 Jee-Hoon Kim1,2†Hyun Kyong Kim1†Heesoo Kim1†Benny K. K. Chan3Seunghyun Kang4*Won Kim1*
Jee-Hoon Kim1,2†Hyun Kyong Kim1†Heesoo Kim1†Benny K. K. Chan3Seunghyun Kang4*Won Kim1*- 1School of Biological Sciences, Seoul National University, Seoul, South Korea
- 2Division of Polar Ocean Science, Korea Polar Research Institute, Incheon, South Korea
- 3Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
- 4Unit of Research for Practical Application, Korea Polar Research Institute, Incheon, South Korea
Introduction
Barnacles are ecologically and economically important species that are common in the intertidal zone and in submerged artificial surfaces, including the bottom of ships, and they are one of the fouling species that cause problems for marine industries (Chan et al., 2009). The dispersal of barnacle larvae by ballast water tanks in ships is the major contributor to the introduction of invasive species, which affect the distributions and the ecosystems of native species around the world (Yamaguchi et al., 2009; Choi et al., 2013). From an application point of view, cement proteins secreted by barnacles which attaching their bases firmly on the substratum are considered as a strong underwater glue (Kamino et al., 1996). Barnacles are present in almost all marine ecosystems, including the intertidal, deep-sea hydrothermal vents, floating objects, turtle, and whales surfaces (Chan and Hǿeg, 2015). Fossils of the whale-specific barnacles can be indirect evidences to study the prehistoric cetacean migration patterns (Buckeridge et al., 2018). The intertidal acorn barnacle, Amphibalanus amphitrite (Darwin, 1854), belonging to the family Balanidae, is a major fouling organism worldwide and present in a huge variety of habitats including ports, estuaries, and mangroves. A. amphitrite is believed to originate from the southwestern Pacific and Indian Oceans, but it has been found worldwide owing to global trade, worldwide industrial shipping, and dispersal through ballast waters (Chen et al., 2014a). A. amphitrite as a model species for larval biology studies because it has a wide distribution and the settlement of the cyprids can be easily performed in the laboratory (Qiu and Qian, 1999).
Several studies have mainly focused on the expression of the proteins or genes in the development and adaptive strategies of barnacle larvae. The transcriptome analysis of different larval stages of A. amphitrite enabled the identification of the possible gene functions required for settlement (Yan et al., 2012). Two other studies have showed the potential adhesion-related genes for cement proteins and proteomes of the developmental stages of A. amphitrite and Tetraclita japonica (Chen et al., 2014b; Lin et al., 2014). These studies have attempted to elucidate the settlement mechanisms at the gene levels. However, understanding the settlement, bioadhesion, and biofouling aspects of barnacles in detail at the gene level will require a genome-wide approach, which is not possible currently owing to the absence of a reference genome (Patrick and David, 2012). Whole-genome sequences of marine crustaceans have recently been analyzed (Huete-Pérez and Quezada, 2013). To date, to the best of our knowledge, there is no draft genome for the entire Balanidae family, which comprises highly evolved sessile barnacles (Burden et al., 2014). Thus, the aim of our study was to report the first de-novo draft genome of A. amphitrite using Pacific Biosciences (PacBio) sequencing for the generation of a comprehensive genome, which will be useful for understanding the cementation, attachment, and different life histories of the barnacles.
Data
The A. amphitrite (Figure 1A) genome size was estimated to be ~481 megabase (Mb) pairs by k-mer analysis using the Jellyfish software (Marçais and Kingsford, 2011) (Figure 1B) and GenomeScope (Vurture et al., 2017) (Table 1). The 56.08 gigabase (Gb) PacBio long-read sequences were assembled into a genome comprising 4,351 contigs totaling 609.7 Mb pairs with an N50 size of 0.24 Mb pairs (Table 1). To further evaluate the correctness of the genome assembly, we aligned the Illumina short-read sequences from whole-genome sequencing data against the genome assembly using the Burrows-Wheeler aligner (BWA v0.7.17) (Li and Durbin, 2009), and mapping statistics were created using Samtools v1.6 (Supplementary Table 1) (Li et al., 2009). We found that 78.1% of the reads were properly aligned to the genome with their mates and 97.1% of the reads were reliably aligned to the genome assembly (Supplementary Table 1). In particular, 16% of the contigs were over 500 kilobase (Kb) in length and only 2% of the contigs were <10 Kb (Figure 1C). The repeat contents in the genome were 27 Mb (4.48%) bases, and most predicted subclasses were simple repeats and LTR elements (Figure 1D). Additionally, the Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis recovered highly conserved metazoa and arthropoda genomes and genes from our draft genome, 93.46% (914/978) in metazoa genome, 94.09% (1,003/1,066) in the arthropoda genome, 85.59% (837/978) in the metazoa gene, and 86.59% (923/1,066) in the arthropoda gene, confirming the completeness of the annotated genes in the assembled genome (Supplementary Figure 1). To further confirm the identity of the present barnacle genome, BLAST was conducted to find the presences of the Settlement-Inducing Protein Complex (SIPC) gene (Dreanno et al., 2006). SIPC is a cuticular glycoprotein which induces gregarious settlement in barnacles. This protein is considered as a keystone protein in barnacle identity. BLAST results show that Protein 008197 is an SIPC gene with Alignment_length 1556, Pct_identity 90%, and E-value 0. The SIPC gene is registered in UniRef. An orthologous analysis of seven species (Supplementary Table 2) showed that A. amphitrite has 8,903 orthologous clusters out of 16,187 orthologous clusters of all species and has 4,285 singletons (Figure 1E). The A. amphitrite genome shared its genome mostly with Daphnia pulex, which is also a crustacean. In total, 704 one-to-one orthologous genes were provided to construct a phylogenetic tree. According to the time-calibrated species time tree, A. amphitrite and D. pulex had diverged 493 million years ago (Figure 1F). Therefore, we suggest that these results are nearly a complete reference genome for A. amphitrite. In addition, this is the first assembled draft for the family Balanidae and subclass Thecostraca at the contig/scaffold level.
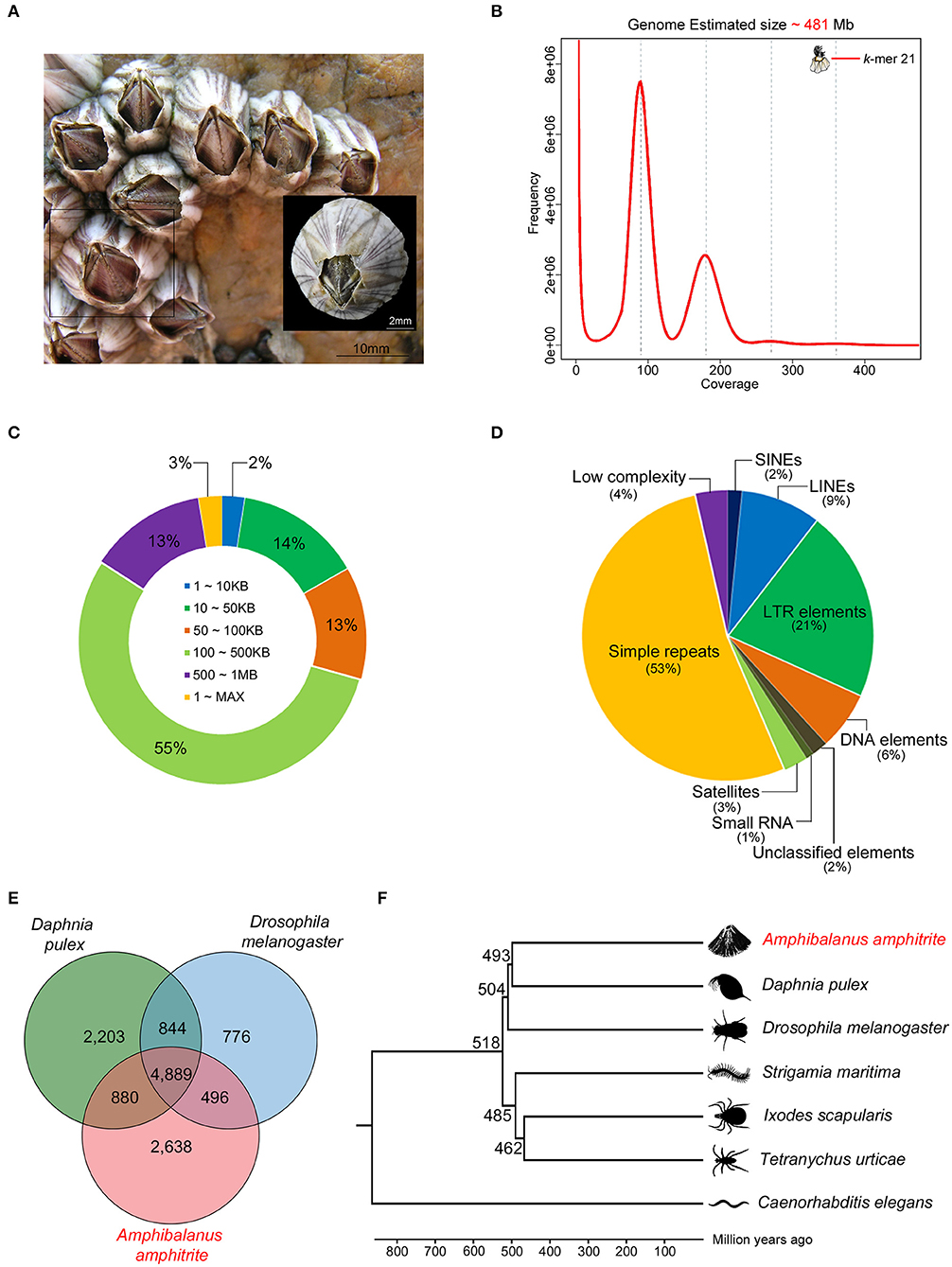
Figure 1. Characteristics of the Amphibalanus amphitrite genome assembly. (A) Photograph of A. amphitrite assemblages with an adult specimen (scale bars: mm); (B) k-mer based genome size estimation; (C) length distributions of the assembled contigs; (D) de-novo repeat predictions and subclass distributions; (E) a Venn diagram of the orthologous gene clusters among the three arthropod lineages; and (F) phylogenetic relationship of A. amphitrite with six other species (estimated divergence time: million years ago).
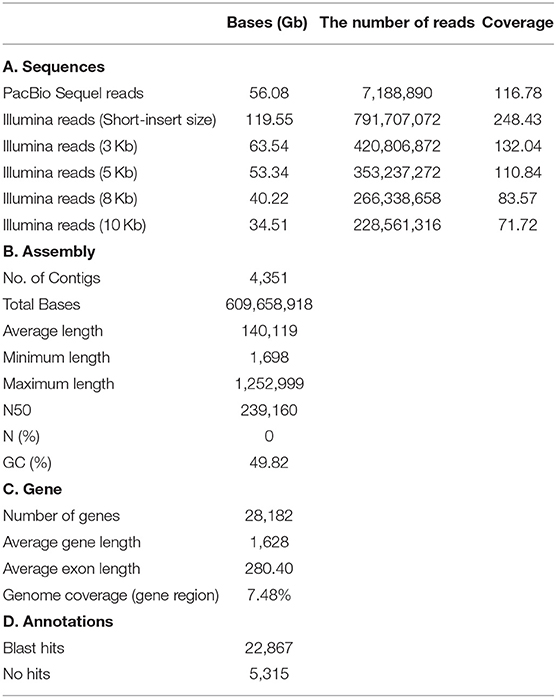
Table 1. Summary of genome assemblies and gene annotations.
Materials and Methods
Sample Collection and Genomic DNA and Transcriptomic RNA Preparation
Specimens of A. amphitrite were collected from Beolpo, Sanyang-eup, Tongyeong-si, Gyeongsangnam-do, Republic of Korea (34°82′N, 128°38′E) on August 12, 2018 (Figure 1A). To obtain high-quality DNA, barnacle species assemblages were carried alive to the laboratory with the molluscan shells attached. Some of them were preserved in RNAlater (Qiagen, Hilden, Germany) to avoid RNA degradation. The total DNAs of A. amphitrite were extracted according to the protocol suggested by Panova et al. (2016). First, two individuals were selected from the barnacle assemblage. To reduce the possibility of contamination of bacteria or algae growing on shell surfaces, the shells of the individuals were rinsed several times in pure water. When live barnacles were rinsed in freshwater, the opercular plates were closed tightly to avoid freshwater entering the mantle, which is a response to situations such as raining in the natural environment. Rinsing live barnacle shells with freshwater will not damage the barnacle somatic body. The opercular plates of each of the individuals were then removed using sterilized tweezers, and the soma was detached without the cirri and trophi. Because the sizes of these individuals vary around 10–20 mm in diameter, and the numerable cells in the tissue samples are low, the soma samples from the two individuals were pooled for better analysis. Total DNAs were extracted from the isolated tissues using the E.Z.N.A. Blood DNA Mini Kit (Omega Bio-Tek, GA, USA). For accurate gene annotation in the draft genome, total RNAs were extracted from the barnacle samples in RNAlater solution using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The quality of the extracted total DNAs and RNAs was investigated using the NanoDrop 1000 spectrometer (Thermo Scientific, DE, USA) and the 2100 Bioanalyzer system (Agilent Technologies, CA, USA).
Genomic DNA Library Preparation, de-novo Genome Sequencing, and Genome Size Estimation
To pass the quality control of the PacBio standard, the two high-quality DNA samples were pooled because of their small somatic body size and lack of cell counts in the soma. These two individuals were collected from the same colony to avoid any population level variations. Eight microgram of the pooled DNA sample was used to prepare the 20 Kb SMRTbell templates. Genomic DNA was sheared using G-tube (Covaris Inc., Woburn, MA, USA) and purified with AMPurePB magnetic beads (Beckman Coulter Inc., Brea, CA, USA). The SMRTbell libraries were sequenced using Sequel sequencing kit 3.0 (included in Sequel Sequencing chemistry 3.0) in the PacBio Sequel (Pacific Biosciences) sequencing platform.
The mate-paired libraries (3, 5, 8, and 10 Kb) were constricted for scaffolding using the Nextera Mate Pair Library preparation kit. Illumina pair-end libraries were also constructed for error correction. Mate-paired and illumina paired-end sequencing was performed using Illumina HiSeq X with paired-end 150 bp (Illumina, San Diego, USA). For RNA sequencing, a transcriptome library was constructed using the TruSeq RNA library preparation kit and sequenced using Illumina HiSeq 4000 (Illumina).
Before genome size estimation, low-quality reads (Q <20) and adapter reads were removed using Trimmomatic (Bolger et al., 2014). Filtration was performed through the Contig Blast to verify the genome assembly. Of the 2,240 scaffolds, 2,086 scaffolds matched those of the A. amphitrite, 97 no-hits, and 57 others. As a result of checking the 57 scaffolds matched different species one by one, both the coverage and the matching rate are considered very low and are regarded as no-hits. Therefore, no scaffolds were suspected of different species sequences. After filtering, the genome size of the remaining reads were estimated based on k-mer analysis. The distribution of the k-mer of 17, 21, and 25 bp was estimated using the JELLYFISH tool (Marçais and Kingsford, 2011). GenomeScope was also used to investigate the characteristics of the genome such as size, heterozygosity rates, and repeat content (Vurture et al., 2017). The genome size was calculated by dividing the number of k-mer by their peak coverage depth.
PacBio Error Correction and de-novo Genome Assembly
The genome of A. amphitrite was fully assembled using the PacBio raw data with the HGAP4 protocol of the SMRT Link Software (v6.0.0.47841), which contains the read-correction step. To ensure assembly integrity, another long-read method assembler Wtdbg2 was also performed. However, Wtdbg2 assembly resulted in more contigs and lower N50 than HGAP4. Thus, HGAP4 was considered as a reliable method of assembly in the present study. To remove the sequence errors, the error correction of the assembled PacBio data was processed using Pilon (v1.21) with Illumina HiSeq short reads (Walker et al., 2014). Purge Haplotigs was used to identify and remove the haplotypic contigs (Roach et al., 2018) because it was confirmed that A. amphitrite has a high heterozygosity rate (Figure 1B). Through the pipeline of Purge Haplotigs, the error corrected PacBio reads were filtered haplotypic contigs and curated contigs were obtained. The curated contigs were scaffolded with mate pairs libraries of various insert sizes (3, 5, 8, and 10 Kb) using SSPACE (Boetzer et al., 2010). After scaffolding, the gaps were filled from the scaffolds using PBJelly (English et al., 2012) and GMcloser (Kosugi et al., 2015). The completeness of the final assembled sequences was assessed by analyzing the BUSCO scores (Simão et al., 2015). The reference BUSCO databases were metazoa_odb9 and arthropoda_odb9.
Gene Prediction and Annotation
The protein-coding genes of A. amphitrite were predicted using two strategies: transcriptome data-based gene prediction and ab initio gene prediction. Before predicting the genes of A. amphitrite, RepeatMasker was performed with RepBase library (release 20140131) to identify the repeats in the genome of A. amphitrite (Tarailo-Graovac and Chen, 2009). For transcriptome data-based gene prediction, transcriptome data were mapped to the assembled genome using Tophat (v.2.0.13) (Trapnell et al., 2009) and these data were used to predict the gene model using Trinity (r20170127) (Grabherr et al., 2011). For ab initio gene prediction, the gene prediction process was followed using the Seqping pipeline (v0.1.33) (Chan et al., 2017). The assembled transcriptome data and genome sequences were used for the training set of AUGUSTUS (v3.2.2) (Stanke and Morgenstern, 2005). MAKER2 (v2.31.8) was used to determine the final gene model based on the two prediction results (Holt and Yandell, 2011). The predicted genes were searched for functional annotation against biological databases [EggNOG (Huerta-Cepas et al., 2015) Uniprot (Apweiler et al., 2016), GO (Ashburner et al., 2000), InterPro (Mitchell et al., 2018), Pfam (Bateman et al., 2000), CDD (Marchler-Bauer et al., 2016), and TIGRFAMs (Haft et al., 2001)] using BLAST (v2.6.0+) (Camacho et al., 2009) with an e-value <1.0E-5.s.
Phylogenomics Analysis
An orthologous analysis was conducted of seven species (Supplementary Table 2) using Orthovenn2 (Xu et al., 2019) which contains high-quality genome information. A phylogenetic tree was constructed using the neighbor-joining tree with a bootstrap value of 500 and a JTT model by MEGA 6 (Tamura et al., 2013).
Deposited Data and Information to the User
The complete sequences and DNA libraries used in the current draft genome assembly for A. amphitrite have been deposited at NCBI under the BioProject accession number PRJNA549550. The sequences of the A. amphitrite draft genome has been deposited at figshare with doi: 10.6084/m9.figshare.8317106. Analyzed results of annotation were deposited at figshare with doi: 10.6084/m9.figshare.8317109.
Data Availability Statement
The datasets generated for this study can be found in the NCBI under the BioProject accession number PRJNA549550, The sequences has been deposited at figshare with doi: 10.6084/m9.figshare.8317106, Analyzed results of annotation were deposited at figshare doi: 10.6084/m9.figshare.8317109.
Author Contributions
WK and SK conceived the study. J-HK and HK performed the bioinformatics analysis. HKK and HK collected the samples and extracted DNA and RNA. SK and J-HK performed the quality control. J-HK, HKK, BC, and HK wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the Collaborative Genome Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Ocean and Fisheries (MOF) (No. 20180430) Korea.
Conflict of Interest
The reviewer, S-YH, declared a shared affiliation, though no other collaboration, with two of the authors, J-HK and SK, to the handling Editor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00465/full#supplementary-material
References
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2016). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkw1099
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bateman, A., Birney, E., Durbin, R., Eddy, S. R., Howe, K. L., and Sonnhammer, E. L. (2000). The Pfam protein families database. Nucleic Acids Res. 28, 263–266. doi: 10.1093/nar/28.1.263
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2010). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buckeridge, J. S., Chan, B. K. K., and Lee, S. W. (2018). Accumulations of fossils of the whale barnacle Coronula bifida Bronn, 1831 (Thoracica: Coronulidae) provides evidence of a late Pliocene cetacean migration route through the Straits of Taiwan. Zool. Stud. 57:54. doi: 10.6620/ZS.2018.57-54
Burden, D. K., Spillmann, C. M., Everett, R. K., Barlow, D. E., Orihuela, B., Deschampls, J. R., et al. (2014). Growth and development of the barnacle Amphibalanus amphitrite: time and spatially resolved structure and chemistry of the base plate. Biofouling 30, 799–812. doi: 10.1080/08927014.2014.930736
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chan, B. K. K., and Hǿeg, J. T. (2015). “Diversity of life styles, sexual systems, and larval development patterns in sessile crustaceans”, in Lifestyles and Feeding Biology, ed H. Martin and W. Les (New York, NY: Oxford University Press), 14–34.
Chan, B. K. K., Prabowo, R. E., and Lee, K. S. (2009). Crustacean Fauna of Taiwan: Barnacles, Volume I - Cirripedia: Thoracica excluding the Pyrgomatidae and Acastinae. Taipei: National Taiwan Ocean University Press.
Chan, K. L., Rosli, R., Tatarinova, T. V., Hogan, M., Firdaus-Raih, M., and Low, E. T. L. (2017). Seqping: gene prediction pipeline for plant genomes using self-training gene models and transcriptomic data. BMC Bioinformatics 18:1. doi: 10.1186/s12859-016-1426-6
Chen, H. N., Tsang, L. M., Chong, V. C., and Chan, B. K. K. (2014a). Worldwide genetic differentiation in the common fouling barnacle, Amphibalanus amphitrite. Biofouling 30, 1067–1078. doi: 10.1080/08927014.2014.967232
Chen, Z. F., Zhang, H., Wang, H., Matsumura, K., Wong, Y. H., Ravasi, T., et al. (2014b). Quantitative proteomics study of larval settlement in the barnacle balanus amphitrite. PLoS ONE 9:e88744. doi: 10.1371/journal.pone.0088744
Choi, K. H., Choi, H. W., Kim, I. H., and Hong, J. S. (2013). Predicting the invasion pathway of Balanus perforatus in Korean seawaters. Ocean Polar Res. 35, 63–68. doi: 10.4217/OPR.2013.35.1.063
Dreanno, C., Matsumura, K., Dohmae, N., Takio, K., Hirota, H., Kirby, R. R., et al. (2006). An α2-macroglobulin-like protein is the cue to gregarious settlement of the barnacle Balanus Amphitrite. Proc. Natl. Acad. Sci. 103, 14396–14401. doi: 10.1073/pnas.0602763103
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Haft, D. H., Loftus, B. J., Richardson, D. L., Yang, F., Eisen, J. A., Paulsen, I. T., et al. (2001). TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res. 29, 41–43. doi: 10.1093/nar/29.1.41
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491. doi: 10.1186/1471-2105-12-491
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2015). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Huete-Pérez, J. A., and Quezada, F. (2013). Genomic approaches in marine biodiversity and aquaculture. Biol. Res. 46, 353–361. doi: 10.4067/S0716-97602013000400007
Kamino, K., Odo, S., and Maruyama, T. (1996). Cement proteins of the acorn barnacle, Megabalanus rosa. Biol. Bull. 190, 403–409. doi: 10.2307/1543033
Kosugi, S., Hirakawa, H., and Tabata, S. (2015). GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments. Bioinformatics 31, 3733–3741. doi: 10.1093/bioinformatics/btv465
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin, H. C., Wong, Y. H., Tsang, L. M., Chu, K. H., Qian, P. Y., and Chan, B. K. K. (2014). First study on gene expression of cement proteins and potential adhesion-related genes of a membranous-based barnacle as revealed from Next-Generation Sequencing technology. Biofouling 30, 169–181. doi: 10.1080/08927014.2013.853051
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2016). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. doi: 10.1093/nar/gkw1129
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2018). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360. doi: 10.1093/nar/gky1100
Panova, M., Aronsson, H. R., Cameron, A., Dahl, P., Godhe, A., Lind, U., et al. (2016). DNA extraction protocols for whole genome sequencing in marine organisms. Methods Mol. Biol. 1452, 13–44. doi: 10.1007/978-1-4939-3774-5_2
Patrick, A. F., and David, M. R. (2012). Genetic Variation In The Acorn Barnacle From Allozymes To Population Genomics. Integr. Comp. Biol. 52, 418–429. doi: 10.1093/icb/ics099
Qiu, J. W., and Qian, P. Y. (1999). Tolerance of the barnacle Balanus amphitrite to salinity and temperature stress: effects of previous experience. Mar. Ecol. Prog. Ser. 188, 123–132. doi: 10.3354/meps188123
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19:460. doi: 10.1186/s12859-018-2485-7
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4–10. doi: 10.1002/0471250953.bi0410s25
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species Nucleic. Acids. Res. 47:W52–W58. doi: 10.1093/nar/gkz333
Yamaguchi, T., Prabowo, R. E., Ohshiro, Y., Shimono, K., Jones, D., Kawai, H., et al. (2009). The introduction to Japan of the Titan barnacle, Megabalanus coccopoma (Darwin, 1854) (Cirripedia: Balanomorpha) and the role of shipping in its translocation. Biofouling 25, 325–333. doi: 10.1080/08927010902738048
Keywords: thecostraca, fouling barnacle, Amphibalanus amphitrite, draft genome, PacBio sequencing
Citation: Kim J-H, Kim HK, Kim H, Chan BKK, Kang S and Kim W (2019) Draft Genome Assembly of a Fouling Barnacle, Amphibalanus amphitrite (Darwin, 1854): The First Reference Genome for Thecostraca. Front. Ecol. Evol. 7:465. doi: 10.3389/fevo.2019.00465
Received: 12 July 2019; Accepted: 20 November 2019;
Published: 06 December 2019.
Edited by:
Liangsheng Zhang, Fujian Agriculture and Forestry University, ChinaReviewed by:
Sagar M. Utturkar, Purdue University, United StatesSun-Yong Ha, Korea Polar Research Institute, South Korea
Copyright © 2019 Kim, Kim, Kim, Chan, Kang and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seunghyun Kang, cy5rYW5nQGtvcHJpLnJlLmty; Won Kim, d29ua2ltQHBsYXphLnNudS5hYy5rcg==
†These authors have contributed equally to this work