 Laura Parducci1*
Laura Parducci1* Inger Greve Alsos2
Inger Greve Alsos2 Per Unneberg3
Per Unneberg3 Mikkel W. Pedersen4
Mikkel W. Pedersen4 Lu Han1,5
Lu Han1,5 Youri Lammers2J. Sakari Salonen6
Youri Lammers2J. Sakari Salonen6 Minna M. Väliranta7
Minna M. Väliranta7 Tanja Slotte8,9
Tanja Slotte8,9 Barbara Wohlfarth10*
Barbara Wohlfarth10*- 1Department of Ecology and Genetics, The Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden
- 2Tromsø Museum, University of Tromsø – The Arctic University of Norway, Tromsø, Norway
- 3Science for Life Laboratory, Department of Cell and Molecular Biology, Uppsala University, Uppsala, Sweden
- 4Department of Zoology, University of Cambridge, Cambridge, United Kingdom
- 5Ancient DNA Laboratory, College of Life Sciences, Jilin University, Changchun, China
- 6Department of Geosciences and Geography, University of Helsinki, Helsinki, Finland
- 7Environmental Change Research Unit (ECRU), Ecosystems and Environment Research Programme, University of Helsinki, Helsinki, Finland
- 8Department of Ecology, Environment and Plant Sciences, Stockholm University, Stockholm, Sweden
- 9Science for Life Laboratory, Solna, Sweden
- 10Department of Geological Sciences and Bolin Centre for Climate Research, Stockholm University, Stockholm, Sweden
The lake sediments of Hässeldala Port in south-east Sweden provide an archive of local and regional environmental conditions ~14.5–9.5 ka BP (thousand years before present) and allow testing DNA sequencing techniques to reconstruct past vegetation changes. We combined shotgun sequencing with plant micro- and macrofossil analyses to investigate sediments dating to the Allerød (14.1–12.7 ka BP), Younger Dryas (12.7–11.7 ka BP), and Preboreal (<11.7 ka BP). Number of reads and taxa were not associated with sample age or organic content. This suggests that, beyond the initial rapid degradation, DNA is still present. The proportion of recovered plant DNA was low, but allowed identifying an important number of plant taxa, thus adding valid information on the composition of the local vegetation. Importantly, DNA provides a stronger signal of plant community changes than plant micro- and plant macrofossil analyses alone, since a larger number of new taxa were recorded in Younger Dryas samples. A comparison between the three proxies highlights differences and similarities and supports earlier findings that plants growing close to or within a lake are recorded by DNA. Plant macrofossil remains moreover show that tree birch was present close to the ancient lake since the Allerød; together with the DNA results, this indicates that boreal to subarctic climatic conditions also prevailed during the cold Younger Dryas interval. Increasing DNA reference libraries and enrichment strategies prior to sequencing are necessary to improve the potential and accuracy of plant identification using the shotgun metagenomic approach.
Introduction
The final stages of the last glacial period were, in the North Atlantic region, characterized by distinct and alternating warmer/colder and wetter/drier climate states before interglacial temperatures were attained (Bjorck et al., 1996; Lowe et al., 2008; Steffensen et al., 2008; Schenk et al., 2018). These marked climatic shifts caused a series of environmental changes that are registered in various geological archives (Blockley et al., 2012; Rasmussen et al., 2014). The sediments of the ancient lake at Hässeldala Port in southern Sweden (Figure 1A–D) provide a detailed record for this time interval and suggest that local and regional responses to climate variability between 14.5 and 9.5 ka BP (thousand years before present) are recorded in multiple types of environmental proxy data (Wohlfarth et al., 2017 and references therein). Hässeldala Port's sediments thus offer an excellent testing ground for DNA analysis, as recently demonstrated in Ahmed et al. (2018), who investigated Archaeal composition changes in these sediments.
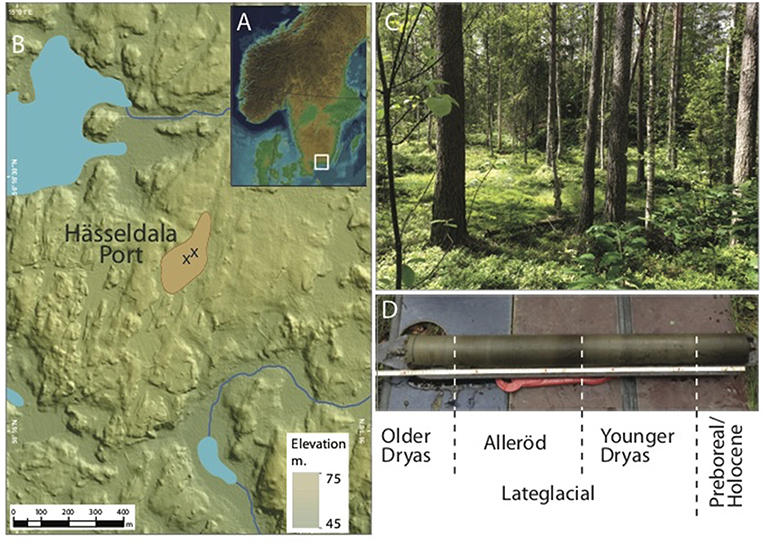
Figure 1. (A) Location of Hässeldala Port in southernmost Sweden. (B) Close-up of the topography around Hässeldala Port. X marks the coring locations; blue = lakes; brown = peatbogs. (C) View over the peatbog at Hässeldala Port. (D) Hässeldala Port's typical Lateglacial/early Holocene sediment record showing the sediments assigned to the Older Dryas (>14.1 ka BP), Allerød (14.1–12.7 ka BP), Younger Dryas (12.7–11.7 ka BP), and Preboreal (<11.7 ka BP) regional pollen zones.
Previous pollen stratigraphic investigations, combined with a high-resolution chronology, identified several climatic shifts between warmer and colder time intervals at Hässeldala Port: the transition from a cold Older Dryas into a warmer Allerød at 14.1 ka BP, the transition from the Allerød into the cold Younger Dryas at 12.7 ka BP, and the transition between the Younger Dryas and the early Holocene (Preboreal) at 11.8–11.7 ka BP (Wohlfarth et al., 2017). The area around Hässeldala Port had become free of stagnant ice some time before 14.5 ka BP. Run off from unstable catchment soils was considerable during the early lake stage and continued during the early Allerød. These unstable soils likely prevented the initial establishment of denser local vegetation. With the start of the Holocene and due to gradual infilling, the lake changed into a peatbog with greatly altered soil conditions. This again had an impact on the type of plants on and around the site.
Over the last decade, environmental DNA (eDNA) studies of lake sediments have added a new dimension to traditional biological proxies (e.g., pollen and plant macrofossil analyses) often used to investigate and reconstruct past vegetation changes and palaeoenvironments (Parducci et al., 2017). We know that in sediments eDNA binds to mineral and organic components (extracellular DNA) and that it is also present within cells in plant and animal remains that become embedded in the sediments (for a review see Nagler et al., 2018). Indeed, PCR (polymerase chain reaction) amplification of short fragments of chloroplast DNA from sediments (metabarcoding) has been successfully used to demonstrate the presence of animals and plants in different palaeoenvironmental settings (e.g. Willerslev et al., 2003, 2014; Parducci et al., 2012; Epp et al., 2015; Alsos et al., 2016; Pedersen et al., 2016).
More recently, eDNA extracted from lake sediments has been directly converted to DNA libraries in order to “shotgun” sequence the entire metagenome present in the sample and to investigate the entire diversity of the taxonomic groups present (Pedersen et al., 2016). Providing that a good fraction of the analyzed taxa is represented in a reference library, metagenomics is a more powerful approach compared to metabarcoding for investigating biodiversity of ancient environments, as it allows all of the fragmented DNA molecules to be sequenced and determined, from microorganisms like bacteria and Archaea (Ahmed et al., 2018) to plants and animals (Pedersen et al., 2016), including humans (Slon et al., 2017). In addition, this approach permits statistical data analyses to detect specific substitutions (C to T and G to A) that are normally present at the ends of ancient DNA fragments, and which indicate their ancient origin (Briggs et al., 2007; Jónsson et al., 2013). Metagenomic data of lake sediment samples, in combination with pollen and plant macrofossil data, resulted in for example detailed palaeoenvironmental reconstructions of the ice-free corridor in North America (Pedersen et al., 2016) providing evidence for new postglacial colonization routes of humans.
Here, we employ low-coverage shotgun sequencing of eDNA and compare the full metagenome data set preserved in the Hässeldala Port record with plant micro- and macrofossil analyses conducted on the same sediment samples. We (1) examine whether sufficient and useful plant DNA information can be retrieved from the lake sediments using the shotgun sequencing technique, (2) discuss the differences between the genomic and micro/macrofossil data sets, (3) explore whether genomic eDNA add valid information on past local and regional vegetation, and (4) assess the new eDNA data set in relation to the marked climatic shifts of the last glacial/interglacial transition.
Materials and Methods
Site Description
Hässeldala Port (56°16′N, 15°01′E; 63 m a.s.l.) in Blekinge province, southeast Sweden is today a peat bog underlain by a distinct Lateglacial lake sediment sequence (Figures 1A–D) (Wohlfarth et al., 2017). Multiple sediment cores have been obtained from this site over the years and studied using a variety of palaeo-environmental and palaeo-climatic proxies (lithostratigraphy, inorganic and organic geochemistry, pollen, diatoms, chironomids, biomarkers, hydrogen isotopes, charcoal) (Wohlfarth et al., 2017 and references therein). These allowed reconstructing temporal changes in lake status, evaporation, local and regional vegetation, and summer temperature, which occurred in response to large-scale hemispheric climatic shifts (Bjorck et al., 1996; Lowe et al., 2008; Steffensen et al., 2008). Hässeldala Port's multi-proxy data set shows that the small lake basin formed >14.5 ka BP and pollen assemblages indicate that the early catchment vegetation was dominated by herbs, shrubs and dwarf-shrubs. This type of vegetation also persisted during the Allerød (14.1–12.7 ka BP) and Younger Dryas (12.7–11.7 ka BP) pollen zones. However, Betula pubescens (tree birch) remains have been found at other sites in Blekinge demonstrating that tree birch has been present regionally before the Holocene (Berglund, 1966; Wohlfarth et al., 2017). With the start of the Holocene, B. pubescens and later also Pinus sylvestris (Scots pine) plant macro remains appear in Hässeldala Port's sediments, testifying to the presence of these trees in the immediate surroundings of the ancient lake. Around 11.8 ka BP, the shallow lake started to transform into minerotrophic mire and a few hundred years later turned into an ombrotrophic bog (Wohlfarth et al., 2017).
Coring and Sub-sampling
Two new sediment cores (#7.4 and #8) were obtained at a distance of ca. 5 m in June 2015 at Hässeldala Port using a Russian corer with a chamber length of 1 m and a diameter of 10 cm (Figures 1B–D). The two cores were taken from the deepest part of the basin in the south-western corner of the bog where several parallel cores (#1-#6) had previously been obtained and analyzed (see Wohlfarth et al., 2017 for a description of all previous studies on these cores). Core #7.4 extends between 340 and 440 cm depth and core #8 between 290 and 430 cm depth (see Table S1 for details). The sediments of cores #7.4 and #8 were described and sub-sampled in August 2015, first for DNA and subsequently for loss-on-ignition (LOI), plant micro and macrofossil analyses.
Samples for DNA Analyses
To minimize the risks for contamination, we took a number of precautions during coring, sampling and DNA analysis. Immediately after collection, the cores were wrapped in plastic and as quickly as possible transported to the cold room at the Department of Geological Sciences (IGV) at Stockholm University where they were stored at 5°C until sub-sampling. The cold storage is in a part of the building where no DNA analyses are being performed. Sub-sampling was conducted in September 2015 in a clean laboratory at IGV using gloves and wearing lab coats and masks. Eight samples and corresponding replicates (16 samples) were taken from each core (total of 32 samples) as representative of the warm (Allerød, Preboreal) and cold (Younger Dryas) time intervals previously described for Hässeldala Port (Wohlfarth et al., 2017). For each sample, we removed the top 1.5 cm of the outer sediment with sterile scalpels, sampled uncontaminated material from the innermost part of the core with a new set of sterile scalpels and placed the sample in sterile tubes that were immediately closed. This procedure was repeated two times for each sample. For an overview of the 16 samples collected and analyzed in cores #7.4 and #8 see Figure 2 and Table 1. The 32 samples were frozen within 2 h after sub-sampling and shipped frozen to the Center for GeoGenetics, University of Copenhagen for molecular analyses. Here, DNA extractions and library constructions were performed in ancient DNA facilities specifically dedicated to eDNA analyses following established criteria and rules during all the steps.
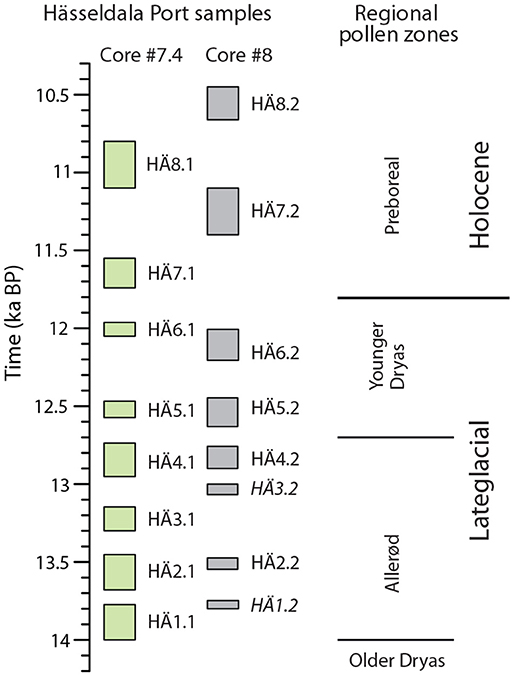
Figure 2. The 16 samples analyzed for eDNA, pollen and plant macrofossil remains in cores #7.4 (green) and #8 (gray) from Hässeldala Port shown on an age scale and in relation to the regional pollen zones. Sample IDs shown in italics represent DNA samples of poor quality, which were excluded from DNA assignments.
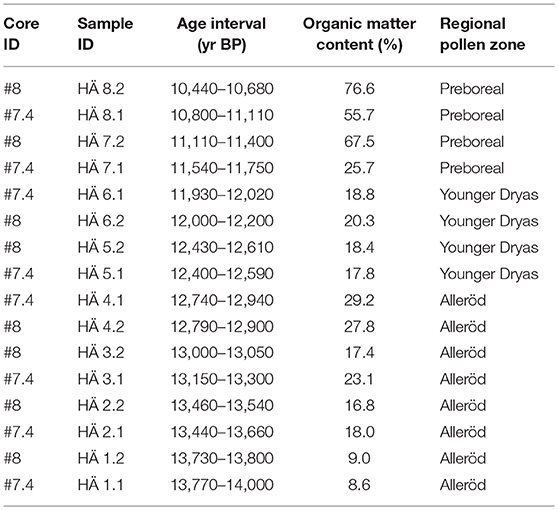
Table 1. The 16 samples analyzed for eDNA, pollen, and plant macrofossil remain in cores #7.4 and #8, their age interval, organic matter content, and their assignment to the regional pollen zones.
DNA Extraction, Libraries Construction, and High-Throughput Sequencing
One gram of wet sediment was added to 3 ml of lysis buffer [68 mM N-lauroylsarcosine sodium salt, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 20 mM EDTA (pH 8.0) with addition of 150 ul 2-mercaptoethanol and 0.3 mM DTT], together with 170 μg of proteinase K, and vortexed vigorously for 2 ×20 s using a FastPrep-24 at speed 4.0 ms−1. An additional 170 μg of proteinase K was added to each sample and incubated, then gently rotated overnight at 37°C. The MOBIO C2 and C3 buffers (MOBIO Laboratories, Carlsbad, CA) were used for removal of inhibitors, following the manufacturer's protocol. The extracts were further purified using phenol:chloroform and up-concentrated using 30 kDa Amicon Ultra-4 centrifugal filters and this step was repeated if more purification was needed. Lastly, all filters were washed twice with 500 ul Qiagen EB buffer and transferred to clean 1.5 ml Eppendorf tube. The 32 samples were extracted in four separate batches with two extraction controls each (40 samples in total). The extracted DNA was quantified using a Quant-iT dsDNA HS assay kit (Invitrogen) on the Qubit 2.0 Fluorometer according to the manufacturer's manual. Aliquots from the DNA extracts were subsequently converted into Illumina double stranded libraries using a NEBNext® DNA Library Prep Master Mix Set for 454 (New England BioLabs) following the manufacturer's protocol with the modifications described in Pedersen et al. (2016). Metagenome libraries were amplified using AmpliTaq Gold (Applied Biosystems) (14–20 cycles), purified using the MinElute PCR Purification kit (Qiagen), quantified on the 2100 BioAnalyzer and then pooled equimolarly. All pooled libraries were then sequenced using Illumina HiSeq 2500 platform (single-end reads). The eight extraction negative controls and the six library negative controls (one for each batch) were also prepared and sequenced as controls for contaminants (46 samples in total).
Bioinformatic Analyses
Sequence Quality Control
We sequenced a total of 1,340,975,627 DNA reads distributed across Hässeldala Port samples and controls. All reads were subjected to the following quality criteria. First, all reads were trimmed for the Illumina 3′ sequencing adapter (parameter setting -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC) using cutadapt v. 1.11 (Martin, 2011). In addition, we used the parameter settings -m 30–trim-n -q 10,10 which were employed to discard reads >30 base pairs (bp), trim trailing N's, and to trim low-quality bases (phred score <= 10) from both ends of the reads. Table S2 summarizes the total number of raw reads and the total number of reads passing quality control filters (48% for samples and 19% for controls). Second, we compared the quality of the raw and trimmed quality filtered reads using FASTQC (Andrews, 2010) version 0.11.5 and found that there were marked difference between the controls and the samples (Figure S1). Eventually, samples HÄ3.1_2, HÄ5.1_2, HÄ6.2_2, HÄ1.2, extraction blank E7 and library blank Blank4 failed to provide sufficient number of sequences. All library and extraction negative controls were markedly different from the “true” samples in terms of read lengths, phred scores and duplicate numbers (Figure S2). The bases sequenced for raw reads and for reads passing quality control filters (QC reads), showed two characteristic features of ancient DNA reads. First, the frequency of C to T substitutions is expected to be elevated at the 5′-end (Briggs et al., 2007); the Hässeldala Port samples display an increase in thymines at the 5′-end in both raw and trimmed reads, whereas all other bases display lower frequencies compared to later positions. Second, the frequency of G to A substitutions is expected to increase starting circa 10 bases from the 3′-end (Briggs et al., 2007); the Hässeldala Port samples display a longer increase in adenines toward the 3′-end. Intermediate positions show less variation than for raw reads (Figure S3). These features indicate the presence of ancient DNA reads in the Hässeldala Port samples, however we cannot conclude on the basis of this information alone that the samples contain DNA damage. We therefore used kPAL (Anvar et al., 2014) to perform a simple reference-free k-mer analysis of the data to identify issues with contamination and poor-quality samples. kPAL counts the number of k-mers of a given length and creates distance matrices based on the differences between k-mer profiles which we then use as input to a PCA using the R function prcomp.
Assessment of Ancient DNA Characteristics
QC reads were mapped with Bowtie2 version 2.2.8 (Langmead and Salzberg, 2012). The reference database consisted of 1,060,223 sequences of unicellular (prokaryote, archaeal, and eukaryote) origin designed to cover as many taxa as possible that are relevant for soil and environmental samples (Ahmed et al., 2018). Bam files were sorted with samtools version 1.3.1 (Li et al., 2009) and duplicate reads were removed with Picard version 2. (http://broadinstitute.github.io/picard) and finally mapped reads with mapping quality <= 25 were filtered out with bamtools version 2.4.0 (Barnett et al., 2011). Table S3 summarizes mapping and filtering statistics for the Hässeldala Port samples. On average 2.3% of the total sample reads mapped against the reference database, of which circa 3.4% remained after removing duplicate reads and mapping quality filtering and were used for taxonomic assignments. Figure S2b shows read length distributions per sample for QC-passed reads and suggests that a third characteristic feature to ancient DNA is visible in the Hässeldala Port samples. In ancient reads, the read length is normally very low and, in our case, it was concentrated to between 30 and 50 bp, all other lengths displayed lower frequencies.
Mapping, Filtering, and Taxonomic Assignment
We determined the taxonomic profiles of all plant DNA in our samples, by constructing a custom reference dataset containing all plastid sequences from the NCBI RefSeq project (2,477 sequences, 29th January 2018) and the plastid and ribosomal sequences from the PhyloNorway project (259 plant species from Norway included; Table S4). The sequence data was mapped against the custom reference dataset using BWA v0.7.5a aln algorithm (Li and Durbin, 2009) and all alignments with a mapping score of 0 (to account for multi-mapped reads) or > = 30 were parsed for downstream quality control. Duplicate reads were removed using samtools v0.1.19 (Li et al., 2009) and low complexity reads (DUST score >1) were removed using PRINSEQ v0.20.4 (Schmieder and Edwards, 2011). In order to remove low-quality hits, the remaining reads were aligned to both the custom reference and the NCBI NT dataset (15th December 2017) with the NCBI-BLAST+ blastn algorithm v2.7.1 (Camacho et al., 2009). Due to a low number of plant sequences and high DNA damage, reads were kept if: (1) they had a similarity score > = 90% and an alignment length > = 90% and (2) they had a better alignment against the custom reference dataset than the NCBI NT dataset (bit score), or, in case of equal bit scores, (3) if the NCBI NT alignment was against a Viridiplantae. The up to 90% similarity level allowed for a maximum of three mismatches on the shortest possible reads (30 bp) and it was used to account for DNA damage, which leads to higher mismatches than expected. The double check against both the custom reference and GenBank nucleotide was needed to remove erroneous hits when using a lower threshold. A custom script was used to determine the last common ancestor for the remaining alignments. In addition, for each mapped read we double-checked that: (1) the assigned taxon was native in the region (Virtuella flora, http://linnaeus.nrm.se/flora/; Mossberg and Stenberg, 2010), (2) that it was likely to be present in the region during the last glacial/interglacial transition based on its current distribution (Hultén and Fries, 1986), and (3) that the reads could possibly be assigned to taxa available in the reference dataset. For each assigned taxon, we also checked for closely related native taxa present in the reference dataset and adjusted the taxonomic identification to a level that we could be certain of. For example, reads assigned to Juncus filiformis are presented as Juncus cf. filiformis because we are certain about the genus-level identification. However, since a full chloroplast reference genome is available only for four of the 25 Juncus species currently occurring in Sweden, we cannot exclude that the same read was shared among all Juncus species. It is also possible that, due to sequence similarity and absence of correct species in the reference dataset, reads were assigned to correct genus but wrong species. This is the case for example for Diplazium dushanense, which is presented as Diplazium sp. because the only species occurring in Sweden, but missing in the reference dataset, is Diplazium sibiricum. Finally, we only retained taxa that were represented by a minimum of two reads and used the maximum read count found in any control (extraction and/or library) as a removal threshold (i.e., we removed taxa that showed higher number of reads in the controls than in the samples). If a read was assigned to taxa that could not have been credibly present at our site based on the modern or ancient flora, we assigned the reads to a higher taxonomical level (either from species to genus or from genus to family), assuming that the first assigned taxon was absent in the reference database. If an assignment at a higher taxonomical level was still not plausible, the taxon was considered a false positive and removed from the dataset. Filtering data is always a trade-off between loosing true positive and keeping false positives (Ficetola et al., 2015; Alsos et al., 2018). We think that our filtering settings removed several true positives as e.g., Ranuculus, although the majority of taxa removed are not native to Scandinavia today and are assumed to be due to artifacts as e.g., sequence similarity, lack of native taxa in database, PCR or sequencing errors or contamination. All data was then parsed to MEGAN (Huson et al., 2007), R and excel for further downstream presentation and statistical analysis.
Loss-on-Ignition (LOI) and Chronology
Contiguous 1-cm thick samples were taken from both cores for LOI analyses. The samples were dried overnight at 105°C and subsequently ignited at 550°C following the procedures described in Heiri et al. (2001) (Table S1). To obtain a timescale for the two new cores, we adopted the core-to-core alignment of Muschitiello et al. (2015) and described in Wohlfarth et al. (2017). This approach is based on a statistical correlation of the LOI% curves of #7.4 and #8 to that of master core #5 (Table S1), which serves as a chronological template for all of Hässeldala's sediment cores (Wohlfarth et al., 2017).
Plant Macro- and Micro-Fossil Analysis
For plant macro- and microfossil analyses 16 sub-samples were taken from the two cores at exactly the same levels as the samples for DNA analysis (Table S1). Our purpose was not to establish a new pollen stratigraphy for cores #7.4 and #8, as these are already available from previous cores (Wohlfarth et al., 2017), but to compare the pollen spectra in the respective samples to the plant macrofossil assemblages and to the DNA results. Prior to sieving for plant macro remains, the sediment samples were soaked in a sodium pyrophosphate [Na4P2O7 ×10H2O] solution over night to disaggregate the clay minerals (Birks, 2002). Samples were sieved using a 250-μm mesh and remains retained on the sieve were identified using a binocular microscope. For microfossil analyses we subsampled 0.5 cm3 of sediment from each of the 16 samples and prepared the samples using standard treatments (HCl, KOH, HF, acetolysis; Moore et al., 1991). The slides were mounted in glycerol, and pollen, spores and coenobia were counted using a light microscope at 400 × magnification. Between 300 and 750 (mean = 575) pollen grains were counted in each sample. Microfossil identification followed Moore et al. (1991) and Reille (1992).
Results
Lithology and Chronology
The lithostratigraphy of sediment cores #7.4 and #8 compares well to that of all previous sediment cores taken from Hässeldala Port (Wohlfarth et al., 2017): fine sands, silt and silty clay in the bottom part; clayey algae gyttja and algae gyttja in the middle part and gyttja and coarse detritus gyttja in the uppermost part (Table S1). The LOI% values of the two cores mirror the changes in sediment organic matter content and are almost identical to those obtained earlier for Hässeldala Port cores (Wohlfarth et al., 2017). The common features of the LOI% curves therefore allow precise statistical alignments to master core #5 (Table S1) for which an excellent chronological framework has been established (Muschitiello et al., 2015; Wohlfarth et al., 2017). Using the statistical alignments described in Muschitiello et al. (2015) and Wohlfarth et al. (2017), we transferred the chronology of core #5 to cores #7.4 and 8 to obtain an age assignment for each of our samples (Table 1). As seen in Figure 2, the ages of several samples in the two cores partly overlap (e.g., HÄ4.1 and HÄ4.2; HÄ5.1 and HÄ5.2), while other samples allow filling the respective age gaps. The age model for both cores combined with the almost identical lithostratigraphy permits combining the data sets of the two cores and presenting the samples in a temporal order (Figure 2 and Table 1). We however clearly differentiate samples from core #7.4 from those of core #8 in all figures.
The statistical alignment of the LOI% data and transferring the age model from core #5 to cores #7.4 and 8, moreover allows us to assign regional pollen zone boundaries to our new cores by adopting the framework presented in Wohlfarth et al. (2017) (Figure 2). Accordingly, none of the samples compares to the regional Older Dryas pollen zone, while samples HÄ1.1–HÄ4.2 compare to the regional Allerød pollen zone, samples HÄ5.1–HÄ6.2 to the regional Younger Dryas pollen zone and samples HÄ7.1–HÄ8.2 to the Preboreal.
Plant Micro and Macrofossils
Microfossil analysis of the 16 samples allowed identifying more taxa than plant macrofossil analysis (45 vs. 22; Table S1). Spores and coenobia of ferns, mosses, algae, and fungi that were counted on the pollen slides, suggest a regional and local presence of these taxa. The two data sets are presented together with the eDNA data in Figures 3A–D. For comparison and convenience, we grouped the identified families and taxa as follows: (1) trees, shrubs and dwarf shrubs; (2) herbs, grasses and aquatics; and (3) ferns, mosses, algae, and fungi. A total of 58 taxa are represented in the plant micro- and macrofossil data sets; of these 36 taxa were identified as pollen, 13 taxa as macrofossils, and 9 taxa were present in both proxies (Figure 4).
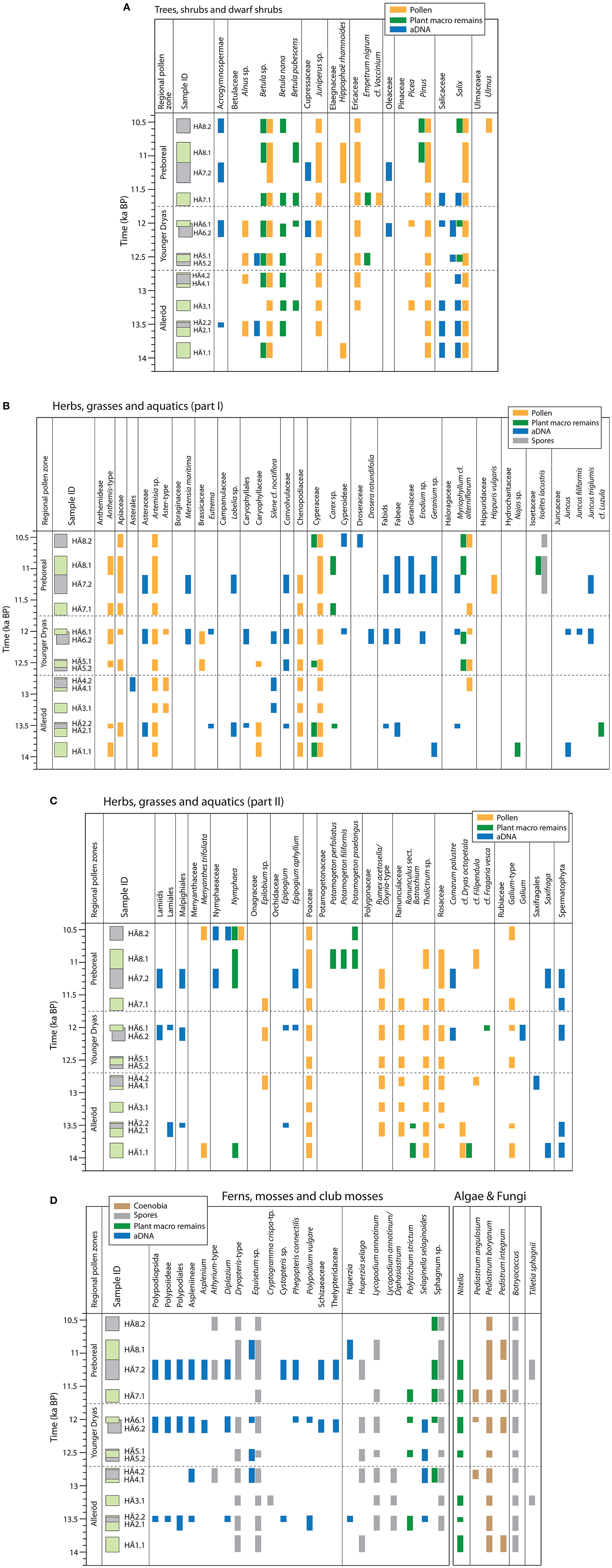
Figure 3. Comparison between the results of micro- and macrofossil analyses and eDNA in Hässeldala Port's sediments shown according to age and in relation to the regional pollen zones. Samples from core #7.4 are shown in green and samples from core #8 in gray. (A) Trees, shrubs, and dwarf shrubs. (B,C) Herbs, grasses, and aquatic plants. (D) Ferns, mosses, algae, and fungi.
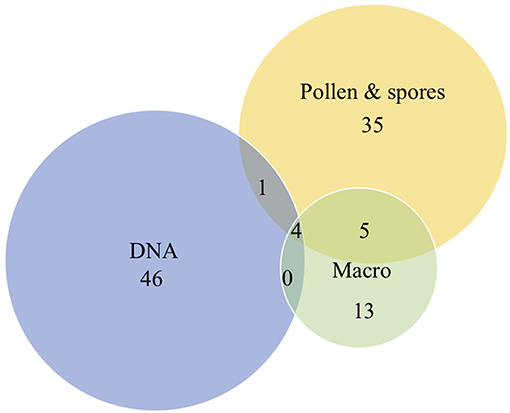
Figure 4. Venn diagram showing the number of plant taxa detected from Hässeldala Port's sediments using micro- and macrofossil and eDNA analyses. See Figure 3 for details on taxa.
Pollen of Betula sp. (undifferentiated) and Pinus were found in all analyzed samples. Pollen of these taxa are transported over long and short distances, which makes it difficult to determine whether they are of local or regional origin. Macroscopic remains of tree Betula (B. pubescens) however suggest that these trees grew close to the ancient lake during the Allerød, Younger Dryas, and Preboreal pollen zones (Table S1). The presence of tree Betula during the Allerød pollen zone has been reported earlier for Blekinge (Berglund, 1966; Wohlfarth et al., 2017), but its occurrence at Hässeldala Port during the Allerød and Younger Dryas pollen zones is novel. Pinus needles and bud scales are however only present in the uppermost samples, which represent the early Holocene. Pollen of shrubs and dwarf shrubs (e.g., Betulaceae, Ericaceae, Juniperus, Salix) occur throughout the record, while macroscopic remains only comprise frequent Betula nana and scarce Empetrum nigrum seeds and Salix sp. bark. Although the pollen record suggests local and regional presence of diverse herb/grass communities, herb/grass macrofossil finds are scarce and only include Cyperaceae/Carex, Luzula, Dryas octopetala, and possibly Fragaria vesca (Table S1). The micro and macrofossil samples analyzed here suggest that the local terrestrial vegetation was dominated by shrubs, dwarf shrubs, herbs and grasses and that tree Betula became established already during the Allerød pollen zone, while Pinus appeared first during the early Holocene.
Aquatic plants are well represented among macrofossils with taxa such as Rancunculus sect. Batrachium, various Potamogeton species, Myriophyllum cf. alterniflorum, Nymphaea, and Isoëtes lacustris (Table S1). Nitella oospores, spores of Botryococcus and Pediastrum coenobia are present in all samples, whereas spores of the fungi Tilletia sphagnii are only present in three samples. Moreover, macroscopic remains of Sphagnum and Polytrichum strictum also occur in several samples, as well as spores of for example Dryopteris-type, Equisetum sp., Huperzia selago, Lycopodium, and Sphagnum.
eDNA
Negative Controls
After DNA extraction none of the eight controls contained measurable DNA amounts. However, after library preparation and sequencing we found that 43,453,148 and 23,290,426 reads passed QC filters in the extraction and library controls, respectively (except for Blank4 that provided no sequences and E7 that had no reads passing quality filters). These reads however, show low quality scores (Figure S1) and do not show the damage patterns typical of ancient reads (Figure S3). In addition, K-mer distribution analyses (PCA of the distance matrix for k = 6, raw data) indicate sequence anomalies, suggesting low data quality due to errors and sequencing biases (Figure S4). Despite our efforts to prevent any contamination, the taxonomic alignment reveals taxa in the controls that originate from commonly known contaminant DNA like human and bacteria. A visual inspection preformed with MEGAN showed that the three most common plant contaminants were rice (Oryza), wheat (Triticum), and maize (Zea), while the rest of the plant reads were assigned to tropical or exotic species (with the exception of only one read assigned to Pinus). These were therefore not considered further in the analyses.
Samples
We generated a total of 1,014,951,5 reads for the Hässeldala Port samples, of which the per-sample read counts lie in the range of 12–62 million (Table S2), except for a few samples that failed to yield sufficient number of high-quality reads (HÄ1.2 and HÄ6.2_2) and those devoid of reads after filtering (HÄ3.1_2 and HÄ5.1_2). Read length distributions per sample for QC-passed reads was concentrated to between 30 and 50 bp and showed no clear association of post-mortem DNA decay (fragmentation and damage) with age (Figures S2), (Figures S3). This suggests that, beyond the initial rapid degradation, eDNA is well-preserved at Hässeldala Port.
On average 2.3% of the total sample reads mapped against the reference database, and of these only 3.4% remained after cleaning and filtering and were used for taxonomic assignments. A general overview of mapped reads passing filters that were assigned to taxa from the three domains of cellular life (Bacteria, Archaea, and Eukaryotes) can be found in Table S5. Of all mapped reads the majority were assigned to Bacteria (79% on average), while 15% was assigned to Eukaryota and <6% to Archaea. These proportions do not necessarily reflect the true abundance of each domain in our samples due to bias and limitations existing in the database. However, given that the alignment with the plant custom reference dataset eventually detected only 1,634 plant reads (see below), we assume the plant percentage to be very low and in the range of 0.1%.
The 1,634 reads obtained matched 122 unique plant taxa, of which 69 were excluded by the following criteria: (1) taxa identified by only one read, (2) taxa present in the controls, (3) taxa exotic to Sweden during the studied time period (Table S6). The excluded taxa are to a large part assigned to tropical plant species (e.g. Musa, Aloe, Bomarea, Cytinus, Ephedra, Cycadales). Out of the remaining 53 taxa, two pairs of reads were assigned to the same taxa resulting in 51 single taxa. Seven of these taxa showed reads in the controls that were however below the removal threshold and were therefore kept. We also excluded sample HÄ3.2 where only one read could be assigned. This resulted in a total of 14 samples containing 51 unique plant taxa assumed to represent true positives. Of these ~22% were assigned at the family level, 31% at the genus level and 22% at the species level. The remaining 25% of the taxa were identified at higher taxonomical levels (order and clade). The high percentage of taxa identified at or below the genus level (53%) is likely due to the inclusion of the local PhyloNorway data set in our reference database for taxonomical assignment.
The largest number of reads (166) was found for the fern Diplazium. The 16 other taxa recorded with 10 or more reads were trees and shrubs such as Salicaceae (27 reads) and Salix (43), the large clade of Acrogymnospermae (31), herbs of Convolvulaceae (54), and Asteraceae (11), aquatic or semi-aquatic plant species e.g., Juncus triglumis (31) and the clubmoss Selaginella selaginoides (10).
Comparison of eDNA, Micro- and Macro-Fossil Data Sets
Overall, we recovered more taxa by DNA (51) than singularly from the micro- (44) and the macro-fossil (22) record (Figure 4). The number of taxa identified at the species level varied in the three data sets between 14 for microfossils (32%), 11 for macrofossils (45%), and 11 for DNA (21%). The three proxies show important differences in respect to identified major plant taxa and only four taxa were detected by all three proxies (Betula sp., Salix, Myriophyllum cf. alterniflorum, and Nymphaea).
Among the trees, shrubs, and dwarf shrubs (Figure 3A), Betula and Salicaceae were recorded by all three proxies, although the taxonomic level of identification varied. Pinus was recorded in the pollen (all periods) and macrofossil (Preboreal) data sets, whereas the one-read DNA record did not pass filtering criteria. Other taxa detected as pollen (e.g., Ulmus, Alnus sp., Hippophaë rhamnoides, Picea, Chenopodiaceae, Poaceae, Apiaceae) were not recorded as macrofossil or DNA. DNA of Cupressaceae was detected and could potentially represent Juniperus, which was identified by pollen. Ericaceae were present in the pollen spectra (some of it was tentatively assigned to Vaccinium) whereas E. nigrum was identified by macrofossils. Oleaceae (family including Fraxinus) and Acrogymnospermae (including Picea, Pinus, and Juniperus) were only recorded by DNA.
Among herbs, grasses and aquatics (Figures 3B–D), Cyperaceae (including Carex sp. and Cyperoideae identified by macroscopic remains and eDNA, respectively), Myriophyllum cf. alterniflorum, and Nymphaea were present in all three proxies. Rosaceae could also be identified by all three proxies, but with assumed different taxa. Ranunculaceae were identified by pollen and macroscopic remains but as different taxa. Isoëtes lacustris was identified both as spores and macroscopic remains. Other herbs are only recorded as pollen, such as e.g., Anthemis-type, Apiaceae, Chenopodiaceae, Hippuris vulgaris, Menyanthes trifoliata, Epilobium sp., Poaceae, and Rumex acetosella. Two taxa appear in the macrofossil record only: Najas sp. and various Potamogeton species. However, eDNA detected families (Asteraceae) that include taxa found as pollen (Artemisia); species or genera (Eutrema, Silene noctiflora, Comarum palustre, Galium) that belong to families identified in the pollen record; and several taxa that show no signal in the micro- and macrofossil records: Boraginaceae (with the species Mertensia maritima), Campanulaceae (with the genus Lobelia), Convolvulaceae, Droseraceae (with the genus Drosera rotundifolia), Geraniaceae (with Erodium and Geranium), Juncaceae (with J. filiformis and J. triglumis), Orchidaceae (with Epipogium and Epipogium aphyllum) the genus Saxifraga, and larger clades like Fabids, Fabeae, Lamiids, and Lamiales. A large number of ferns without identifiable spores were also identified exclusively by eDNA (Schizaeaceae, Thelypteridaceae, and several taxa within the Polypodiaceae family). Moreover, eDNA could detect a few club mosses (Huperzia, Selaginella selaginoides), but none of the algae species Pediastrum and Botryococcus that were identified by microfossil analysis (Figure 3D), although these were present in the database.
Except for a few taxa—trees/shrubs growing close to the lake (Betula and Salix), some aquatic plants (Myriophyllum alterniflorum, Nymphaea) and some ferns (Equisetum)—we found limited overlap between the three proxies (Figure 4). However, several of the taxa that were recorded by eDNA now allow complementing information regarding the local vegetation, especially in respect to herbs/grasses/aquatics (M. maritima, Eutrema, Lobelia, S. noctiflora, D. rotundifolia, Erodium, Geranium, J. filiformis, J. triglumis, E. aphyllum, C. palustre, Saxifraga) and a variety of ferns and mosses (Figures 3B–D). Taken together, the eDNA results allow us to increase the total number of identified taxa from 67 (pollen and plant macro remains) to more than 100.
Taxa Representation During the Regional Pollen Zones
The diversity of plant DNA taxa was compared to the regional pollen zones (Allerød, Younger Dryas, and Preboreal) earlier identified at Hässeldala Port, which correspond to warmer/colder/warmer time intervals (Wohlfarth et al., 2017). The eDNA of some tree and shrub species, such as Betula sp. and Salicaceae/Salix, was present in Allerød and Younger Dryas samples, in accordance with the pollen and macrofossil data (Figure 3A), which suggests that boreal to subarctic climate conditions also prevailed during the Younger Dryas pollen zone.
The proportion of taxa detected in all pollen zones was generally lower for eDNA (31%) than for pollen (59%) or macrofossils (42%) (Figure 5). The eDNA data set however suggests that a higher number of new taxa became established during Younger Dryas, while the pollen and macrofossil data indicate that new plants became established only during the Preboreal. We also found a higher number of taxa and reads per sample in Younger Dryas samples (average 17.3 and 103.8, respectively), than in Allerød (average 7.8 and 34, respectively) and Preboreal (11.5 and 49.3, respectively) samples (Figure 5). This may be a mapping artifact because our reference library is biased toward cold tolerant species. It is however notable that all taxa, except for two of the new species that appeared during Younger Dryas, persisted into the Preboreal.
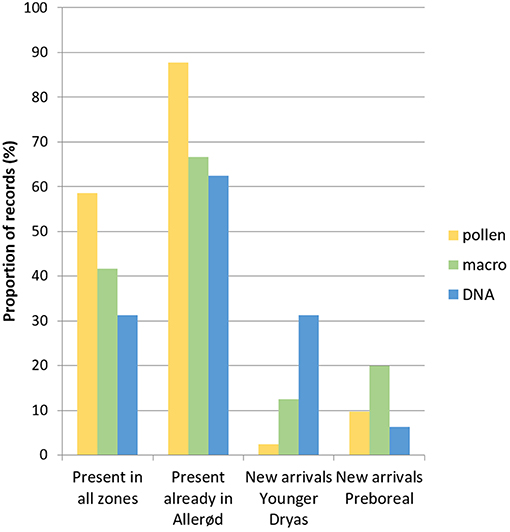
Figure 5. Presences and arrival of taxa (in percentage) detected in pollen, macrofossils and ancient DNA analyses.
Discussion
At Hässeldala Port, the metagenomic data allowed to identify a limited number of plant reads, but sufficient to identify a larger number of taxa than either micro- or macro-fossils alone and therefore increased the overall information about past floras.
The eDNA Record
eDNA signals were mainly derived from terrestrial taxa. These likely represent common and dominant taxa that were growing close to the lake as they have the strongest signal (Alsos et al., 2018). Aquatic taxa, such as Juncaceae, Nymphaceae, Myriophyllum alternifolium may have been less common or had a scattered presence at Hässeldala, since aquatic taxa have usually a higher chance to be detected due to less taphonomical restrictions (Alsos et al., 2018).
The Shotgun Sequencing Approach
Starting from more than a billion reads only very few reads (<2,000) could eventually be assigned to the likely local plant taxa. Thus, the majority of reads did not match any plants in our reference library and could not be used in our study. Similarly, in two recent genomic studies on sediments only a small fraction of the reads that aligned to the database could be eventually assigned to plants using the shotgun sequencing approach. Slon et al. (2017) shotgun sequenced sediments from seven caves and aligned between 4.9 and 21% of the reads to the database and only a small proportion of these (1–2%) aligned to plant sequences (Viridiplantae). Pedersen et al. (2016) aligned and assigned to plant taxa only 2,596 out of 1 billion aligned reads. In our study, more than 78% of the mapped reads (2.3% of the total) were assigned to Bacteria, circa 15% to Eukaryotes and more than 5% to Archaea (see also Ahmed et al., 2018).
Overall these results suggest that the genomic data is complementary to micro and macrofossil records and potentially holds many more taxa than those detected in this study. Thus, if whole community reconstructions are the goal of a study, genomics is a practical approach for exploring a large number of taxa, from large animals like mammoth, to bacteria, algae, and archaea. However, the number of plant taxa we found in each sample was less than seven for the majority of the samples, which is lower than the 20–60 taxa detected with PCR-based metabarcoding approaches that are generally used to target plants (Pansu et al., 2015; Parducci et al., 2015; Alsos et al., 2016, 2018; Sjögren et al., 2016; Niemeyer et al., 2017; Zimmermann et al., 2017). The PCR-based method however, may be biased due to differences in the amplification of different taxa. For example, Cyperaceae is normally underrepresented in metabarcoding studies probably due to a longer than average fragment size (Alsos et al., 2018). Our hope using the shotgun sequencing approach was to overcome this bias and provide data that also allow for more quantitative interpretations. However, we note that Cyperaceae pollen or macro remains were found in many samples, but Cyperaceae DNA was only found in two samples. As metabarcoding is cheaper and requires less bioinformatic work than the shotgun sequencing approach, we conclude that, if vegetation reconstruction alone is the goal of a study, metabarcoding is currently a more useful and cost-efficient method.
We think that several issues currently limit the shotgun sequencing approach. The first is the lack of sufficiently curated plant reference genomes. For mapping we here used plastid DNA sequences present at NCBI in combination with plastid DNA and ribosomal DNA derived from the PhyloNorway data set. However, we did not check the nuclear genomes of the plants present in NCBI, which could have contributed with more information. At the moment there is little suitable reference material available in the NCBI database for plant nuclear genomes and the majority of the sequences come from economically important crop species like maize, rice, soya and potato. The lack of reference genome sequence data for non-commercial plant species as those analyzed in our study, substantially reduces the potential for success of the bioinformatic analyses with metagenomic data. In addition, metagenomic data can vary in content across samples from the same or similar environments (Quince et al., 2017), as it was also seen in our two cores which were located only few meters from each other, but reported in several instances a different taxa composition (e.g., Hä2.1/Hä2.2, Hä7.1/Hä7.2, or Hä8.1/Hä8.2 in Table S6). This complicates the detection of biologically meaningful differences between samples of different ages.
The second issue is methodological and may be related to the protocols used for extraction and library construction. These often allow only a small percentage of eukaryote reads (and in particular plant reads) to be extracted and sequenced as compared to archaeal and bacterial reads. The problem can be partially circumvented by using new developed DNA extraction methods for sediments like the phosphate-based protocols developed by Taberlet et al. (2012); Zinger et al. (2016); a disadvantage is however a strong detriment of the final DNA amount obtained. More recent advances in high-throughput DNA sequencing technologies allow circumventing the problem more efficiently. Today it is possible to target-enrich specific genomic DNA regions via hybridization capture prior to sequencing (Schmid et al., 2017; Slon et al., 2017). This allows generating larger data sets for multiple DNA target loci in parallel with high efficiency and improving substantially the quality of the taxonomical assignments after sequencing. The target capture sequencing technology coupled with shotgun sequencing seems a good alternative and a powerful strategy for molecular ecologists who want to work with taxa for which few genomic resources are available.
A third issue to consider is the properties of the sediments in relation to DNA preservation. It is very difficult to assess the time frame for which DNA molecules are preserved in lake sediments. We know, however that local environmental conditions may be more important than other factors, including time (Allentoft et al., 2012). Cold, dry and anoxic environments and a neutral or slightly alkaline pH represent optimal conditions for DNA preservation. The degree of preservation may also be a function of water depth, with the poorest preservation resulting from decay in shallow and relatively turbulent water and/or high algae content (Rich, 1989; Alsos et al., 2018). Even if current studies seem to show little success with eDNA extracted from below bogs (Pedersen personal communication), at Hässeldala Port we did not find a clear correlation between sample age and post-mortem decay (damage and fragmentation), suggesting that, beyond the initial rapid degradation, DNA molecules were well-preserved. We found on the other hand, a high variation among samples both in number of reads and in number of taxa detected. This observation compares to plant macrofossil studies, which can show large variations in number and type of taxa recovered (Birks, 2013).
Finally, because of contamination risks, we employed a very cautious approach during DNA read assignment; this further limited the power of the shotgun sequencing approach. To avoid misidentifications, we used 90% of the sequences matching to at least two different reference database entries occurring at least two times in a sample. We therefore excluded several taxa that were assigned by only one read. The reason for using a lower percentage threshold (90%) was that we did not have access to a complete reference database. Indeed, a shotgun read can originate from any part of the genome, and since it is a whole molecule and not an amplified fragment as in the metabarcoding approach, the DNA damage patterns become a more serious issue for identifications. By limiting to a 90% match we included also some damaged sequences. The successive GenBank comparison was done to make sure to exclude sequences originating from non-plant sources (most likely bacteria).
The fact that our extraction controls did not contain any measurable DNA but still yielded reads after sequencing analyses highlights the challenges we are still facing for decontaminating laboratory reagents and consumables (Champlot et al., 2010). Mostly, background contamination comes from reagents used in the laboratory since the controls contained mainly exotic plant species and not much of the north European taxa found in the samples. We therefore find our samples to contain a mixture of authentic ancient DNA as well as low-background contamination of modern human, bacterial and plant taxa from the reagents which were eventually removed from the dataset.
Proxy Comparison
We found limited overlap between the three proxies (Figure 4). This is not surprising since pollen reflect the local and regional vegetation as well as long-distance transportation, while the plant macrofossil and eDNA records register the local vegetation in and around the site. It is also a positive result since a larger number of taxa could be identified using the three proxies. Some major taxa detected by pollen (e.g., Pinus, Ulmus, Chenopodiaceae, Poaceae, Apiaceae), some assigned to long-distance transport, were not recorded either by the macrofossil or eDNA data sets, confirming that pollen does not contribute to eDNA in lake sediments (Parducci et al., 2015; Alsos et al., 2016; Pedersen et al., 2016; Sjögren et al., 2016; Clarke et al., 2018), likely because only few pollen grains per species are present in each sediment sample (in our case 623 on average Table S1) and each pollen grain contains only two or three cells.
Surprisingly, some taxa as the algae Pediastrum and Botryococcus, the clubmosse Lycopodium, the moss Sphagnum, the dwarf shrub Empetrum and some aquatic plant species (Potamogeton) were identified by both micro- and macrofossil analyses, but not by eDNA even though they were present in our reference database. This may relate to differences in preservation conditions for different taxa and at different locations and may also explain the variations we observed across samples in our two cores located only a few meters apart. There is still limited knowledge regarding the taphonomy of DNA in sediments (Yoccoz et al., 2012; Alsos et al., 2018; Edwards et al., 2018). Importantly, all of the three proxies only detect a fraction of the total diversity of the past vegetation (Allen and Huntley, 1999; Drake and Burrows, 2012; Birks and Birks, 2016; Alsos et al., 2018). Thus, a complete overlap can never be expected.
One strange observation was Juniperus, as this genus is present in our database by several Cupressaceae species, including Juniperus communis. However, 19 reads were assigned to Cupressus chengiana (a tree endemic to China) and were therefore presented as Cupressaceae. We have no explanation why it was not assigned to J. communis and we suggest therefore to consider this record with caution.
Climate and Plant Diversity
All three proxies show a subset of species that are only recorded during warmer periods, as has also been observed in previous studies from the same site using pollen and macrofossils (Wohlfarth et al., 2006, 2017). Surprisingly, eDNA is the only proxy to show a high number of new taxa appearing during the Younger Dryas pollen zone (Figure 5). Even more puzzling is that most of these new taxa persist into the Preboreal period. There is only one new taxon detected by pollen appearing in Younger Dryas (Brassicaceae), and this does not persist into the Preboreal. Macrofossils detect far less species, but notably, the three new species appearing in Younger Dryas (E. nigrum, Salix, and Myriophyllum cf. alterniflorum) persist also into the Preboreal period. Thus, cold adapted species may indeed have survived for some time after the start of the Holocene warming due to a lag response to climate change (Alexander et al., 2018). These results confirm previous studies suggesting that in lake sediments eDNA is more efficient in recording persistence of species as compared to macrofossils (Alsos et al., 2016), and may help to better understand how climate change affected not only colonization but also the survival and persistence of plant species.
The appearance of new DNA taxa during a cold period may be related to an over representation of cold-tolerant species in the reference library and/or better preservation of DNA in sediments under cold conditions, or also to the presence of clays in the sediments to which DNA can more easily bind and remain protected by microbial enzymatic activity. During the Younger Dryas the ancient lake was likely stratified, ice-covered and anoxic large parts of the year, which may have also favored DNA preservation in the sediments. The area around Hässeldala Port became ice free some time before 14.5 ka BP. Sediment lithology, geochemistry and the low organic matter content of the basal sediments (Table 1) suggest unstable catchment soils and higher run-off during the early lake stage (Kylander et al., 2013). These unstable soils likely prevented the initial establishment of denser local vegetation. Catchment erosion gradually decreased during the Alleröd and was low during the Younger Dryas (Kylander et al., 2013). Lower organic matter content during the Younger Dryas is therefore related to lower lake organic productivity and not to catchment run-off. With the start of the Holocene and due to infilling, the lake gradually transformed into dry mire and subsequently into a dry shrub heathland as shown by diatom and insect studies, respectively, and this change led to a shift in soil conditions from alkaline to more acid (Wohlfarth et al., 2017). This again had an impact on the type of plants on and around the site. Furthermore, water depth and lake status changes may have played an important role for plant establishment and survival, or the fact that smaller plants did not have to compete with taller plants during the cold Younger Dryas and could expand more easily. An alternative hypothesis is that boreal-subarctic conditions also prevailed during the Younger Dryas, as also recently proposed by Schenk et al. (2018).
Our knowledge regarding the local impact of the distinct climatic shifts that characterized the end of the last glacial period is still limited and depends largely on the choice and interpretation of available proxies. Wohlfarth et al. (2017) for example had suggested, based on their multi-proxy study of Hässeldala Port's sedimentary record, that climatic shifts had a major impact on the site's environment affecting plant, diatom, and chironomid communities. In contrast, Schenk et al. (2018) recently noted that summer temperatures did not change markedly between Allerød and Younger Dryas, but that Younger Dryas summers were distinctly drier and winter temperatures significantly lower as compared to the Allerød. A re-evaluation of Hässeldala Port's chironomid-based summer temperature record (Schenk et al., personal communication) moreover shows that Younger Dryas summer temperatures were not significantly different from Allerød summer temperatures. Our new plant macrofossil and metagenomic data sets confirm these observations, as they show no change in plant community response or taxa composition between warmer (Allerød) and colder (Younger Dryas) time intervals, but the presence of a boreal-subarctic vegetation.
Conclusions
We present the full metagenome data set preserved in the Hässeldala Port lake sediment record and compare it to the plant micro- and macrofossil datasets analyzed in the same sediment samples. Our hypothesis was that the shotgun metagenomic approach allows retrieving complementary information to the micro- and macro-fossil record, thus providing details about plant community changes in relation to the climatic shifts that occurred at the transition from a glacial to an interglacial climate state. The genomic approach however, permitted only a small percentage of reads to map against our database (2.3%) and of these, plant reads were a small percentage (ca 0.1% of the total). We did not find clear associations between the number of reads/taxa, sample age and sediment organic content, suggesting that, beyond the initial rapid degradation, eDNA in the Hässeldala Port sediments was present and overall well-preserved.
Overall, we found: (1) limited overlap between proxies (but more taxa could be in common at different taxonomic levels), (2) highest diversity in number of reads and number of read/sample in the colder period (Younger Dryas) with no direct association with climate, (3) no association between DNA post-mortem decay (damage and fragmentation) and sample age or changes in organic matter content in the sediments, (4) several taxa detected only by eDNA.
Although the proportion of plant DNA was very low in the analyzed samples, the shotgun metagenomic approach added a considerable number of new taxa to those identified as pollen or plant macrofossils and thus increased the overall information about past floral assemblages (from 67 taxa identified by pollen and plant macro remains to more than 100).
The combined eDNA and plant macrofossil data sets indicate that tree Betula (B. pubescens) grew close to the ancient lake during the Allerød, Younger Dryas, and Preboreal pollen zones and that boreal to subarctic climate conditions and not arctic conditions prevailed during the Younger Dryas pollen zone. Comparisons between the pollen and eDNA data sets also confirm earlier observations that eDNA in lake sediments reflects local plant occurrences only. Moreover, the eDNA data set suggests that a higher number of new taxa became already established during Younger Dryas while the pollen and macrofossil data indicate that new plants became established only during the Preboreal.
Shotgun sequencing, though less time-consuming is still quite expensive, and for the moment, metabarcoding seems to be a more cost-efficient approach for the detection of plants from eDNA samples extracted from lake sediments. In the future, coupling target capture technology with shotgun metagenomics could significantly enhance the ability to investigate past genomic diversity in lake sediments, particularly as full-genome reference databases are being built up. So far, the performance of these combined methodologies has seen limited investigations with ancient lake sediments and we think it has a potential for becoming an important tool for future investigations of plant biodiversity changes in lake sediments, in combination with complementary pollen and plant macrofossil analyses.
Author Contributions
LP, TS, and BW designed the research. LH, BW, and LP organized and performed the coring. LH, PU, MP, MV, and JS performed the lab research. LP, TS, BW, PU, MP, YL, IA, MV, and JS analyzed the data. LP, BW, IA, and MP wrote the paper with final contributions from MV, JS, PU, YL, LH, and TS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Frederik Schenk for help during field work, Eske Willerslev (University of Copenhagen, Denmark) for being able to use the facilities of the Center of Geo-Genetics during DNA extraction and library preparation; Engy Ahmed for help during coring, for loss-on-ignition analyses of cores #7.4 and #8, and for preparing the DNA samples at the Center of Geo-Genetics; Francesco Muschitiello for creating the age models for cores #7.4 and #8. The present study was supported by SFO grants from the Faculty of Science, Stockholm University (#5.1.2-3222-14) to BW, LP, and TS; Bioinformatics Long-term Support (WABI) to BW, LP, and TS; the Swedish Research Council (grant no. 2013- D0568401), Formas (Dnr 2016-01236), and the Carl Triggers' Foundation (grant no. 14:371) to LP. PU was supported by a grant from the Knut and Alice Wallenberg Foundation to the Wallenberg Advanced Bioinformatics Infrastructure. Bioinformatics Infrastructure, IA by grants from the the Research council of Norway (226134/F50), and JS by grants from the Academy of Finland (projects 1278692 and 1310649).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00189/full#supplementary-material
References
Ahmed, E., Parducci, L., Unneberg, P., Ågren, R., Schenk, F., Rattray, J. E., et al. (2018). Archaeal community changes in Lateglacial lake sediments: evidence from ancient DNA. Quat. Sci. Rev. 181, 19–29. doi: 10.1016/j.quascirev.2017.11.037
Alexander, J. M., Chalmandrier, L., Lenoir, J., Burgess, T. I., Essl, F., Haider, S., et al. (2018). Lags in the response of mountain plant communities to climate change. Glob. Chang. Biol. 24, 563–579. doi: 10.1111/gcb.13976
Allen, J. R. M., and Huntley, B (1999). Estimating past floristic diversity in montane regions from macrofossil assemblages. J. Biogeogr. 26, 55–73. doi: 10.1046/j.1365-2699.1999.00284.x
Allentoft, M. E., Collins, M., Harker, D., Haile, J., Oskam, C. L., Hale, M. L., et al. (2012). The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils. Proc. R. Soc. B Biol. Sci. 279, 4724–4733. doi: 10.1098/rspb.2012.1745
Alsos, I. G., Lammers, Y., Yoccoz, N. G., Jørgensen, T., Sjögren, P., Gielly, L., et al. (2018). Plant DNA metabarcoding of lake sediments: how does it represent the contemporary vegetation. PLoS ONE 13:e0195403. doi: 10.1371/journal.pone.0195403
Alsos, I. G., Sjögren, P., Edwards, M. E., Landvik, J. Y., Gielly, L., Forwick, M., et al. (2016). Sedimentary ancient DNA from Lake Skartjørna, Svalbard: assessing the resilience of arctic flora to Holocene climate change. Holocene 26, 1–16. doi: 10.1177/0959683615612563
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed May 5, 2017).
Anvar, S. Y., Khachatryan, L., Vermaat, M., van Galen, M., Pulyakhina, I., Ariyurek, Y., et al. (2014). Determining the quality and complexity of next-generation sequencing data without a reference genome. Genome Biol. Evol. 15:555. doi: 10.1186/s13059-014-0555-3
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P., and Marth, GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27,1691–1692. doi: 10.1093/bioinformatics/btr174
Berglund, B. E. (1966). Late-Quaternary vegetation in eastern Blekinge, south-eastern Sweden. Opera Botanica, 12, 5–180.
Birks, H. H. (2002). “Plant macrofossils,” in Tracking Environmental Change Using Lake Sediments, Vol. 3, ed J. P. Smol, H. J. B. Birks, W. M. Last, R. S. Bradley, and K. Alverson (Dordrecht: Springer) 49–74.
Birks, H. J., and Birks, H. H. (2016). How have studies of ancient DNA from sediments contributed to the reconstruction of Quaternary floras? New Phytol. 209, 499–506. doi: 10.1111/nph.13657
Birks, H. J. B. (2013). “Plant macrofossil introduction,” in Encyclopedia of Quaternary Science, eds H. P. Mock and S. A. Elias (Amsterdam: Elsevier), 593–612. doi: 10.1016/B978-0-444-53643-3.00203-X
Bjorck, S., Kromer, B., Johnsen, S., Bennike, O., Hammarlund, D., Lemdahl, G., et al. (1996). Synchronised terrestrial-atmospheric deglacial records around the North Atlantic. Science 274, 1155–1160. doi: 10.1126/science.274.5290.1155
Blockley, S. P. E., Lane, C. S., Hardiman, M., Rasmussen, S. O., Seierstad, I. K., Steffensen, J. P., et al. (2012). Synchronisation of palaeoenvironmental records over the last 60,000 years, and an extended INTIMATE1 event stratigraphy to 48,000 b2k. Quat. Sci. Rev. 36, 2–10. doi: 10.1016/j.quascirev.2011.09.017
Briggs, A. W., Stenzel, U., Johnson, P. L., Green, R. E., Kelso, J., Prüfer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621. doi: 10.1073/pnas.0704665104
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Champlot, S., Berthelot, C., Pruvost, M., Bennett, E. A., Grange, T., and Geigl, E. M. (2010). An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE 5:e13042. doi: 10.1371/journal.pone.0013042
Clarke, C. L., Edwards, M. E., Brown, A. G., Gielly, L., Lammers, Y., Heintzman, P. D., et al. (2018). Holocene floristic diversity and richness in northeast Norway revealed by sedimentary ancient DNA (sedaDNA) and pollen. Boreas 12:299. doi: 10.1111/bor.12357
Drake, H., and Burrows, C. J. (2012). The influx of potential macrofossils into Lady Lake, north Westland, New Zealand. N.Z. J. Bot. 18, 257–274. doi: 10.1080/0028825X.1980.10426924
Edwards, M. E., Alsos, I. G., Yoccoz, N. G., Coissac, E., Goslar, T., Gielly, L., et al. (2018). Metabarcoding of modern soil DNA gives a highly local vegetation signal in Svalbard tundra. Holocene 28, 2006–2016. doi: 10.1177/0959683618798095
Epp, L. S., Gussarova, G., Boessenkool, S., Olsen, J., Haile, J., Schrøder-Nielsen, A., et al. (2015). Lake sediment multi-taxon DNA from North Greenland records early post-glacial appearance of vascular plants and accurately tracks environmental changes. Quat. Sci. Rev. 117, 152–163. doi: 10.1016/j.quascirev.2015.03.027
Ficetola, G. F., Pansu, J., Bonin, A., Coissac, E., Giguet-Covex, C., De Barba, M., et al. (2015). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556. doi: 10.1111/1755-0998.12338
Heiri, O., Lotter, A. F., and Lemcke, G. (2001). Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results. J. Paleolimnol. 25, 101–110. doi: 10.1023/A:1008119611481
Hultén, E., and Fries, M. (1986). Atlas of North European Vascular Plants North of the Tropic of Cancer. Königstein: Koeltz Scientific Books.
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L., and Orlando, L. (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Kylander, M. E., Klaminder, J., Wohlfarth, B., and Löwemark, L. (2013). Geochemical responses to paleoclimatic changes in southern Sweden since the late glacial: The Hässeldala Port lake sediment record. J. Paleolimnol. 50, 57–70. doi: 10.1007/s10933-013-9704-z
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lowe, J. J., Rasmussen, S. O., Björck, S., Hoek, W. Z., Steffensen, J. P., Walker, M. J. C., et al. (2008). Synchronisation of palaeoenvironmental events in the North Atlantic region during the Last Termination: a revised protocol recommended by the INTIMATE group. Quat. Sci. Rev. 27, 6–17. doi: 10.1016/j.quascirev.2007.09.016
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mossberg, B., and Stenberg, L. (2010). Den nya Nordiska Flora (The new Nordens Flora). Stockholm: Wahlström & Widstrand.
Muschitiello, F., Pausata, F. S. R., Watson, J. E., Smittenberg, R. H., Salih, A. A. M., Brooks, S. J., et al. (2015). Fennoscandian freshwater control on Greenland hydroclimate shifts at the onset of the Younger Dryas. Nat. Commun. 6:8939. doi: 10.1038/ncomms9939
Nagler, M., Insam, H., Pietramellara, G., and Ascher-Jenull, J. (2018). Extracellular DNA in natural environments: features, relevance and applications. Appl. Microbiol. Biotechnol. 116, 67–14. doi: 10.1007/s00253-018-9120-4
Niemeyer, B., Epp, L. S., Stoof-Leichsenring, K. R., Pestryakova, L. A., and Herzschuh, U. (2017). A comparison of sedimentary DNA and pollen from lake sediments in recording vegetation composition at the Siberian treeline. Mol. Ecol. Resour. 17, e46–e62. doi: 10.1111/1755-0998.12689
Pansu, J., Giguet-Covex, C., Ficetola, G. F., Gielly, L., Boyer, F., Zinger, L., et al. (2015). Reconstructing long-term human impacts on plant communities: an ecological approach based on lake sediment DNA. Mol. Ecol. 24, 1485–1498. doi: 10.1111/mec.13136
Parducci, L., Bennett, K. D., Ficetola, G. F., Alsos, I. G., Suyama, Y., Wood, J. R., et al. (2017). Ancient plant DNA in lake sediments. New Phytol. 214, 924–942. doi: 10.1111/nph.14470
Parducci, L., Jørgensen, T., Tollefsrud, M. M., Elverland, E., Alm, T., Fontana, S. L., et al. (2012). Glacial survival of boreal trees in northern Scandinavia. Science 335, 1083–1086. doi: 10.1126/science.1216043
Parducci, L., Väliranta, M., Salonen, J. S., Ronkainen, T., Matetovici, I., Fontana, S. L., et al. (2015). Proxy comparison in ancient peat sediments: pollen, macrofossil and plant DNA. Philos. Trans. R. Soc. B Biol. Sci. 370:20130382. doi: 10.1098/rstb.2013.0382
Pedersen, M. W., Ruter, A., Schweger, C., Friebe, H., Staff, R. A., Kjeldsen, K. K., et al. (2016). Postglacial viability and colonization in North America's ice-free corridor. Nature 53, 45–49. doi: 10.1038/nature19085
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844. doi: 10.1038/nbt.3935
Rasmussen, S. O., Birks, H. H., Blockley, S. P. E., Brauer, A., Hajdas, I., Hoek, W. Z., et al. (2014). Dating, synthesis, and interpretation of palaeoclimatic records of the Last Glacial cycle and model-data integration: advances by the INTIMATE (INTegration of Ice-core, MArine and TErrestrial records) COST Action ES0907. Quat. Sci. Rev. 106, 1–13. doi: 10.1016/j.quascirev.2014.10.031
Rich, F. J. (1989). A review of the taphonomy of plant remains in lacustrine sediments. Rev. Palaeobot. Palynol. 58, 33–46. doi: 10.1016/0034-6667(89)90055-9
Schenk, F., Väliranta, M., Muschitiello, F., Tarasov, L., Heikkilä, M, Björck, S., et al. (2018). Warm summers during the Younger Dryas cold reversal. Nat. Commun. 9:1634. doi: 10.1038/s41467-018-04071-5
Schmid, S., Genevest, R., Gobet, E., Suchan, T., Sperisen, C., Tinner, W., et al. (2017). HyRAD-X, a versatile method combining exome capture and RAD sequencing to extract genomic information from ancient DNA. Methods Ecol. Evol. 8, 1374–1388. doi: 10.1111/2041-210X.12785
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. doi: 10.1093/bioinformatics/btr026
Sjögren, P., Edwards, M. E., Gielly, L., Langdon, C. T., Croudace, I. W., Merkel, M. K., et al. (2016). Lake sedimentary DNA accurately records 20th Century introductions of exotic conifers in Scotland. New Phytol. 213, 929–941. doi: 10.1111/nph.14199
Slon, V., Hopfe, C., Weiß, C. L., Mafessoni, F., de la Rasilla, M., Lalueza-Fox, C., et al. (2017). Neandertal and Denisovan DNA from Pleistocene sediments. Science 53:eaam9695. doi: 10.1126/science.aam9695
Steffensen, J. P., Andersen, K. K., Bigler, M., Clausen, H. B., Dahl-Jensen, D., Fischer, H., et al. (2008). High-resolution Greenland ice core data show abrupt climate change happens in few years. Science 321, 680–684. doi: 10.1126/science.1157707
Taberlet, P., Prud'Homme, S. M., Campione, E., Roy, J., Miquel, C., Shehzad, W., et al. (2012). Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies. Mol. Ecol. 21, 1816–1820. doi: 10.1111/j.1365-294X.2011.05317.x
Willerslev, E., Davison, J., Moora, M., Zobel, M., Coissac, E., Edwards, M. E., et al. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51. doi: 10.1038/nature12921
Willerslev, E., Hansen, A. J., Binladen, J., Brand, T. B., Gilbert, M. T., Shapiro, B., et al. (2003). Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300, 791–795. doi: 10.1126/science.1084114
Wohlfarth, B., Blaauw, M., Davies, S. M., Andersson, M., Wastegård, S., Hormes, A., et al. (2006). Constraining the age of lateglacial and early Holocene pollen zones and tephra horizons in southern Sweden with bayesian probability methods. J. Quaternary Sci. 21, 321–334. doi: 10.1002/jqs.996
Wohlfarth, B., Muschitiello, F. L., Greenwood, S., Andersson, A., Kylander, M., Smittenberg, R. H., et al. (2017). Hässeldala - a key site for Last Termination climate events in northern Europe. Boreas 46, 143–161. doi: 10.1111/bor.12207
Yoccoz, N. G., Bråthen, K. A., Gielly, L., Haile, J., Edwards, M. E., Goslar, T., et al. (2012). DNA from soil mirrors plant taxonomic and growth form diversity. Mol. Ecol. 21, 3647–3655. doi: 10.1111/j.1365-294X.2012.05545.x
Zimmermann, H. H., Raschke, E., Epp, L. S., Stoof-Leichsenring, K. R., Schirrmeister, L., Schwamborn, G., et al. (2017). The history of tree and shrub taxa on Bol'shoy Lyakhovsky Island (New Siberian Archipelago) since the Last Interglacial uncovered by sedimentary ancient DNA and pollen data. Genes (Basel) 8:273. doi: 10.3390/genes8100273
Keywords: environmental DNA, ancient DNA, shotgun sequencing (metagenomics), pollen, macrofossils remains, lake sediments
Citation: Parducci L, Alsos IG, Unneberg P, Pedersen MW, Han L, Lammers Y, Salonen JS, Väliranta MM, Slotte T and Wohlfarth B (2019) Shotgun Environmental DNA, Pollen, and Macrofossil Analysis of Lateglacial Lake Sediments From Southern Sweden. Front. Ecol. Evol. 7:189. doi: 10.3389/fevo.2019.00189
Received: 04 December 2018; Accepted: 08 May 2019;
Published: 20 June 2019.
Edited by:
Gael Le Roux, UMR5245 Laboratoire Ecologie Fonctionnelle et Environnement (ECOLAB), FranceReviewed by:
Patrick Rioual, Institute of Geology and Geophysics (CAS), ChinaCharline Giguet-Covex, UMR5204 Environnements, DYnamiques et TErritoires de la Montagne (EDYTEM), France
Copyright © 2019 Parducci, Alsos, Unneberg, Pedersen, Han, Lammers, Salonen, Väliranta, Slotte and Wohlfarth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Parducci, bGF1cmEucGFyZHVjY2lAZWJjLnV1LnNl; Barbara Wohlfarth, YmFyYmFyYS53b2hsZmFydGhAZ2VvLnN1LnNl