 Sabrina Simon1*
Sabrina Simon1* Harald Letsch2
Harald Letsch2 Sarah Bank3
Sarah Bank3 Thomas R. Buckley4,5
Thomas R. Buckley4,5 Alexander Donath6
Alexander Donath6 Shanlin Liu7
Shanlin Liu7 Ryuichiro Machida8
Ryuichiro Machida8 Karen Meusemann6,9,10Bernhard Misof6Lars Podsiadlowski6
Karen Meusemann6,9,10Bernhard Misof6Lars Podsiadlowski6 Xin Zhou7
Xin Zhou7 Benjamin Wipfler11
Benjamin Wipfler11 Sven Bradler3*
Sven Bradler3*- 1Biosystematics Group, Wageningen University and Research, Wageningen, Netherlands
- 2Department of Botany and Biodiversity Research, University of Vienna, Vienna, Austria
- 3Department of Animal Evolution and Biodiversity, Johann-Friedrich-Blumenbach Institute for Zoology and Anthropology, University of Göttingen, Göttingen, Germany
- 4New Zealand Arthropod Collection, Manaaki Whenua–Landcare Research, Auckland, New Zealand
- 5School of Biological Sciences, The University of Auckland, Auckland, New Zealand
- 6Center for Molecular Biodiversity Research, Zoological Research Museum Alexander Koenig, Bonn, Germany
- 7Beijing Advanced Innovation Center for Food Nutrition and Human Health, College of Plant Protection, China Agricultural University, Beijing, China
- 8Sugadaira Research Station, Mountain Science Center, University of Tsukuba, Nagano, Japan
- 9Evolutionary Biology and Ecology, Institute for Biology I (Zoology), University of Freiburg, Freiburg, Germany
- 10Australian National Insect Collection, Commonwealth Scientific and Industrial Research Organisation National Research Collections Australia, Canberra, ACT, Australia
- 11Center for Taxonomy and Evolutionary Research, Zoological Research Museum Alexander Koenig, Bonn, Germany
Phasmatodea comprises over 3,000 extant species and stands out as one of the last remaining insect orders for which a robust, higher-level phylogenetic hypothesis is lacking. New research suggests that the extant diversity is the result of a surprisingly recent and rapid radiation that has been difficult to resolve with standard Sanger sequence data. In order to resolve the early branching events of stick and leaf insects, we analyzed transcriptomes from 61 species, including 38 Phasmatodea species comprising all major clades and 23 outgroup taxa, including all other Polyneoptera orders. Using a custom-made ortholog set based on reference genomes from four species, we identified on average 2,274 orthologous genes in the sequenced transcriptomes. We generated various sub-alignments and performed maximum-likelihood analyses on several representative datasets to evaluate the effect of missing data and matrix composition on our phylogenetic estimates. Based on our new data, we are able to reliably resolve the deeper nodes between the principal lineages of extant Phasmatodea. Among Euphasmatodea, we provide strong evidence for a basal dichotomy of Aschiphasmatodea and all remaining euphasmatodeans, the Neophasmatodea. Within the latter clade, we recovered a previously unrecognized major New World and Old World lineage, for which we introduce the new names Oriophasmata tax. nov. (“Eastern phasmids”) and Occidophasmata tax. nov. (“Western phasmids”). Occidophasmata comprise Diapheromerinae, Pseudophasmatinae, and Agathemera, whereas all remaining lineages form the Oriophasmata, including Heteropterygidae, Phylliinae, Bacillus, Lonchodidae (Necrosciinae + Lonchodinae), Clitumninae, Cladomorphinae, and Lanceocercata. We furthermore performed a divergence time analysis and reconstructed the historical biogeography for stick and leaf insects. Phasmatodea either originated in Southeast Asia or in the New World. Our results suggest that the extant distribution of Phasmatodea is largely the result of dispersal events in a recently and rapidly diversified insect lineage rather than the result of vicariant processes.
Introduction
Exploring large-scale patterns of species diversity in time and space is a major research goal for evolutionary biology. Understanding the evolutionary processes generating these patterns requires broad comparative investigations of phenotypic attributes across taxa combined with well-corroborated phylogenies.
In recent years, numerous large-scale, highly resolved phylogenies for several insect lineages have been published and these have led to workable classifications for nearly all major groups traditionally referred to as insect orders, for instance, major groups of Polyneoptera (Wipfler et al., 2019), Blattodea (Evangelista et al., 2019), true bugs (Heteroptera) and other hemipteroid lineages (Johnson et al., 2018), Hymenoptera (Peters et al., 2017), Coleoptera (Zhang et al., 2018), Lepidoptera (Breinholt et al., 2018), and dipteran lineages (Pauli et al., 2018). Stick and leaf insects (Phasmatodea) however, stand out as one of the last remaining insect lineages traditionally referred to as orders for which a robust higher-level phylogenetic hypothesis is still lacking, as highlighted in numerous textbooks (e.g., Grimaldi and Engel, 2005; Tilgner, 2009; Beutel et al., 2014; Gullan and Cranston, 2014). Most recently, phasmatodean systematics were described as “muddled” and “burdened with much paraphyly and polyphyly” by Engel et al. (2016). This shortcoming largely impedes the study of evolutionary patterns within this group and yet, stick insects are an emerging model system in evolutionary biology (Brand et al., 2018, for overview, see also Bradler and Buckley, 2018).
Stick and leaf insects form a mesodiverse group of large, mostly nocturnal terrestrial herbivores with a mainly tropical and subtropical distribution. They exhibit impressive forms of camouflage directed against visually hunting predators by imitation of various parts of plants, such as twigs, bark and leaves (Figure 1). Over 3,100 described species are distributed across nearly 500 genera (Bradler, 2015; Bradler and Buckley, 2018). The relative large number of genera, of which around 175 are monotypic, reflects the degree of disparity regarding the morphological diversity found in Phasmatodea.

Figure 1. Various members of Neophasmatodea: (A–D): Occidophasmata; (E–I): Oriophasmata. (A), couple of Pseudosermyle phalangiphora, Diapheromerinae (Mexico); (B), couple of Oreophoetes peruana, Diapheromerinae (Peru); (C), female of Metriophasma diocles, Pseudophasmatinae (Panama); (D), couple of Peruphasma schultei, Pseudophasmatinae (Peru); (E), female of Carausius morosus, Lonchodinae (India); (F), female of Heteropteryx dilatata, Heteropterygidae (Malaysia); (G), couple of Eurycantha calcarata, Lonchodinae (New Guinea); (H), female of Extatosoma tiaratum, Lanceocercata (Australia); (I), couple of Diapherodes gigantea, Cladomorphinae (Grenada). Photos by Christoph Seiler, Altlussheim, Germany.
Over recent decades, the debate dealing with phasmatodean systematics was mostly devoid of an explicit phylogenetic basis. Instead, unmethodical classifications and numerous artificial groupings at various taxonomic ranks (e.g., Bradley and Galil, 1977; Zompro, 2004; Brock and Marshall, 2011) resulted in a highly chaotic taxonomy with incorrectly appointed taxa still recognized in the current Phasmida Species File database (Brock et al., 2017).
This situation has now been improved by numerous phylogenetic analyses of molecular datasets being published (Whiting et al., 2003; Buckley et al., 2009, 2010; Bradler et al., 2014, 2015; Goldberg et al., 2015; Robertson et al., 2018). However, datasets initially were too restricted to allow reliable conclusions for broader phasmatodean systematics. For instance, Whiting et al. (2003) and Buckley et al. (2009) did not recover monophyletic groups, such as Heteropterygidae, that have now been strongly supported as monophyletic with increased taxon sampling (Bradler et al., 2015; Goldberg et al., 2015; Robertson et al., 2018). Even the analyses of mitochondrial genomes (Kômoto et al., 2011, 2012; Tomita et al., 2011; Zhou et al., 2017) did not allow for any general systematic conclusion due to their non-representative, negligible taxon sampling.
In other cases, geographical distribution rather than morphological similarity appeared to reflect the evolutionary relationships among stick and leaf insects. Thus, phylogenetic inferences of the phasmatodean faunas of New Caledonia, New Zealand, the Mascarene Archipelago and Madagascar (Buckley et al., 2009, 2010; Bradler et al., 2015; Robertson et al., 2018) rendered several traditional groupings as polyphyletic.
In summary, the published studies based on Sanger-sequencing approaches have resulted in some sound topologies in regard to shallower nodes, but were unable to resolve the deeper nodes of the presumed rapid radiation of Phasmatodea (Bradler et al., 2014). Therefore, much of the early evolutionary history, such as the relationships between families, subfamilies, and some perpetually enigmatic taxa such as Agathemera, was difficult to address in the past and is still a matter of debate (Bradler and Buckley, 2018).
In order to illuminate these previously unresolved deep nodes of phasmatodean phylogeny and to test previous conflicting topologies, we conducted a phylogenomic study based on 27 novel transcriptomes of a representative set of stick and leaf insects and combined these with previously published transcriptomes of 11 phasmatodeans and 23 outgroup polyneopterans (Misof et al., 2014; Wipfler et al., 2019). We estimated divergence times for our new phylogeny using a validated set of fossils as calibration points (Bradler et al., 2015) and reconstructed the historical biogeography using explicit models. Whereas in the past phylobiogeographic research addressing Phasmatodea was restricted to specific geographic areas such as Australia, New Caledonia, and New Zealand (Buckley et al., 2010), our present study is the first to formally infer the historical biogeography for the whole order, albeit with the shortcoming of lacking some African taxa.
Methods
Dataset Generation
Our dataset comprised 38 species representing all major clades of Phasmatodea and 23 outgroup species including all other Polyneoptera orders (Supplementary Table S1). Data was derived from transcriptomes, except for the termite Zootermopsis nevadensis for which we used the official gene set obtained from a whole genome project (Terrapon et al., 2014). For most species, we sampled the RNA from the head and thorax of adult specimens (see Supplementary Table S1.1). Further detailed information (e.g., sex, collection date, collector, etc.) can be found at the National Center for Biotechnology Information (NCBI) under the 1KITE umbrella project and the respective BioSample number (Supplementary Table S1.1). RNA extraction and cDNA library preparation, transcriptome sequencing (HiSeq 2000 platform with 150 bp paired-end (PE) reads), and de novo assembly (SOAPdenovo-Trans-31kmer; Xie et al., 2014) were conducted at the Beijing Genomics Institute (BGI) Shenzhen and are described in detail by Peters et al. (2017). Transcriptome quality assessment, removal of contaminants and submission to the NCBI Sequence Read Archive (SRA) as well as to the Transcriptome Shotgun Assembly (TSA) database were conducted as described in Peters et al. (2017) and under the 1KITE umbrella project (Supplementary Tables S1.1, S1.2).
The phylogenomic pipeline is described in detail in Evangelista et al. (2019) (Supplementary Material). Briefly, for the identification of orthologous transcripts we used the custom-made ortholog set, specifically designed for Polyneoptera taxa and comprising 3,247 orthologous genes/groups (OGs) for the four reference species Ephemera danica, Ladona fulva, Zootermopsis nevadensis, and Rhodnius prolixus. Transcripts of each query taxon were assigned to these ortholog groups (OGs) with Orthograph v.0.5.4 (Petersen et al., 2017) with following settings: max-blast-searches = 50; blast-max-hits = 50; extend-orf = 1; substitute-u-with = X. We identified on average 2,274 OGs in the transcriptomes (minimum: 637; maximum: 2,861) (Supplementary Table S2). OGs were aligned applying the L-INS-i algorithm of MAFFT v.7.221 (Katoh and Standley, 2013) at the translational (amino-acid) level. Each multiple sequence alignment (MSA) was quality assessed and outliers were removed using the procedure outlined by Misof et al. (2014) but using the -addfragments algorithm implemented in MAFFT. Subsequently, we excluded the sequences of three reference species Ephemera danica, Ladona fulva, and Rhodnius prolixus since we aimed to include only Polyneoptera taxa for the phylogenetic inference, and removed columns resulting in only gaps. Subsequently, MSAs of nucleotides (nt) corresponding to the amino-acid (aa) MSAs were generated with a modified version of the software PAL2NAL (Suyama et al., 2006; see Misof et al., 2014). We further used the Pfam-A database release 28.0 (Punta et al., 2012) in conjunction with the software pfam_scan.pl v.1.5 and HMMER (Eddy, 2011; http://hmmer.org/), Domain-identification-v1.3 and Domain-parser-v1.4.1-dist, to identify regions in the MSAs annotated as protein clans, families, single domains or non-annotated regions (so called voids, see also Bank et al., 2017). In parallel, we identified putative alignment ambiguities or randomized MSA sections within each aa MSA with Aliscore v.1.2 (Misof and Misof, 2009) (options -e -r) and combined this information with the results from the protein domain identification step to generate a supermatrix. We followed two approaches to create matrices for the phylogenetic analyses: (1) we used MARE v.0.1.2-rc (Misof et al., 2013) to remove taxa or partitions with lowest average information content (IC) from the aa supermatrix, yielding a selected optimal subset (SOS) (for rationale see also Meusemann et al., 2010) and defining all phasmatodeans as taxon constraints; thus, they were not dropped from the matrix (AASOS: 61 taxa, 837,567 aa positions, 2,773 partitions); (2) we only kept partitions with IC > 0 as identified by MARE removing partitions with an IC = 0 from the aa and nt supermatrix. Thus, we further increased data coverage by including only data blocks, i.e., that contained sequence information for at least one representative of specified taxonomic groups (Supplementary Table S3) resulting in a decisive aa dataset (sensu Dell'Ampio et al., 2014; AAdecisive: 61 taxa, 387,987 aa positions, 388 partitions). The specified taxonomic groups for Phasmatodea represented uncontroversial monophyletic groups according to previously published studies. For the corresponding decisive nt dataset we subsequently evaluated whether or not our datasets have evolved under globally stationary, reversible, and homogeneous (SRH) conditions with SymTest v.2.0.47 (Ho and Jermiin, 2004) (see for rationale also Evangelista et al., 2019). We applied the in SymTest implemented Bowker Test on the 1st, 2nd, and 3rd codon position separately, on the 1st + 2nd, and on all codon positions. Further downstream analyses were performed on the decisive nucleotide dataset keeping the 2nd codon position only (NTdecisive: 61 taxa, 387,987 nt positions, 388 partitions), as this showed smaller among-lineage heterogeneity compared to the other nt datasets (see Supplementary Figure S1).
For the two amino-acid datasets, we additionally evaluated the coverage with respect to pairwise sequence coverage of unambiguous data using AliStat v.1.6 (https://github.com/thomaskf/AliStat) (see also Misof et al., 2014; Wong et al., 2017). Details are provided in Supplementary Figure S2.
Phylogenetic Analyses
Prior to the tree inference, we optimized our partitioning scheme and searched for the best-scoring substitution models for the two aa datasets (AASOS and AAdecisive) by using PartitionFinder v.2.0.0 (prerelease 10) (http://www.robertlanfear.com/partitionfinder/; Lanfear et al., 2014, 2016) in combination with RaxML v.8.2.4 (Stamatakis, 2014) (options –rcluster –rcluster-max 6000 (for AASOS) –rcluster-max 2000 (for AAdecisive) –rcluster-percent 100 -q -p 24 –weights 1,1,0,1 –all-states –min-subset-size 100). We further restricted the PartitionFinder search to 11 amino-acid substitution models as these are the most selected models for empirical studies on Hexapoda (Misof et al., 2014; Peters et al., 2017; Pauli et al., 2018), namely LG+G, WAG+G, DCMUT+G, JTT+G, BLOSUM62+G, LG+G+F, WAG+G+F, DCMUT+G+F, JTT+G+F, BLOSUM62+G+F, LG4X (Yang, 1994; Gu et al., 1995; Müller and Vingron, 2000; Whelan and Goldman, 2001; Veerassamy et al., 2003; Kosiol and Goldman, 2005; Le and Gascuel, 2008; Soubrier et al., 2012).
In order to find the best-scoring substitution model for each partition of the nt dataset (NTdecisive), we applied ModelFinder as implemented in IQ-TREE v.1.5.1 (Kalyaanamoorthy et al., 2017) (options –m TESTNEWONLY –gmedian). Please note that the boundaries of the partitions identified on amino-acid level are equivalent to the boundaries we kept for the decisive nucleotide dataset.
Phylogenetic relationships were inferred under the maximum likelihood (ML) optimality criterion as implemented in IQ-TREE v.1.3.11 and v.1.4.4 (Nguyen et al., 2015; Chernomor et al., 2016) and by using the best-scoring amino-acid substitution matrix for each partition (option–spp). We performed 50 independent ML tree searches with a random start tree for all three datasets, taking the median for each rate category (–gmedian) and an increased number of unsuccessful iterations before stopping (–numstop 200). Note that for each independent tree search, there were in total 100 initial random trees generated. To assess the number of unique topologies present within the 50 inferred trees, we used the software UniqueTree v.1.9. For each dataset, the 50 independently inferred tree showed all the same, i.e., one unique topology. However, there were topological differences between these three topologies, see discussion below.
Branch support was estimated via non-parametric bootstrapping of 100 bootstraps alignments in IQ-TREE and mapped onto the ML tree with the best log-likelihood. To assess the minimum number of replicates needed for a reliable estimation of bootstrap support, we subsequently used the “autoMRE” bootstrap convergence criterion (Pattengale et al., 2010) as implemented in RAxML with default settings. Bootstrap convergence was reached after 50 replicates for all three datasets in all tests.
Analyses of Phylogenetic Signal
In addition to the non-parametric bootstrap support, we determined support for the deeper phylogenetic relationships with the aid of Four-cluster Likelihood Mapping (FcLM) (Strimmer and von Haeseler, 1997), see Simon et al. (2018) and Supplementary Information in Misof et al. (2014) for details on the strategy. We applied the FcLM analyses on the decisive amino-acid dataset (AAdecisive) and tested all major relationships of the Phasmatodea lineages (see Supplementary Figure S3). For all 17 splits/relationships in question, we defined four groups and included only partitions for which at least one representative species of each of the four addressed groups was present. Taxa that did not address a particular hypothesis were discarded from the alignment (see Supplementary Table S4 for included species, group definitions, and number of drawn quartets). For two incongruent nodes based on the tree inferences (see section Results and Discussion), we additionally checked for confounding signal due to among-lineage heterogeneity, non-random substitution processes and/or distribution of missing data using the FcLM approach with permuted datasets with phylogenetic signal destroyed. FcLM analyses were performed using IQ-TREE v.1.4.2 (Nguyen et al., 2015).
Divergence Time Estimations
For divergence time estimations, we used five fossils (Table 1) representing the oldest known fossils for the respective groups. The reported calibration points thus provide a lower limit to the age of each group. We modeled all fossil constraints as an exponential distribution with a rigid lower bound equal to the age of the fossils and a soft upper bound so that 95% of the distribution lies between the age of the fossil and the end of the Triassic (201.3 million years ago [mya], fossil 1), the end of the Jurassic (144.9 mya, fossil 2), and the end of the Paleogene (66.4 mya, fossils 3–5). For the root, a rather conservative prior estimate of the divergence date was specified, using a uniform distribution with rigid lower and upper bound indicated by the begin of the Permian (298.8 mya) and the end of the Jurassic. For the divergence date inference, we compiled a reduced version of the decisive amino-acid dataset (AAdecisive) only containing sites with unambiguous data for at least 95% of the 61 taxa (AAdecisive95) comprising 31,298 aa positions. To reduce computational effort, we chose an unpartitioned dating analysis. Divergence time estimates were performed in the program BEAST v.1.8.2 (Drummond et al., 2012) with a JTT+G+I substitution model, an uncorrelated lognormal relaxed clock, and a Yule tree prior. The JTT+G+I substitution model was determined in ModelFinder as implemented in IQ-TREE as best-scoring model of the available models implemented in BEAST for the reduced unpartitioned dataset. Markov Chain Monte Carlo analyses were conducted on the fixed topology obtained by the IQ-TREE ML analyses. Four separate runs for 50 million generations were conducted and sampled every 5,000 generations. The first 10 million generations of each run were discarded as burn-in as indicated by the software Tracer v.1.7.6 (Rambaut et al., 2018), which was also used to inspect convergence and mixing of parameters and the effective sample sizes (ESS). Post-burn-in samples were combined across all four runs to summarize parameter estimates and were used to construct a maximum clade credibility tree with median node heights in TreeAnnotator v.1.8.2 (Drummond et al., 2012). All BEAST analyses were conducted using the CIPRES portal (Miller et al., 2015).
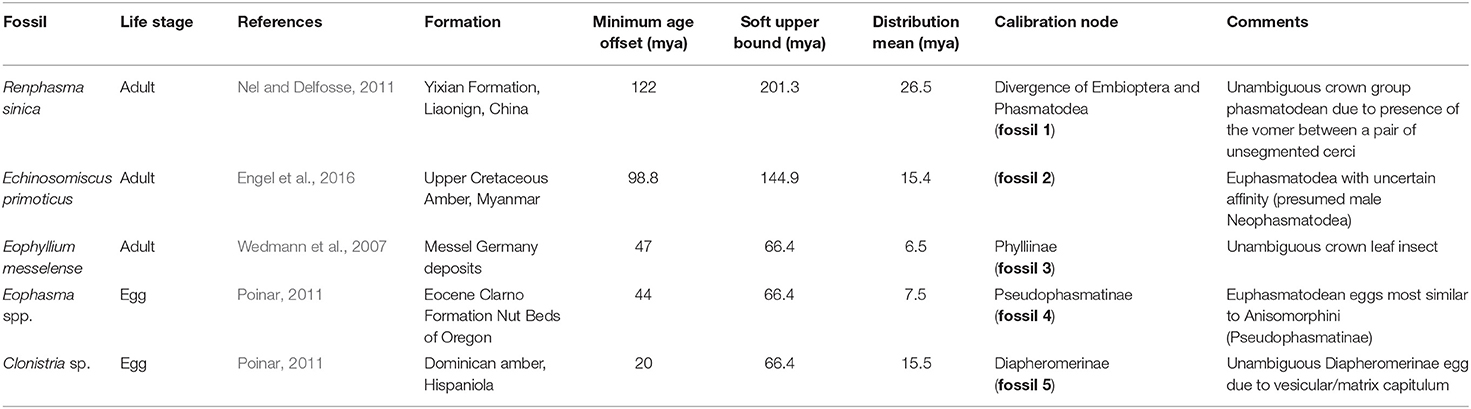
Table 1. Fossil Calibrations used in the divergence time estimate (modified from Robertson et al., 2018), fossils numbered according to Figure 2.
Ancestral Range Reconstruction
For the reconstruction of ancestral ranges, we determined five areas according to the current distribution of the 38 extant phasmatodean species and following Wallace's zoogeographical regions by Holt et al. (2013): New World (Nearctic + Neotropical), Palearctic, Madagascan, Australian (including Oceanian), and Oriental. Based on the dated phylogenetic tree and allowing a maximum of two areas to be occupied, biogeographic models were compared using the maximum likelihood approach implemented in the R package BioGeoBEARS v.1.1.2 (Matzke, 2013, 2018). To cover a variety of different settings concerning dispersal, extinction, sympatry, and vicariance, we conducted the analysis using three models, namely, the Dispersal-Extinction-Cladogenesis model (DEC) and the likelihood interpretations of the models DIVA and BayArea (DIVALIKE and BAYAREALIKE). BayArealike features a parameter for widespread sympatry, DEC one for subset sympatry, and DIVALIKE one for widespread vicariance. Each model was also run with the +J parameter accounting for jump dispersal and founder-event speciation (Matzke, 2014). The best fitting model was assessed with the Likelihood-Ratio-Test (LRT), the Akaike Information criterion (AIC) and corrected AIC (AICc).
Results and Discussion
Phylogenetic Relationships
Overall, our phylogenomic reconstruction received strong support in all analyses and significantly alters previous ideas regarding the evolutionary history of Phasmatodea, but at the same time corroborates clades that appeared well-supported in past molecular studies, both within and outside Phasmatodea (see Figure 3 for comparison). All trees were rooted with the clade Zoraptera + Dermaptera according to Wipfler et al. (2019). Previously observed topologies by Wipfler et al. (2019) were fully corroborated, e.g., Plecoptera as sister group to a polyneopteran clade comprising Orthoptera, Blattodea, Mantodea, Phasmatodea, Embioptera, Mantophasmatodea, and Grylloblattodea. We also received in all three tree inferences support for Dictyoptera (Mantodea + Blattodea) as sister group to Xenonomia (Grylloblattodea + Mantophasmatodea) + Eukinolabia (Embiotera + Phasmatodea) (Figure 2; Supplementary Figures S4, S5). The only notable topological differences between the three phylogenetic analyses were observed within Embioptera in regard to the position of Rhagadochir, and within Phasmatodea in regard to the positions of the two phasmatodean taxa Agathemera and Heteropteryx (see discussion below). Results of all FcLM analyses testing all major nodes connecting Phasmatodea are mainly compatible with the ML tree reconstructions showing that they are robust against taxon sampling (Supplementary Figure S6). There are only two exceptions, which are further discussed and evaluated in Supplementary Data Sheet 1.
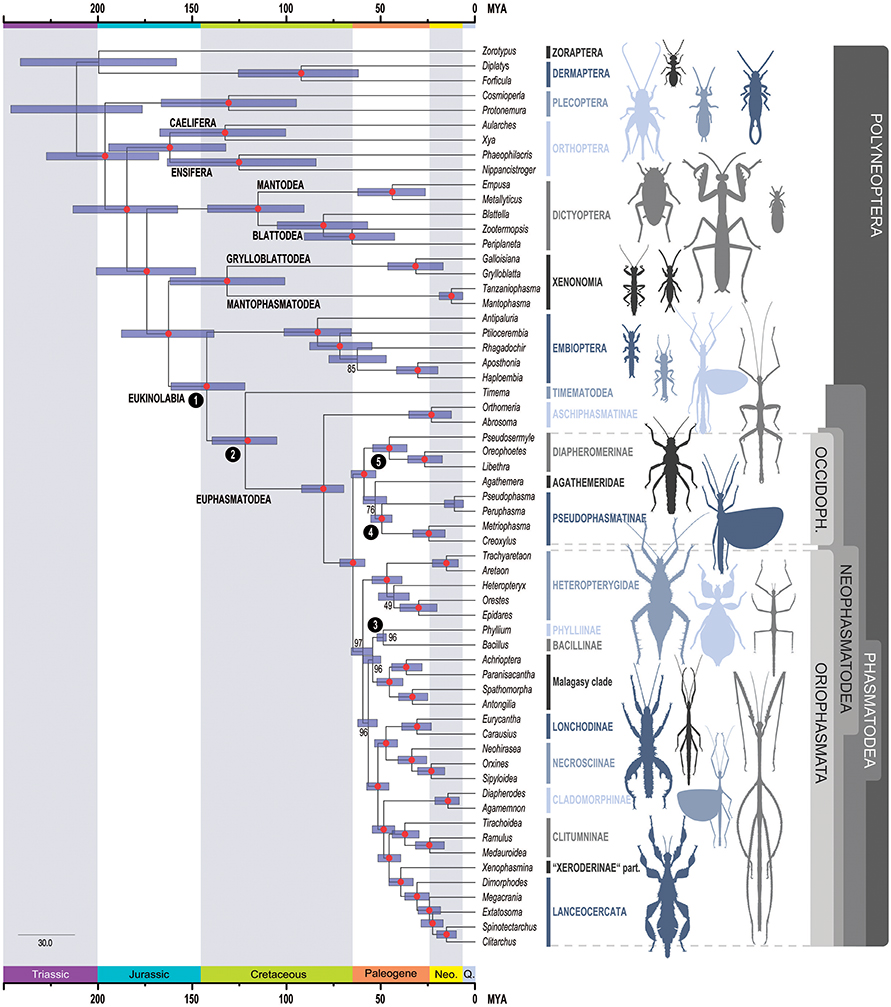
Figure 2. Time-calibrated phylogeny of Phasmatodea. Inferred phylogenetic relationships based on dataset AAdecisive (387,987 aa positions, 205 metapartitions). Colored circles represent bootstrap support derived from 100 BS. Node dates (posterior mean) were inferred using the dataset AAdecisive reduced to sites containing >95% data completeness, and five fossil calibrations. Error bars represent 95% confidence intervals. Fossils used for calibrations are indicated as numbers in black circles at nodes: 1: Nel and Delfosse (2011), 2: Wedmann et al. (2007), 3: Sellick (1994), 4, 5: Poinar (2011) (see also Table 1). OCCIDOPH., Occidophasmata; Neo., Neogene; Q., Quaternary.
Within Phasmatodea, the species-poor Timema (= Timematodea) with 21 described species in western North America and species-rich Euphasmatodea (over 3,000 described species with worldwide distribution) form the basal sister groups, as has been demonstrated in a plethora of studies before (for an overview see Bradler and Buckley, 2018). The Euphasmatodea are divided into Aschiphasmatodea and Neophasmatodea as suggested by Engel et al. (2016). Our study thus confirms the idea of Aschiphasmatodea (= Aschiphasmatinae) forming the sister group to all remaining euphasmatodeans, which originally stems from the cladistic analysis of morphological characters by Tilgner (2002). In the past, this was controversially discussed (Bradler et al., 2003), but also recovered most recently by Robertson et al. (2018).
The subdivision of Neophasmatodea gives rise to an Old World and New World clade of stick and leaf insects, which was previously unsuspected. For these well-supported major clades we introduce the new rank-free taxon names Oriophasmata tax. nov. (“Eastern phasmids,” Old World) and Occidophasmata tax. nov. (“Western phasmids,” New World). There has never been any taxonomic scheme or morphological character presented that suggested these clades, in consequence, neither have there ever been alternative robust phylogenetic hypotheses postulated for their major subgroups that would question this novel assumptions.
The New World clade Occidophasmata comprises the two species-rich and dominant stick insect groups of South and North America, the Diapheromerinae and Pseudophasmatinae (= Pseudophasmatidae by some authors), which were originally assigned to the two traditional suborders “Areolatae” (Pseudophasmatinae) and “Anareolatae” (Diapheromerinae), based on the presence or absence of a triangular field at the apex of the tibiae (Günther, 1953). While all previous phylogenetic studies already demonstrated that the traditional subdivision of Phasmatodea into “Areolatae” and “Anareolatae” is meaningless, there is not a single morphological character known to support a close relationship of Diapheromerinae and Pseudophasmatinae. Yet, this clade receives support from a biogeographical point of view considering that we have seen in the past that biogeography generally plays a more important role for the evolutionary history of stick insects than morphological similarity (Buckley et al., 2009; Bradler et al., 2015). This New World clade also comprises Agathemera, a wingless South American taxon with a stout body form and eight described species from Chile and Argentina (Domínguez et al., 2009, Vera et al., 2012). The phylogenetic position of Agathemera has been an evolutionary enigma. Based on morphological evidence, Agathemera was repeatedly placed as sister group to all remaining Euphasmatodea (Bradler, 2000, 2009; = Neophasmatidae sensu Bradler, 2003; = Verophasmatodea sensu Zompro, 2004; Klug and Bradler, 2006; Klug, 2008; Friedemann et al., 2012). However, this assumption and consequently the monophyly of Verophasmatodea have never been supported by molecular studies, which place Agathemera as a subordinate taxon, albeit with unstable position among stick insects (Whiting et al., 2003; Buckley et al., 2009; Bradler et al., 2014, 2015; Goldberg et al., 2015; Büscher et al., 2018). Most recently, Agathemera has been found to be the sister group to Pseudophasmatinae including Heteronemiini and Prisopidini (Robertson et al., 2018), thus basically re-erecting the Pseudophasmatinae that included all these taxa as originally proposed by Günther (1953). Our results partially support this view here by a placement of Agathemera as sister to Pseudophasmatinae in the trees inferred from the datasets AAdecisive (Figure 2) and NTdecisive (Supplementary Figure S4), yet this is one of the lesser supported clades (BS = 76 and BS = 94, respectively), and our analysis does not include members of Prisopodini and Heteronemiini. In contrast, trees inferred from the dataset AASOS support Agathemera as sister group to a clade comprising Pseudophasmatinae and Diapheromerinae (Supplementary Figure S5). However, further FcLM analyses in combination with permutation tests show that the position of Agathemera as sister to Pseudophasmatinae in the trees inferred from the datasets AAdecisive could not be explained by confounding signal. In contrast, the position of Agathemera as sister group to a clade comprising Pseudophasmatinae and Diapheromerinae inferred by the dataset AASOS may be caused by the presence of confounding signal (e.g., heterogeneous amino-acid site composition, non-stationary substitution processes, or non-random distribution of missing data). For details refer to Supplementary Material Data Sheet 1.
Furthermore, we recovered a subdivision of Pseudo-phasmatinae (or Pseudophasmatidae) into Pseudophasmatinae (Peruphasma, Pseudophasma) and Xerosomatinae (Creoxylus, Metriophasma). Within Diapheromerinae, the subdivision into Oreophoetini (Libethra, Oreophoetes) and Diapheromerini (Pseudosermyle) is supported (Robertson et al., 2018).
The Oriophasmata comprise all remaining neophasmatodean taxa with Heteropterygidae as sister group to the rest. Within Heteropterygidae, the Obriminae (Aretaon, Trachyaretaon) and Dataminae (Epidares, Orestes) (traditionally referred to as Obrimini and Datamini, see Bradler and Buckley, 2018) were recovered, and Heteropteryx (representing the third subgroup Heteropteryginae or, traditionally, Heteropterygini) as sister to Dataminae in the trees inferred from the dataset AAdecisive (Figure 2). The latter clade, Dataminae + Heteropteryginae, gained extremely low support (BS = 49), but was repeatedly recovered before (Büscher et al., 2018; Robertson et al., 2018). In contrast, Bradler et al. (2015) obtained a topology Dataminae + (Heteropteryginae + Obriminae), which was also favored based on phylogenetic analysis of morphological characters by Bradler (2009) and also in our study supported by the trees inferred from the datasets NTdecisive and AASOS (Supplementary Figures S4, S5). The third possible topology Heteropteryginae + (Obriminae + Dataminae), as proposed by Zompro (2004), was favored by one previous molecular phylogeny (Goldberg et al., 2015) but was not recovered by any of our phylogenetic inferences. Additional FcLM analyses in combination with permutation tests revealed that the inferred relationship by the dataset AASOS (Heteropteryx and Obriminae being closest relatives) is strongly biased by confounding signal. The FcLM analyses further supported a sister group relationship of Heteropteryx with Dataminae which is less prone to confounding signal in the dataset AAdecisive as well as AASOS (for details, please refer to Supplementary Material Data Sheet 1). However, given the low number of possible drawn quartets (88) and the low support observed in the present analysis for a sister group relationship of Heteropteryx with Dataminae, the internal phylogeny of Heteropterygidae must still be considered an open question.
Within the remaining Oriophasmata, Bacillus + Phyllium form an early branch together with the Malagasy clade of stick insects. The subordinate phylogenetic placement of Phylliinae, the true leaf insects (represented here by Phyllium philippinicum), within these Old World Euphasmatodea, is in contrast to recent molecular phylogenies that favor Phylliinae to be a rather early diverging lineage among Euphasmatodea (Kômoto et al., 2011; Bradler et al., 2015; Robertson et al., 2018). Again, our finding corroborates the view of Günther (1953) who considered Phylliinae to be just one subordinate subfamily of many within Phasmatodea. A close affinity between Phylliinae and members of Bacillinae has never been postulated before, yet we received moderate support (AAdecisive: BS = 96; NTdecisive: BS = 80; AAsos: BS = 100) for a sister group relationship of Phyllium and the European Bacillus. However, the FcLM analyses and further permutation tests of the two amino-acid datasets (AAdecisive, AAsos) revealed that the tree reconstructions might be biased by confounding signal (for details refer to Supplementary Material Data Sheet 1). Nevertheless, this grouping receives support from a biogeographical point of view: Although the extant Phylliinae have a predominantly Asian distribution, the fossil record provides evidence of a European origin of leaf insects with the 47-million-years-old Eophyllium, the extinct sister group of all remaining leaf insects, described from Grube Messel in Germany (Wedmann et al., 2007).
A monophyletic Malagasy clade comprises Anisacanthidae (Paranisacantha), Achriopterini (Achrioptera), Antongiliinae (Antongilia), and Phasmatidae taxa incertae sedis (Spathomorpha), with the former two and the latter two forming sister taxa in accordance with previous results (Bradler et al., 2015; Goldberg et al., 2015; Büscher et al., 2018; Robertson et al., 2018; Glaw et al., 2019). Closely related Malagasy stick insects were only recently suggested but not recovered as monophyletic when certain African taxa were included that are lacking in the present analysis (Bradler et al., 2015; Robertson et al., 2018).
One major radiation within Oriophasmata comprises Lonchodidae (Lonchodinae + Necrosciinae, Robertson et al., 2018) that contain over 1,000 described species, thus accounting for one third of the described extant phasmatodean diversity. The sister lineage of Lonchodidae contains taxa that have been recovered multiple times before as monophyletic groups including Lanceocercata and its sister taxon Stephanacridini, Cladomorphinae, Medaurini (Medauroidea), Clitumnini (Ramulus), and Pharnaciini (Tirachoidea) (Buckley et al., 2009, 2010; Bradler et al., 2014, 2015; Büscher et al., 2018; Robertson et al., 2018), however, with a slightly different topology in our tree based on transcriptomes.
Monophyly of Lanceocercata with Dimorphodes as sister group to all remaining Lanceocercata appears to be undisputed (Buckley et al., 2009, 2010; Bradler et al., 2014, 2015; Büscher et al., 2018; Robertson et al., 2018). In previous studies, Stephanacridini were usually observed as sister taxon to Lanceocercata. Here, we obtain Xenophasmina as sister to Lanceocercata, a taxon currently considered to be a member of the Xeroderinae. Since Xeroderinae are an obviously polyphyletic assemblage (Buckley et al., 2009; Bradler et al., 2015), Xenophasmina must be considered as a taxon incertae sedis that might be a member of Stephanacridini, which needs to be corroborated in subsequent analyses.
Surprisingly, we infer monophyletic Clitumninae as erected by Hennemann and Conle (2008), to be comprised of Clitumnini, Medaurini, and Pharnaciini, as well as to be more closely related to Lanceocercata than the Neotropical Cladomorphinae. This is in sharp contrast to numerous previous results that concordantly inferred polyphyletic Clitumninae and suggested a well-supported lineage of Cladomorphinae + (Stephanacridini + Lanceocercata) (Buckley et al., 2009, 2010; Bradler et al., 2014, 2015; Büscher et al., 2018; Robertson et al., 2018). This disagreement needs to be further addressed in future analyses by adding missing taxa such as Stephanacridini and members of Gratidiini that were demonstrated to be closely related to Medaurini and Clitumnini in the past (Bradler et al., 2014, 2015; Büscher et al., 2018; Robertson et al., 2018).
Divergence Time Estimation
According to our analyses, extant Phasmatodea started to diversify in the Mesozoic with the split between Timema and Euphasmatodea lying in the Early Cretaceous (95% confidence interval [CI]: 105.1–139.4 mya; mean: 121.8 mya, Figure 2), with the oldest currently known fossils assigned unambiguously to stick insects being from the Mesozoic of China, ~128 mya (Wang et al., 2014). This estimation is older than previous estimates (see Figure 3 for comparison), for instance, ~103 mya (CI: 85.52–122.12 mya, Bradler et al., 2015) or ~95 mya (Buckley et al., 2009). The overall divergence time estimates for the early splits of stick and leaf insects are older than in previous studies, for instance, ~61.26 mya (CI: 51.04–75.43 mya) for the earliest node within Euphasmatodea according to Bradler et al. (2015), and indicate a diversification of Euphasmatodea around 80.3 mya (CI: 69.6–92.0 mya), which constitutes the split between Aschiphasmatodea and Neophasmatodea in our study. Neophasmatodea further radiated into Orio- and Occidophasmata around 64.8 mya (CI: 58.4–71.8 mya). Radiation of Occidophasmata in North and South America began ~58.9 mya (CI: 52.6–65.7 mya), with Agathemera being a separate, old genus since about 52.9 mya (CI for the split from Pseudophasmatinae: 46.8–59.4 mya).
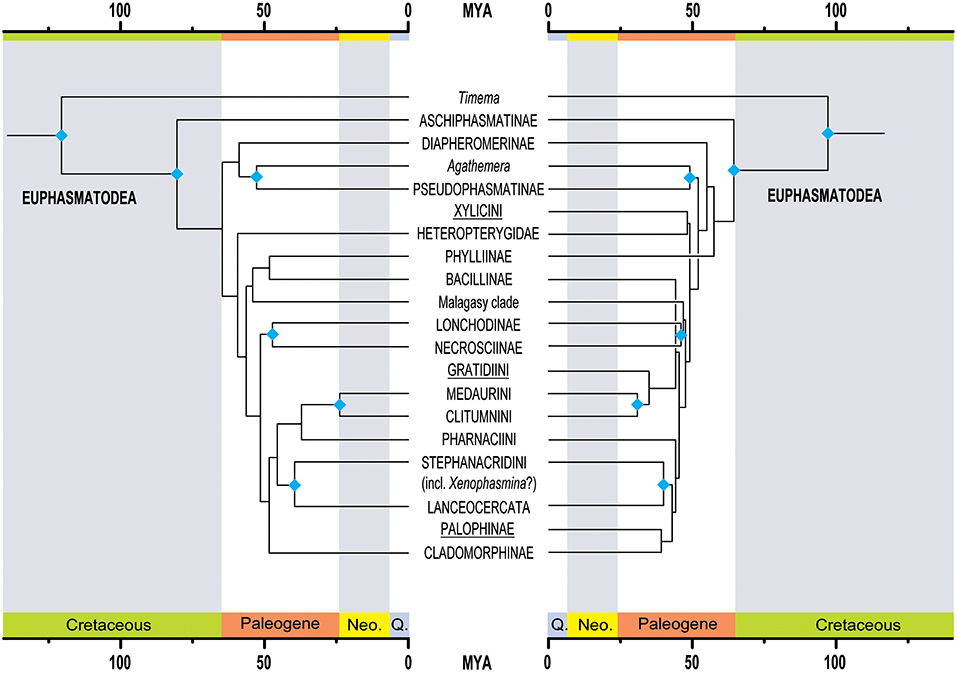
Figure 3. Comparison of topology and divergence time estimations between the herein presented new phylogeny of Phasmatodea (left side) to the most recent and comprehensive phylogeny based on Sanger sequence data by Robertson et al. (2018). The underlined taxa are African lineages included in the study from Robertson et al. (2018) but missing in the present study. The blue rhomb symbols indicate taxa supported in both studies. Neo., Neogene; Q., Quaternary.
Within Oriophasmata, radiation started approximately at the same time, namely, 59.5 mya (CI: 54.2–65.6 mya), when the ground-dwelling Heteropterygidae split off from the remainder of this clade. The start of Heteropterygidae radiation is estimated at ~46.8 mya (CI: 38.7–54.6 mya), which largely confirms the date recovered by Robertson et al. (2018) who gave a mean of 46.4 mya (between 53 and 40 mya, see Figure 3).
The Malagasy clade radiation started between 38.2 and 52.1 mya (mean: ~46.8 mya) with the split from its sister group occurring around 56.6 mya (CI: 51.8–62.2 mya), which is most probably much younger than the separation of Madagascar from any significant landmass (65–96 mya; Vences et al., 2009). This corroborates the previous assumption that stick insects colonized this fragmentary island long after it became isolated, between 38 and 51 mya (Bradler et al., 2015).
Further significant radiation dates within Oriophasmata include Lonchodidae at 47.3 mya (CI: 41.1–53.3 mya), constituting the split between Lonchodinae and Necrosciinae, and Clitumninae at 37.3 mya (CI: 29.7–44 mya). The Neotropical Cladomorphinae diverged from the Australasian stick insects between 42.6 and 54.5 mya (mean: 48.4 mya). This lies within the dates for the rifting of Antarctica from Australia and South America, which was implicated as the origin for the split of the New World Cladomorphinae from their Australasian sister taxon (Buckley et al., 2010). However, the biogeographic reconstructions favor an orientally distributed ancestor (see below).
The Lanceocercata radiation is relatively young, starting around 30.6 mya (CI: 24.3–37.3 mya), which is in the range of previous estimates (or younger) spanning from 32 mya (CI: 29.4–37.5 mya, Buckley et al., 2009) over ~40 mya (Bradler et al., 2015) up to 51.76 mya (36.23–69.78 mya, Buckley et al., 2010), the latter based on the arthropod molecular clock rates suggested by Brower (1994), i.e., without fossil calibrations.
In summary, our novel divergence time estimation generally supports the previous molecular clock analyses that suggested the extant radiation of stick and leaf insects to be rather young, having occurred largely after the Cretaceous–Tertiary (K-T) boundary (~66 mya) (Buckley et al., 2009; Bradler et al., 2015; Robertson et al., 2018), although some early splits and some lower 95% confidence interval boundaries fall into the Late Cretaceous. The reason for the successful, rather recent radiation of Euphasmatodea is not yet fully understood. It has been partly explained by the co-evolution of phasmatodeans with angiosperms (Archibald and Bradler, 2015) although similar ecological associations such as predator-prey interactions encountered by plant-mimicry were already found in Cretaceous stick insects living in gymnosperm forests (Wang et al., 2014). Other studies suggested that horizontal gene transfer of pectinase genes from gut bacteria into the genome of stick insects (Shelomi et al., 2016) or that the development of hard-shelled seed-like eggs (Robertson et al., 2018) were key innovations explaining the recent success of Euphasmatodea. These properties surely enhanced their ability to mimic and feed on angiosperm plants, and most probably a combination of these traits might be accountable for the impressive euphasmatodean diversification.
Historical Biogeography
Our biogeographical model selection analysis under the maximum likelihood optimization criterion in BioGeoBEARS resulted in DIVALIKE +J being the best fitting model and, generally, showed a clear preference for the three models incorporating the J parameter indicating the significance of potential jump dispersal and founder events (see Supplementary Table S5 for details on the results).
The ancestral geographic range at the root of Phasmatodea was equivocally reconstructed to be New World (Nearctic + Neotropical) and Oriental, and must be considered unresolved (Figure 4). The recent report of Burmese amber fossils interpreted as mid-Cretaceous members of Timematodea (Chen et al., 2018) would further support an Oriental origin of Phasmatodea, but a critical re-evaluation of this finding is still pending. Every following node is clearly resolved. However, while the ancestral region of Aschiphasmatinae and Neophasmatodea is depicted as Oriental according to the best-fitting model DIVALIKE +J, the estimations calculated by the models DEC and BAYAREALIKE (with and without parameter J) show the node as unresolved (see Supplementary Data Sheet 2). Similarly, the ancestral range of Neophasmatodea is Oriental according to the best-fitting model, but unresolved for some of the other models, whereas the origin of Oriophasmata is unambiguously reconstructed as Oriental and that of Occidophasmata Nearctic/Neotropical.
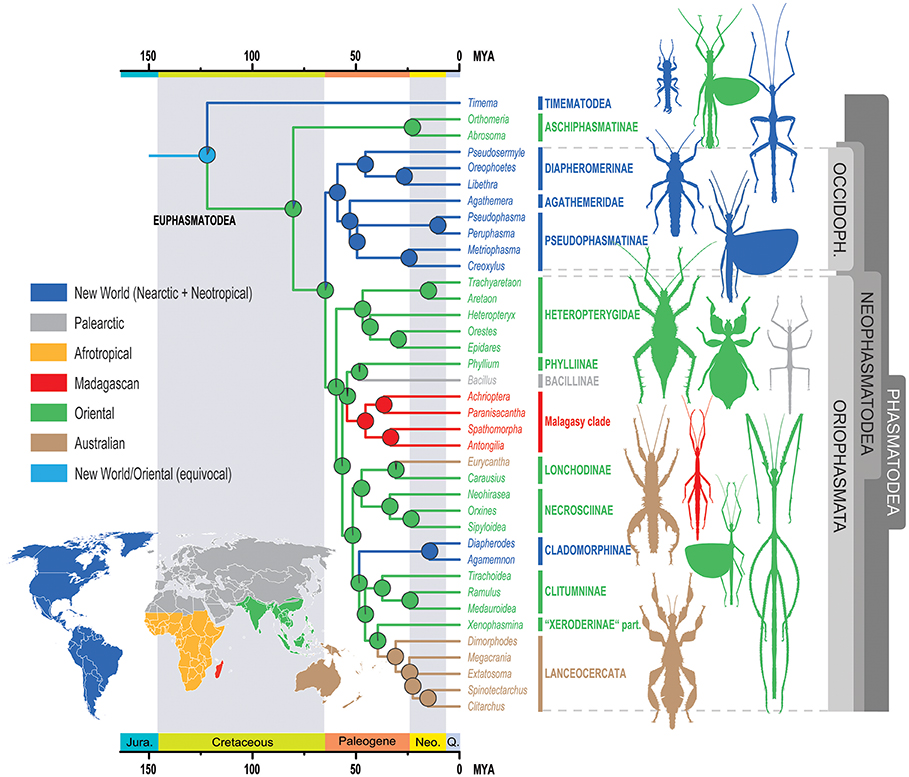
Figure 4. Ancestral range estimates of Phasmatodea for the BioGeoBEARS DIVALIKE+J model. The nodal pie charts show the relative probability of the geographic ranges according to the in-figure color code. OCCIDOPH., Occidophasmata, Jura., Jurassic, Neo., Neogene, Q., Quaternary.
Beyond this ambiguity at the base of the tree, reconstruction of the historical biogeography appears to be rather straightforward. On account of the young age of the recovered phasmatodean lineages, their geographic distribution must be explained by dispersal events rather than vicariant processes. A few major colonization events out of Southeast Asia can be observed: A single colonization of Madagascar, although this might have occurred via Africa (Bradler et al., 2015) with the African taxa in question lacking in our study. Furthermore, Southern Europe was colonized once (Bacillus) by a lineage probably also including the leaf insects (Phylliinae) with a potential European origin (see above). The Cladomorphinae of Central and South America also originated in Southeast Asia and can be interpreted as the result of a transoceanic, most probably trans-Pacific dispersal event, or via Antarctica which was connected to Australia and South America. Ancient long-distance dispersals in eastward direction across the Pacific have been reported for terrestrial arthropods before, for instance in Amaurobioides coastal spiders (Araneae; Ceccarelli et al., 2016) and metalmark moths (Lepidoptera: Choreutidae, Rota et al., 2016). Transoceanic crossings repeatedly played a pivotal role for stick and leaf insect distribution (Buckley et al., 2009, 2010), with oceanic distances covered as far as from Australia to the Mascarene Archipelago (Bradler et al., 2015). Above-water dispersal might have occurred via animals rafting on mats of vegetation (King, 1962) or as eggs that are exceptionally robust in phasmatodeans and have been shown to survive extended periods floating in sea water (Kobayashi et al., 2014) and even passing the digestive tract of insectivorous birds (Suetsugu et al., 2018).
Conclusion and Outlook
Our study confirms the power of phylogenomic approaches for inferring evolutionary relationships that have been difficult to assess in the past by yielding a well-supported topology at the base of the tree of life of stick and leaf insects. We provide strong evidence for resolving the deep phylogenetic nodes among all major lineages of Phasmatodea, and were furthermore able to date the individual divergence events and reconstruct their biogeographic history. Our study provides a substantial basis for establishing a natural classification of the stick and leaf insects and for further developing the role of phasmatodeans as emerging model systems in evolutionary research. Future studies need to address minor but crucial taxonomic problems that still await revelation such as the phylogenetic placement of the Southeast Asian Stephanacridini, the African Gratidiini, Bacillinae, and Palophinae, and the Neotropical Heteronemiini, by for instance, applying DNA enrichment methods in order to generate phylogenetically informative data sets that can be combined with those generated in the present study.
Data Availability
The transcriptome data will be made available via NCBI GenBank BioProject ID PRJNA183205 (https://www.ncbi.nlm.nih.gov/bioproject/183205). Supplementary data files are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.65492qt (Simon et al., 2019).
Author Contributions
SBr, SS, BW, and TB designed the study. SBr, TB, BW, and RM collected or provided samples. AD, SL, LP, XZ, KM, and SS assembled and processed the transcriptomes. AD, BM, LP, KM, and SS developed scripts, datasets, and programs. SS phylogenetically analyzed the transcriptomes and performed topology tests. HL conducted age divergence analysis. SBa performed biogeography reconstruction. SBr generated the figures. SBr, SS, SBa, and TB wrote the manuscript. BW and HL added to manuscript editing. All authors approved the final version of the manuscript.
Funding
This study was supported by the German Science Foundation (DFG grants BR 2930/2-1, 2930/3-1, BR 2930/4-1, and BR 2930/5-1 to SBr, and WI 4324/1-1 to BW).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The presented data are the result of the collaborative efforts of the 1KITE consortium and the authors extend their gratitude to all those who assisted with any aspect of this project, from specimen collection, to molecular lab work, to bioinformatics, and all our analyses. Thank you to all who assisted with specimen acquisition, including: Lars Möckel, Hans Pohl, Dieter Schulten, Rene Koehler, Reinhard Predel, Adriana Marvaldi as well as Julia Goldberg and Nicolas Cliquennois who assisted during field work on Madagascar, Ondrej Hlinka for High-Computing Performance assistance, and other colleagues from BGI-Shenzhen who contributed to sample and data curation and management. Thank you to BGI-Shenzhen for supporting and funding this study, through the 1KITE initiative. We thank Christoph Seiler for providing photographs and Bernd Baumgart for technical assistance with the graphics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00345/full#supplementary-material
References
Archibald, S. B., and Bradler, S. (2015). Stem-group stick insects (Phasmatodea) in the early Eocene at McAbee, British Columbia, Canada, and Republic, Washington, United States of America. Can. Entomol. 147, 744–753. doi: 10.4039/tce.2015.2
Bank, S., Sann, M., Mayer, C. U., Meusemann, K., Donath, A, Podsiadlowski, L., et al. (2017). Transcriptome and target DNA enrichment sequence data provide new insights into the phylogeny of vespid wasps (Hymenoptera: Aculeata: Vespidae). Mol. Phyl. Evol. 116, 213–226. doi: 10.1016/j.ympev.2017.08.020
Beutel, R. G., Friedrich, F., Ge, S.-Q., and Yang, X.-K. (2014). Insect Morphology and Phylogeny. Berlin: Walter de Gruyter GmbH.
Bradler, S. (2000). On the systematic position of Agathemera Stal 1875 within the Phasmatodea (Insecta). Zoology 103(Suppl. III):99.
Bradler, S. (2003). “Phasmatodea, Gespestschrecken,” in Lehrbuch der Speziellen Zoologie, Bd. I, Wirbellose Tiere, 5. Teil Insecta, ed H. H. Dathe (Heidelberg; Berlin: Spektrum). 251–261.
Bradler, S. (2009). Phylogenie der Stab- und Gespenstschrecken (Phasmatodea). Spec. Phyl. Evol. 2, 3–139. doi: 10.17875/gup2009-710
Bradler, S. (2015). Der Phasmatodea Tree of Life: Überraschendes und Ungeklärtes in der Stabschrecken-evolution. Entomologie heute 27, 1–23.
Bradler, S., and Buckley, T. R. (2018). “Biodiversity of Phasmatodea,” in Insect Biodiversity: Science and Society, Vol. II, eds R. G. Foottit and P. H. Adler (Hoboken, NJ: Wiley-Blackwell, 281–313.
Bradler, S., Cliquennois, N., and Buckley, T. R. (2015). Single origin of Mascarene stick insects: ancient radiation on sunken islands? BMC Evol. Biol. 15:196. doi: 10.1186/s12862-015-0478-y
Bradler, S., Robertson, J. A., and Whiting, M. F. (2014). A molecular phylogeny of Phasmatodea with emphasis on Necrosciinae, the most species-rich subfamily of stick insects. Sys. Ent. 39, 205–222. doi: 10.1111/syen.12055
Bradler, S., Whiting, M. F., and Klug, R. (2003). Basal diversification and the evolution of wings in stick insects (Phasmatodea). Entomol. Abh. 61, 132–133.
Bradley, J. C., and Galil, B. S. (1977). The taxonomic arrangement of the Phasmatodea with keys to the subfamilies and tribes. Proc. Entomol. Soc. Wash. 79, 176–208.
Brand, P., Lin, W., and Johnson, B. R. (2018). The draft genome of the invasive walking stick, Medauroidea extradendata, reveals extensive lineage-specific gene family expansions of cell wall degrading enzymes in Phasmatodea. G3 8, 1403–1408. doi: 10.1534/g3.118.200204
Breinholt, J. W., Lemmon, A., Lemmon, E., Xiao, L., Earl, C., and Kawahara, A.Y. (2018). Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for Anchored Phylogenomics. Syst. Biol. 67, 78–93. doi: 10.1093/sysbio/syx048
Brock, P. D., Büscher, T. H., and Baker, E. (2017). “Phasmida species file online: phasmida species file version 5.0/5.0,” in Species 2000 and ITIS Catalogue of Life, eds Y. Roskov, G. Ower, T. Orrell, D. Nicolson, N. Bailly, P. M. Kirk, T. Bourgoin, R. E. DeWalt, W. Decock, E. van Nieukerken, J. Zarucchi, and L. Penev (Leiden: Species 2000; Naturalis). Available online at: www.catalogueoflife.org/col (accessed April 20, 2019).
Brock, P. D., and Marshall, J. (2011). Order Phasmida Leach. 1815. In: Zhang, Z.-Q. (Ed.) Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148:198. doi: 10.11646/zootaxa.3148.1.36
Brower, A. V. Z. (1994). Rapid morphological radiation and convergence in geographical races of the butterfly, Heliconius erato, inferred from patterns of mitochondrial DNA evolution. Proc. Nat. Acad. Sci. U.S.A. 91, 6491–6495. doi: 10.1073/pnas.91.14.6491
Buckley, T. R., Attanayake, D., and Bradler, S. (2009). Extreme convergence in stick insect evolution: phylogenetic placement of the Lord Howe Island tree lobster. Proc. R. Soc. Lond. B 276, 1055–1062. doi: 10.1098/rspb.2008.1552
Buckley, T. R., Attanayake, D., Nylander, J. A. A., and Bradler, S. (2010). The phylogenetic placement and biogeographical origins of the New Zealand stick insects (Phasmatodea). Sys. Ent. 35, 207–225. doi: 10.1111/j.1365-3113.2009.00505.x
Büscher, T. H., Buckley, T. R., Grohmann, C., Gorb, S. N., and Bradler, S. (2018). The evolution of tarsal adhesive microstructures in stick and leaf insects (Phasmatodea). Front. Ecol. Evol. 6:69. doi: 10.3389/fevo.2018.00069
Ceccarelli, F. S., Opell, B. D., Haddad, C. R., Raven, R. J., Soto, E. M., and Ramírez, M. J. (2016). Around the world in eight million years: Historical biogeography and evolution of the spray zone spider Amaurobioides (Araneae: Anyphaenidae). PLoS ONE 11:e0163740. doi: 10.1371/journal.pone.0163740
Chen, S., Deng, S.-.W., Shih, C., Zhang, W.-W., Zhang, P., Ren, D., et al. (2018). The earliest timematids in Burmese amber reveal diverse tarsal pads of stick insects in the mid-Cretaceous. Ins. Sci. 26:945–957. doi: 10.1111/1744-7917.12601
Chernomor, O., von Haeseler, A., and Minh, B. Q. (2016). Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008. doi: 10.1093/sysbio/syw037
Dell'Ampio, E., Meusemann, K., Szucsich, N. U., Peters, R. S., Meyer, B., Borner, J., et al. (2014). Decisive data sets in phylogenomics: lessons from studies on the phylogenetic relationships of primarily wingless insects. Mol. Biol. Evol. 31, 239–249. doi: 10.1093/molbev/mst196
Domínguez, M. C., San-Blas, G., Agrain, F., Roig-Juñent, S. A., Scollo, A. M., and Debandi, G. O. (2009). Cladistic, biogeographic and environmental niche analysis of the species of Agathemera Stål (Phasmatida, Agathemeridae). Zootaxa 2308, 43–57. doi: 10.11646/zootaxa.2308.1.3
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Engel, M. S., Wang, B., and Alqarni, A. S. (2016). A thorny, ‘anareolate' stick-insect (Phasmatidae s.l.) in Upper Cretaceous amber from Myanmar, with remarks on diversification times among Phasmatodea. Cret. Res. 63, 45–53. doi: 10.1016/j.cretres.2016.02.015
Evangelista, D. A., Wipfler, B., Béthoux, O., Donath, A., Fujita, M., Kohli, M. K., et al. (2019). An integrative phylogenomic approach illuminates the evolutionary history of cockroaches and termites (Blattodea). Proc. R. Soc. Lond. B 286:20182076. doi: 10.1098/rspb.2018.2076
Friedemann, K., Wipfler, B., Bradler, S., and Beutel, R. G. (2012). On the head morphology of Phyllium and the phylogenetic relationships of Phasmatodea (Insecta). Acta Zool. 93, 184–199. doi: 10.1111/j.1463-6395.2010.00497.x
Glaw, F., Hawlitschek, O., Dunz, A., Goldberg, J., and Bradler, S. (2019). When giant stick insects play with colors: Molecular phylogeny of the Achriopterini and description of two new splendid species (Phasmatodea: Achrioptera) from Madagascar. Front. Ecol. Evol. 7:105. doi: 10.3389/fevo.2019.00105
Goldberg, J., Bresseel, J., Constant, J., Kneubühler, B., Leubner, F., Michalik, P., et al. (2015). Extreme convergence in egg-laying strategy across insect orders. Sci. Rep. 5:7825. doi: 10.1038/srep07825
Grimaldi, D., and Engel, M. (2005). Evolution of the Insects. Cambridge: Cambridge University Press.
Gu, X., Fu, Y. X., and Li, W. H. (1995). Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 12, 546–557.
Gullan, P. J., and Cranston, P. S. (2014). The Insects: An Outline of Entomology, 4th Edn. Oxford: Wiley-Blackwell.
Günther, K. (1953). Über die taxonomische Gliederung und geographische Verbreitung der Insektenordnung der Phasmatodea. Beitr. Entomol. 3, 541–563.
Hennemann, F. H., and Conle, O. V. (2008). Revision of oriental Phasmatodea: the tribe Pharnaciini, Günther, 1953, including the description of the world's longest insect, and a survey of the family Phasmatidae Gray, 1835 with keys to the subfamilies and tribes (Phasmatodea: “Anareolatae”: Phasmatidae). Zootaxa 1906, 1–316. doi: 10.11646/zootaxa.1906.1.1
Ho, S. Y., and Jermiin, L. S. (2004). Tracing the decay of the historical signal in biological sequence data. Syst. Biol. 53, 623–637. doi: 10.1080/10635150490503035
Holt, B. G., Lessard, J. P., Borregaard, M. K., Fritz, S. A., Araújo, M. B., Dimitrov, D., et al. (2013). An update of Wallace's zoogeographic regions of the world. Science 339, 74–78. doi: 10.1126/science.1228282
Johnson, K. P., Dietrich, C. H., Friedrich, F., Beutel, R. G., Wipfler, B., Peters, R. S., et al. (2018). Phylogenomics and the evolution of hemipteroid insects. Proc. Nat. Acad. Sci. U.S.A. 115, 12775–12780. doi: 10.1073/pnas.1815820115
Kômoto, N., Yukuhiro, K., and Tomita, S. (2012). Novel gene rearrangements in the mitochondrial genome of a webspinner, Aposthonia japonica (Insecta: Embioptera). Genome 55, 222–233. doi: 10.1139/g2012-007
Kômoto, N., Yukuhiro, K., Ueda, K., and Tomita, S. (2011). Exploring the molecular phylogeny of phasmids with whole mitochondrial genome sequences. Mol. Phyl. Evol. 58, 43–52. doi: 10.1016/j.ympev.2010.10.013
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
King, W. (1962). The occurrence of rafts for dispersal of land animals into the West Indies. Q. J. Florida. Acad. Sci. 25, 45–52.
Klug, R. (2008). Anatomie des Pterothorax der Phasmatodea, Mantophasmatodea und Embioptera und seine Bedeutung für die Phylogenie der Polyneoptera (Insecta). Spec. Phyl. Evol. 1, 171–217.
Klug, R., and Bradler, S. (2006). The pregenital abdominal musculature in phasmids and its implications for the basal phylogeny of Phasmatodea (Insecta: Polyneoptera). Org. Div. Evol. 6, 171–184. doi: 10.1016/j.ode.2005.08.004
Kobayashi, S., Usui, R., Nomoto, K., Ushirokita, M., Denda, T., and Izawa, M. (2014). Does egg dispersal occur via the ocean in the stick insect genus Megacrania (Phasmida: Phasmatidae)? Ecol. Res. 29, 1025–1032. doi: 10.1007/s11284-014-1188-4
Kosiol, C., and Goldman, N. (2005). Different versions of the Dayhoff rate matrix. Mol. Biol. Evol. 22, 193–199. doi: 10.1093/molbev/msi005
Lanfear, R., Calcott, B., Kainer, D., Mayer, C., and Stamatakis, A. (2014). Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14:82. doi: 10.1186/1471-2148-14-82
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2016). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Matzke, N. J. (2013). Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front. Biogeogr. 5, 242–248. doi: 10.21425/F55419694
Matzke, N. J. (2014). Model selection in historical biogeography reveals that founder-event speciation is a crucial process in Island Clades. Syst. Biol. 63, 951–970. doi: 10.1093/sysbio/syu056
Matzke, N. J. (2018). BioGeoBEARS: BioGeography with Bayesian (and likelihood) Evolutionary Analysis with R Scripts. version 1.1.1. San Francisco, CA: GitHub. Available online at: https://github.com/nmatzke/BioGeoBEARS
Meusemann, K., von Reumont, B. M., Simon, S., Roeding, F., Strauss, S., Kück, P., et al. (2010). A phylogenomic approach to resolve the arthropod tree of life. Mol. Biol. Evol. 27, 2451–2464. doi: 10.1093/molbev/msq130
Miller, M. A., Schwartz, T., Pickett, B.E., He, S., Klem, E.B., Scheuermann, R.H., et al. (2015). A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evol. Bioinform. 11, 43–48. doi: 10.4137/EBO.S21501
Misof, B., Liu, S., Meusemann, K., Peters, R. S., Donath, A., Mayer, C., et al. (2014). Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767. doi: 10.1126/science.1257570
Misof, B., Meyer, B., von Reumont, B. M., Kück, P., Misof, K., and Meusemann, K. (2013). Selecting informative subsets of sparse supermatrices increases the chance to find correct trees. BMC Bioinform. 14:348. doi: 10.1186/1471-2105-14-348
Misof, B., and Misof, K. (2009). A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 58, 21–34. doi: 10.1093/sysbio/syp006
Müller, T., and Vingron, M. (2000). Modeling amino acid replacement. J. Comput. Biol. 7, 761–776. doi: 10.1089/10665270050514918
Nel, A., and Delfosse, E. (2011). A new Chinese Mesozoic stick insect. Act. Pal. Pol. 56, 429–432. doi: 10.4202/app.2009.1108
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R. P., Moret, B. M. E., and Stamatakis, A. (2010). How many bootstrap replicates are necessary? J. Comput. Biol. 17, 337–354. doi: 10.1089/cmb.2009.0179
Pauli, T., Burt, T. O., Meusemann, K., Bayless, K., Donath, A., Podsiadlowski, L., et al. (2018). New data, same story: phylogenomics does not support Syrphoidea (Diptera: Syrphidae, Pipunculidae). Syst. Entomol. 43, 447–459. doi: 10.1111/syen.12283
Peters, R. S., Krogmann, L., Mayer, C., Donath, A., Gunkel, S., Meusemann, K., et al. (2017). Evolutionary history of the Hymenoptera. Curr. Biol. 27, 1–6. doi: 10.1016/j.cub.2017.01.027
Petersen, M., Meusemann, K., Donath, A., Dowling, D., Liu, S., Peters, R. S., et al. (2017). Orthograph: a versatile tool for mapping coding nucleotide sequences to clusters of orthologous genes. BMC Bioinform. 18:111. doi: 10.1186/s12859-017-1529-8
Poinar, G. O. (2011). A walking stick, Clonistria dominicana n. sp. (Phasmatodea: Diapheromeridae) in Dominican amber. Hist. Biol. 23, 223–226. doi: 10.1080/08912963.2010.522405
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Robertson, J. A., Bradler, S., and Whiting, M. F. (2018). Evolution of oviposition techniques in stick and leaf insects (Phasmatodea). Front. Ecol. Evol. 6:216. doi: 10.3389/fevo.2018.00216
Rota, J., Peña, C., and Miller, S. E. (2016). The importance of long-distance dispersal and establishment events in small insects: historical biogeography of metalmark moths (Lepidoptera, Choreutidae). J. Biogeogr. 43, 1254–1265. doi: 10.1111/jbi.12721
Sellick, J. T. C. (1994). Phasmida (stick insects) eggs from the Eocene of Oregon. Paleaontology 37, 913–921.
Shelomi, M., Danchin, E. G., Heckel, D., Wipfler, B., Bradler, S., Zhou, X., et al. (2016). Horizontal gene transfer of pectinases from bacteria preceded the diversification of stick and leaf insects. Sci. Rep. 6:26388. doi: 10.1038/srep26388
Simon, S., Blanke, A., and Meusemann, K. (2018). Reanalyzing the Palaeoptera problem–the origin of insect flight remains obscure. Arthropod Struc. Dev. 47, 328–338. doi: 10.1016/j.asd.2018.05.002
Simon, S., Letsch, H., Bank, S., Buckley, T. R., Donath, A., Liu, S., et al. (2019). Data from: old world and new world phasmatodea: phylogenomics resolve the evolutionary history of stick and leaf insects. Dryad Digital Repository. doi: 10.5061/dryad.65492qt
Soubrier, J., Steel, M., Lee, M. S., Der Sarkissian, C., Guindon, S., Ho, S. Y., et al. (2012). The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 29, 3345–3358. doi: 10.1093/molbev/mss140
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Strimmer, K., and von Haeseler, A. (1997). Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. U.S.A. 94, 6815–6819. doi: 10.1073/pnas.94.13.6815
Suetsugu, K., Funaki, S., Takahashi, A., Ito, K., and Yokoyama, T. (2018). Potential role of bird predation in the dispersal of otherwise flightless stick insects. Ecology 99, 1504–1506. doi: 10.1002/ecy.2230
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Terrapon, N., Li, C., Robertson, H. M., Ji, L., Meng, W., Booth, W., et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat. Comm. 5:3636. doi: 10.1038/ncomms4636
Tilgner, E. H. (2002). Systematics of Phasmida, (dissertation), University of Georgia, Athens, GA, United States.
Tilgner, E. H. (2009). “Phasmida (Stick and leaf insects),” in Encyclopedia of Insects, 2nd Edn, eds V. H. Resh and R. T. Cardé (Amsterdam; Boston; London; New York; Oxford; Paris; San Diego; San Francisco; Singapore; Sydney; Tokyo: Elsevier, 765–766.
Tomita, S., Yukuhiro, K., and Kômoto, N. (2011). The mitochondrial genome of a stick insect Extatosoma tiaratum (Phasmatodea) and the phylogeny of polyneopteran insects. J. Ins. Biotech. Sericol. 80, 79–88. doi: 10.11416/jibs.80.3_079
Veerassamy, S., Smith, A., and Tillier, E. R. (2003). A transition probability model for amino acid substitutions from blocks. J. Comput. Biol. 10, 997–1010. doi: 10.1089/106652703322756195
Vences, M., Wollenberg, K. C., Vieites, D. R., and Lees, D. C. (2009). Madagascar as a model region of species diversification. TREE 24, 456–465. doi: 10.1016/j.tree.2009.03.011
Vera, A., Pastenes, L., Veloso, C., and Méndez, M. A. (2012). Phylogenetic relationships in the genus Agathemera (Insecta: Phasmatodea) inferred from the genes 16S, COI and H3. Zool. J. Linn. Soc. 165, 63–72. doi: 10.1111/j.1096-3642.2011.00802.x
Wang, M., Béthoux, O., Bradler, S., Jacques, F., Cui, Y., and Ren, D. (2014). Under cover at pre-angiosperm times: a cloaked phasmatodean insect from the Early Cretaceous Jehol biota. PLoS ONE 9:e91290. doi: 10.1371/journal.pone.0091290
Wedmann, S., Bradler, S., and Rust, J. (2007). The first fossil leaf insect: 47 million years of specialized cryptic morphology and behavior. Proc. Nat. Acad. Sci. U.S.A. 104, 565–569. doi: 10.1073/pnas.0606937104
Whelan, S., and Goldman, N. (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699. doi: 10.1093/oxfordjournals.molbev.a003851
Whiting, M. F., Bradler, S., and Maxwell, T. (2003). Loss and recovery of wings in stick insects. Nature 421, 264–267. doi: 10.1038/nature01313
Wipfler, B., Letsch, H., Frandsen, P. B., Kaplie, P., Mayer, C., Bartel, D., et al. (2019). Evolutionary history of Polyneoptera and its implications for our understanding of early winged insects. Proc. Nat. Acad. Sci. U.S.A. 116, 3024–3029. doi: 10.1073/pnas.1817794116
Wong, T.K., Kalyaanamoorthy, S., Meusemann, K., Yeates, D., Misof, B., and Jermiin, L. (2017). AliStat Version 1.3. Canberra, ACT: Commonwealth Scientific and Industrial Research Organisation (CSIRO).
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., et al. (2014). SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Yang, Z. (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314. doi: 10.1007/BF00160154
Zhang, S.-Q., Che, L.-H., Li, Y., Liang, D., Pang, H., Slipinski, A., et al. (2018). Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Comm. 9:205. doi: 10.1038/s41467-017-02644-4
Zhou, Z., Guan, B., Chai, J., and Che, X. (2017). Next-generation sequencing data used to determine the mitochondrial genomes and a preliminary phylogeny of Verophasmatodea insects. J. Asia-Pacific Ent. 20, 713–719. doi: 10.1016/j.aspen.2017.04.012
Keywords: phasmids, transcriptomes, historical biogeography, Polyneoptera, Euphasmatodea
Citation: Simon S, Letsch H, Bank S, Buckley TR, Donath A, Liu S, Machida R, Meusemann K, Misof B, Podsiadlowski L, Zhou X, Wipfler B and Bradler S (2019) Old World and New World Phasmatodea: Phylogenomics Resolve the Evolutionary History of Stick and Leaf Insects. Front. Ecol. Evol. 7:345. doi: 10.3389/fevo.2019.00345
Received: 23 May 2019; Accepted: 28 August 2019;
Published: 07 October 2019.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Jesús A. Ballesteros, University of Wisconsin-Madison, United StatesMarco Gottardo, University of Siena, Italy
Copyright © 2019 Simon, Letsch, Bank, Buckley, Donath, Liu, Machida, Meusemann, Misof, Podsiadlowski, Zhou, Wipfler and Bradler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Bradler, c2JyYWRsZUBnd2RnLmRl; Sabrina Simon, aW5mb0BzYWJyaW5hLXNpbW9uLmNvbQ==