 Hidetoshi Shimodaira
Hidetoshi Shimodaira Yoshikazu Terada
Yoshikazu Terada- 1Graduate School of Informatics, Kyoto University, Kyoto, Japan
- 2Mathematical Statistics Team, RIKEN Center for Advanced Intelligence Project, Tokyo, Japan
- 3Graduate School of Engineering Science, Osaka University, Osaka, Japan
Selective inference is considered for testing trees and edges in phylogenetic tree selection from molecular sequences. This improves the previously proposed approximately unbiased test by adjusting the selection bias when testing many trees and edges at the same time. The newly proposed selective inference p-value is useful for testing selected edges to claim that they are significantly supported if p > 1−α, whereas the non-selective p-value is still useful for testing candidate trees to claim that they are rejected if p < α. The selective p-value controls the type-I error conditioned on the selection event, whereas the non-selective p-value controls it unconditionally. The selective and non-selective approximately unbiased p-values are computed from two geometric quantities called signed distance and mean curvature of the region representing tree or edge of interest in the space of probability distributions. These two geometric quantities are estimated by fitting a model of scaling-law to the non-parametric multiscale bootstrap probabilities. Our general method is applicable to a wider class of problems; phylogenetic tree selection is an example of model selection, and it is interpreted as the variable selection of multiple regression, where each edge corresponds to each predictor. Our method is illustrated in a previously controversial phylogenetic analysis of human, rabbit and mouse.
1. Introduction
A phylogenetic tree is a diagram showing evolutionary relationships among species, and a tree topology is a graph obtained from the phylogentic tree by ignoring the branch lengths. The primary objective of any phylogenetic analysis is to approximate a topology that reflects the evolution history of the group of organisms under study. Branches of the tree are also referred to as edges in the tree topology. Given a rooted tree topology, or a unrooted tree topology with an outgroup, each edge splits the tree so that it defines the clade consisting of all the descendant species. Therefore, edges in a tree topology represent clades of species. Because the phylogenetic tree is commonly inferred from molecular sequences, it is crucial to assess the statistical confidence of the inference. In phylogenetics, it is a common practice to compute confidence levels for tree topologies and edges. For example, the bootstrap probability (Felsenstein, 1985) is the most commonly used confidence measure, and other methods such as the Shimodaira-Hasegawa test (Shimodaira and Hasegawa, 1999) and the multiscale bootstrap method (Shimodaira, 2002) are also often used. However, these conventional methods are limited in how well they address the issue of multiplicity when there are many alternative topologies and edges. Herein, we discuss a new approach, selective inference (SI), that is designed to address the issue of multiplicity.
For illustrating the idea of selective inference, we first look at a simple example of 1-dimensional normal random variable Z with unknown mean θ ∈ ℝ and variance 1:
Observing Z = z, we would like to test the null hypothesis H0 : θ ≤ 0 against the alternative hypothesis H1 : θ > 0. We denote the cumulative distribution function of N(0, 1) as Φ(x) and define the upper tail probability as . Then, the ordinary (i.e., non-selective) inference leads to the p-value of the one-tailed z-test as
What happens when we test many hypotheses at the same time? Consider random variables Zi ~ N(θi, 1), i = 1, …, Kall, not necessarily independent, with null hypotheses θi ≤ 0, where Ktrue hypotheses are actually true. To control the number of falsely rejecting the Ktrue hypotheses, there are several multiplicity adjusted approaches such as the family-wise error rate (FWER) and the false discovery rate (FDR). Instead of testing all the Kall hypotheses, selective inference (SI) allows for Kselect hypotheses with zi > ci for constants ci specified in advance. This kind of selection is very common in practice (e.g., publication bias), and it is called as the file drawer problem by Rosenthal (1979). Instead of controlling the multiplicity of testing, SI alleviates it by reducing the number of tests. The mathematical formulation of SI is easier than FWER and FDR in the sense that hypotheses can be considered separately instead of simultaneously. Therefore, we simply write z > c by dropping the index i for one of the hypotheses. In selective inference, the selection bias is adjusted by considering the conditional probability given the selection event, which leads to the following p-value (Fithian et al., 2014; Tian and Taylor, 2018)
where p(z) of Equation (2) is divided by the selection probability . In the case of c = 0, this corresponds to the two-tailed z-test, because the selection probability is and p(z, c) = 2p(z). For significance level α (we use α = 0.05 unless otherwise stated), it properly controls the type-I error conditioned on the selection event as P(p(Z, c) < α ∣ Z > c, θ = 0) = α, while the non-selective p-value violates the type-I error as . The selection bias can be very large when (i.e., c ≫ 0), or Kselect ≪ Kall.
Selective inference has been mostly developed for inferences after model selection (Taylor and Tibshirani, 2015; Tibshirani et al., 2016), particularly variable selection in regression settings such as lasso (Tibshirani, 1996). Recently, Terada and Shimodaira (2017) developed a general method for selective inference by adjusting the selection bias in the approximately unbiased (AU) p-value computed by the multiscale bootstrap method (Shimodaira, 2002, 2004, 2008). This new method can be used to compute, for example, confidence intervals of regression coefficients in lasso (Figure 1). In this paper, we apply this method to phylogenetic inference for computing proper confidence levels of tree topologies (dendrograms) and edges (clades or clusters) of species. As far as we know, this is the first attempt to consider selective inference in phylogenetics. Our selective inference method is implemented in software scaleboot (Shimodaira, 2019) working jointly with CONSEL (Shimodaira and Hasegawa, 2001) for phylogenetics, and it is also implemented in a new version of pvclust (Suzuki and Shimodaira, 2006) for hierarchical clustering, where only edges appeared in the observed tree are “selected” for computing p-values. Although our argument is based on the rigorous theory of mathematical statistics in Terada and Shimodaira (2017), a self-contained illustration is presented in this paper for the theory as well as the algorithm of selective inference.
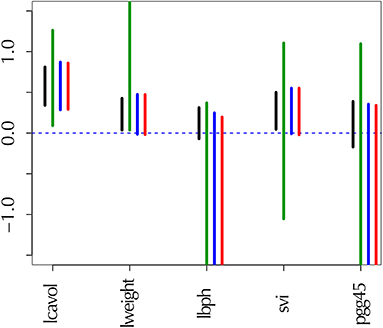
Figure 1. Confidence intervals of regression coefficients for selected variables by lasso; see section 6.8 for details. All intervals are computed for confidence level 1 − α at α = 0.01. (Black) the ordinary confidence interval . (Green) the selective confidence interval under the selected model. (Blue) the selective confidence interval under the selection event that variable j is selected. (Red) the multiscale bootstrap version of selective confidence interval under the selection event that variable j is selected.
Phylogenetic tree selection is an example of model selection. Since each tree can be specified as a combination of edges, tree selection can be interpreted as the variable selection of multiple regression, where edges correspond to the predictors of regression (Shimodaira, 2001; Shimodaira and Hasegawa, 2005). Because all candidate trees have the same number of model parameters, the maximum likelihood (ML) tree is obtained by comparing log-likelihood values of trees (Felsenstein, 1981). In order to adjust the model complexity by the number of parameters in general model selection, we compare Akaike Information Criterion (AIC) values of candidate models (Akaike, 1974). AIC is used in phylogenetics for selecting the substitution model (Posada and Buckley, 2004). There are several modifications of AIC that allow for model selection. These include the precise estimation of the complexity term known as Takeuchi Information Criterion (Burnham and Anderson, 2002; Konishi and Kitagawa, 2008), and adaptations for incomplete data (Shimodaira and Maeda, 2018) and covariate-shift data (Shimodaira, 2000). AIC and all these modifications are derived for estimating the expected Kullback-Leibler divergence between the unknown true distribution and the estimated probability distribution on the premise that the model is misspecified. When using regression model for prediction purpose, it may be sufficient to find only the best model which minimizes the AIC value. Considering random variations of dataset, however, it is obvious in phylogenetics that the ML tree does not necessarily represent the true history of evolution. Therefore, Kishino and Hasegawa (1989) proposed a statistical test whether two log-likelihood values differ significantly (also known as Kishino-Hasegawa test). The log-likelihood difference is often not significant, because its variance can be very large for non-nested models when the divergence between two probability distributions is large; see Equation (26) in section 6.1. The same idea of model selection test whether two AIC values differ significantly has been proposed independently in statistics (Linhart, 1988) and econometrics (Vuong, 1989). Another method of model selection test (Efron, 1984) allows for the comparison of two regression models with an adjusted bootstrap confidence interval corresponding to the AU p-value. For testing which model is better than the other, the null hypothesis in the model selection test is that the two models are equally good in terms of the expected value of AIC on the premise that both models are misspecified. Note that the null hypothesis is whether the model is correctly specified or not in the traditional hypothesis testing methods including the likelihood ratio test for nested models and the modified likelihood ratio test for non-nested models (Cox, 1962). The model selection test is very different from these traditional settings. For comparing AIC values of more than two models, a multiple comparisons method is introduced to the model selection test (Shimodaira, 1998; Shimodaira and Hasegawa, 1999), which computes the confidence set of models. But the multiple comparisons method is conservative by nature, leading to more false negatives than expected, because it considers the worst scenario, called the least favorable configuration. On the other hand, the model selection test (designed for two models) and bootstrap probability (Felsenstein, 1985) lead to more false positives than expected when comparing more than two models (Shimodaira and Hasegawa, 1999; Shimodaira, 2002). The AU p-value mentioned earlier has been developed for solving this problem, and we are going to upgrade it for selective inference.
2. Phylogenetic Inference
For illustrating phylogenetic inference methods, we analyze a dataset consisting of mitochondrial protein sequences of six mammalian species with n = 3, 414 amino acids (n is treated as sample size). The taxa are labeled as 1 = Homo sapiens (human), 2 = Phoca vitulina (seal), 3 = Bos taurus (cow), 4 = Oryctolagus cuniculus (rabbit), 5 = Mus musculus (mouse), and 6 = Didelphis virginiana (opossum). The dataset will be denoted as . The software package PAML (Yang, 1997) was used to calculate the site-wise log-likelihoods for trees. The mtREV model (Adachi and Hasegawa, 1996) was used for amino acid substitutions, and the site-heterogeneity was modeled by the discrete-gamma distribution (Yang, 1996). The dataset and evolutionary model are similar to previous publications (Shimodaira and Hasegawa, 1999; Shimodaira, 2001, 2002), thus allowing our proposed method to be easily compared with conventional methods.
The number of unrooted trees for six taxa is 105. These trees are reordered by their likelihood values and labeled as T1, T2, …, T105. T1 is the ML tree as shown in Figure 2 and its tree topology is represented as (((1(23))4)56). There are three internal branches (we call them as edges) in T1, which are labeled as E1, E2, and E3. For example, E1 splits the six taxa as {23|1456} and the partition of six taxa is represented as -++---, where +/- indicates taxa 1, …, 6 from left to right and ++ indicates the clade {23} (we set - for taxon 6, since it is treated as the outgroup). There are 25 edges in total, and each tree is specified by selecting three edges from them, although not all the combinations of three edges are allowed.
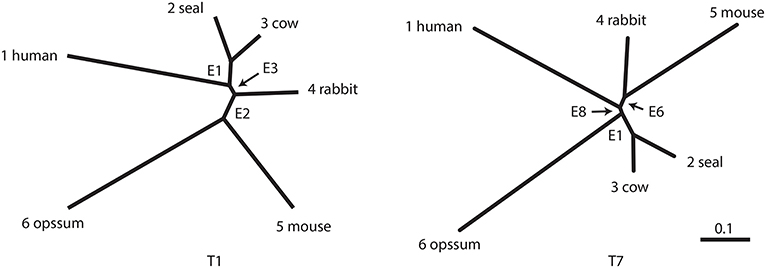
Figure 2. Examples of two unrooted trees T1 and T7. Branch lengths represent ML estimates of parameters (expected number of substitutions per site). T1 includes edges E1, E2, and E3 and T7 includes E1, E6, and E8.
The result of phylogenetic analysis is summarized in Table 1 for trees and Table 2 for edges. Three types of p-values are computed for each tree as well as for each edge. BP is the bootstrap probability (Felsenstein, 1985) and AU is the approximately unbiased p-value (Shimodaira, 2002). Bootstrap probabilities are computed by the non-parametric bootstrap resampling (Efron, 1979) described in section 6.1. The theory and the algorithm of BP and AU will be reviewed in section 3. Since we are testing many trees and edges at the same time, there is potentially a danger of selection bias. The issue of selection bias has been discussed in Shimodaira and Hasegawa (1999) for introducing the method of multiple comparisons of log-likelihoods (also known as Shimodaira-Hasegawa test) and in Shimodaira (2002) for introducing AU test. However, these conventional methods are only taking care of the multiplicity of comparing many log-likelihood values for computing just one p-value instead of many p-values at the same time. Therefore, we intend to further adjust the AU p-value by introducing the selective inference p-value, denoted as SI. The theory and the algorithm of SI will be explained in section 4 based on the geometric theory given in section 3. After presenting the methods, we will revisit the phyloegnetic inference in section 4.3.
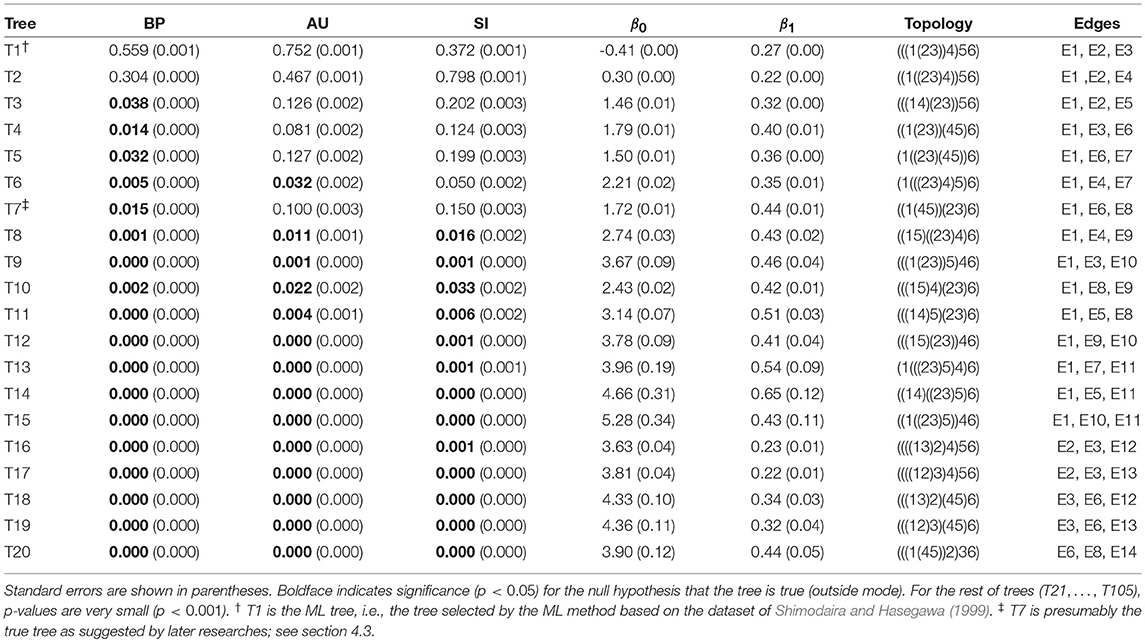
Table 1. Three types of p-values (BP, AU, SI) and geometric quantities (β0, β1) for the best 20 trees.
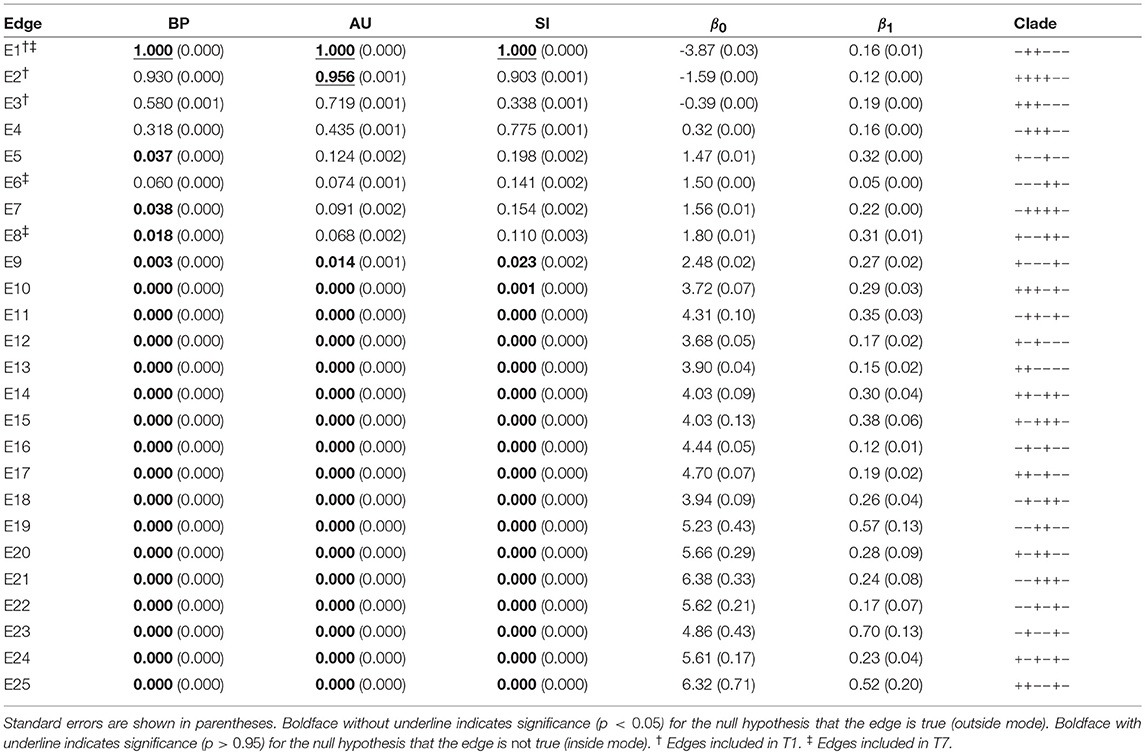
Table 2. Three types of p-values (BP, AU, SI) and geometric quantities (β0, β1) for all the 25 edges of six taxa.
For developing the geometric theory in sections 3 and 4, we formulate tree selection as a mathematical formulation known as the problem of regions (Efron et al., 1996; Efron and Tibshirani, 1998). For better understanding the geometric nature of the theory, the problem of regions is explained below for phylogenetic inference, although the algorithm is simple enough to be implemented without understanding the theory. Considering the space of probability distributions (Amari and Nagaoka, 2007), the parametric models for trees are represented as manifolds in the space. The dataset (or the empirical distribution) can also be represented as a “data point” X in the space, and the ML estimates for trees are represented as projections to the manifolds. This is illustrated in the visualization of probability distributions of Figure 3A using log-likelihood vectors of models (Shimodaira, 2001), where models are simply indicated as red lines from the origin; see section 6.2 for details. This visualization may be called as model map. The point X is actually reconstructed as the minimum full model containing all the trees as submodels, and the Kullback-Leibler divergence between probability distributions is represented as the squared distance between points; see Equation (27). Computation of X is analogous to the Bayesian model averaging, but based on the ML method. For each tree, we can think of a region in the space so that this tree becomes the ML tree when X is included in the region. The regions for T1, T2, and T3 are illustrated in Figure 3B, and the region for E2 is the union of these three regions.
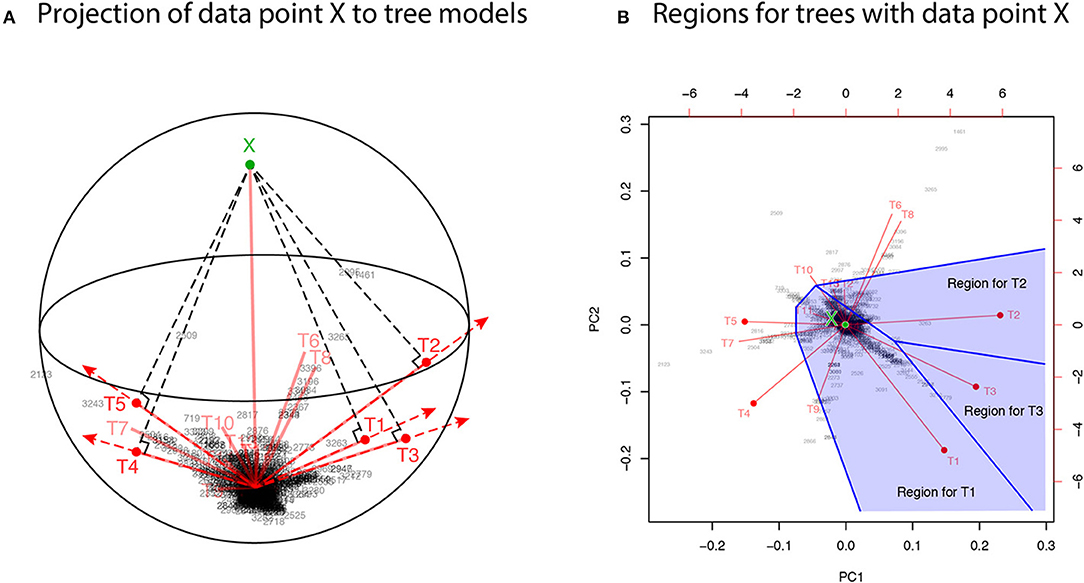
Figure 3. Model map: Visualization of ML estimates of probability distributions for the best 15 trees. The origin represents the star-shaped tree topology (obtained by reducing the internal branches to zero length). Sites of amino acid sequences t = 1, …, n (black numbers) and probability distributions for trees T1, …, T15 (red segments) are drawn by biplot of PCA. Auxiliary lines are drawn by hand. (A) 3-dimensional visualization using PC1, PC2, and PC3. The reconstructed data point X is also shown (green point). The ML estimates are represented as the end points of the red segments (shown by red points only for the best five trees), and they are placed on the sphere with the origin and X being placed at the poles. (B) The top-view of model map. Regions for the best three trees Ti, i = 1, 2, 3 (blue shaded regions) are illustrated; Ti will be the ML tree if X is included in the region for Ti.
In Figure 3A, X is very far from any of the tree models, suggesting that all the models are wrong; the likelihood ratio statistic for testing T1 against the full model is 113.4, which is highly significant as (Shimodaira, 2001, section 5). Instead of testing whether tree models are correct or not, we test whether models are significantly better than the others. As seen in Figure 3B, X is in the region for T1, meaning that the model for T1 is better than those for the other trees. For convenience, observing X in the region for T1, we state that T1 is supported by the data. Similarly, X is in the region for E2 that consists of the three regions for T1, T2, T3, thus indicating that E2 is supported by the data. Although T1 and E2 are supported by the data, there is still uncertainty as to whether the true evolutionary history of lineages is depicted because the location of X fluctuates randomly. Therefore, statistical confidence of the outcome needs to be assessed. A mathematical procedure for statistically evaluating the outcome is provided in the following sections.
3. Non-Selective Inference for the Problem of Regions
3.1. The Problem of Regions
For developing the theory, we consider (m + 1)-dimensional multivariate normal random vector Y, m ≥ 0, with unknown mean vector μ ∈ ℝm+1 and the identity variance matrix Im+1:
A region of interest such as tree and edge is denoted as , and its complement set is denoted as . There are Kall regions , i = 1, …, Kall, and we simply write for one of them by dropping the index i. Observing Y = y, the null hypothesis is tested against the alternative hypothesis . This setting is called problem of regions, and the geometric theory for non-selective inference for slightly generalized settings (e.g., exponential family of distributions) has been discussed in Efron and Tibshirani (1998) and Shimodaira (2004). This theory allows arbitrary shape of without assuming a particular shape such as half-space or sphere, and only requires the expression (29) of section 6.3.
The problem of regions is well described by geometric quantities (Figure 4). Let be the projection of y to the boundary surface defined as
and β0 be the signed distance defined as for and for ; see Figures 4A,B, respectively. A large β0 indicates the evidence for rejecting , but computation of p-value will also depend on the shape of . There should be many parameters for defining the shape, but we only need the mean curvature of at , which represents the amount of surface bending. It is denoted as β1 ∈ ℝ, and defined in (30).
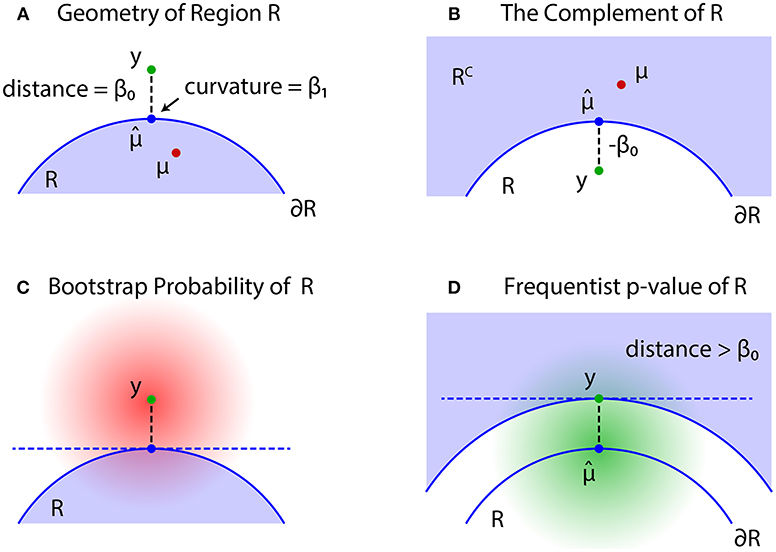
Figure 4. Problem of regions. (A) β0 > 0 when , then select the null hypothesis . (B) β0 ≤ 0 when , then select the null hypothesis . (C) The bootstrap distribution of (red shaded distribution). (D) The null distribution of (green shaded distribution).
Geometric quantities β0 and β1 of regions for trees (T1, …, T105) and edges (E1, …, E25) are plotted in Figure 5, and these values are also found in Tables 1, 2. Although the phylogenetic model of evolution for the molecular dataset is different from the multivariate normal model (4) for y, the multiscale bootstrap method of section 3.4 estimates β0 and β1 using the non-parametric bootstrap probabilities (section 6.1) with bootstrap replicates for several values of sample size n′.
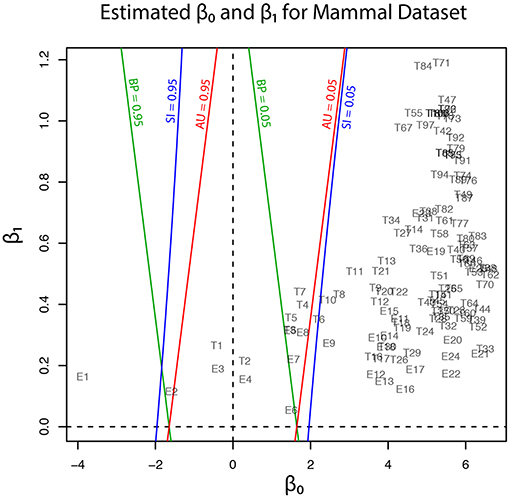
Figure 5. Geometric quantities of regions (β0 and β1) for trees and edges are estimated by the multiscale bootstrap method (section 3.4). The three types of p-value (BP, AU, SI) are computed from β0 and β1, and their contour lines are drawn at p = 0.05 and 0.95.
3.2. Bootstrap Probability
For simulating (4) from y, we may generate replicates Y* from the bootstrap distribution (Figure 4C)
and define bootstrap probability (BP) of as the probability of Y* being included in the region :
can be interpreted as the Bayesian posterior probability , because, by assuming the flat prior distribution π(μ) = constant, the posterior distribution μ|y ~ Nm+1(y, Im + 1) is identical to the distribution of Y* in (5). An interesting consequence of the geometric theory of Efron and Tibshirani (1998) is that BP can be expressed as
where ≃ indicates the second order asymptotic accuracy, meaning that the equality is correct up to with error of order ; see section 6.3.
For understanding the formula (7), assume that is a half space so that is flat and β1 = 0. Since we only have to look at the axis orthogonal to , the distribution of signed distance is identified as (1) with β0 = z. The bootstrap distribution for (1) is Z* ~ N(z, 1), and bootstrap probability is expressed as . Therefore, we have . For general with curved , the formula (7) adjusts the bias caused by β1. As seen in Figure 4C, becomes smaller for β1 > 0 than β1 = 0, and BP becomes smaller.
BP of is closely related to BP of . From the definition,
The last expression also implies that the signed distance and the mean curvature of is −β0 and −β1, respectively; this relation is also obtained by reversing the sign of v in (29).
3.3. Approximately Unbiased Test
Although may work as a Bayesian confidence measure, we would like to have a frequentist confidence measure for testing against . The signed distance of Y is denoted as β0(Y), and consider the region {Y ∣ β0(Y) > β0} in which the signed distance is larger than the observed value β0 = β0(y). Similar to (2), we then define an approximately unbiased (AU) p-value as
where the probability is calculated for as illustrated in Figure 4D. The shape of the region {Y ∣ β0(Y) > β0} is very similar to the shape of ; the difference is in fact only . Let us think of a point y′ with signed distance −β0 (shown as y in Figure 4B). Then we have
where the last expression is obtained by substituting (−β0, β1) for (β0, β1) in (8). This formula computes AU from (β0, β1). An intuitive interpretation of (10) is explained in section 6.4.
In non-selective inference, p-values are computed using formula (10). If , the null hypothesis is rejected and the alternative hypothesis is accepted. This test procedure is approximately unbiased, because it controls the non-selective type-I error as
and the rejection probability increases as μ moves away from , while it decreases as μ moves into .
Exchanging the roles of and also allows for another hypothesis testing. AU of is obtained from (9) by reversing the inequality as . This is also confirmed by substituting (−β0, −β1), i.e., the geometric quantities of , for (β0, β1) in (10) as
If or equivalently , then we reject and accept .
3.4. Multiscale Bootstrap
In order to estimate β0 and β1 from bootstrap probabilities, we consider a generalization of (5) as
for a variance σ2 > 0, and define multiscale bootstrap probability of as
where indicates the probability with respect to (13).
Although our theory is based on the multivariate normal model, the actual implementation of the algorithm uses the non-parametric bootstrap probabilities in section 6.1. To fill the gap between the two models, we consider a non-linear transformation fn so that the multivariate normal model holds at least approximately for and . An example of fn is given in (25) for phylogenetic inference. Surprisingly, a specification of fn is not required for computing p-values, but we simply assume the existence of such a transformation; this property may be called as “bootstrap trick.” For phylogenetic inference, we compute the non-parametric bootstrap probabilities by (24) and substitute these values for (14) with σ2 = n/n′.
For estimating β0 and β1, we need to have a scaling law which explains how depends on the scale σ. We rescale (13) by multiplying σ−1 so that has the variance σ2 = 1. y and are now resaled by the factor σ−1, which amounts to signed distance and mean curvature β1σ (Shimodaira, 2004). Therefore, by substituting for (β0, β1) in (7), we obtain
For better illustrating how depends on σ2, we define
We can estimate β0 and β1 as regression coefficients by fitting the linear model (16) in terms of σ2 to the observed values of non-parametric bootstrap probabilities (Figure 6). Interestingly, (10) is rewritten as by formally letting σ2 = −1 in the last expression of (16), meaning that AU corresponds to n′ = −n. Although σ2 should be positive in (15), we can think of negative σ2 in . See section 6.5 for details of model fitting and extrapolation to negative σ2.
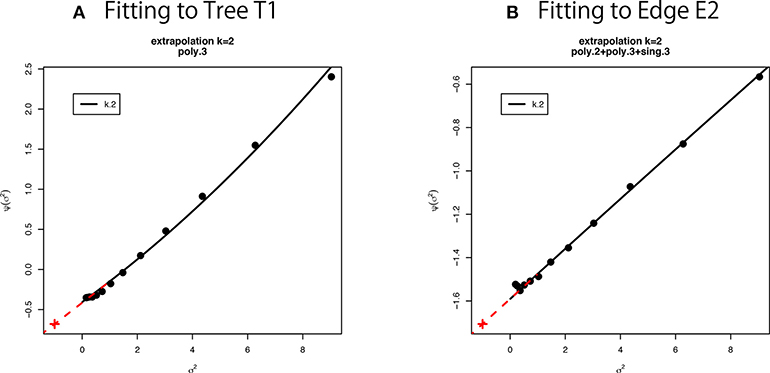
Figure 6. Multiscale bootstrap for (A) tree T1 and (B) edge E2. is computed by the non-parametric bootstrap probabilities for several σ2 = n/n′ values, then β0 and β1 are estimated as the intercept and the slope, respectively. See section 6.5 for details.
4. Selective Inference for the Problem of Regions
4.1. Approximately Unbiased Test for Selective Inference
In order to argue selective inference for the problem of regions, we have to specify the selection event. Let us consider a selective region so that we perform the hypothesis testing only when . Terada and Shimodaira (2017) considered a general shape of , but here we treat only two special cases of and ; see section 6.6. Our problem is formulated as follows. Observing Y = y from the multivariate normal model (4), we first check whether or . If and we are interested in the null hypothesis , then we may test it against the alternative hypothesis . If and we are interested in the null hypothesis , then we may test it against the alternative hypothesis . In this paper, the former case (, and so β0 > 0) is called as outside mode, and the latter case (, and so β0 ≤ 0) is called as inside mode. We do not know which of the two modes of testing is performed until we observe y.
Let us consider the outside mode by assuming that , where β0 > 0. Recalling that in section 1, we divide by the selection probability to define a selective inference p-value as
From the definition, , because for β0 > 0. This p-value is computed from (β0, β1) by
where is obtained by substituting (0, β1) for (β0, β1) in (8). An intuitive justification of (18) is explained in section 6.4.
For the outside mode of selective inference, p-values are computed using formula (18). If , then reject and accept . This test procedure is approximately unbiased, because it controls the selective type-I error as
and the rejection probability increases as μ moves away from , while it decreases as μ moves into .
Now we consider the inside mode by assuming that , where β0 ≤ 0. SI of is obtained from (17) by exchanging the roles of and .
For the inside mode of selective inference, p-values are computed using formula (20). If , then reject and accept . Unlike the non-selective p-value , is not equivalent to , because . For convenience, we define
so that SI′ > 1 − α implies . In our numerical examples of Figure 5, Tables 1, 2, SI′ is simply denoted as SI. We do not need to consider (21) for BP and AU, because and from (8) and (12).
4.2. Shortcut Computation of SI
We can compute SI from BP and AU. This will be useful for reanalyzing the results of previously published researches. Let us write and . From (7) and (10), we have
We can compute SI from β0 and β1 by (18) or (20). More directly, we may compute
4.3. Revisiting the Phylogenetic Inference
In this section, the analytical procedure outlined in section 2 is used to determine relationships among human, mouse, and rabbit. The question is: Which of mouse or human is closer to rabbit? The traditional view (Novacek, 1992) is actually supporting E6, the clade of rabbit and mouse, which is consistent with T4, T5, and T7. Based on molecular analysis, Graur et al. (1996) strongly suggested that rabbit is closer to human than mouse, thus supporting E2, which is consistent with T1, T2, and T3. However, Halanych (1998) criticized it by pointing out that E2 is an artifact caused by the long branch attraction (LBA) between mouse and opossum. In addition, Shimodaira and Hasegawa (1999) and Shimodaira (2002) suggested that T7 is not rejected by multiplicity adjusted tests. Shimodaira and Hasegawa (2005) showed that T7 becomes the ML tree by resolving the LBA using a larger dataset with more taxa. Although T1 is the ML tree based on the dataset with fewer taxa, T7 is presumably the true tree as indicated by later researches. With these observations in mind, we retrospectively interpret p-values in Tables 1, 2.
The results are shown below for the two test modes (inside and outside) as defined in section 4.1. The extent of multiplicity and selection bias depends on the number of regions under consideration, thus these numbers are considered for interpreting the results. The numbers of regions related to trees and edges are summarized in Table 3; see section 6.7 for details.
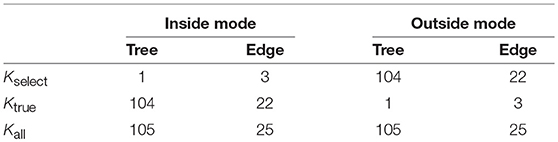
Table 3. The number of regions for trees and edges. The number of taxa is N = 6.
In inside mode, the null hypothesis is tested against the alternative hypothesis for (i.e., β0 ≤ 0). This applies to the regions for T1, E1, E2, and E3, and they are supported by the data in the sense mentioned in the last paragraph of section 2. When H0 is rejected by a test procedure, it is claimed that is significantly supported by the data, indicating H1 holds true. For convenience, the null hypothesis H0 is said like E1 is not true, and the alternative hypothesis H1 is said like E1 is true; then rejection of H0 implies that E1 is true. This procedure looks unusual, but makes sense when both and are regions with nonzero volume. Note that selection bias can be very large in the sense that Kselect/Kall ≈ 0 for many taxa, and non-selective tests may lead to many false positives because Ktrue/Kall ≈ 1. Therefore selective inference should be used in inside mode.
In outside mode, the null hypothesis is tested against the alternative hypothesis for (i.e., β0 > 0). This applies to the regions for T2, …, T105, and E4, …, E25, and they are not supported by the data. When H0 is rejected by a test procedure, it is claimed that is rejected. For convenience, the null hypothesis is said like T9 is true, and the alternative hypothesis is said like T9 is not true; rejection of H0 implies that T9 is not true. This is more or less a typical test procedure. Note that selection bias is minor in the sense that Kselect/Kall ≈ 1 for many taxa, and non-selective tests may result in few false positives because Ktrue/Kall ≈ 0. Therefore selective inference is not much beneficial in outside mode.
In addition to p-values for some trees and edges, estimated geometric quantities are also shown in the tables. We confirm that the sign of β0 is estimated correctly for all the trees and edges. The estimated β1 values are all positive, indicating the regions are convex. This is not surprising, because the regions are expressed as intersections of half spaces at least locally (Figure 3B).
Now p-values are examined in inside mode. (T1, E3) BP, AU, SI are all p ≤ 0.95. This indicates that T1 and E3 are not significantly supported. There are nothing claimed to be definite. (E1) BP, AU, SI are all p > 0.95, indicating E1 is significantly supported. Since E1 is associated with the best 15 trees T1, …, T15, some of them are significantly better than the rest of trees T16, …, T105. Significance for edges is common in phylogenetics as well as in hierarchical clustering (Suzuki and Shimodaira, 2006). (E2) The results split for this presumably wrong edge. AU > 0.95 suggests E2 is significantly supported, whereas BP, SI ≤ 0.95 are not significant. AU tends to violate the selective type-I error, leading to false positives or overconfidence in wrong trees/edges, whereas SI is approximately unbiased for the selected hypothesis. This overconfidence is explained by the inequality AU > SI (meant SI′ here) for , which is obtained by comparing (12) and (20). Therefore SI is preferable to AU in inside mode. BP is safer than AU in the sense that BP < AU for β1 > 0, but BP is not guaranteed for controlling type-I error in a frequentist sense. The two inequalities (SI, BP < AU) are verified as relative positions of the contour lines at p = 0.95 in Figure 5. The three p-values can be very different from each other for large β1.
Next p-values are examined in outside mode. (T2, E4, E6) BP, AU, SI are all p ≥ 0.05. They are not rejected, and there are nothing claimed to be definite. (T8, T9, …, T105, E9,…, E25) BP, AU, SI are all p < 0.05. These trees and edges are rejected. (T7, E8) The results split for these presumably true tree and edge. BP < 0.05 suggests T7 and E8 are rejected, whereas AU, SI ≥ 0.05 are not significant. AU is approximately unbiased for controlling the type-I error when H0 is specified in advance (Shimodaira, 2002). Since BP < AU for β1 > 0, BP violates the type-I error, which results in overconfidence in non-rejected wrong trees. Therefore BP should be avoided in outside mode. Inequality AU < SI can be shown for by comparing (10) and (18). Since the null hypothesis is chosen after looking at , AU is not approximately unbiased for controlling the selective type-I error, whereas SI adjusts this selection bias. The two inequalities (BP < AU < SI) are verified as relative positions of the contour lines at p = 0.05 in Figure 5. AU and SI behave similarly (Note: Kselect/Kall ≈ 1), while BP is very different from AU and SI for large β1. It is arguable which of AU and SI is appropriate: AU is preferable to SI in tree selection (Ktrue = 1), because the multiplicity of testing is controlled as . The FWER is multiplied by Ktrue ≥ 1 for edge selection, and SI does not fix it either. For testing edges in outside mode, AU may be used for screening purpose with a small α value such as α/Ktrue.
5. Conclusion
We have developed a new method for computing selective inference p-values from multiscale bootstrap probabilities, and applied this new method to phylogenetics. It is demonstrated through theory and a real-data analysis that selective inference p-values are in particular useful for testing selected edges (i.e., clades or clusters of species) to claim that they are supported significantly if p > 1 − α. On the other hand, the previously proposed non-selective version of approximately unbiased p-values are still useful for testing candidate trees to claim that they are rejected if p < α. Although we focused on phylogenetics, our general theory of selective inference may be applied to other model selection problems, or more general selection problems.
6. Remarks
6.1. Bootstrap Resampling of Log-Likelihoods
Non-parametric bootstrap is often time consuming for recomputing the maximum likelihood (ML) estimates for bootstrap replicates. Kishino et al. (1990) considered the resampling of estimated log-likelihoods (RELL) method for reducing the computation. Let be the dataset of sample size n, where xt is the site-pattern of amino acids at site t for t = 1, …, n. By resampling xt from with replacement, we obtain a bootstrap replicate of sample size n′. Although n′ = n for the ordinary bootstrap, we will use several n′ > 0 values for the multiscale bootstrap. The parametric model of probability distribution for tree Ti is pi(x; θi) for i = 1, …, 105, and the log-likelihood function is . Computation of the ML estimate is time consuming, so we do not recalculate for bootstrap replicates. Define the site-wise log-likelihood at site t for tree Ti as
so that the log-likelihood value for tree Ti is written as . The bootstrap replicate of the log-likelihood value is approximated as
where is the number of times xt appears in . The accuracy of this approximation as well as the higher-order term is given in Equations (4) and (5) of Shimodaira (2001). Once , i = 1, …, 105, are computed by (23), its ML tree is T* with .
The non-parametric bootstrap probability of tree Ti is obtained as follows. We generate B bootstrap replicates , b = 1, …, B. In this paper, we used B = 105. For each , the ML tree T*b is computed by the method described above. Then we count the frequency that Ti becomes the ML tree in the B replicates. The non-parametric bootstrap probability of tree Ti is computed by
The non-parametric bootstrap probability of a edge is computed by summing BP(Ti, n′) over the associated trees.
An example of the transformation mentioned in section 3.4 is
where with and Vn is the variance matrix of . According to the approximation (23) and the central limit theorem, (13) holds well for sufficiently large n and n′ with m = 104 and σ2 = n/n′. It also follows from the above argument that , and thus the variance of log-likelihood difference is
which gives another insight into the visualization of section 6.2, where the variance can be interpreted as the divergence between the two models; see Equation (27). This approximation holds well when the two predictive distributions , are not very close to each other. When they are close to each other, however, the higher-order term ignored in (26) becomes dominant, and there is a difficulty for deriving the limiting distribution of the log-likelihood difference in the model selection test (Shimodaira, 1997; Schennach and Wilhelm, 2017).
6.2. Visualization of Probability Models
For representing the probability distribution of tree Ti, we define from (22) for i = 1, …, 15. The idea behind the visualization of Figure 3 is that locations of ξi in ℝn will represent locations of in the space of probability distributions. Let DKL(pi||pj) be the Kullback-Leibler divergence between the two distributions. For sufficiently small , the squared distance in ℝn approximates n times Jeffreys divergence
for non-nested models (Shimodaira, 2001, section 6). When a model p0 is nested in pi, it becomes . We explain three different visualizations of Figure 7. There are only minor differences between the plots, and the visualization is not sensitive to the details.
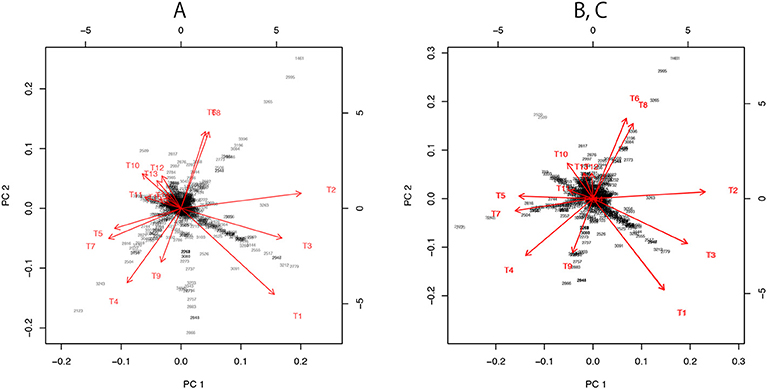
Figure 7. Three versions the visualization of probability distributions for the best 15 trees drawn using different sets of models. (A) Only the 15 bifurcating trees. (B) 15 bifurcating trees + 10 partially resolved trees + 1 star topology. This is the same plot as Figure 3B. (C) 15 bifurcating trees + 1 star topology. Note that (B,C) are superimposed, since their plots are almost indistinguishable.
For dimensionality reduction, we have to specify the origin c ∈ ℝn and consider vectors ai: = ξi−c. A naive choice would be the average . By applying PCA without centering and scaling (e.g., prcomp with option center=FALSE, scale=FALSE in R) to the matrix (a1, …, a15), we obtain the visualization of ξi as the axes (red arrows) of biplot in Figure 7A.
For computing the “data point” X in Figure 3, we need more models. Let tree T106 be the star topology with no internal branch (completely unresolved tree), and T107, …, T131 be partially resolved tree topologies with only one internal branch corresponding to E1, …, E25, whereas T1, …, T105 are fully resolved trees (bifurcating trees). Then define ηi: = ξ106+i, i = 0, …, 25. Now we take c = η0 for computing ai = ξi − η0 and bi = ηi − η0. There is hierarchy of models: η0 is the submodel nested in all the other models, and η1, η2, η3, for example, are submodels of ξ1 (T1 includes E1, E2, E3). By combining these non-nested models, we can reconstruct a comprehensive model in which all the other models are nested as submodels (Shimodaira, 2001, Equation 10 in section 5). The idea is analogous to reconstructing the full model y = β1x1 + ··· + β25x25 + ϵ of multiple regression from submodels y = β1x1 + ϵ, …, y = β25x25 + ϵ. Thus we call it as “full model” in this paper, and the ML estimate of the full model is indicated as the data point X; it is also said “super model” in Shimodaira and Hasegawa (2005). Let and , then the vector for the full model is computed approximately by
For the visualization of the best 15 trees, we may use only b1, …, b11, because they include E1 and two more edges from E2, …, E11. In Figures 3, 7B, we actually modified the above computation slightly so that the star topology T106 is replaced by T107, the partially resolved tree corresponding to E1 (T107 is also said star topology by treating clade (23) as a leaf of the tree), and the 10 partially resolved trees for E2, …, E11 are replaced by those for (E1,E2), …, (E1,E11), respectively; the origin becomes the maximal model nested in all the 15 trees, and X becomes the minimal full model containing all the 15 trees. Just before applying PCA in Figure 7B, a1, …, a15 are projected to the space orthogonal to aX, so that the plot becomes the “top-view” of Figure 3A with aX being at the origin.
In Figure 7C, we attempted a even simpler computation without using ML estimates for partially resolved trees. We used B = (a1, …, a15) and , and taking the largest 10 singular values for computing the inverse in (28). The orthogonal projection to aX is applied before PCA.
6.3. Asymptotic Theory of Smooth Surfaces
For expressing the shape of the region , we use a local coordinate system (u, v) ∈ ℝm + 1 with u ∈ ℝm, v ∈ ℝ. In a neighborhood of y, the region is expressed as
where h is a smooth function; see Shimodaira (2008) for the theory of non-smooth surfaces. The boundary surface is expressed as v = −h(u), u ∈ ℝm. We can choose the coordinates so that y = (0, β0) (i.e., u = (0, …, 0) and v = β0), and h(0) = 0, ∂h/∂ui|0 = 0, i = 1, …, m. The projection now becomes the origin , and the signed distance is β0. The mean curvature of surface at is now defined as
which is interpreted as the trace of the hessian matrix of h. When is convex at least locally in the neighborhood, all the eigenvalues of the hessian are non-negative, leading to β1 ≥ 0, whereas concave leads to β1 ≤ 0. In particular, β1 = 0 when is flat (i.e., h(u) ≡ 0).
Since the transformation depends on n, the shape of the region actually depends on n, although the dependency is implicit in the notation. As n goes larger, the standard deviation of estimates, in general, reduces at the rate n−1/2. For keeping the variance constant in (4), we actually magnifying the space by the factor n1/2, meaning that the boundary surface approaches flat as n → ∞. More specifically, the magnitude of mean curvature is of order . The magnitude of and higher order derivatives is , and we ignore these terms in our asymptotic theory. For keeping μ = O(1) in (4), we also consider the setting of “local alternatives,” meaning that the parameter values approach a origin on the boundary at the rate n−1/2.
6.4. Bridging the Problem of Regions to the Z-Test
Here we explain the problem of regions in terms of the z-test by bridging the multivariate problem of section 3 to the 1-dimensional case of section 1.
Ideal p-values are uniformly distributed over p ∈ (0, 1) when the null hypothesis holds. In fact, for as indicated in (11). The statistic may be called pivotal in the sense that the distribution does not change when moves on the surface. Here we ignore the error of , and consider only the second order asymptotic accuracy. From (10), we can write , where the notation such as β0(Y) and β1(Y) indicates the dependency on Y. Since β1(Y) ≃ β1(y) = β1, we treat β1(Y) as a constant. Now we get the normal pivotal quantity (Efron, 1985) as for . More generally, it becomes
Let us look at the z-test in section 1, and consider substitutions:
The 1-dimensional model (1) is now equivalent to (31). The null hypothesis is also equivalent: . We can easily verify that AU corresponds to p(z), because , which is expected from the way we obtained (31) above. Furthermore, we can derive SI from p(z, c). First verify that the selection event is equivalent: . Finally, we obtain SI as .
6.5. Model Fitting in Multiscale Bootstrap
We have used thirteen σ2 values from 1/9 to 9 (equally spaced in log-scale). This range is relatively large, and we observe a slight deviation from the linear model in Figure 6. Therefore we fit other models to the observed values of as implemented in scaleboot package (Shimodaira, 2008). For example, poly.k model is , and sing.3 model is . In Figure 6A, poly.3 is the best model according to AIC (Akaike, 1974). In Figure 6B, poly.2, poly.3, and sing.3 are combined by model averaging with Akaike weights. Then β0 and β1 are estimated from the tangent line to the fitted curve of at σ2 = 1. In Figure 6, the tangent line is drawn as red line for extrapolating to σ2 = −1. Shimodaira (2008) and Terada and Shimodaira (2017) considered the Taylor expansion of at σ2 = 1 as a generalization of the tangent line for improving the accuracy of AU and SI.
In the implementation of CONSEL (Shimodaira and Hasegawa, 2001) and pvclust (Suzuki and Shimodaira, 2006), we use a narrower range of σ2 values (ten σ−2 values: 0.5, 0.6, …, 1.4). Only the linear model is fitted there. The estimated β0 and β1 should be very close to those estimated from the tangent line described above. An advantage of using wider range of σ2 in scaleboot is that the standard error of β0 and β1 will become smaller.
6.6. General Formula of Selective Inference
Let be regions for the null hypothesis and the selection event, respectively. We would like to test the null hypothesis against the alternative conditioned on the selection event . We have considered the outside mode in (18) and the inside mode in (20). For a general case of , Terada and Shimodaira (2017) gave a formula of approximately unbiased p-value of selective inference as
where geometric quantities β0, β1 are defined for the regions . We assumed that and are expressed as (29), and two surfaces are nearly parallel to each other with tangent planes differing only . The last assumption always holds for (18), because and are identical and of course parallel to each other.
Here we explain why we have considered the special case of for phylogenetic inference. First, we suppose that the selection event satisfies , because a reasonable test would not reject H0 unless . Note that implies . Therefore, leads to
where is obtained from (33) by letting for . The p-value becomes smaller as grows, and gives the smallest p-value, leading to the most powerful selective test. Therefore the choice is preferable to any other choice of selection event satisfying . This kind of property is mentioned in Fithian et al. (2014) as the monotonicity of selective error in the context of “data curving.”
Let us see how these two p-values differ for the case of E2 by specifying and . In this case, the two surfaces may not be very parallel to each other, thus violating the assumption of , so we only intend to show the potential difference between the two p-values. The geometric quantities are , , ; the p-values are calculated using more decimal places than shown. SI of E2 conditioned on selecting T1 is
and it is very different from SI of E2 conditioned on selecting E2
where is shown in Table 2. As you see, is easier to reject H0 than .
6.7. Number of Regions for Phylogenetic Inference
The regions , i = 1, …, Kall correspond to trees or edges. In inside and outside modes, the number of total regions is Kall = 105 for trees and Kall = 25 for edges when the number of taxa is N = 6. For general N ≥ 3, they grow rapidly as for trees and for edges. Next consider the number of selected regions Kselect. In inside mode, regions with are selected, and the number is counted as Kselect = 1 for trees and Kselect = N − 3 = 3 for edges. In outside mode, regions with are selected, and thus the number is Kall minus that for inside mode; Kselect = Kall − 1 = 104 for trees and Kselect = Kall − (N − 3) = 22 for edges. Finally, consider the number of true null hypotheses, denoted as Ktrue. The null hypothesis holds true when in inside mode and in outside mode, and thus Ktrue is the same as the number of regions with in inside mode and in outside mode (These numbers do not depend on the value of y by ignoring the case of ). Therefore, Ktrue = Kall − Kselect for both cases.
6.8. Selective Inference of Lasso Regression
Selective inference is considered for the variable selection of regression analysis. Here, we deal with prostate cancer data (Stamey et al., 1989) in which we predict the level of prostate-specific antigen (PSA) from clinical measures. The dataset is available in the R package ElemStatLearn (Halvorsen, 2015). We consider a linear model to the log of PSA (lpsa), with 8 predictors such as the log prostate weight (lweight), age, and so on. All the variables are standardized to have zero mean and unit variance.
The goal is to provide the valid selective inference for the partial regression coefficients of the selected variables by lasso (Tibshirani, 1996). Let n and p be the number of observations and the number of predictors. is the set of selected variables, and represents the signs of the selected regression coefficients. We suppose that regression responses are distributed as where μ ∈ ℝn and τ > 0. Let ei be the ith residual. Resampling the scaled residuals σei (i = 1, …, n) with several values of scale σ2, we can apply the multiscale bootstrap method described in section 4 for the selective inference in the regression problem. Here, we note that the target of the inference is the true partial regression coefficients:
where X ∈ ℝn×p is the design matrix. We compute four types of intervals with confidence level 1 − α = 0.95 for selected variable j. is the non-selective confidence interval obtained via t-distribution. is the selective confidence interval under the selected model proposed by Lee et al. (2016) and Tibshirani et al. (2016), which is computed by fixedLassoInf with type=“full” in R package selectiveInference (Tibshirani et al., 2017). By extending the method of , we also computed , which is the selective confidence interval under the selection event that variable j is selected. These three confidence intervals are exact, in the sense that
Note that the selection event of variable j, i.e., can be represented as a union of polyhedra on ℝn, and thus, according to the polyhedral lemma (Lee et al., 2016; Tibshirani et al., 2016), we can compute a valid confidence interval . However, this computation is prohibitive for p > 10, because all the possible combinations of models with variable j are considered. Therefore, we compute its approximation by the multiscale bootstrap method of section 4 with much faster computation even for larger p.
We set λ = 10 as the penalty parameter of lasso, and the following model and signs were selected:
The confidence intervals are shown in Figure 1. For adjusting the selection bias, the three confidence intervals of selective inference are longer than the ordinary confidence interval. Comparing and , the latter is shorter, and would be preferable. This is because the selection event of the latter is less restrictive as ; see section 6.6 for the reason why larger selection event is better. Finally, we verify that approximates very well.
Author Contributions
HS and YT developed the theory of selective inference. HS programmed the multiscale bootstrap software and conducted the phylogenetic analysis. YT conducted the lasso analysis. HS wrote the manuscript. All authors have approved the final version of the manuscript.
Funding
This research was supported in part by JSPS KAKENHI Grant (16H02789 to HS, 16K16024 to YT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors appreciate the feedback from the audience of seminar talk of HS at Department of Statistics, Stanford University. The authors are grateful to Masami Hasegawa for his insightful comments on phylogenetic analysis of mammal species.
References
Adachi, J., and Hasegawa, M. (1996). Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol. 42, 459–468. doi: 10.1007/BF02498640
Akaike, H. (1974). A new look at the statistical model identification. IEEE Trans. Autom. Cont. 19, 716–723. doi: 10.1109/TAC.1974.1100705
Amari, S.-I., and Nagaoka, H. (2007). Methods of Information Geometry, Translations of Mathematical Monographs, Vol. 191. Providence, RI: American Mathematical Society.
Burnham, K. P., and Anderson, D. R. (2002). Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach, 2nd Edn. New York, NY: Springer-Verlag.
Cox, D. R. (1962). Further results on tests of separate families of hypotheses. J. R. Stat. Soc. Ser. B (Methodol.). 24, 406–424. doi: 10.1111/j.2517-6161.1962.tb00468.x
Efron, B. (1979). Bootstrap methods: another look at the jackknife. Ann. Stat. 7, 1–26. doi: 10.1214/aos/1176344552
Efron, B. (1984). Comparing non-nested linear models. J. Am. Stat. Assoc. 79, 791–803. doi: 10.1080/01621459.1984.10477096
Efron, B. (1985). Bootstrap confidence intervals for a class of parametric problems. Biometrika 72, 45–58. doi: 10.1093/biomet/72.1.45
Efron, B., Halloran, E., and Holmes, S. (1996). Bootstrap confidence levels for phylogenetic trees. Proc. Natl. Acad. Sci. U.S.A. 93, 13429–13434. doi: 10.1073/pnas.93.23.13429
Efron, B., and Tibshirani, R. (1998). The problem of regions. Ann. Sta. 26, 1687–1718. doi: 10.1214/aos/1024691353
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Fithian, W., Sun, D., and Taylor, J. (2014). Optimal inference after model selection. arXiv:1410.2597.
Graur, D., Duret, L., and Gouy, M. (1996). Phylogenetic position of the order lagomorpha (rabbits, hares and allies). Nature 379:333. doi: 10.1038/379333a0
Halanych, K. M. (1998). Lagomorphs misplaced by more characters and fewer taxa. Syst. Biol. 47, 138–146. doi: 10.1080/106351598261085
Halvorsen, K. (2015). ElemStatLearn: data sets, functions and examples from the book: “the elements of statistical learning, data mining, inference, and prediction” by trevor hastie, robert tibshirani and jerome friedman. R package. Available online at: https://CRAN.R-project.org/package=ElemStatLearn
Kishino, H., and Hasegawa, M. (1989). Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 29, 170–179. doi: 10.1007/BF02100115
Kishino, H., Miyata, T., and Hasegawa, M. (1990). Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 30, 151–160. doi: 10.1007/BF02109483
Konishi, S., and Kitagawa, G. (2008). Information Criteria and Statistical Modeling. New York, NY: Springer Science & Business Media. doi: 10.1007/978-0-387-71887-3
Lee, J. D., Sun, D. L., Sun, Y., and Taylor, J. E. (2016). Exact post-selection inference, with application to the lasso. Ann. Stat. 44, 907–927. doi: 10.1214/15-AOS1371
Novacek, M. J. (1992). Mammalian phytogeny: shaking the tree. Nature 356, 121–125. doi: 10.1038/356121a0
Posada, D., and Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808. doi: 10.1080/10635150490522304
Rosenthal, R. (1979). The file drawer problem and tolerance for null results. Psychol. Bull. 86, 638–641. doi: 10.1037/0033-2909.86.3.638
Schennach, S. M., and Wilhelm, D. (2017). A simple parametric model selection test. J. Am. Stat. Assoc. 112, 1663–1674. doi: 10.1080/01621459.2016.1224716
Shimodaira, H. (1997). Assessing the error probability of the model selection test. Ann. Inst. Stat. Math. 49, 395–410. doi: 10.1023/A:1003140609666
Shimodaira, H. (1998). An application of multiple comparison techniques to model selection. Ann. Inst. Stat. Math. 50, 1–13. doi: 10.1023/A:1003483128844
Shimodaira, H. (2000). Improving predictive inference under covariate shift by weighting the log-likelihood function. J. Stat. Plan. Inference 90, 227–244. doi: 10.1016/S0378-3758(00)00115-4
Shimodaira, H. (2001). Multiple comparisons of log-likelihoods and combining nonnested models with applications to phylogenetic tree selection. Commun. Stat. Theory Methods 30, 1751–1772. doi: 10.1081/STA-100105696
Shimodaira, H. (2002). An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508. doi: 10.1080/10635150290069913
Shimodaira, H. (2004). Approximately unbiased tests of regions using multistep-multiscale bootstrap resampling. Ann. Stat. 32, 2616–2641. doi: 10.1214/009053604000000823
Shimodaira, H. (2008). Testing regions with nonsmooth boundaries via multiscale bootstrap. J. Stat. Plan. Inference 138, 1227–1241. doi: 10.1016/j.jspi.2007.04.001
Shimodaira, H. (2019). Scaleboot: Approximately Unbiased p-Values via Multiscale Bootstrap. R package version 1.0-0. Available online at: https://CRAN.R-project.org/package=scaleboot
Shimodaira, H., and Hasegawa, M. (1999). Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116. doi: 10.1093/oxfordjournals.molbev.a026201
Shimodaira, H., and Hasegawa, M. (2001). CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247. doi: 10.1093/bioinformatics/17.12.1246
Shimodaira, H., and Hasegawa, M. (2005). “Assessing the uncertainty in phylogenetic inference,” in Statistical Methods in Molecular Evolution, Statistics for Biology and Health, ed R. Nielsen (New York, NY: Springer), 463–493. doi: 10.1007/0-387-27733-1_17
Shimodaira, H., and Maeda, H. (2018). An information criterion for model selection with missing data via complete-data divergence. Ann. Inst. Stat. Math. 70, 421–438. doi: 10.1007/s10463-016-0592-7
Stamey, T., Kabalin, J., Johnstone, I., Freiha, F., Redwine, E., and Yang, N. (1989). Prostate specific antigen in the diagnosis and treatment of adenocarcinoma of the prostate II. Radical prostatectomy treted patients. J. Urol. 16, 1076–1083. doi: 10.1016/S0022-5347(17)41175-X
Suzuki, R., and Shimodaira, H. (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542. doi: 10.1093/bioinformatics/btl117
Taylor, J., and Tibshirani, R. (2015). Statistical learning and selective inference. Proc. Natl. Acad. Sci. U.S.A. 112, 7629–7634. doi: 10.1073/pnas.1507583112
Terada, Y., and Shimodaira, H. (2017). Selective inference for the problem of regions via multiscale bootstrap. arXiv:1711.00949.
Tian, X., and Taylor, J. (2018). Selective inference with a randomized response. Ann. Stat. 46, 679–710. doi: 10.1214/17-AOS1564
Tibshirani, R. (1996). Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.). 58, 267–288. doi: 10.1111/j.2517-6161.1996.tb02080.x
Tibshirani, R., Taylor, J., Lockhart, R., and Tibshirani, R. (2016). Exact post-selection inference for sequential regression procedures. J. Amer. Stat. Assoc. 111, 600–620. doi: 10.1080/01621459.2015.1108848
Tibshirani, R., Tibshirani, R., Taylor, J., Loftus, J., and Reid, S. (2017). SelectiveInference: Tools for Post-Selection Inference. R package version 1.2.4. Available online at: https://CRAN.R-project.org/package=selectiveInference
Vuong, Q. H. (1989). Likelihood ratio tests for model selection and non-nested hypotheses. Econometrica 57, 307–333. doi: 10.2307/1912557
Yang, Z. (1996). Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 11, 367–372. doi: 10.1016/0169-5347(96)10041-0
Keywords: statistical hypothesis testing, multiple testing, selection bias, model selection, Akaike information criterion, bootstrap resampling, hierarchical clustering, variable selection
Citation: Shimodaira H and Terada Y (2019) Selective Inference for Testing Trees and Edges in Phylogenetics. Front. Ecol. Evol. 7:174. doi: 10.3389/fevo.2019.00174
Received: 31 January 2019; Accepted: 30 April 2019;
Published: 24 May 2019.
Edited by:
Rodney L. Honeycutt, Pepperdine University, United StatesReviewed by:
Carlos Garcia-Verdugo, Jardín Botánico Canario “Viera y Clavijo”, SpainMichael S. Brewer, University of California, Berkeley, United States
Copyright © 2019 Shimodaira and Terada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hidetoshi Shimodaira, c2hpbW9AaS5reW90by11LmFjLmpw