 Paula Vizoso
Paula Vizoso Soledad Francisca Undurraga
Soledad Francisca Undurraga Juan Velozo
Juan Velozo- 1Facultad de Ciencias, Center of Plant Propagation and Conservation (CEPROVEG), Universidad Mayor, Santiago, Chile
- 2Departamento de Genética Molecular y Microbiología, Facultad de Ciencias Biológicas, FONDAP Center for Genome Regulation, Millennium Institute for Integrative Biology, Pontificia Universidad Católica de Chile, Santiago, Chile
Introduction
Quillaja saponaria (Quillay) is an endemic Chilean tree species within the Quillajaceae family, from the Fabales order that is adapted to grow under dry temperate conditions and poor soils (Villagran and Hinojosa, 1997; Luebert, 2014). Its high tri-terpenoid saponin content makes it an economically attractive species. Quillay contains amphipathic glycosides called saponins that are used by the pharmaceutical and cosmetic industries (Hostettmann and Marston, 1995; Guo et al., 1998). Their harvest has conventionally been done using natural Quillay populations as the raw material, damaging natural ecosystems (Santelices and Bobadilla, 1997). This unsustainable overexploitation of native Quillaja forest has compelled the establishment of new plantations that would satisfy an increased demand for Q. saponaria products (Donoso et al., 2011). In this study, we present the first draft of Quillay's chloroplast genome sequence (cpDNA) and functional annotation. Our study provides information for Quillay germplasm characterization, to address genetic diversity and gene flow from parental trees through uniparental mode of chloroplast inheritance (Daniell et al., 2016), which may contribute to the long-term goal of selection for superior saponin-producing trees amenable for commercial plantations.
Materials and Methods
Plant Materials, DNA Extraction, Library Preparation, and Sequencing
We collected young leaves from a Quillay clone with high saponin content. Genomic DNA was extracted by a modified Healey's protocol (Healey et al., 2014). The integrity of genomic DNA was verified using 1% agarose gel electrophoresis, and its concentration was quantified with a Picogreen Assay. Total DNA samples with concentrations >30 ng/μL−1 were chosen for Illumina sequencing. Library construction and Illumina (Illumina, CA, USA) sequencing were done at GenomaMayor (University Mayor, Santiago, Chile) and the Cornell University Biotechnology Resource (BRC–Ithaca, NY, USA). The DNA was used to prepare a library with NEBNext dsDNA Fragmentase according to the manufacturer's instructions (M0348- New England Biolabs). Three libraries were sequenced in Illumina MiSeq and Illumina HiSeq 2500 platforms to generate 250 bp paired-end reads. Reads were filtered against adapter contamination or low-quality values (quality value ≤ 5) using FastQCv.0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). In order to obtain chloroplast specific reads, we performed a BLAST search using NCBI-blast version 2.2.31 + against 441 sequenced plastomes of the Fabales order, and plastome models of C. quinoa (CM008430), G. max (NC_007942), L. japonicus (NC_002694), P. vulgaris (NC_009259), P. trichocarpa (NC_009143), and A. thaliana (NC_000932). The e-value cut-off was 10−6 and 90% identity.
Plastome Assembly, Annotation and Analysis
De-novo assembly was processed with the software SOAPdenovo2 (parameters: kmers35, rd_len_cutof = 200) (Luo et al., 2012), ABySS version 1.9.0 (parameters: K = 16 k = 96) (Simpson et al., 2012), and SPAdes version 3.1.0 (Bankevich et al., 2012) with k-mers sizes of 21, 33, 55, and 77 for several runs of the de-novo assembly to increase accuracy. Before the assembly reconciliation (Zimin et al., 2008), the assemblies were evaluated and scaffolded using CAMSA (Aganezov and Alekseyev, 2017). The workflow consisted of sequential assembly steps with the highest similarity 90% and decreased the percentage of length fraction of 100 bp over read and contig length between assemblers. The scaffolding process was performed using a guided reference (from 441 plastomes) to join gradually every super-contig to form the scaffold of the plastome. The assembled contigs were pooled and ordered against 441 plastomes downloaded from Genbank using the Burrows-Wheeler Alignment tool BWA (Li and Durbin, 2010) to obtain super-contigs. To close gaps between super-contigs and reduce ambiguous bases, we re-mapped reads to candidate scaffold using BWA. Only reads that aligned to a sequence with at least 98% identity and with non-zero mapping quality were considered to close gaps. Ambiguous bases were corrected by visual inspection of the alignment. Any positions with low coverage or low-quality base calls were checked manually. To reduce the number of false positive or false negative variant calls, we used only calls at a position with a minimum 90% depth of coverage threshold. Then the consensus sequence was exported using a minimum coverage threshold of 1X. At positions where the threshold of low coverage was not met, the scaffold was trimmed and joined by minor manual adjustment. This approach has been used to assemble several large and complex genomes (Gajer et al., 2004; Card et al., 2014; Olson et al., 2015). To identify simple sequence repeat (SSR) loci, we used the MISA microsatellite finder tool (Beier et al., 2017).
Our gene annotation process was based on cumulative bioinformatic evidence. To generate high-quality annotation, we curated gene annotations using Dual Organellar GenoMe Annotator “DOGMA” (Wyman et al., 2004) and GeSeq (Tillich et al., 2017) using default parameters to predict protein-coding genes by HMMER profile search and ARAGORN v1.2.38 (Laslett and Canback, 2004). tRNA genes were annotated with tRNAscan-SE v2.0(Lowe and Eddy, 1997), and BLAST searches were used to annotate ribosomal RNA (rRNA), tRNA and DNA genes conserved at embryophyte chloroplasts (Wommack et al., 2008). AUGUSTUS software was used to validate putative genes, using the Arabidopsis thaliana chloroplast genome as a reference (Sato et al., 1999). The coding region sequences (CDS) were translated into aminoacids using a standard codon table. The functions of the predicted genes were annotated by BLAST2GOPRO (B2G) (Conesa et al., 2005) with NR, SwissProt, InterProScan, KEGG, COG, and Gene Ontology (GO) methods. Finally, GenomeVx (Conant and Wolfe, 2008) software was used to draw the circular map of the chloroplast genome.
Results and Discussion
To obtain a high-quality assembly of quillay plastome, we developed a pipeline that included several stages of validation based at the information available from chloroplast genomes from several related species, as plastomes have low mutation rates with high conservation in their structure and gene content (Jansen et al., 2007). From a total of 13,6 million reads obtained in the sequencing of Quillay complete genome, a 1,512,173 chloroplast specific reads (766,658 paired reads and 783,579 single reads) were obtained for de-novo assembly. As an independent control for the read selection, we applied the approach of mapping reads to 441 plastomes. The top ten plastomes with the highest coverage can be found in Supplementary Table 1. Alignments revealed that the greatest number of conserved positions and highest coverage were obtained with the Chenopodium quinoa ecotype Real Blanca chloroplast (CM008430), with a 90% and 1,239X average coverage.
The progressive analysis of de-novo genome assembly and the assembly reconciliation resulted in 97 contigs. We obtained a contig N50 was 9,536 bp with a minimum contig size of 2,045 bp, a maximum contig size of 50,454 and a 36.04% G+C content. From the results obtained of the mapped reads to several plastomes, de-novo Quillay contigs were arranged using the complete plastome of Quinoa (Hong et al., 2017) and C. chuniana. Interestingly, Quinoa and Quillay species had traits related to biotic-abiotic stress and saponin production (Gómez-Caravaca et al., 2012; Shin et al., 2015). These results could be favorable to the identification of cpSSRs (Ebert and Peakall, 2009), since the regions flanking them are strongly conserved, facilitating the breeding selection of individuals and, the identification and conservation of valuable traits found in Quillaja varieties.
Finally, the scaffolding process using a guided reference, resulted in eleven super-contigs. To close sequence gaps and, to improve accuracy in sparsely covered regions from super-contigs, we mapped reads to the putative scaffold. The read coverage was adequate to detect any miss-assemblies (Supplementary Figure 1). The first draft of the Quillay plastome, consisting of one scaffold chloroplast genome sequence with a total length of 132,854 bp (Figure 1), corresponding, on average, to a 2,417 × coverage of the assembled genome size. This study also shows that it is possible to assemble high-quality complete chloroplast genomes from whole genome shotgun sequencing datasets. We identified 10 SSR loci, 112 mononucleotide motifs with variable length, which ranged between 10 and 25 nucleotides. We also found 10 dinucleotide, 15 trinucleotide, and 5 tetranucleotide SSRs. The most common SSRs were A or T mononucleotide repeats.
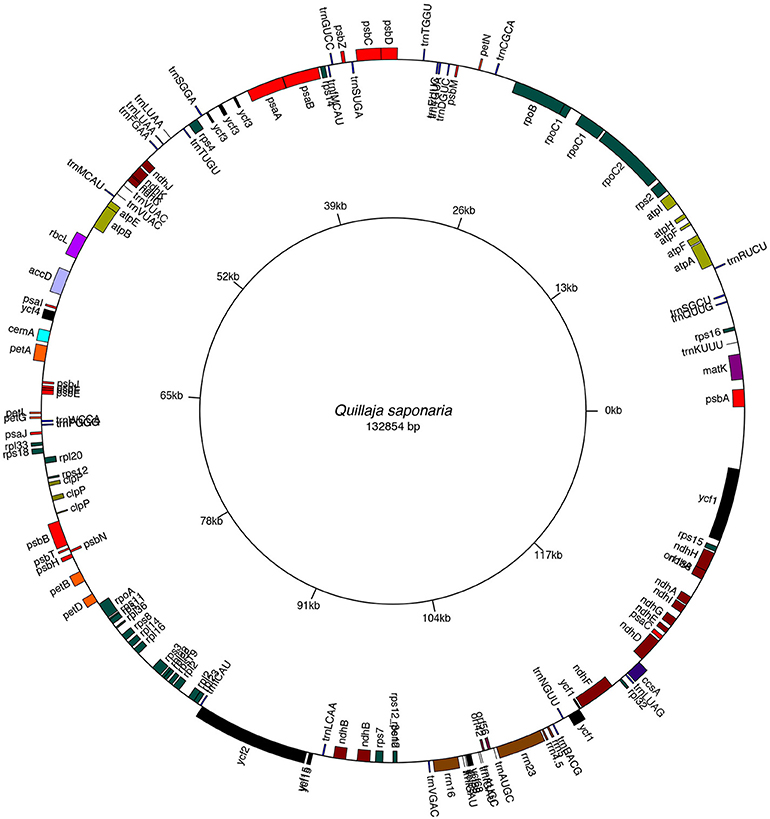
Figure 1. Gene map of Quillaja saponaria cpDNA. Genes depicted within the map are transcribed in a clockwise direction. Genes on the outer part of the map are transcribed counterclockwise. Plastome map was drawn with the OrganellarGenomeDRAW (OGDRAW) program. See GenBank accession no MH880827.
We identified approximately 139 putative ORFs that represent 112 of the 120–130 genes comprising the plant plastomes (Rogalski et al., 2015), 76% of them were completely B2G annotated (GOmapping, INTERPROSCAN, and Blast Hits). We assigned the Enzyme Code to 17 putative genes that matched Blast2Go functions. INTERPROSCAN profiles or domains were found in 100% of the sequences. For genes with low sequence identity, we used manual annotation with translated amino acid sequences of the chloroplast/bacterial genetic code. Ninety percent sequences were assigned GO ID. Among them, about 49 genes encoded proteins of photosystem I and II as well as for other photosynthesis categories. We also found 26 genes for ribosomal proteins (large and small subunit), 5 genes coding for RNA polymerases, 4 ribosomal RNAs genes, 13 genes coding for proteins with unknown function, 33 genes coding for tRNAs, one-carbon metabolism (cemA), one RNA processing gene (matK), one fatty acid synthesis gene (accD) and, three genes coding for proteolysis protein (clpP). As shown in Table 1, we list the genes and functional groups identified in the Quillay cpDNA.

Table 1. Genes identified in the Quillaja saponaria chloroplast genome.
The gene content and organization of a Quillay chloroplast was compared with a closely related species of the Fabaceae family, Cercis chuniana. Comparison of the intergenic spacer in trnD-trnT genes showed differences related to the presence of trnY-trnE genes between both genes (trnY-trnE genes absent in C. chuniana). An additional difference is absence of the trnG and infA genes at Quillaja, also reported in Legumes (Magee et al., 2010), Rosids (Millen, 2001) and Solanales (Wicke et al., 2011). Comparison analysis of three plastomes shows positional differences next to IRb regions. These are related to rbcL, atpB, and atpE genes, which are different in Quinoa and Quillay but are highly conserved between Quillay and C. chuniana (Supplementary Figure 2). The rest of the overall structure resembles the majority of 441 plant cp genomes evaluated.
Even though a direct relationship between the plastome-encoded genes and saponin biosynthesis has not yet been established, it has been shown that Quillay trees significantly reduce their photosynthesis rate, stomatal conductance, and transpiration under restricted irrigation conditions (Donoso et al., 2011). In addition, environmental changes, such as season, soil fertility, light, and water availability alter biomass, and saponin content. The lowest concentration of saponin has been found during the winter, and the highest in autumn, suggesting that abiotic factors may play a major role in the regulation of saponin production (Copaja et al., 2003; Szakiel et al., 2011). Furthermore, chloroplasts also have a critical role in plant immunity, as a site for the production for salicylic acid and jasmonic acid (Nomura et al., 2012; de Costa et al., 2013) demonstrated an overproduction of triterpene saponins in cell suspensions treated with methyl jasmonate (MeJA) in both P. ginseng and P. Notoginseng, and an enhancement of saponin content using different light and UV radiation treatments in P. ginseng and Quillaja. This result suggests that chloroplasts can act as environmental sensors increasing accumulation of triterpene saponins as a part of the defense response. Our results can provide valuable information related to molecular markers and facilitate the identification of valuable traits and genetic variants that can be subsequently used for breeding programs.
Links to the Deposited Data
The draft genome sequence and gene models of Quillay are available at NCBI genome database with the BioProject number PRJNA415043. The high-throughput sequencing data for genome assembly is available at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under the accession number SRR6297481. The source accession for this DNA sample is SAMN07812699. The first draft chloroplast genome sequence with all genes annotated has been submitted to GenBank MH880827.
Author Contributions
SFU and JV conceived this study. PV performed data analyses. SFU and JV supervised the research. PV and SFU wrote the article.
Funding
This work was supported by Fondo de Fomento al Desarrollo Científico y Tecnológico (Fondef–project IT15I10086) to JV Conicyt, Santiago, Chile.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00104/full#supplementary-material
References
Aganezov, S. S., and Alekseyev, M. A. (2017). Open Access CAMSA : a tool for comparative analysis and merging of scaffold assemblies. BMC Bioinformatics 18(Suppl. 15):496. doi: 10.1186/s12859-017-1919-y
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Card, D. C., Schield, D. R., Reyes-Velasco, J., Fujita, M. K., Andrew, A. L., Oyler-McCance, S. J., et al. (2014). Two low coverage bird genomes and a comparison of reference-guided versus de novo genome assemblies. PLoS ONE 9:e106649. doi: 10.1371/journal.pone.0106649
Conant, G. C., and Wolfe, K. H. (2008). GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862. doi: 10.1093/bioinformatics/btm598
Conesa, A., Götz, S., García-gómez, J. M., Terol, J., Talón, M., Genómica, D., et al. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Copaja, S. V., Blackburn, C., and Carmona, R. (2003). Variation of saponin contents in Quillaja saponica molina. Wood Sci. Technol. 37, 103–108. doi: 10.1007/s00226-002-0150-8
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
de Costa, F., Yendo, A. C. A., Fleck, J. D., Gosmann, G., and Fett-Neto, A. G. (2013). Accumulation of a bioactive triterpene saponin fraction of Quillaja brasiliensis leaves is associated with abiotic and biotic stresses. Plant Physiol. Biochem. 66, 56–62. doi: 10.1016/j.plaphy.2013.02.003
Donoso, S., Peña, K., Pacheco, C., Luna, G., and Aguirre, A. (2011). Respuesta fisiológica y de crecimiento en plantas de Quillaja saponaria y Cryptocarya alba sometidas a restricción hídrica. Bosque 32, 187–195. doi: 10.4067/S0717-92002011000200009
Ebert, D., and Peakall, R. (2009). A new set of universal de novo sequencing primers for extensive coverage of non-coding chloroplast DNA: new opportunities for phylogenetic studies and cpSSR discovery. Mol. Ecol. Resour. 9, 777–783. doi: 10.1111/j.1755-0998.2008.02320.x
Gajer, P., Schatz, M., and Salzberg, S. L. (2004). Automated correction of genome sequence errors. Nucleic Acids Res. 32, 562–569. doi: 10.1093/nar/gkh216
Gómez-Caravaca, A. M., Iafelice, G., Lavini, A., Pulvento, C., Caboni, M. F., and Marconi, E. (2012). Phenolic compounds and saponins in quinoa samples (Chenopodium quinoa Willd.) grown under different saline and non-saline irrigation regimens. J. Agric. Food Chem. 60, 4620–4627. doi: 10.1021/jf3002125
Guo, S., Kenne, L., Lundgren, L., Ronnberg, B., and Sundquist, B. (1998). Triterpenoid saponins from Quillaja saponaria. Phytochemistry 48, 175–180.
Healey, A., Furtado, A., Cooper, T., and Henry, R. (2014). Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10:21. doi: 10.1186/1746-4811-10-21
Hong, S.-Y., Cheon, K.-S., Yoo, K.-O., Lee, H.-O., Cho, K.-S., Suh, J.-T., et al. (2017). Complete chloroplast genome sequences and comparative analysis of chenopodium quinoa and C. album. Front. Plant Sci. 8:e1696. doi: 10.3389/fpls.2017.01696
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Claude, W., Leebens-mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Luebert, F. (2014). Taxonomy and distribution of the genus Quillaja molina (Quillajaceae). Feddes Repert. 124, 157–162. doi: 10.1002/fedr.201400029
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Magee, A. M., Aspinall, S., Rice, D. W., Cusack, B. P., Sémon, M., Perry, A. S., et al. (2010). Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 20, 1700–1710. doi: 10.1101/gr.111955.110
Millen, R. S. (2001). Many parallel losses of infa from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell Online 13, 645–658. doi: 10.1105/tpc.13.3.645
Nomura, H., Komori, T., Uemura, S., Kanda, Y., Shimotani, K., Nakai, K., et al. (2012). Chloroplast-mediated activation of plant immune signalling in Arabidopsis. Nat. Commun. 3, 910–926. doi: 10.1038/ncomms1926
Olson, N. D., Lund, S. P., Colman, R. E., Foster, J. T., and Zook, J. M. (2015). Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front. Genet. 6:235. doi: 10.3389/fgene.2015.00235
Rogalski, M., do Nascimento Vieira, L., Fraga, H. P., and Guerra, M. P. (2015). Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 6:586. doi: 10.3389/fpls.2015.00586
Santelices, R., and Bobadilla, C. (1997). Arraigamiento de estacas de Quillaja sapolnaria Mol. y Peumus boldus Mol. Bosque 18, 77–85.
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., and Tabata, S. (1999). Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290.
Shin, B. K., Kwon, S. W., and Park, J. H. (2015). Chemical diversity of ginseng saponins from Panax ginseng. J. Ginseng Res. 39, 287–298. doi: 10.1016/j.jgr.2014.12.005
Simpson, J. T., Wong, K., Jackman, S. D., Durbin, R., Salzberg, S. L., Phillippy, A. M., et al. (2012). ABySS : a parallel assembler for short read sequence data structures ABySS : a parallel assembler for short read sequence data. Genome Res. 1117–1123. doi: 10.1101/gr.089532.108
Szakiel, A., Paczkowski, C., and Henry, M. (2011). Influence of environmental biotic factors on the content of saponins in plants. Phytochem. Rev. 10, 493–502. doi: 10.1007/s11101-010-9164-2
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Villagran, C., and Hinojosa, F. (1997). Historia de 10s bosques del sur de Sudamerica, 11 : analisis fitogeografico. History of the forests of southern South America, 11 : phytogeographical analysis. Rev. Chil. Hist. Nat. 70, 241–267.
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wommack, K. E., Bhavsar, J., and Ravel, J. (2008). Metagenomics: read length matters. Appl. Environ. Microbiol. 74, 1453–1463. doi: 10.1128/AEM.02181-07
Wyman, S., Jansen, R., and Boore, J. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Keywords: quillay, plastome, genome, saponins, chloroplasts
Citation: Vizoso P, Undurraga SF and Velozo J (2019) Chloroplast Genome of the Soap Bark Tree Quillaja saponaria. Front. Ecol. Evol. 7:104. doi: 10.3389/fevo.2019.00104
Received: 17 May 2018; Accepted: 15 March 2019;
Published: 05 April 2019.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Humira Sonah, Laval University, CanadaPerla Hamon, Institut de Recherche Pour le Développement (IRD), France
Copyright © 2019 Vizoso, Undurraga and Velozo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Velozo, anVhbi52ZWxvem9AdW1heW9yLmNs