 Li-Ying Feng1†
Li-Ying Feng1† Li-Zhi Gao
Li-Zhi Gao- 1Institution of Genomics and Bioinformatics, South China Agricultural University, Guangzhou, China
- 2Plant Germplasm and Genomics Research Center, Germplasm Bank of Wild Species in Southwest China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
- 3Yunnan Institute of Tropical Crops, Xishuangbanna, China
Rubber tree is the only commercialized natural resource to produce high-quality natural rubber with unique physical and chemical properties. They currently foster in Southeast Asia with marked morphological and productive differences with wild germplasms native to the Amazonian basin of South America. Here, we report complete chloroplast (cp) genomes of six cultivated and six wild accessions of H. brasiliensis using Illumina paired-end sequencing platform. The 12 H. brasiliensis cp genomes ranged from 161,168 to 161,254 bp. All 12 cp genomes displayed a typical quadripartite structure, which consisted of a pair of IR regions (26,787–26,804 bp) separated by a LSC region (89,216–89,284 bp) and a SSC region (18,370–18,377 bp). Phylogenomic analysis revealed that cultivated and wild rubber trees failed formed separate clades. However, we observed that wild rubber trees possessed more variable sites and ∼2.8-fold higher level of nucleotide variation than cultivated rubber trees despite a short domestication history. We drew a comprehensive map of genomic variation across rubber tree plastomes, exhibiting that the density of genomic variants in wild rubber trees was slightly higher than that detected in cultivated ones. The obtainability of genomic variation across cp genomes will provide useful information for better conserving and utilizing rubber tree germplasms.
Introduction
Chloroplasts are key organelles that play a key role in plant cell for photosynthesis and biochemical pathways, including the biosynthesis of starch, fatty acid, pigmengts, and amino acids (Raman and Park, 2015; Dong et al., 2016; Xu et al., 2017). In angiosperms, chloroplast (cp) genomes contain a circular DNA ranging from 72 to 217 kb in size (Sugiura, 1992, 1995; Shi et al., 2012), consisting of two large inverted repeat (IR) regions, a large-single-copy (LSC) region and a small-single-copy (SSC) region (Bendich, 2004; Raubeson and Jansen, 2005; Yang et al., 2010, 2017). Moreover, cp genomes usually exhibit maternal inheritance, enabling the conservation of gene content and genome structure (Sasaki et al., 2003; Parks et al., 2009). A recent study reported that the entire plastome is transcribed in photosynthetic green plants, and that this pattern originated from prokaryotic cyanobacteria – ancestor of the chloroplast genomes that diverged about 1 billion years ago (Shi et al., 2016). Concatenating sequences from a large number of chloroplast genes may overcome the problem of multiple substitutions that results in the loss of phylogenetic information between chloroplast lineages, and thus, can reduce gene-sampling errors due to substitutions. Owing to their small sizes, relatively conserved genome structure, gene content, gene order and simple complexity compared to mitochondrial and particularly nuclear genomes, the cp genomes have recently provided valuable information for taxonomic classification, phylogenetic reconstruction and adaptive evolution as a result of sequence divergence among plant species (e.g., Huang et al., 2014; Zhang et al., 2016; Gao et al., 2019). Given decreased costs to generate increasingly large amount of genome sequences using the Next Generation Sequencing (NGS) platform, up to now (Mardis, 2008), approximately 3,869 plant cp genomes have been sequenced and deposited in the National Center for Biotechnology Information (NCBI), including the cp genome of rubber tree, Hevea brasiliensis (Wild.) Muell. Arg (Tangphatsornruang et al., 2011).
H. brasiliensis is a diploid plant (2n = 2x = 36), a member of the family Euphorbiaceae, which was most commonly known as the rubber tree (Leitch et al., 1998; Lau et al., 2016). It is a cross-pollinated tropical economical tree that grows to 30–40 m tall and can live up to 100 years in the wild (Priyadarshan and Clément-Demange, 2004). The domestication of the rubber tree, which is native to the Amazon basin in South America, began in 1896 and then spread to Southeast Asia with the relocation of H. brasiliensis seedlings (Chan, 2000). Over 100 year of traditional breeding has greatly increased rubber productivity from its wild populations (Priyadarshan and Clément-Demange, 2004). Nevertheless, although artificial selection has led to a slow growth in rubber productivity, it is even more immediately essential to raise new H. brasiliensis varieties with desired agronomic traits. Rubber trees produce natural rubber that is still required for the world’s high-performance engineering components, such as heavy tires. Thus, there will be huge potential to be exploited through genetic breeding programs to help enlarge genomic diversity, important for the generation of more environmentally resilient and high-yielding varieties (Sneller et al., 1997). With this regard, long-standing efforts to obtain high-quality reference nuclear genome assemblies (Rahman et al., 2013; Lau et al., 2016; Tang et al., 2016; Pootakham et al., 2017; Liu et al., 2020) and the mitochondrial genome (Shearman et al., 2014) have long put to apply for genomics-assisted selection to make use of the unexploited reservoir of novel alleles from the wild.
In this study, we sequenced, assembled and characterized the cp genomes of six cultivated and six wild rubber trees using the next-generation sequencing platform. We comprehensively characterized the organization, gene content, intraspecific diversity and structural variants across the 12 cp genomes of rubber tree. The obtained results may largely help to improve our understanding of cp genome evolution of rubber tree and will provide abundant genetic resources to assist the exploration of the precious H. brasiliensis wild germplasms for future rubber tree genetic improvement program.
Materials and Methods
Plant Material Sampling
We selected a panel of world-wide representative rubber trees, including six cultivated and six wild accessions, respectively (Table 1). The six wild H. brasiliensis accessions were originally collected from the Amazon River Basin of Brazil; three were collected from Acre, while the three other were sampled from Rondônia. The six H. brasiliensis cultivars were bred in Indonesia, Malaysia, Brazil, India, Vietnam and China, respectively (Table 1). Fresh leaves of H. brasiliensis germplasms were sampled and immediately dried with silica gel for subsequent experiments.
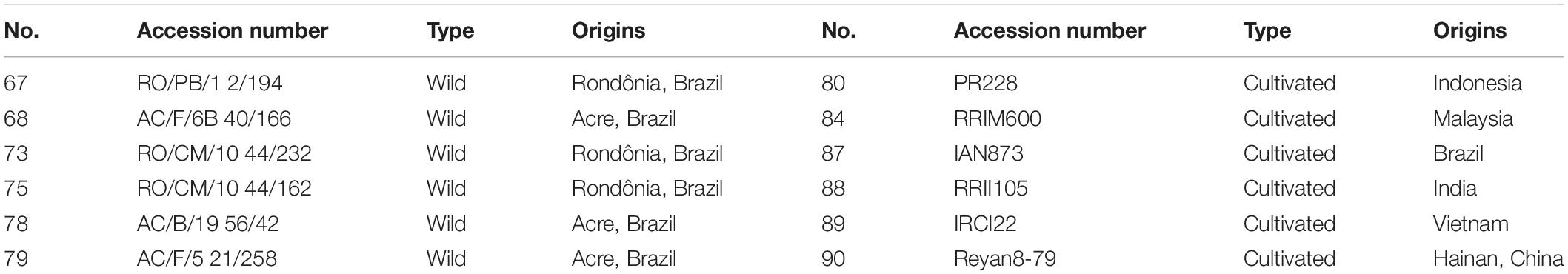
Table 1. Summary of the cultivated and wild rubber tree germplasms in this study.
DNA Extraction, Genome Sequencing, and Assembly
Total genomic DNA for each sample was extracted from ∼100 mg dried leaves using the Mag-MK Plant Genomic DNA extraction kit (Sangon Biotech, CA). The DNA concentration was quantified using Life Invitrogen Qubit® 3.0 (Life Invitrogen, United States), and DNA concentration >30 ng μL–1 was finally determined using Bioanalyzer 2100 (Agilent Technologies). Short-insert paired-end sequencing libraries were generated according to the Illumina standard protocol. Genome sequencing was performed on Hiseq 2500 sequencing platform (Illumina, San Diego, California, United States). Approximately 5.0 GB of raw data were generated with read length of pair-end 100 bp. First, raw reads were trimmed to obtain the high-quality clean data by removing adaptor sequences and low-quality bases with Q-value ≤20 using CLC-quality trim tool1. The cp genome reads were isolated from the mixed DNA of nucleus, mitochondria and chloroplast based on the previously published rubber tree cp genome sequence (Tangphatsornruang et al., 2011). Then, the filtered reads were assembled into contigs using CLC genome assembler v4.06, which were aligned (≥90% similarity and query coverage) and ordered according to the earlier reported cp genome (Tangphatsornruang et al., 2011). Finally, the draft cp genome for each rubber tree was assembled by Geneious 9.0.5 software2, and genomic regions with ambiguous alignments were trimmed manually and considered as gaps, which were filled by sequencing fragments yielded by Polymerase Chain Reaction (PCR) using Geneious assembly software.
Genome Annotation and Whole Genome Comparisons
Initial gene annotation of the 12 rubber tree cp genomes was performed by Organellar Genome Annotator (Wyman et al., 2004). The annotation was manually corrected for start/stop codons and intron/exon orders. BLASTX and BLASTN searches were employed to accurately annotate protein-coding genes and identify the location of the ribosomal RNA (rRNA) and transfer RNA (tRNA) genes. The annotation results were manually checked and codon positions were adjusted by comparing to homologous genes especially from those closely related cp genomes in the public database; boundaries between introns and exons, positions of start and stop codons for protein-coding genes were accurately identified. The cp genome map was drawn using Genome Vx software (Conant and Wolfe, 2008), and cp genome sequences of the 12 rubber trees were deposited in GenBank. The shrinkage and expansion of the IR region is detected by online software IRscope (Amiryousefi et al., 2018). The comparative analysis between genomes was carried out using mVISTA (Frazer et al., 2004). The average pair wise sequence divergence within large single copy (LSC), small single copy (SSC) and inverted repeat (IR) regions was calculated between the cultivated and wild H. brasiliensis cp genomes using MEGA v6.0 (Tamura et al., 2013). The complete cp genome sequences of the four other spurge plastomes, including Euphorbia esula, Jatropha curcas, Manihot esculenta, and Ricinus communis, were downloaded from GenBank, which were further compared with the 12 wild and cultivated H. brasiliensis cp genomes generated in this study.
Repeat Sequence Annotation
Simple sequence repeats including SSRs were predicted using MISA (Thiel et al., 2003) with the parameters set to 10 repeat units ≥10 for mononucleotide SSRs, six repeat units ≥6 for dinucleotide, five repeat units ≥5 for trinucleotide, four repeat units ≥4 for tetranucleotide, and three repeat units ≥3 for pentanucleotide and hexanucleotide SSRs, respectively.
Detection of Genomic Structural Variants
To assess levels of genomic diversity variable and parsimony-informative base sites and nucleotide diversity (Pi) were calculated for the cultivated and wild H. brasiliensis cp genomes using DnaSP version 6.1 (Rozas et al., 2017). To examine microstructural variants among the H. brasiliensis cp genomes, numbers and positions of insertions/deletions (indels) and SNP across the 12 cp genome sequences were detected using Mummer3. The number of SNPs and Indels were identified using BCFtools (Li, 2011). Circos was used to display the detected chloroplast structural variants (Krzywinski et al., 2009).
Phylogenomic Analyses
The phylogenomic analyses were performed for complete cp genome sequences of the 12 cultivated and wild H. brasiliensis accessions using J. curcasas as outgroup. Neighbor-joining (NJ) analysis was executed using MAFFT version 7.221 (Katoh et al., 2005), while Maximum likelihood (ML) analyses of the six Euphorbiaceae species using Gossypium barbadense as outgroup were conducted using MEGA 7.0 (Kumar et al., 2016). RAxML (Stamatakis et al., 2008) searches relied on the general time reversible (GTR) model of nucleotide substitution with the gamma model of rate heterogeneity. Non-parametric bootstrapping test was completed using 500 replicates as implemented in the “fast bootstrap” algorithm of RAxML (Stamatakis et al., 2008). The aligned data matrices are available upon request.
Results and Discussion
Chloroplast Genome Sequencing and Assembly
We employed the Illumina short-read technology with paired-end libraries on the HiSeq2000 sequencing platform to resequence them. For each H. brasiliensis accession, ∼5 GB raw reads were generated with an average length of ∼100 bp (Liu et al., 2020). Paired-end reads were mapped to the published cp genome of H. brasiliensis (Tangphatsornruang et al., 2011) (GenBank Accession Number: HQ285842). Gaps were validated using PCR-based experiments and then sequenced on ABI sequencing platform. All 12 cp genome sequences were deposited into GenBank with the following accession numbers: KY363216, KY363217, KY363218, KY363219, KY363220, KY363221, KY363222, KY3632223, KY419133, KY419134, KY419135, and KY419136.
Characterization of cp Genome Features
Nucleotide sequences of the 12 H. brasiliensis cp genomes ranged from 161,168 bp (IRCI22) to 161,254 bp (AC/F/6B 40/166) (Figure 1 and Table 2). All 12 cp genomes displayed a typical quadripartite structure, which consisted of a pair of IR regions (26,787–26,804 bp) separated by a LSC region (89,216–89,284 bp) and a SSC region (18,370–18,377 bp). The GC content was almost identical each other among the 12 H. brasiliensis cp genomes. GC contents for eight (67,68,78,79,80,84,87, and 88), two (73 and 75), and two (89 and 90) cp genomes were 35.72, 35.71, and 35.73%, respectively, with an average GC content of 35.72%. If duplicated genes in IR regions were counted only once, the 12 H. brasiliensis cp genomes harbored 112 genes in the same order, including 78 protein-coding genes and 34 RNA genes. Overall, the genomic structure, including gene number and order, were well-conserved across the 12 cp genomes (Tables 2, 3). Among these 112 unique genes, 15 (trnA-UGC, trnI-GAU, trnG-GCC, trnK-UUU, trnV-UAC, trnL-UAA, rpl2, rpl16, petD, petB, rpoC1, atpF, ndhA, ndhB, and rps16) had one intron, while three (ycf3, clpP, and rps12) contained two introns (Table 3). The rps12 gene with 3′ end exon and intron located in the IR regions, and the 5′ end exon in the LSC region. Furthermore, matK was located within the intron of trnK-UUU. Overlaps of adjacent genes were found in the chloroplast genomes, for example, atpE-atpB had a 4-bp intersecting region, and psbD-psbC had a 53-bp overlapping region. Unusual initiator codons were observed in rps19 with TGA and ndhD with TGG. Of the 61 tRNAs encoded 20 amino acids, 30 tRNAs were encoded in the rubber chloroplast genome, and the remaining 31 tRNAs may be encoded in the nuclear genome.
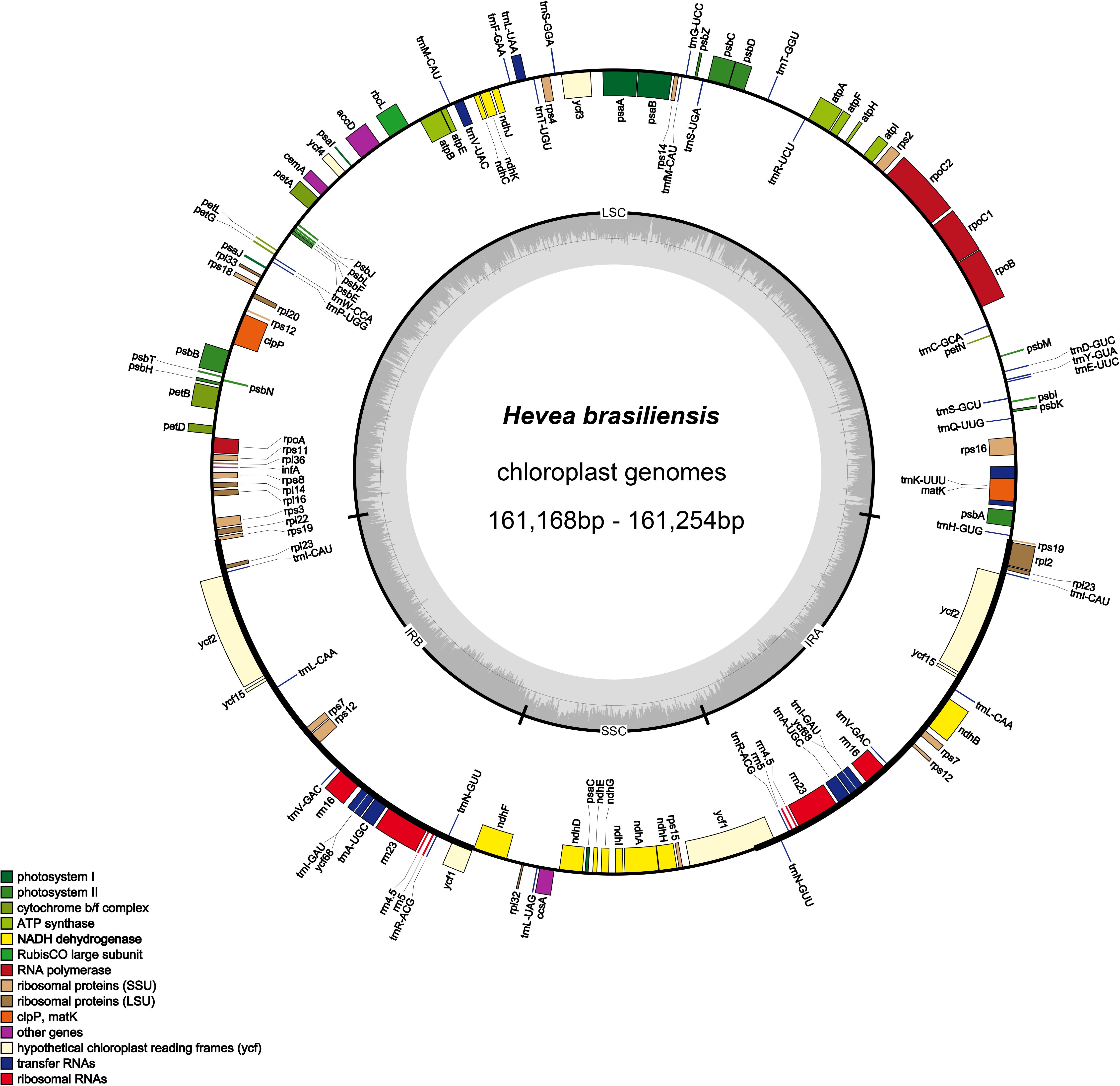
Figure 1. Gene map of the H. brasiliensis chloroplast genomes. Genes lying outside of the outer circle are transcribed in the clockwise direction whereas genes inside are transcribed in the counterclockwise direction. Genes belonging to different functional groups are color-coded. Area dashed darker gray in the inner circle indicates GC content while the lighter gray corresponds to AT content of the genome.
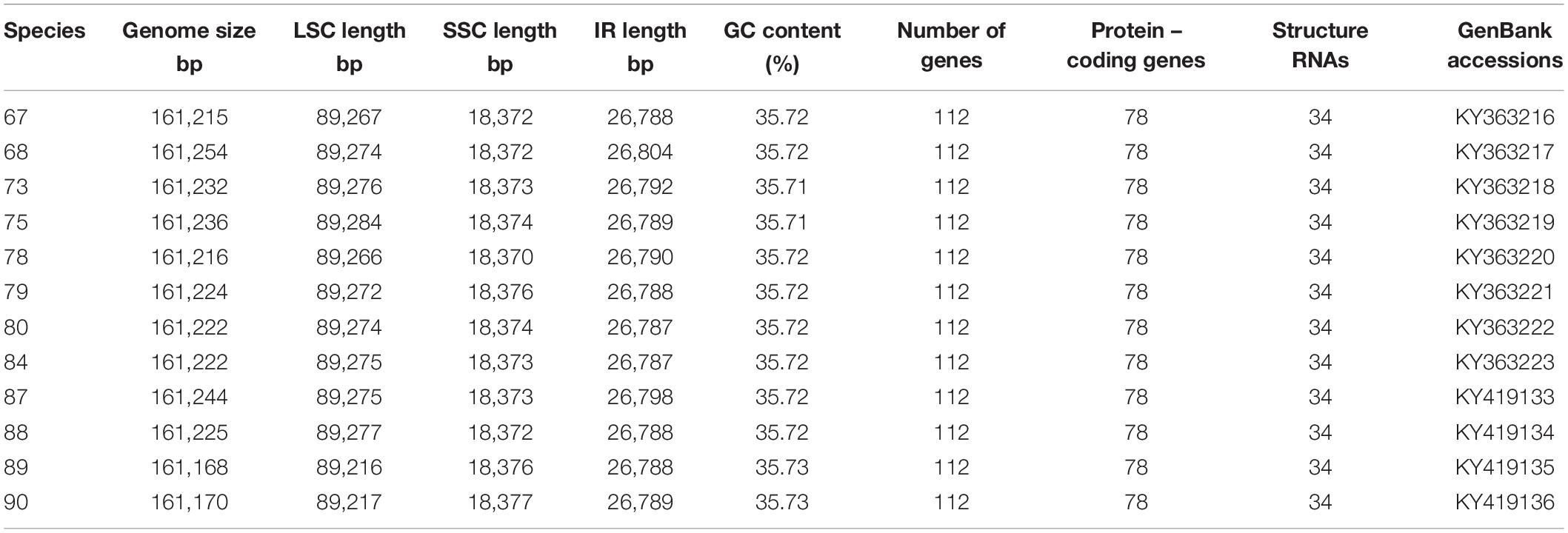
Table 2. Statistic of chloroplast genome features of H. brasiliensis germplasms.
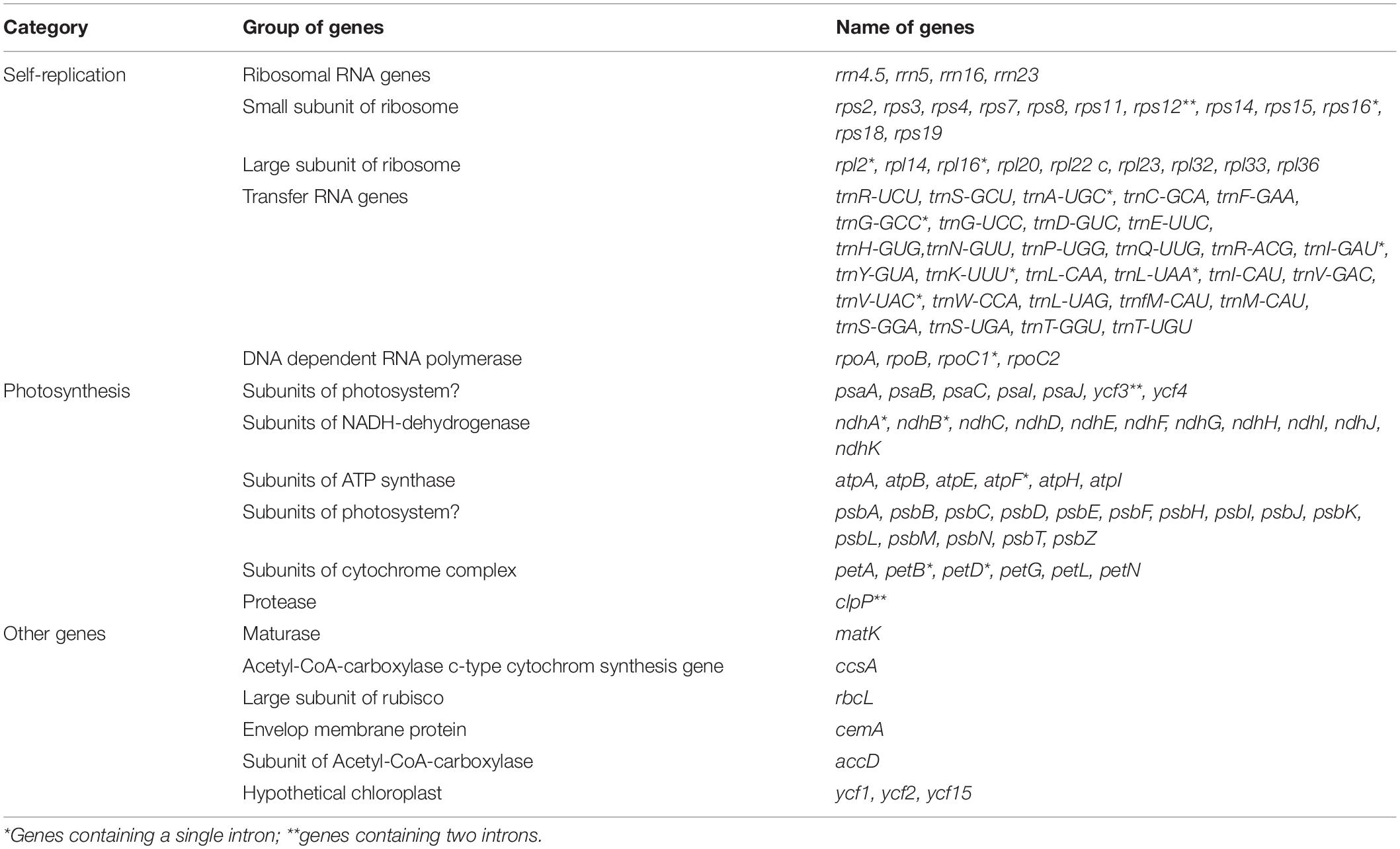
Table 3. Genes contained in the cultivated and wild rubber tree chloroplast genomes.
Repeat Sequences in the 12 H. brasiliensis cp Genomes
Chloroplast simple sequence repeats (cpSSRs) are usually applied to characterizing genetic diversity and performing phylogenetic analyses (Flannery et al., 2006; Yang et al., 2011; Jiao et al., 2012). In total, we detected 105–113 SSRs in the examined H. brasiliensis cp genomes (Figure 2A and Supplementary Table S1A). Our results showed slightly more SSRs in wild rubber tree cp genomes than cultivated rubber three cp genomes. It is clear that mononucleotides were the most abundant while tetranucleotides were the lowest across these cp genomes (Figure 2A and Supplementary Table S1A). Mononucleotide repeat (A/T) was the most abundant, ranging from 90 to 97 across the examined H. brasiliensis cp genomes (Figure 2B and Supplementary Table S1B).
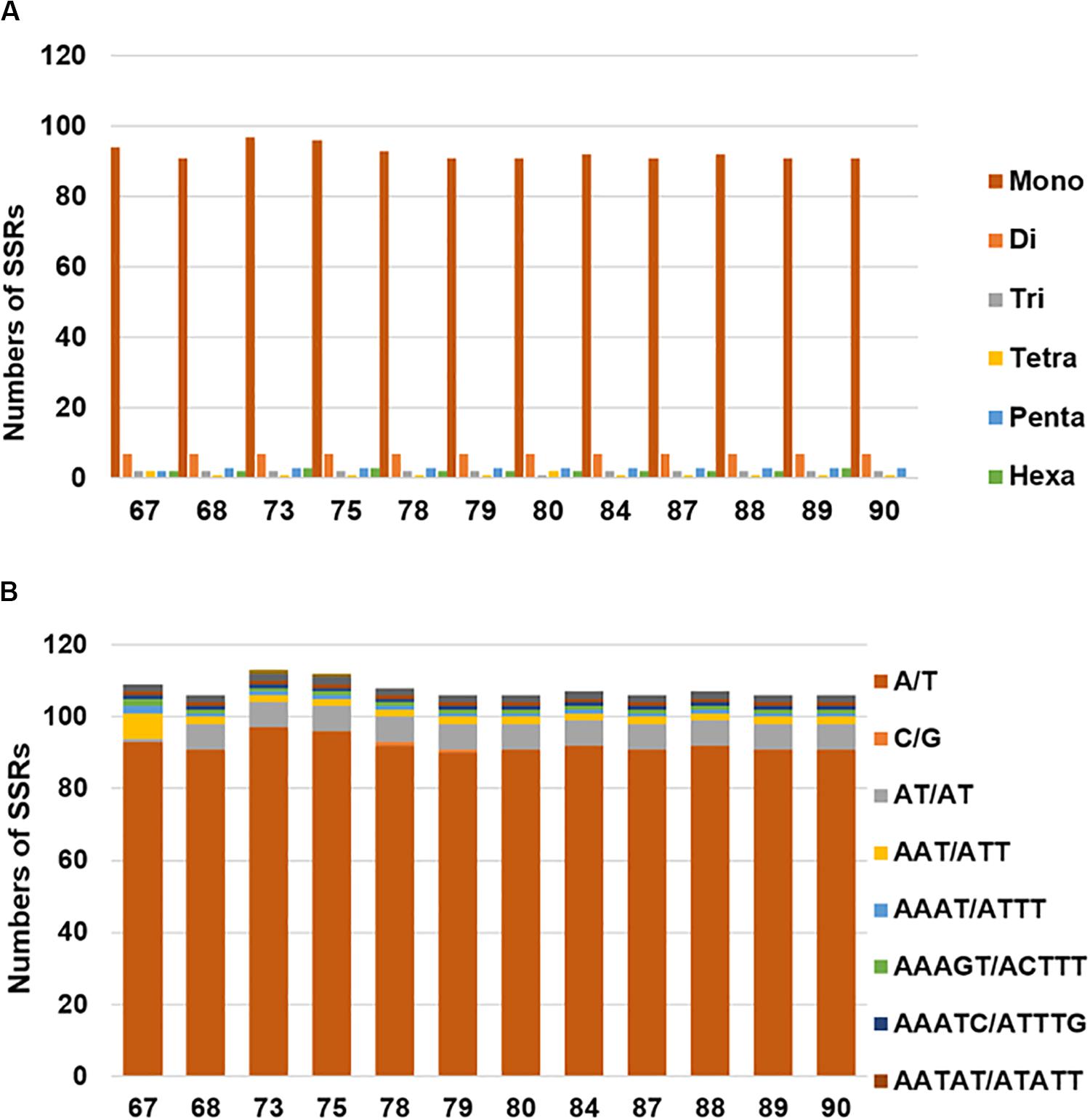
Figure 2. Number and distribution of simple sequence repeats (SSRs) in the 12 H. brasiliensis cp genomes. (A) Number of SSR types in the 12 H. brasiliensis chloroplast genomes; (B) distribution of SSRs in the 12 H. brasiliensis chloroplast genomes.
Genomic Structural Variation Between Cultivated and Wild Rubber Tree Plastomes
We assessed levels of nucleotide variation between cultivated and wild rubber tree plastomes. Our results showed that wild rubber trees possessed more variable sites and ∼2.8-fold higher level of nucleotide variation than cultivated rubber trees (Table 4). This finding highly supports our former population genomic analysis based on nuclear genome resequencing of these rubber tree germplasms (Liu et al., 2020). With these 12 cultivated and wild rubber tree cp genomes at hand, we drew a comprehensive map of their precise genomic variation on the basis of microstructural variants. Using the wild rubber tree cp genome (67) (Table 1) as a reference, we totally identified 725 SNPs and 493 InDels (Figure 3 and Supplementary Table S2). Our results showed that the wild rubber tree (78) from Acre, Brazil harbored the largest number (193) of genomic variants, while rubber tree cultivar (Reyan8-79) had the fewest number of genomic variants (91). We interestingly observed that the density of genomic variants was as high as ∼3.86/Kb in wild rubber trees, which is slightly larger than ∼3.71/Kb detected in cultivated rubber trees. It is clear that genomic variants differently occurred across cultivated and wild rubber tree cp genomes, further supporting the observed patterns of genomic variation detected in the genus Oryza (Gao et al., 2019). In gene regions, such as rpoC1, there were a number of genomic variants detected in wild rubber trees, while cultivated rubber trees showed no genomic variation.

Table 4. Levels of nucleotide variation in the cultivated and wild H. brasiliensis cp genomes.
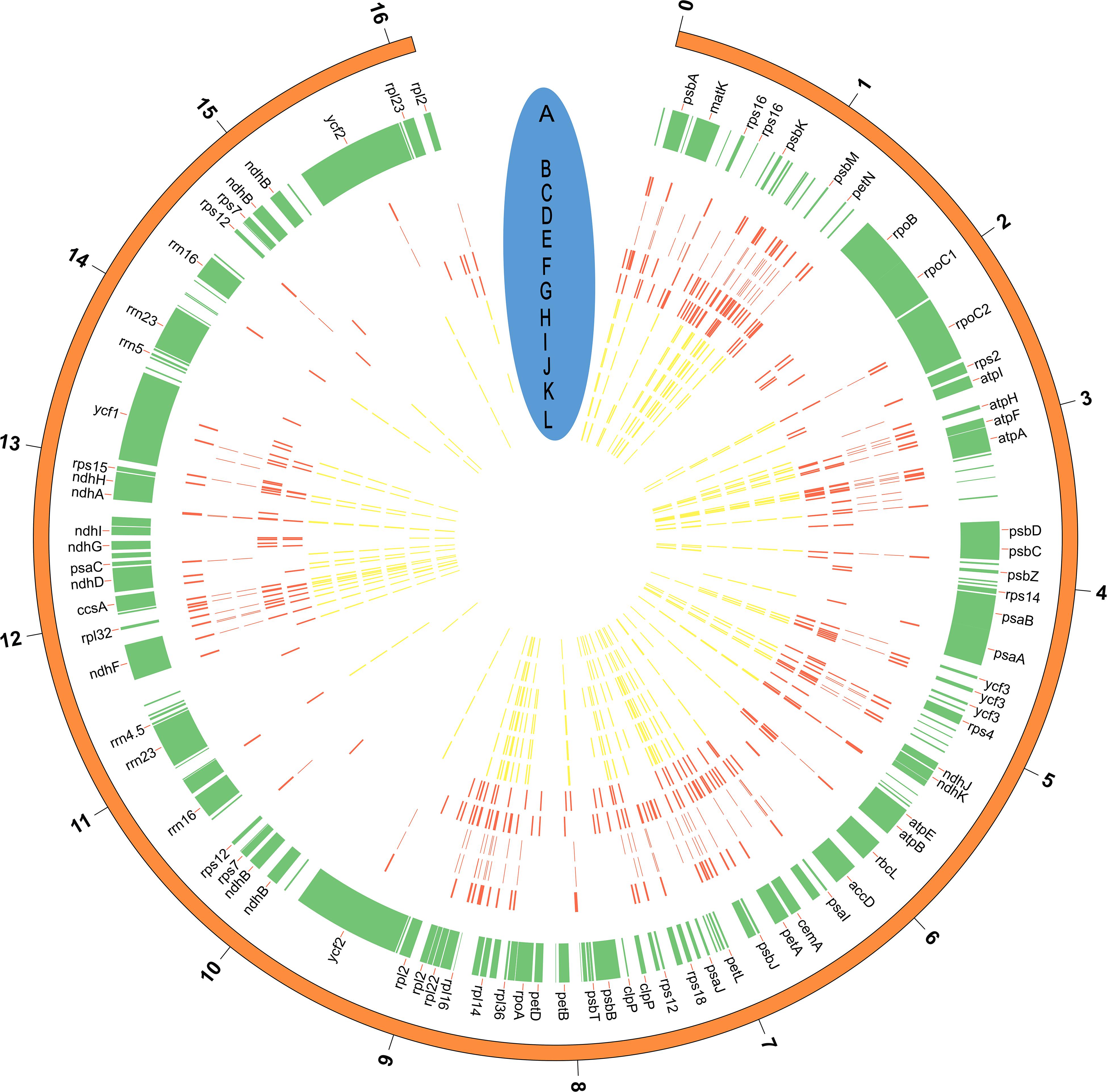
Figure 3. The distribution of genomic variants (SNPs and InDels) across the rubber tree chloroplast genomes. (A) The tag for each gene located in the reference cp genome (67) (Table 1); (B–L) positions of genomic variants identified in the rubber tree cp genomes, representing 68,73,75,78,79,80,84,87,88,89, and 90 (Table 1), respectively. Wild rubber trees are colored in red, while cultivated rubber trees are colored in yellow. The outside distance unit is 10 Kb. Note that not all of cp genes are illustrated in the figure owning to the space limitation.
Expansion and Contraction Among H. brasiliensis and the Other Spurge Plastomes
Although the cp genome size, structure, gene order and gene number are well-conserved, the junctions between IR regions and single-copy (SC) boundary regions were considered as a primarily mechanism causing the length variation of angiosperm cp genomes (Huang et al., 2014; Liu et al., 2018). To elucidate the potential expansion and/or contraction of IR regions, we assessed the variation in the IR/LSC and IR/SSC boundary regions across the six spurge plastomes, including cultivated and wild H. brasiliensis plastomes. The junctions between IR and SSC regions were highly conserved between the cultivated and wild H. brasiliensis plastomes. The genes (ycf1 and rps19-rpl2-rpl22-trnH) were located within the junctions of SSC/IR and LSC/IR regions (Figure 4). Two copies of the ycf1 gene crossed SSC/IRa and SSC/IRb. Compared to the relatively fixed location of the ycf1 gene across all cp genomes, the LSC/IR boundary regions were seemingly variable. The rps19 gene in M. esculenta and H. brasiliensis overlapped the LSC/IRb region with 186-bp and 96-bp located at the IRb region, the rpl22 gene in R. communis spanned the LSC/IRb region with 26-bp located at the IRb region, and the intergenic spacer of rps19-rps12 extended 18-bp or 71-bp to the LSC region in E. esula and J. curcas, respectively. The junction of LSC/IRa regions was located in the intergenic spacer of rps19-trnH (R. communis, M. esculenta and H. brasiliensis), rpl2-trnH (J. curcas), and rpl2-rps19 (E. esula). We observed that the distances between trnH and IRa region varied from 1 to 198-bp.
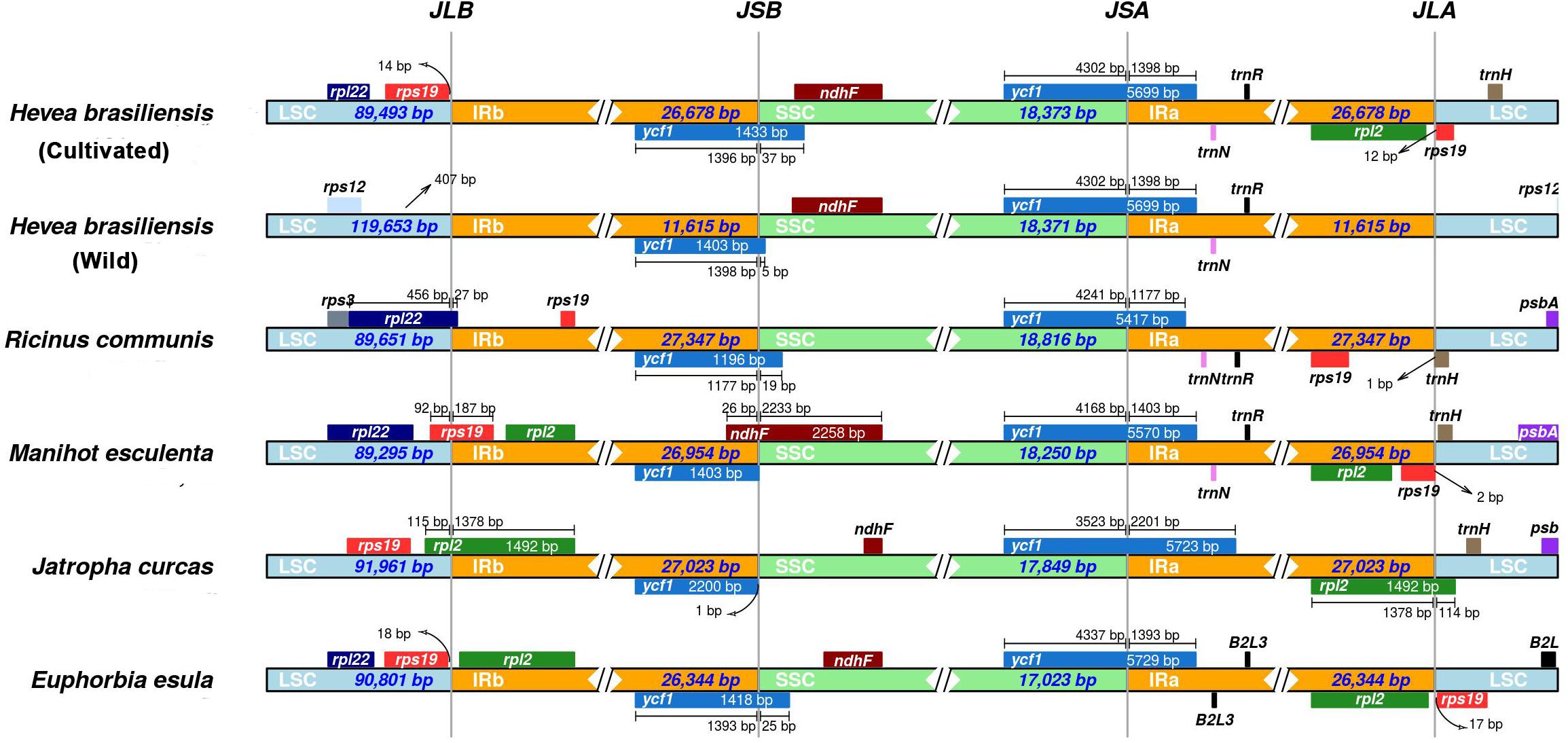
Figure 4. Comparison of border distances between adjacent genes and junctions of LSC, SSC, and two IR regions among chloroplast genomes of the five spurge species.
Comparative Analyses Among H. brasiliensis and the Other Spurge Plastomes
Pairwise chloroplast genomic alignments among six spurge plastomes showed a relatively high degree of synteny conservation. The R. communis cp genome was used as a reference for plotting the overall sequence identity of the cp genomes of the six spurge plastomes (Figure 5). Our results revealed relatively high sequence identities. The LSC and SSC regions exhibited less similarity than the two IR regions across all Euphorbiaceae species. All the rRNA genes were highly conserved and similar to other plant chloroplast genomes (Huang et al., 2014). In addition, non-coding regions displayed greater sequence divergence than protein-coding regions. These highly divergent regions contained ccsA, ndhA, rbcL, rpoC2, rpl33, ycf3, atpI-atpH, psbM-petN, and petA-psbJ (Figure 5). Such hotspot regions could be developed as molecular markers and species barcoding for future phylogenetic analyses and species identification in the family Euphorbiaceae.
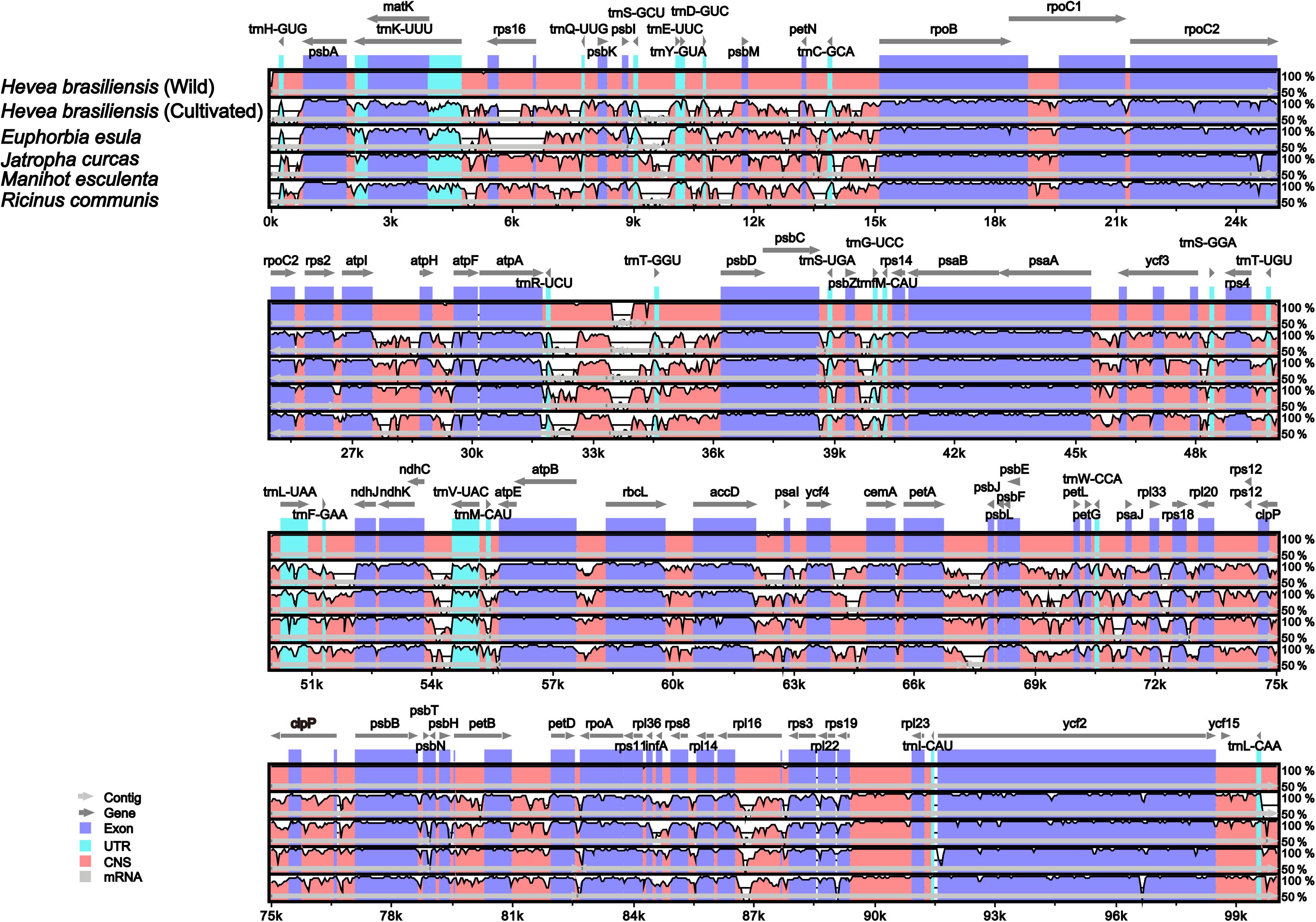
Figure 5. Alignment of the six chloroplast genome sequences. VISTA-based identity plot shows sequence identity among the six Euphorbiaceae species using Hevea brasiliensis cp genome (67) as a reference. The vertical scale designates the percentage of identity, ranging from 50 to 100%. The horizontal axis indicates the coordinates within the chloroplast genome. Genome regions are color-coded as exons, introns, and conserved non-coding sequences (CNS).
Phylogenomic Analysis of Cultivated and Wild H. brasiliensis Germplasms
To determine evolutionary relationships of cultivated and wild rubber trees, we compared all 12 cp genomes sequences using J. curcas as outgroup. Using neighbor-joining (NJ) method our results showed that cultivated and wild rubber trees failed to form separate clades (Figure 6A), further supporting our former phylogenomic analysis based on nuclear genome resequencing (Liu et al., 2020). We further investigated phylogenetic relationships of the six Euphorbiaceae species established by utilizing complete cp genomes using Gossypium barbadense as outgroup. Phylogenetic analyses of the aligned data matrix using Maximum Likelihood (ML) suggested that that cultivated and wild rubber trees formed together with 100% bootstrap supports, which were further grouped with M. esculenta with a full bootstrap support (Figure 6B). Thus, the ML tree has well-resolved phylogenetic relationships of these spurge species with ≥95% bootstrap values, which is fairly congruent with former phylogenetic analyses based on complete chloroplast and nuclear genomes (Tangphatsornruang et al., 2011; Rahman et al., 2013; Tang et al., 2016; Pootakham et al., 2017; Liu et al., 2020).
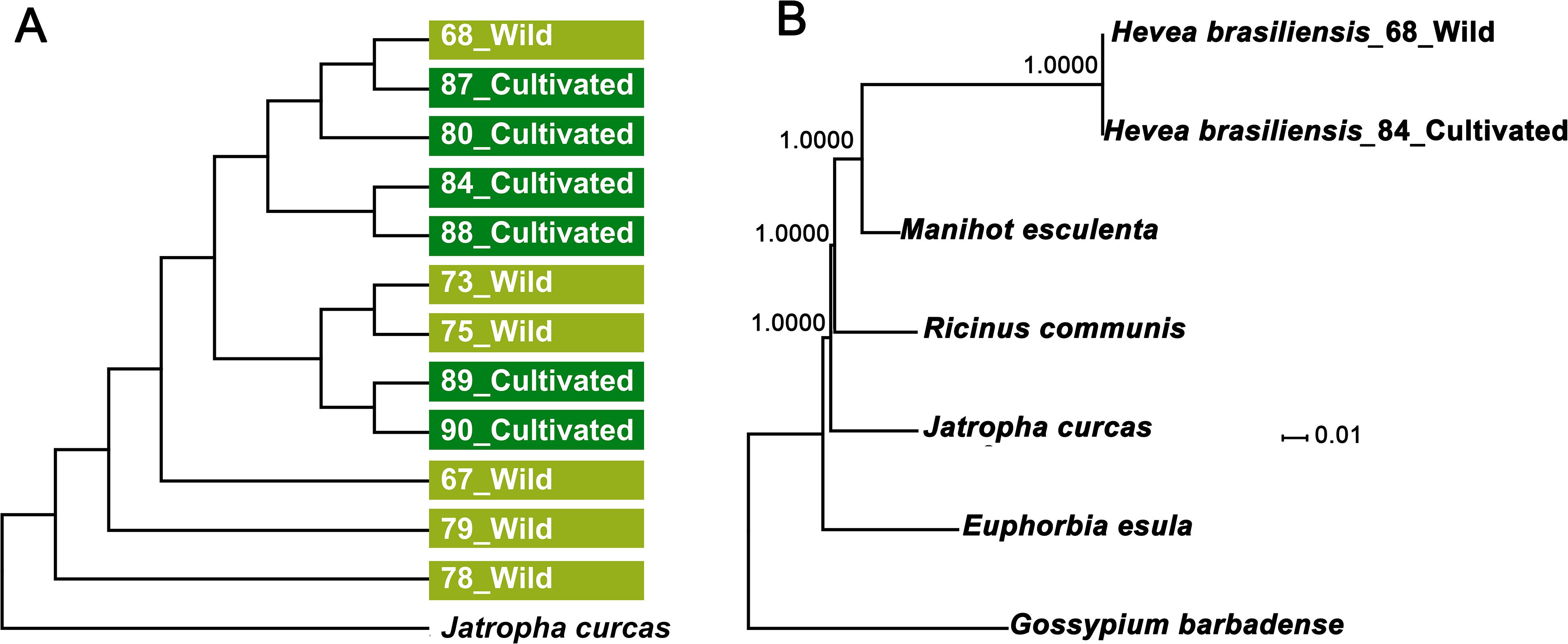
Figure 6. Phylogenetic analyses of the 12 H. brasiliensis accessions and the five Euphorbiaceae species. (A) Phylogenetic tree of the 12 H. brasiliensis accessions inferred from complete nucleotide sequences of the chloroplast genomes; (B) ML phylogenetic tree of the five Euphorbiaceae species.
Conclusion
In the present study, we present the 12 complete chloroplast genomes of cultivated and wild H. brasiliensis. We characterized the microstructural variation in the five spurge plastomes, including H. brasiliensis, Euphorbia esula, Jatropha curcas, Manihot esculenta, and Ricinus communis. We show that the H. brasiliensis cp genome differed from the other spurge plastomes with large inversions. We find that cp genomes of wild rubber trees are more variable than cultivated rubber tree cp genomes. The detection of precise genomic variation across rubber tree cp genomes further reveals more genomic variants occurred in wild rubber trees than cultivated rubber trees. Further efforts are still needed to obtain a comprehensive map of chloroplast genomic variation in cultivated and wild rubber trees in a broad geographic range, which will help to better conserve and utilize rubber tree germplasms.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
L-ZG: conceptualization, visualization, supervision, and funding acquisition. L-YF, C-WG, and H-BW: formal analysis. JL and G-HL: investigation. JL: resources. L-YF: data curation. L-YF and JL: writing – original draft preparation. L-ZG, L-YF, and JL: writing – review and editing. G-HL: project administration. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Yunnan Innovation Team Project (to L-ZG), Key Research and Development Project of Guangdong Province (2020B020217002), and MOST of China (2018YFD1000502).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00237/full#supplementary-material
TABLE S1A | Type of SSRs in the 12 H. brasiliensis chloroplast genomes.
TABLE S1B | Distribution of SSRs in the 12 H. brasiliensis chloroplast genomes.
TABLE S2 | Nucleotide variation across the 12 H. brasiliensis chloroplast genomes.
Footnotes
- ^ http://www.clcbio.com/products/clc-assemblycell/
- ^ http://www.geneious.com
- ^ http://www.tigr.org/software/mummer/
References
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Bendich, A. J. (2004). Circular chloroplast chromosomes: the grand illusion. Plant Cell 16, 1661–1666. doi: 10.1105/tpc.160771
Conant, G. C., and Wolfe, K. H. (2008). GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862. doi: 10.1093/bioinformatics/btm598
Dong, W., Xu, C., Li, D., Jin, X., Li, R., Lu, Q., et al. (2016). Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 4:e2699. doi: 10.7717/peerj.2699
Flannery, M. L., Mitchell, F. J., Coyne, S., Kavanagh, T. A., Burke, J. I., Salamin, N., et al. (2006). Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 113, 1221–1231. doi: 10.1007/s00122-006-0377-0
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273. doi: 10.1093/nar/gkh458
Gao, L. Z., Liu, Y., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2:278. doi: 10.1038/s42003-019-0531-2
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. Mol. Biol. Evol. 14:151. doi: 10.1186/1471-2148-14-151
Jiao, Y., Jia, H. M., Li, X. W., Chai, M. L., Jia, H. J., Chen, Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genom. 13:201. doi: 10.1186/1471-2164-13-201
Katoh, K., Kuma, K., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lau, N., Makita, Y., Kawashima, M., Taylor, T. D., Kondo, S., Othman, A. S., et al. (2016). The rubber tree genome shows expansion of gene family associated with rubber biosynthesis. Sci. Rep. 6:28594. doi: 10.1038/srep28594
Leitch, A., Lim, K., Leitch, I., O’Neill, M., Chye, M., and Low, F. (1998). Molecular cytogenetic studies in rubber, Hevea brasiliensis Muell. arg. (Euphorbiaceae). Génome 41, 464–467. doi: 10.1139/g98-012
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Liu, J., Shi, C., Shi, C., Li, W., Zhang, Q., Zhang, Y., et al. (2020). The chromosome-based rubber tree genome provides new insights into spurge genome evolution and rubber biosynthesis. Mol. Plant. 13, 336–350. doi: 10.1016/j.molp.2019.10.017
Liu, L., Wang, Y., He, P., Li, P., Lee, J., Soltis, D. E., et al. (2018). Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 19:235. doi: 10.1186/s12864-018-4633-x
Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402. doi: 10.1146/annurev.genom.9.081307.164359
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Pootakham, W., Sonthirod, C., Naktang, C., Ruang-Areerate, P., Yoocha, T., Sangsrakru, D., et al. (2017). De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci. Rep. 7:41457. doi: 10.1038/srep41457
Priyadarshan, P. M., and Clément-Demange, A. (2004). Breeding Hevea rubber: formal and molecular genetics. Adv. Genet. 52:51. doi: 10.1016/S0065-2660(04)52003-5
Rahman, A. Y. A., Usharraj, A. O., Misra, B. B., Thottathil, G. P., Jayasekaran, K., Feng, Y., et al. (2013). Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genom. 14:75. doi: 10.1186/1471-2164-14-75
Raman, G., and Park, S. (2015). Analysis of the Complete Chloroplast Genome of a Medicinal Plant, Dianthus superbus var. longicalyncinus, from a Comparative Genomics Perspective. PLoS One 10:e141329. doi: 10.1371/journal.pone.0141329
Raubeson, L. A., and Jansen, R. K. (2005). Chloroplast genomes of plants, Plant diversity and evolution: genotypic and phenotypic variation in higher plants. Divers. Evol. Plants Genotypic Phenotypic Var. High. Plants 3, 26–45. doi: 10.1079/9780851999043.0045
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sasaki, T., Yukawa, Y., Miyamoto, T., Obokata, J., and Sugiura, M. (2003). Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol. Biol. Evol. 20, 1028–1035. doi: 10.1093/molbev/msg098
Shearman, J. R., Sangsrakru, D., Ruang-Areerate, P., Sonthirod, C., Uthaipaisanwong, P., Yoocha, T., et al. (2014). Assembly and analysis of a male sterile rubber tree mitochondrial genome reveals DNA rearrangement events and a novel transcript. BMC Plant Biol. 14:45. doi: 10.1186/1471-2229-14-45
Shi, C., Hu, N., Huang, H., Gao, J., Zhao, Y. J., and Gao, L. Z. (2012). An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS One 7:e31468. doi: 10.1371/journal.pone.0031468
Shi, C., Wang, S., Xia, E. H., Jiang, J. J., Zeng, F. C., Yao, Q. Y., et al. (2016). Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 6:30135. doi: 10.1038/srep30135
Sneller, C. H., Miles, J. W., and Hoyt, J. M. (1997). Agronomic performance of soybean plant introductions and their genetic similarity to elite lines. Crop Sci. 37, 1595–1600. doi: 10.2135/cropsci1997.0011183X003700050032x
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Sugiura, M. (1992). The chloroplast genome. Plant. Mol. Biol. 19, 149–168. doi: 10.1007/978-94-011-2656-4_10
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, C., Yang, M., Fang, Y., Luo, Y., Gao, S., Xiao, X., et al. (2016). The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants. 2:16073. doi: 10.1038/nplants.2016.73
Tangphatsornruang, S., Uthaipaisanwong, P., Sangsrakru, D., Chanprasert, J., Yoocha, T., Jomchai, N., et al. (2011). Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 475, 104–112. doi: 10.1016/j.gene.2011.01.002
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare. L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xu, C., Dong, W., Li, W., Lu, Y., Xie, X., Jin, X., et al. (2017). Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 8:15. doi: 10.3389/fpls.2017.00015
Yang, A. H., Zhang, J. J., Yao, X. H., and Huang, H. W. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98, 123–126. doi: 10.3732/ajb.1000532
Yang, J., Yue, M., Niu, C., Ma, X. F., and Li, Z. H. (2017). Comparative analysis of the complete chloroplast genome of four endangered herbals of Notopterygium. Genes 8:124. doi: 10.3390/genes8040124
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS One 5:e12762. doi: 10.1371/journal.pone.0012762
Keywords: Hevea brasiliensis, chloroplast genome, rubber tree, comparative genomics, phylogenetic analysis
Citation: Feng L-Y, Liu J, Gao C-W, Wu H-B, Li G-H and Gao L-Z (2020) Higher Genomic Variation in Wild Than Cultivated Rubber Trees, Hevea brasiliensis, Revealed by Comparative Analyses of Chloroplast Genomes. Front. Ecol. Evol. 8:237. doi: 10.3389/fevo.2020.00237
Received: 24 March 2020; Accepted: 30 June 2020;
Published: 17 July 2020.
Edited by:
Liangsheng Zhang, Zhejiang University, ChinaReviewed by:
Shaojun Dai, Northeast Forestry University, ChinaXiayan Liu, Northwest A&F University, China
Copyright © 2020 Feng, Liu, Gao, Wu, Li and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Zhi Gao, TGdhb2dlbm9taWNzQDE2My5jb20=; bGdhb0BtYWlsLmtpYi5hYy5jbg==
†These authors have contributed equally to this work