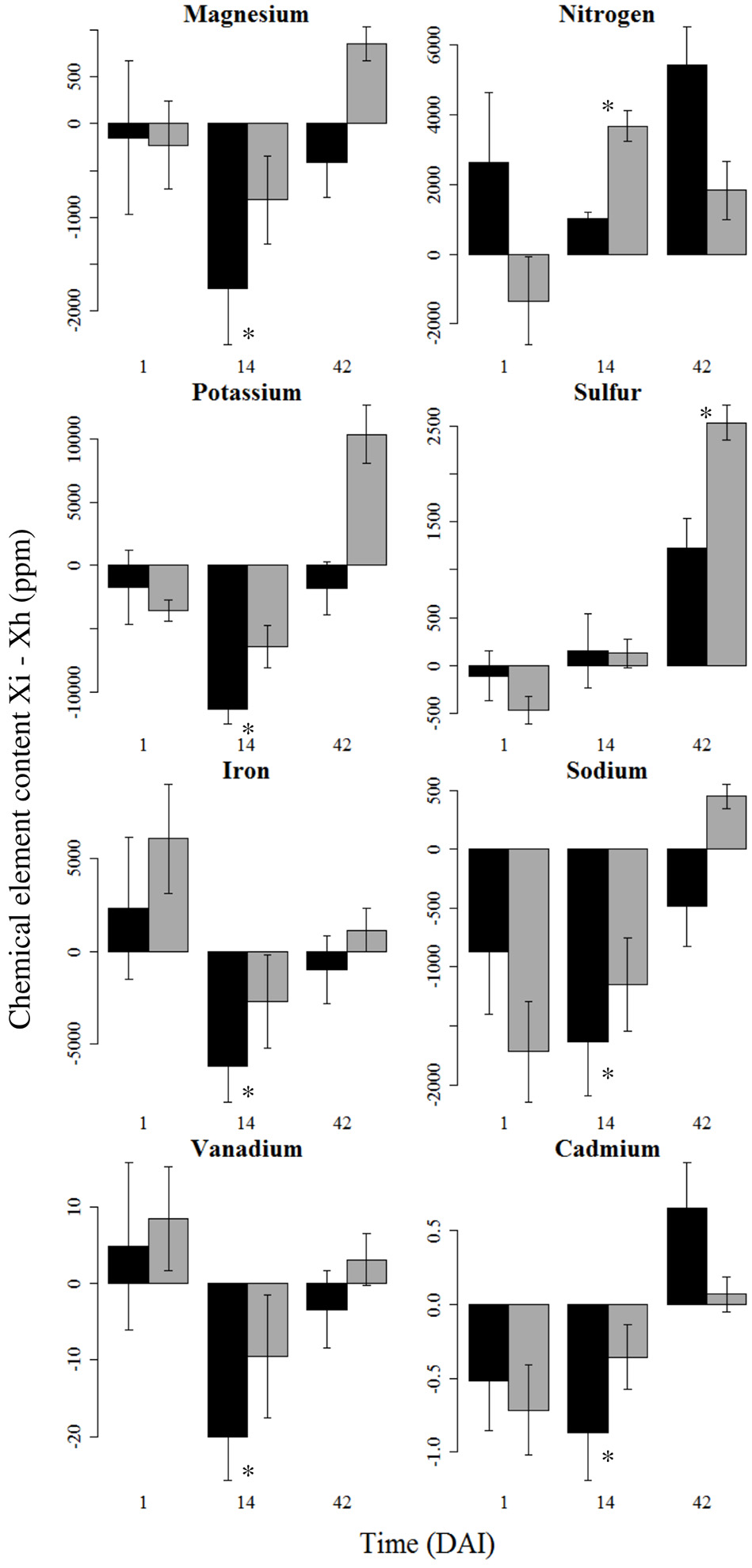
Variation of root chemical element content between healthy and infested plants. Mean differential content (± se) of the macro- (Mg, N, K, S), microelements (Fe, Na, V), and heavy metal (Cd) at 1, 14, and 42 days after infestation (DAI) that were significantly impacted by the plant treatment (i.e., healthy vs. infested), are represented in this figure. Chemical element content is expressed as the difference between content of infested plants (Xi) and content of healthy plants (Xh). Asterisks show significant differences (P < 0.05) between healthy and infested plants at a given time. High and low initial soil microbial diversities are represented in black and gray bars respectively. Chemical elements are sorted out in the same order as in Table S6, with only the significant chemical elements remaining.
Relationships between root compounds and microbial communities
At the peak of herbivory (i.e., 14 DAI), the DIABLO method confirmed the significant differences between infested and healthy plants (CER = 0, P = 0.002) with the three score plots showing similar patterns (Figure 8). The axis 1 of microbiome data was positively correlated to axes 1 of metabolite and element data (r = 0.86 and 0.95, respectively) while metabolite data and element data were also positively correlated (r = 0.84). Infested plants seemed to be associated to (i) four bacterial genera (Clostridium, Paenibacillus, Pseudomonas, and Stenotrophomonas) and one fungal genus (Torulaspora), that were overexpressed, (ii) trehalose (CPO) and neoglucobrassicin (GSL), also overexpressed, (iii) nitrogen but to a lesser extent. The remaining AAs, CPOs, GSLs and seven elements were underexpressed in infested plants.
Figure 8
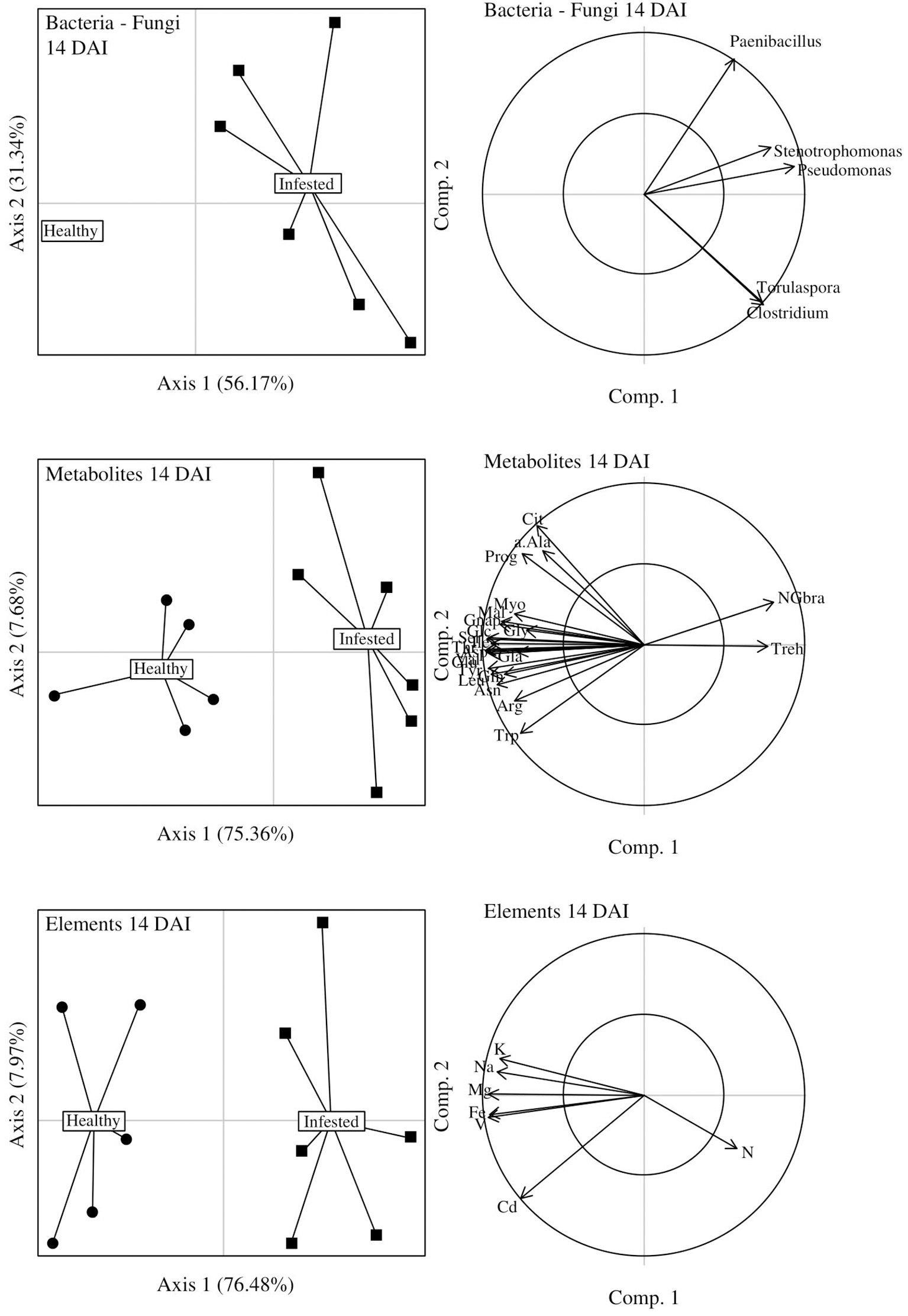
DIABLO graphical outputs at 14 days after infestation (DAI). Score plots of microbiome, metabolite and chemical element datasets are positioned on the left side while the correlation circles associated to each of these score plots are positioned on the right side of the figure. Correlations between axes 1 (axes 2) of microbiome and metabolite datasets: 0.86 (0.86); of microbiome and chemical element datasets: 0.95 (0.60); of metabolite and element datasets: 0.84 (0.77).
At 42 DAI, the discrimination between infested and healthy plants was also significant (CER = 0, P = 0.002) and axis 1 of microbiome data was highly positively correlated to axis 1 of combined metabolite and element data (r = 0.95), confirmed by similar patterns on the two score plots (Figure 9). At the end of herbivory, infested plants seemed to be associated to (i) four bacterial genera (Clostridium, Paenibacillus, Pseudomonas, Stenotrophomonas) and three fungal genera (Ajellomyces, Filobasidiella, Torulaspora), which were overexpressed and (ii) SMCSO (AA), glycerate (CPO), glucobrassicin and neoglucobrassicin (GSLs), and sulfur. Conversely, one bacterial and one fungal genus, as well as four metabolites were underexpressed in these infested plants.
Figure 9
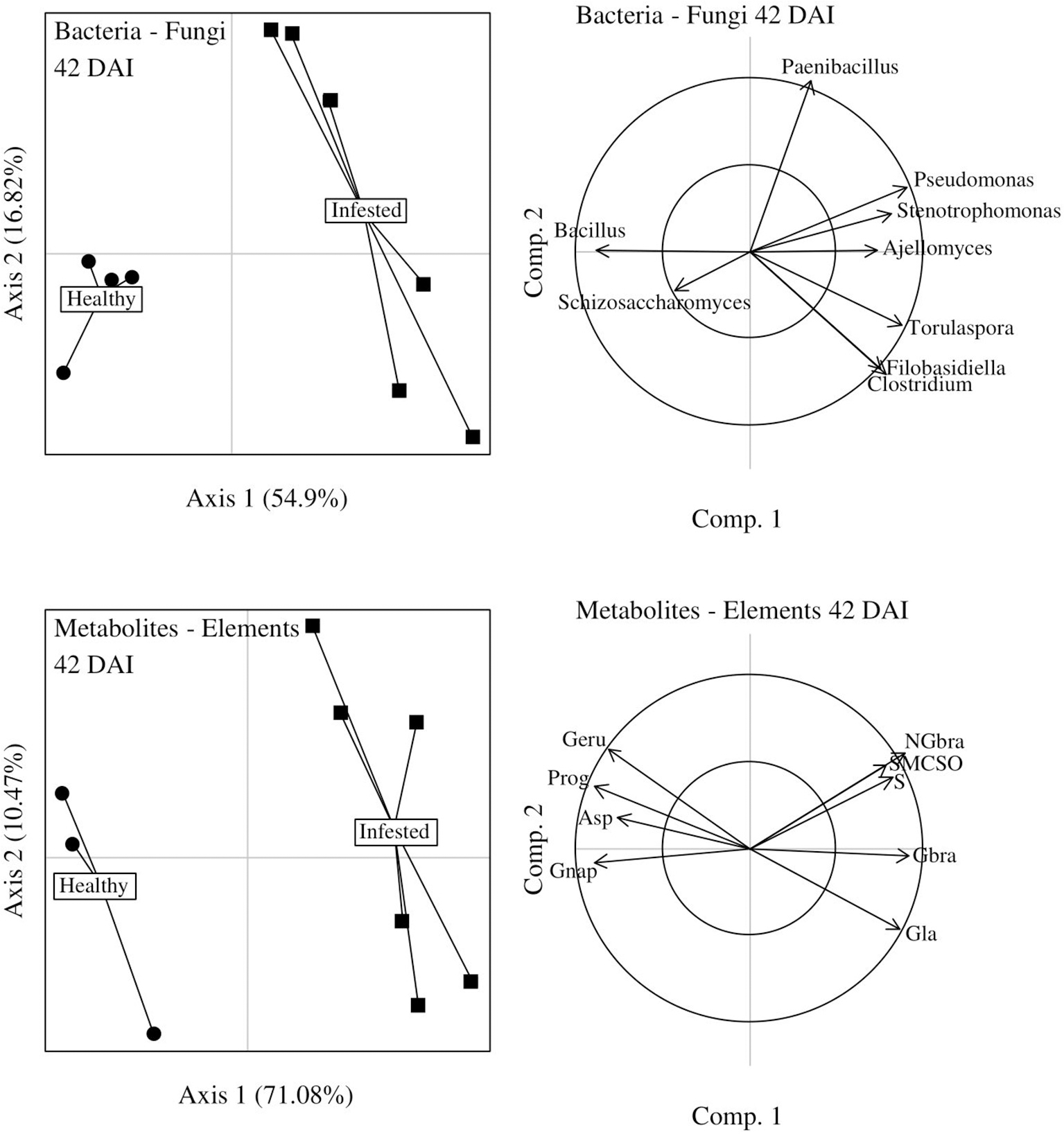
DIABLO graphical outputs at 42 days after infestation (DAI). Score plots of microbiome dataset and combined metabolite and chemical element datasets are positioned on the left side while the correlation circles associated to each of these score plots are positioned on the right side of the figure. Correlations between axes 1 (axes 2) of microbiome and metabolite/chemical element datasets: 0.95 (0.90).
Discussion
Our study showed that infestation of OSR by a belowground herbivore increased root bacterial alpha-diversity, while it had no effect on rhizosphere bacteria and very little on fungi. Interestingly, herbivory was associated with an increase of γ-Proteobacteria and Firmicutes (and five of their most dominant affiliated genera: Pseudomonas, Stenotrophomonas and Bacillus, Clostridium, Paenibacillus respectively) as well as Ajellomyces and Filobasidiella from the fungal phyla of Ascomycota and Basidiomycota. Trehalose, sulfur-containing metabolites and sulfur contents increased in infested plants and could explain the variations observed in bacterial phyla and genera. Herbivory seemed to have only a short-term negative effect on the richness and diversity of the microbial communities, which were both restored when the perturbation ended. The chemical composition of roots matched this restoration. Initial soil microbial diversity itself had little impact on microbial communities of the root and its rhizosphere and did not significantly influence root chemistry.
Herbivory influences microbial community diversity and composition
Our study showed that root herbivory decreased bacterial richness and diversity in roots at the peak of herbivory but globally increased both at the end of herbivory. Opposite trends were found in grassland plant communities, where plant diversity decreased after the end of herbivory (e.g., Collins et al., 1998; Pucheta et al., 1998) but microbial communities may be hard to compare with plant communities. Further from the roots, bacterial and fungal communities of the rhizosphere were respectively not affected and lightly affected by herbivory in our study. In other studies on the putative influence of herbivory on bacterial communities, herbivory marginally increased the abundance of soil nitrifying bacteria and archeae (Le Roux et al., 2008) and, (in contradiction with our own results) Kong et al. (2016) found that white fly herbivory significantly decreased bacterial richness in the rhizosphere of pepper. Differences between these two earlier studies may be due to differences in the biological models studied but also to the nature and length of the perturbation and type of experiments. Compared to bacteria, we observed that fungal communities were very variable and only lightly influenced by herbivory. These results are consistent with previous studies showing that fungi are quite resistant to herbivory, such as leaf mining in Ageratina altissima (Asteraceae), which did not influence the communities of endophytes colonizing leaves (Christian et al., 2016).
Roots and rhizosphere microbial communities of healthy OSR were mainly composed of bacterial taxa Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria (more specifically of Bacillus, Paenibacillus, and Pseudomonas at the genera level), while fungal taxa were mainly Ascomycota, Chytridiomycota and Basidiomycota. Our results on bacteria are in line with those of Bulgarelli et al. (2012) and Bodenhausen et al. (2013) in Arabidopsis thaliana. Regarding fungi, Ascomycota and Basidiomycota accounted for 96% of the community in Microthlaspi spp. populations, another brassicaceous plant (Glynou et al., 2018). A recent study on OSR showed that Proteobacteria and Actinobacteria as well as Ascomycota and Basidiomycota were the most abundant bacterial and fungal phyla respectively in the roots and rhizosphere, however with different dominant genera (Gkarmiri et al., 2017). While the phylum of Chytridiomycota was only present in roots in the previous study, it was part of the rhizosphere composition in ours. These differences in phyla and genera might be due to the use of isotopic labeling by Gkarmiri et al. (2017), representing only the active portion (i.e., assimilating labeled photosynthetates) of the plant communities. Furthermore, many drivers have been shown to shape plant microbial communities: plant root exudates (Turner et al., 2013; Tsunoda and van Dam, 2017); plant development (de Campos et al., 2013; Chaparro et al., 2014; Wagner et al., 2016); accession (Micallef et al., 2009); genotype (Wagner et al., 2016); species (Miethling et al., 2000); soil (Vandenkoornhuyse et al., 2015); and agricultural management practices (Hartmann et al., 2015; Rathore et al., 2017). It is therefore not surprising to find some differences in our study.
Root herbivory increased the abundance of γ-Proteobacteriain the roots and rhizosphere, and that ofFirmicutes in the rhizosphere but it decreased abundance of α-Proteobacteria in both compartments and δ-Proteobacteria and Bacteroidetes in the roots only. These changes were accompanied by an increased abundance of Bacillus, Paenibacillus, Pseudomonas, and Stenotrophomonas. However, some of these phyla in infested plants at 42 DAI reached back a stable state similar to that found in healthy plants at 42 DAI, hence highlighting the resilience of microbial communities after the end of insect herbivory. These results are consistent with those of Kong et al. (2016) on whitefly herbivory on pepper showing an increase in the abundance of γ-Proteobacteria and Stenotrophomonas in the rhizosphere and with those of Kim et al. (2016) where aphid herbivory increased the abundance of Paenibacillus. According to Card et al. (2015), Bacillus, Paenibacillus, Pseudomonas, and Stenotrophomas are either inhibitors or antagonists of plant-pathogenic bacteria or fungi, while Bacillus and Paenibacillus are known to be also entomopathogenous (Monnerat et al., 2009; Grady et al., 2016; Zhao et al., 2017).
Herbivory also affected fungal phyla and genera but to a lesser extent than bacterial taxa: it decreased the abundance of Chytridiomycota in the rhizosphere and increased the abundance of Ajellomyces (slightly) and Filobasidiella (strongly) in the roots. Tkacz et al. (2015) demonstrated an increase of a Chytridiomycota in the rhizosphere of Brassica rapa over time, while this phylum progressively colonized the roots during plant development, to finally dominate fungal communities (Lebreton, personal communication). Most fungi are oligotrophic and grow slowly, with limited carbon sources (Ho et al., 2017). The lesser effect of herbivory on fungal communities observed in our study might be due to the timeframe of our experiment, probably too short compared to the growth rate of fungi, which is based on their utilization of complex trophic resources.
Herbivory influences root metabolite and chemical element composition
Herbivory tended to decrease root AAs and CPOs, which is consistent with the results of Hopkins et al. (1999), van Leur et al. (2008), and Lachaise et al. (2017). Herbivory increased glycerate and trehalose, a sugar assumed to play a role in plant defenses against aphid infestation (Singh et al., 2011). Sucrose was not affected by herbivory, contrary to the study of Pierre et al. (2012). S-methyl cysteine sulfoxide (SMCSO), a toxic compound detectable in brassicaceous crops (Edmands et al., 2013) was increased by herbivory. We therefore hypothesize that both SMCSO and trehalose might play a role in OSR defense against root herbivory.
Our study showed that herbivory also modulated GSLs by increasing two indolyl (glucobrassicin and neoglucobrassicin) but decreasing four aliphatic and one aromatic GSLs consistently with van Dam and Raaijmakers (2006). This is also coherent with the same decrease of total GSL contents found by van Dam and Raaijmakers (2006) and Lachaise et al. (2017). A recent study demonstrated the detrimental effect of glucobrassicin on aboveground generalist and specialist herbivores: a lower larval weight and faster development time associated with an increased mortality (Santolamazza-Carbone et al., 2017). However, in another study, aliphatic GSLs affected larval weight in generalists but not in specialist insects (Arany et al., 2008). Glucosinolates are generally considered as defensive compounds against herbivores but their impact on insect life history traits appear very species specific and their influence on microbial communities is difficult to predict.
Herbivory increased sulfur and nitrogen contents. The increase of sulfur content in root tissues probably corresponded to higher amounts of indolyl GSLs (glucobrassicin and neoglucobrassicin) and SMCSO, which are all sulfur-containing metabolites. The increase in nitrogen could be linked to insect frass (Kagata and Ohgushi, 2011). These authors demonstrated that there was more nitrogen excreted by larva than ingested and they suggested that the excess of nitrogen might originate from the plant organic nitrogen (e.g., AAs or proteins), which could not be digested by the insect. These observations could explain our results, especially since the nitrogen increased occurred only at 14 DAI, the peak of herbivory when the CRF was still at the larval stage.
Relationship between bacterial genera and root chemistry in response to herbivory: focus on dominant genera and compounds increased by herbivory
Root herbivory—obviously—generates root degradation, which creates habitat spatial heterogeneity and as a consequence could modify microbial communities. Changes in microbial diversity and composition following herbivory is often hypothesized to be based on modifications of the plant chemistry, such as plant exudates (Kim et al., 2016; Kong et al., 2016). However, variations in root metabolite and rhizodeposits occur during the plant's life cycle. In addition, bacteria and fungi are known to differ in their ability to use organic compounds (Boer et al., 2005). In our study, we focused on the vegetative stage of OSR, a stage when fungal communities show only limited changes (Mougel et al., 2006).
Increased trehalose following herbivory was associated with an overexpression ofStenotrophomonas andPseudomonas. Stenotrophomonas appears to produce trehalose as an osmoprotective substance (Wolf, 2002) while Pseudomonas is able to use this compound as a carbon source for its growth (De La Fuente et al., 2007) as well as other sugars found in root exudates, a trait that makes this genus quite competitive in microbial communities (Lugtenberg et al., 1999). A modification of plant microbial communities mediated by plant chemistry was also found in Kim et al. (2016) where bacteria such as Paenibacillus were able to grow on medium-based root exudates, which came from aphid-infested plants. Moreover, glycerate was also associated with microbial changes under herbivory. This could be explained by the fact that glycerate-derived compounds are usually accumulated by microorganisms under abiotic stress and this process could be similar for biotic stress (see review on abiotic stress by Empadinhas and da Costa, 2011). We suggest that the root chemistry disturbed by CRF herbivory might be beneficial to colonization of microorganisms such as Pseudomonas, exhibiting competitive traits to exploit trophic resources (Ho et al., 2017).
Following herbivory, enhancement of sulfur-containing compounds (i.e., SMCSO, glucobrassicin, neoglucobrassicin, sulfur) was associated to an overexpression ofBacillus, Paenibacillus, Pseudomonas, andStenotrophomonas. Aziz et al. (2016) demonstrated that plant biomass loss caused by aboveground herbivory was lessened in presence of Bacillus. This decrease seemed mediated by an increase of indolyl GSLs. The fact that these bacteria are associated to sulfur-containing compounds might be linked to their potential role in the sulfur cycle. Pseudomonas for example dominates the arylsulfatase bacterial communities in the OSR rhizosphere (Cregut et al., 2009) and this enzyme (produced by microorganisms in order to mineralize organic sulfur) is present both in the roots and in the rhizosphere of this plant (Knauff et al., 2003). A plant undergoing herbivory could send external signals to recruit bacteria, which could stimulate the synthesis of plant defense compounds (e.g., GSLs). An increase of trehalose in the roots and rhizosphere via root exudates (Paterson et al., 2007) could represent an external signal, attracting beneficial symbionts and promoting bacterial growth. These bacteria could then enhance enzymatic activities (e.g., arylsulfatase) allowing sulfur mineralization and stimulation of GSL production.
Following herbivory, nitrogen was also associated with microorganisms but to a lesser extent. Nitrogen is a limiting factor for plant growth because of its low bioavailability in the soil, since eukaryotes are not able to fix atmospheric nitrogen (Grady et al., 2016). To have access to nitrogen sources, plants rely on nitrogen-fixing microorganisms (i.e., diazotroph). Clostridium and Paenibacillus are free-living diazotrophic species (Choudhary and Varma, 2016; Grady et al., 2016) while a nitrogen fixing trait was recently discovered in the genome of a Pseudomonas species (Yan et al., 2008). We suggest that plants stressed by herbivory might be able to select better microorganisms (i.e., in terms of fitness and cost in nutrients, such as carbohydrates, to the plant), which will provide more nitrogen and enable them to better resist this perturbation.
Soil microbial diversity did not influence the plant-microorganism interactions
Initial soil diversity had respectively little and no effect on root bacterial and fungal alpha-diversities. Only rhizosphere bacterial and fungal diversities were impacted by soil microbial diversity, but not by herbivory. When evaluating abiotic and biotic stresses (Kissoudis et al., 2014), some studies established a positive relationship between an ecosystem stability-resilience and its diversity, while others found opposite relationships or none at all (Griffiths and Philippot, 2013). In our study, the resilience of microbial communities did not seem to come from their initial diversity: microbial results were globally similar at high and low initial diversities and plant chemistry was not impacted by these levels of initial diversity either, as found by Lachaise et al. (2017). We hypothesize that our “low” microbial diversity condition might still have been too rich to properly assess the effect of soil diversity and that a stronger dilution may be necessary in future studies.
Conclusion
In summary, herbivory led to root chemical changes, involving carbohydrates and sulfur-containing compounds, which partly shaped belowground microbial communities and particularly the phyla of γ-Proteobacteria and Firmicutes and a couple of their affiliated genera. It indicates that a plant suffering from herbivory emits either defensive and/or nutritive compounds that influence the recruitment of soil microorganisms by the rhizosphere and the roots.
Our results encourage the determination of the precise identity and functions of microorganisms responding to herbivory, using more accurate primers and metatranscriptomic approaches, and the understanding of the feedback-loop existing between these identified microorganisms and the plant chemistry modified by herbivory. This future research could represent the next step toward the transition from correlation to causation in order to develop sustainable and innovative plant protection strategies.
Statements
Author contributions
MO, CM, and A-MC conceived and designed research. LL and A-YG-E prepared the soil used in the experiment. MO, CP, and VC conducted plant harvest. JL and CM conducted respectively the molecular and bioinformatics analyses. MO performed metabolomics analysis, advised by NM. MO did the statistical analyses and interpreted data, advised by LL, MH, AO, and DP. MO wrote the manuscript, which was commented and approved by all authors.
Funding
This work was supported by grants from the Plant Health and Environment division of the French National Institute for Agricultural Research (INRA) (AAP2015) and a grant from Rennes Métropole for CM (300 01003).
Acknowledgments
The authors thank the GenoScreen platform for sequencing analyses and the Metabolic and Metabolomics Profiling Platform (P2M2) for metabolomics analyses. We are most grateful to PLATIN' (Plateau d'Isotopie de Normandie) core facility for all element analysis used in this study. We also thank Christophe Lunel for his help during the plant sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2018.00091/full#supplementary-material
- AA
Amino Acid
- CPO
Carbohydrate, Polyol, and Organic Acid
- CRF
Cabbage Root Fly
- DAI
Days After Infestation
- dbRDA
distance-based Redundancy Discriminant Analysis
- DIABLO
Data Integration Analysis for Biomarker discovery using Latent cOmponents
- FDR
False Discovery Rate
- GSL
Glucosinolate
- OSR
Oilseed Rape
- RDA
Redundancy Analysis
- SMCSO
S-Methyl Cysteine Sulfoxide.
Abbreviations
References
1
AhujaI.RohloffJ.BonesA. M. (2010). Defence mechanisms of Brassicaceae: implications for plant-insect interactions and potential for integrated pest management. A review. Agron. Sustain. Dev.30, 311–348. 10.1051/agro/2009025
2
AndersS.McCarthyD. J.ChenY.OkoniewskiM.SmythG. K.HuberW.et al. (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc.8, 1765–1786. 10.1038/nprot.2013.099
3
AranyA. M.de JongT. J.KimH. K.van DamN. M.ChoiY. H.VerpoorteR.et al. (2008). Glucosinolates and other metabolites in the leaves of Arabidopsis thaliana from natural populations and their effects on a generalist and a specialist herbivore. Chemoecology18, 65–71. 10.1007/s00049-007-0394-8
4
AzizM.NadipalliR. K.XieX.SunY.SurowiecK.ZhangJ.-L.et al. (2016). Augmenting sulfur metabolism and herbivore defense in Arabidopsis by bacterial volatile signaling. Front. Plant Sci.7:458. 10.3389/fpls.2016.00458
5
BodenhausenN.HortonM. W.BergelsonJ. (2013). Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE8:e56329. 10.1371/journal.pone.0056329
6
Boer Wd FolmanL. B.SummerbellR. C.BoddyL. (2005). Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev.29, 795–811. 10.1016/j.femsre.2004.11.005
7
BoltonM. D. (2009). Primary metabolism and plant defense—fuel for the fire. Mol. Plant. Microbe Interact.22, 487–497. 10.1094/MPMI−22-5-0487
8
BulgarelliD.Garrido-OterR.MünchP. C.WeimanA.DrögeJ.PanY.et al. (2015). Structure and Function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe17, 392–403. 10.1016/j.chom.2015.01.011
9
BulgarelliD.RottM.SchlaeppiK.Ver Loren van ThemaatE.AhmadinejadN.AssenzaF.et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature488, 91–95. 10.1038/nature11336
10
CardS. D.HumeD. E.RoodiD.McGillC. R.MillnerJ. P.JohnsonR. D. (2015). Beneficial endophytic microorganisms of Brassica – A review. Biol. Control90, 102–112. 10.1016/j.biocontrol.2015.06.001
11
ChaparroJ. M.BadriD. V.VivancoJ. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J.8, 790. 10.1038/ismej.2013.196
12
ChoudharyD. K.VarmaA. (eds.) (2016). Microbial-mediated Induced Systemic Resistance in Plants. Singapore: Springer. Available online at: http://link.springer.com/10.1007/978-981-10-0388-2 [Accessed November 29, 2016].
13
ChristianN.SullivanC.VisserN. D.ClayK. (2016). Plant host and geographic location drive endophyte community composition in the face of perturbation. Microb. Ecol.72, 621–632. 10.1007/s00248-016-0804-y
14
CingolaniA. M.Noy-MeirI.DíazS. (2005). Grazing effects on rangeland diversity: a synthesis of contemporary models. Ecol. Appl.15, 757–773. 10.1890/03-5272
15
CollinsS. L.KnappA. K.BriggsJ. M.BlairJ. M.SteinauerE. M. (1998). Modulation of diversity by grazing and mowing in native tallgrass prairie. Science280, 745–747.
16
CregutM.PiuttiS.VongP.-C.Slezack-DeschaumesS.CrovisierI.BenizriE. (2009). Density, structure, and diversity of the cultivable arylsulfatase-producing bacterial community in the rhizosphere of field-grown rape and barley. Soil Biol. Biochem.41, 704–710. 10.1016/j.soilbio.2009.01.005
17
DawsonL. A.GraystonS. J.MurrayP. J.RossJ. M.ReidE. J.TreonisA. M. (2004). Impact of Tipula paludosa larvae on plant growth and the soil microbial community. Appl. Soil Ecol.25, 51–61. 10.1016/S0929-1393(03)00099-4
18
de CamposS. B.YounJ.-W.FarinaR.JaenickeS.JünemannS.SzczepanowskiR.et al. (2013). Changes in root bacterial communities associated to two different development stages of canola (Brassica napus L. var oleifera) Evaluated through next-generation sequencing technology. Microb. Ecol.65, 593–601. 10.1007/s00248-012-0132-9
19
De La FuenteL.MavrodiD. V.ThomashowL. S.WellerD. M. (2007). Utilization of trehalose, benzoate, valerate, and seed and root exudates by genotypes of 2,4-diacetylphloroglucinol producing Pseudomonas fluorescens. Soil Biol. Biochem.39, 2712–2722. 10.1016/j.soilbio.2007.04.029
20
EdmandsW. M. B.GooderhamN. J.HolmesE.MitchellS. C. (2013). S-Methyl-L-cysteine sulphoxide: the Cinderella phytochemical?Toxicol. Res.2, 11–22. 10.1039/C2TX20030A
21
EmpadinhasN.da CostaM. S. (2011). Diversity, biological roles and biosynthetic pathways for sugar-glycerate containing compatible solutes in bacteria and archaea: sugar-glycerate containing compatible solutes. Environ. Microbiol.13, 2056–2077. 10.1111/j.1462-2920.2010.02390.x
22
Fürstenberg-HäggJ.ZagrobelnyM.BakS. (2013). Plant defense against insect herbivores. Int. J. Mol. Sci.14, 10242–10297. 10.3390/ijms140510242
23
GkarmiriK.MahmoodS.EkbladA.AlströmS.HögbergN.FinlayR. (2017). Identifying the active microbiome associated with roots and rhizosphere soil of oilseed rape. Appl. Environ. Microbiol.83:e01938–e01917. 10.1128/AEM.01938-17
24
GlynouK.NamB.ThinesM.Maciá-VicenteJ. G. (2018). Facultative root-colonizing fungi dominate endophytic assemblages in roots of nonmycorrhizal Microthlaspi species. New Phytol.217, 1190–1202. 10.1111/nph.14873
25
GradyE. N.MacDonaldJ.LiuL.RichmanA.YuanZ.-C. (2016). Current knowledge and perspectives of Paenibacillus: a review. Microb. Cell Factories15:203. 10.1186/s12934-016-0603-7
26
GravotA.DittamiS. M.RousvoalS.LuganR.EggertA.CollénJ.et al. (2010). Diurnal oscillations of metabolite abundances and gene analysis provide new insights into central metabolic processes of the brown alga Ectocarpus siliculosus. New Phytol.188, 98–110. 10.1111/j.1469-8137.2010.03400.x
27
GraystonS. J.DawsonL. A.TreonisA. M.MurrayP. J.RossJ.ReidE. J.et al. (2001). Impact of root herbivory by insect larvae on soil microbial communities. Eur. J. Soil Biol.37, 277–280. 10.1016/S1164-5563(01)01098-6
28
GriffithsB. S.PhilippotL. (2013). Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol. Rev.37, 112–129. 10.1111/j.1574-6976.2012.00343.x
29
GriffithsD. W.BirchA. N. E.Macfarlane-SmithW. H. (1994). Induced changes in the indole glucosinolate content of oilseed and forage rape (Brassica napus) plants in response to either turnip root fly (Delia floralis) larval feeding or artificial root damage. J. Sci. Food Agric.65, 171–178.
30
HartmannM.FreyB.MayerJ.MäderP.WidmerF. (2015). Distinct soil microbial diversity under long-term organic and conventional farming. ISME J.9, 1177–1194. 10.1038/ismej.2014.210
31
HervéM. R. (2016a). Aide-Mémoire de Statistique Appliquée à la Biologie. Construire son Étude et Analyser les résultats à L'aide du Logiciel R. Version Finale. Available online at: http://mirror.psu.ac.th/pub/cran/doc/contrib/Herve-Aide-memoire-statistique.pdf [Accessed May 27, 2016].
32
HervéM. R. (2016b). RVAideMemoire: Diverse Basic Statistical and Graphical Functions. Available online at: https://CRAN.R-project.org/package=RVAideMemoire.
33
HervéM. R.DelourmeR.GravotA.MarnetN.BerardoccoS.CorteseroA. M. (2014). Manipulating feeding stimulation to protect crops against insect pests?J. Chem. Ecol.40, 1220–1231. 10.1007/s10886-014-0517-y
34
HervéM. R.NicolèF.Lê CaoK.-A. (2018). Multivariate Analysis of multiple datasets: a practical guide for chemical ecology. J. Chem. Ecol.44, 215–234. 10.1007/s10886-018-0932-6
35
HoA.LonardoD. P. D.BodelierP. L. E. (2017). Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol.93:fix006. 10.1093/femsec/fix006
36
HoaglandD. R.ArnonD. I. (1950). The Water-Culture Method for Growing Plants without Soil. Berkeley, CA: College of Agriculture, University of California.
37
HolW. H.de BoerW.TermorshuizenA. J.MeyerK. M.SchneiderJ. H.van DamN. M.et al. (2010). Reduction of rare soil microbes modifies plant-herbivore interactions. Ecol. Lett.13, 292–301. 10.1111/j.1461-0248.2009.01424.x
38
HopkinsR. J.BirchA. N. E.GriffithsD. W.MorrisonI. M.McKinlayR. G. (1995). Changes in the dry matter, sugar, plant fibre and lignin contents of swede, rape and kale roots in response to turnip root fly (Delia floralis) larval damage. J. Sci. Food Agric.69, 321–328.
39
HopkinsR. J.GriffithsD. W.McKinlayR. G.BirchA. N. E. (1999). The relationship between cabbage root fly (Delia radicum) larval feeding and the freeze-dried matter and sugar content of Brassica roots. Entomol. Exp. Appl.92, 109–117.
40
IvesA. R.CarpenterS. R. (2007). Stability and diversity of ecosystems. Science317, 58–62. 10.1126/science.1133258
41
JohnsonS. N.RasmannS. (2015). Root-feeding insects and their interactions with organisms in the rhizosphere. Annu. Rev. Entomol. 60, 517–535. 10.1146/annurev-ento-010814-020608
42
JubaultM.HamonC.GravotA.LariagonC.DelourmeR.BouchereauA.et al. (2008). Differential regulation of root arginine catabolism and polyamine metabolism in clubroot-susceptible and partially resistant arabidopsis genotypes. Plant Physiol.146, 2008–2019. 10.1104/pp.108.117432
43
KagataH.OhgushiT. (2011). Ingestion and excretion of nitrogen by larvae of a cabbage armyworm: the effects of fertilizer application. Agric. For. Entomol.13, 143–148. 10.1111/j.1461-9563.2010.00502.x
44
KatayamaN.SilvaA. O.KishidaO.UshioM.KitaS.OhgushiT. (2014). Herbivorous insect decreases plant nutrient uptake: the role of soil nutrient availability and association of below-ground symbionts. Ecol. Entomol.39, 511–518. 10.1111/een.12125
45
KimB.SongG. C.RyuC.-M. (2016). Root exudation by aphid leaf infestation recruits root-associated Paenibacillus spp. to lead plant insect susceptibility. J. Microbiol. Biotechnol.26, 549–557. 10.4014/jmb.1511.11058
46
KissoudisC.van de WielC.VisserR. G. F.van der LindenG. (2014). Enhancing crop resilience to combined abiotic and biotic stress through the dissection of physiological and molecular crosstalk. Front. Plant Sci.5:207. 10.3389/fpls.2014.00207
47
KnauffU.SchulzM.SchererH. W. (2003). Arylsufatase activity in the rhizosphere and roots of different crop species. Eur. J. Agron.19, 215–223. 10.1016/S1161-0301(02)00035-7
48
KongH. G.KimB. K.SongG. C.LeeS.RyuC.-M. (2016). Aboveground whitefly infestation-mediated reshaping of the root microbiota. Front. Microbiol.7:1314. 10.3389/fmicb.2016.01314
49
Lê VanA.QuaiserA.DuhamelM.Michon-CoudouelS.DufresneA.VandenkoornhuyseP. (2017). Ecophylogeny of the endospheric root fungal microbiome of co-occurring Agrostis stolonifera. PeerJ5, e3454. 10.7717/peerj.3454
50
LachaiseT.OurryM.LebretonL.Guillerm-ErckelboudtA.-Y.LinglinJ.PatyC.et al. (2017). Can soil microbial diversity influence plant metabolites and life history traits of a rhizophagous insect? A demonstration in oilseed rape. Insect Sci.24, 1045–1056. 10.1111/1744-7917.12478
51
Le RouxX.PolyF.CurreyP.CommeauxC.HaiB.NicolG. W.et al. (2008). Effects of aboveground grazing on coupling among nitrifier activity, abundance and community structure. ISME. J. 2, 221–232. 10.1038/ismej.2007.109
52
LuganR.NiogretM.-F.KervazoL.LarherF. R.KopkaJ.BouchereauA. (2009). Metabolome and water status phenotyping of Arabidopsis under abiotic stress cues reveals new insight into ESK1 function. Plant Cell Environ.32, 95–108. 10.1111/j.1365-3040.2008.01898.x
53
LugtenbergB. J.KravchenkoL. V.SimonsM. (1999). Tomato seed and root exudate sugars: composition, utilization by Pseudomonas biocontrol strains and role in rhizosphere colonization. Environ. Microbiol.1, 439–446.
54
MaillardA. (2016). Adaptations des Végétaux à un Environnement Minéral Fluctuant : Mécanismes Impliqués et Interactions Entre Nutriments. Caen: Université de Caen Normandie.
55
MaillardA.DiquélouS.BillardV.Laîn,éP.GarnicaM.PrudentM.et al. (2015). Leaf mineral nutrient remobilization during leaf senescence and modulation by nutrient deficiency. Front. Plant Sci.6:317. 10.3389/fpls.2015.00317
56
MicallefS. A.ShiarisM. P.Colon-CarmonaA. (2009). Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J. Exp. Bot.60, 1729–1742. 10.1093/jxb/erp053
57
MiethlingR.WielandG.BackhausH.TebbeC. C. (2000). Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33. Microb. Ecol.40, 43–56. 10.1007/s002480000021
58
MilchunasD. G.SalaO. E.LauenrothW. K. (1988). A generalized model of effects of grazing by large herbivores on grassland community structure. Am. Nat.132, 87–106. 10.1086/284839
59
MonneratR. G.SoaresC. M.CapdevilleG.JonesG.MartinsÉ. S.PraçaL.et al. (2009). Translocation and insecticidal activity of Bacillus thuringiensis living inside of plants. Microb. Biotechnol.2, 512–520. 10.1111/j.1751-7915.2009.00116.x
60
MougelC.OffreP.RanjardL.CorberandT.GamaleroE.RobinC.et al. (2006). Dynamic of the genetic structure of bacterial and fungal communities at different developmental stages of Medicago truncatula Gaertn. cv. Jemalong line J5. New Phytol.170, 165–175. 10.1111/j.1469-8137.2006.01650.x
61
Neveu Bernard-GriffithsN. (1998). Sélection de L'Hôte chez Trybliographa rapae W. (Hymenoptera: Figitidae), Parasitoïde de la Mouche du Chou Delia radicum L. (Diptera: Anthomyiidae) ; Perspectives D'application en Lutte Biologique.
62
NishidaR. (2014). Chemical ecology of insect–plant interactions: ecological significance of plant secondary metabolites. Biosci. Biotechnol. Biochem.78, 1–13. 10.1080/09168451.2014.877836
63
PangestiN.PinedaA.DickeM.van LoonJ. J. A. (2015a). Variation in plant-mediated interactions between rhizobacteria and caterpillars: potential role of soil composition. Plant Biol.17, 474–483. 10.1111/plb.12265
64
PangestiN.WeldegergisB. T.LangendorfB.van LoonJ. J. A.DickeM.PinedaA. (2015b). Rhizobacterial colonization of roots modulates plant volatile emission and enhances the attraction of a parasitoid wasp to host-infested plants. Oecologia178, 1169–1180. 10.1007/s00442-015-3277-7
65
PatersonE.GebbingT.AbelC.SimA.TelferG. (2007). Rhizodeposition shapes rhizosphere microbial community structure in organic soil. New Phytol.173, 600–610. 10.1111/j.1469-8137.2006.01931.x
66
PierreP. S.DugravotS.CorteseroA.-M.PoinsotD.RaaijmakersC. E.HassanH. M.et al. (2012). Broccoli and turnip plants display contrasting responses to belowground induction by Delia radicum infestation and phytohormone applications. Phytochemistry73, 42–50. 10.1016/j.phytochem.2011.09.009
67
PinedaA.KaplanI.BezemerT. M. (2017). Steering Soil Microbiomes to Suppress Aboveground Insect Pests. Trends Plant Sci.22, 770–778. 10.1016/j.tplants.2017.07.002
68
PlassartP.TerratS.ThomsonB.GriffithsR.DequiedtS.LelievreM.et al. (2012). Evaluation of the ISO standard 11063 DNA extraction procedure for assessing soil microbial abundance and community structure. PLoS ONE7:e44279. 10.1371/journal.pone.0044279
69
PonzioC.PapazianS.AlbrectsenB. R.DickeM.GolsR. (2017). Dual herbivore attack and herbivore density affect metabolic profiles of Brassica nigra leaves: plant metabolome during dual insect attack. Plant Cell Environ.40, 1356–1367. 10.1111/pce.12926
70
PuchetaE.CabidoM.DíazS.FunesG. (1998). Floristic composition, biomass, and aboveground net plant production in grazed and protected sites in a mountain grassland of central Argentina. Acta Oecol.19, 97–105.
71
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna Available online at: https://www.R-project.org/.
72
RathoreR.DowlingD. N.ForristalP. D.SpinkJ.CotterP. D.BulgarelliD.et al. (2017). Crop Establishment practices are a driver of the plant microbiota in winter oilseed rape (Brassica napus). Front. Microbiol.8:1489. 10.3389/fmicb.2017.01489
73
RichardsonA. E.BareaJ.-M.McNeillA. M.Prigent-CombaretC. (2009). Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil321, 305–339. 10.1007/s11104-009-9895-2
74
Santolamazza-CarboneS.VelascoP.CarteaM. E. (2017). Resistance to the cabbage root fly, Delia radicum (Diptera, Anthomyiidae), of turnip varieties (Brassica rapa subsp. rapa). Euphytica213:274. 10.1007/s10681-017-2069-z
75
SchnitzerS. A.KlironomosJ. N.HilleRisLambersJ.KinkelL. L.ReichP. B.XiaoK.et al. (2011). Soil microbes drive the classic plant diversity–productivity pattern. Ecology92, 296–303. 10.1890/10-0773.1
76
SinghB. K.MillardP.WhiteleyA. S.MurrellJ. C. (2004). Unravelling rhizosphere–microbial interactions: opportunities and limitations. Trends Microbiol.12, 386–393. 10.1016/j.tim.2004.06.008
77
SinghV.LouisJ.AyreB. G.ReeseJ. C.ShahJ. (2011). TREHALOSE PHOSPHATE SYNTHASE11-dependent trehalose metabolism promotes Arabidopsis thaliana defense against the phloem-feeding insect Myzus persicae: trehalose signaling in plant defense. Plant J.67, 94–104. 10.1111/j.1365-313X.2011.04583.x
78
TerratS.ChristenR.DequiedtS.LelièvreM.NowakV.RegnierT.et al. (2012). Molecular biomass and MetaTaxogenomic assessment of soil microbial communities as influenced by soil DNA extraction procedure: Soil DNA extraction impact on bacterial diversity. Microb. Biotechnol.5, 135–141. 10.1111/j.1751-7915.2011.00307.x
79
TilmanD.DowningJ. A. (1994). Biodiversity and stability in grasslands. Nature367, 363–365.
80
TkaczA.CheemaJ.ChandraG.GrantA.PooleP. S. (2015). Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J.9, 2349. 10.1038/ismej.2015.41
81
TreonisA. M.GraystonS. J.MurrayP. J.DawsonL. A. (2005). Effects of root feeding, cranefly larvae on soil microorganisms and the composition of rhizosphere solutions collected from grassland plants. Appl. Soil Ecol.28, 203–215. 10.1016/j.apsoil.2004.08.004
82
TsunodaT.van DamN. M. (2017). Root chemical traits and their roles in belowground biotic interactions. Pedobiologia65, 58–67. 10.1016/j.pedobi.2017.05.007
83
TurnerT. R.JamesE. K.PooleP. S. (2013). The plant microbiome. Genome Biol.14:209. 10.1186/gb-2013-14-6-209
84
van DamM.RaaijmakersC. E. (2006). Local and systemic induced responses to cabbage root fly larvae (Delia radicum) in Brassica nigra and B. oleracea. Chemoecology16, 17–24. 10.1007/s00049-005-0323-7
85
van GeemM.HarveyJ. A.CorteseroA. M.RaaijmakersC. E.GolsR. (2015). Interactions between a belowground herbivore and primary and secondary root metabolites in wild cabbage. J. Chem. Ecol.41, 696–707. 10.1007/s10886-015-0605-7
86
van LeurH.RaaijmakersC. E.van DamN. M. (2008). Reciprocal interactions between the cabbage root fly (Delia radicum) and two glucosinolate phenotypes of Barbarea vulgaris. Entomol. Exp. Appl.128, 312–322. 10.1111/j.1570-7458.2008.00722.x
87
van RuijvenJ.BerendseF. (2010). Diversity enhances community recovery, but not resistance, after drought: diversity promotes community recovery. J. Ecol.98, 81–86. 10.1111/j.1365-2745.2009.01603.x
88
VandenkoornhuyseP.QuaiserA.DuhamelM.Le VanA.DufresneA. (2015). The importance of the microbiome of the plant holobiont. New Phytol.206, 1196–1206. 10.1111/nph.13312
89
WagnerM. R.LundbergD. S.del RioT. G.TringeS. G.DanglJ. L.Mitchell-OldsT. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun.7, 12151. 10.1038/ncomms12151
90
WolfA. (2002). Stenotrophomonas rhizophila sp. nov., a novel plant-associated bacterium with antifungal properties. Int. J. Syst. Evol. Microbiol. 52, 1937–1944. 10.1099/ijs.0.02135-0
91
YanY.YangJ.DouY.ChenM.PingS.PengJ.et al. (2008). Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proc. Natl. Acad. Sci. U.S.A.105, 7564–7569. 10.1073/pnas.0801093105
92
ZhaoY.GaoZ.TianB.BiK.ChenT.LiuH.et al. (2017). Endosphere microbiome comparison between symptomatic and asymptomatic roots of Brassica napus infected with Plasmodiophora brassicae. PLOS ONE12:e0185907. 10.1371/journal.pone.0185907
Summary
Keywords
Delia radicum, Brassica napus, soil microbial diversity, rhizosphere and root microbial communities, herbivory, metabolites, chemical elements
Citation
Ourry M, Lebreton L, Chaminade V, Guillerm-Erckelboudt A-Y, Hervé M, Linglin J, Marnet N, Ourry A, Paty C, Poinsot D, Cortesero A-M and Mougel C (2018) Influence of Belowground Herbivory on the Dynamics of Root and Rhizosphere Microbial Communities. Front. Ecol. Evol. 6:91. doi: 10.3389/fevo.2018.00091
Received
05 March 2018
Accepted
07 June 2018
Published
26 June 2018
Volume
6 - 2018
Edited by
Philip G. Hahn, University of Montana, United States
Reviewed by
Nurmi Pangesti, Netherlands Institute of Ecology (NIOO-KNAW), Netherlands; Natalie Susan Christian, Indiana University Bloomington, United States
Updates
Copyright
© 2018 Ourry, Lebreton, Chaminade, Guillerm-Erckelboudt, Hervé, Linglin, Marnet, Ourry, Paty, Poinsot, Cortesero and Mougel.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Mougel christophe.mougel@inra.fr
This article was submitted to Behavioral and Evolutionary Ecology, a section of the journal Frontiers in Ecology and Evolution
†These authors have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.