 Elizabeth M. A. Kern
Elizabeth M. A. Kern Taeho Kim
Taeho Kim Joong-Ki Park
Joong-Ki Park- Division of EcoScience, Ewha Womans University, Seoul, South Korea
In recent decades, great strides have been made using various kinds of data including molecular sequences to clarify the phylogenetic relationships of nematodes. Although SSU rDNA sequences are among the most widely used markers in nematode phylogeny, there are areas within Nematoda that do not seem amenable to resolution by this single nuclear gene. The use of the complete mitochondrial genome (mitogenome) is an alternative and powerful molecular method for inferring phylogenies and is frequently used in nematodes. Here we present an overview of the contributions of mitogenome phylogenies in the phylum Nematoda. The nematode mitogenome has several practical strengths as a phylogenetic marker, and has yielded well-supported results for clades which were not well resolved using other approaches. We discuss places where nematode mitogenomes have sometimes produced alternative tree topologies compared to nuclear gene phylogenies, review to what extent mitochondrial gene order is useful in nematode phylogenetics, and make recommendations for promising future areas of research.
Introduction
Well-reconstructed phylogenies are essential to interpreting much of biological research. In the phylum Nematoda, inferring accurate phylogenies can provide significant insights into the evolution and diversity of this remarkable group. Nematodes constitute one of the most common, ecologically diverse, and speciose animal groups in the world. They are found in almost every type of terrestrial and aquatic environment, from the tropics to the polar regions and from hot deserts to deep sea trenches, and their diverse lifestyles include free-living forms and invertebrate-, plant- and vertebrate-parasitic species (Golden, 1971; Poinar, 1991). Many nematode species are of great economic importance or medical interest, such as plant-parasitic nematodes that affect global crop and timber yields (Ruehle, 1967; Sikora et al., 2005) and major livestock and human parasites (Anderson, 2000). Other species have served as models for biological research (i.e., Caenorhabditis elegans), or biomonitoring (Semprucci et al., 2015). The number of nematode species has been estimated at 1 million, of which about 27,000 species have been described (Hugot et al., 2001).
During the last two decades of nematode phylogenetics, the use of molecular data has led to dramatic revision of early taxonomies which relied heavily on a limited number of “key morphological characters” or overemphasized the importance of ecological features (Blaxter et al., 1998; De Ley and Blaxter, 2002). Great strides have been made using SSU rDNA data (i.e., the sequence of the small subunit RNA gene, also known as 18S rDNA) to establish a phylum-wide phylogeny for nematodes, which has considerably reduced the confusion arising from reliance on morphological characters alone (Blaxter et al., 1998; Holterman et al., 2006; van Megen et al., 2009). Other contributions have come from LSU rDNA (28S rDNA) or mitochondrial cytochrome c oxidase subunit 1 (cox1) sequences, and in the future we may even see whole nuclear genome sequences become widely used (Kumar et al., 2012). At present, SSU rDNA sequences are readily available and have been publicly archived for a large number of nematode species. However, there are branches in the nematode phylogenetic tree that have not been well resolved using SSU rDNA (or other single gene sequences). Despite the many advantages of using nuclear rDNA sequences, they are not suitable for all types of phylogenetic research; for example, their highly conserved nature does not always make them adequate for distinguishing among closely related species. Furthermore, nematode phylogenies using nuclear rDNA markers are very sensitive to sequence alignment due to length heterogeneity across taxa, a limiting factor that often hinders a robust phylogeny, especially when assessing deep relationships (Smythe et al., 2006). Recent phylogenomic analyses of whole genomes or transcriptome information have produced results that are largely consistent with the relationships among major nematode clades supported by SSU phylogenies (Blaxter and Koutsovoulos, 2015; Smythe et al., 2019). These whole genome and transcriptome studies have contributed to our understanding of deep node nematode phylogeny, but have been limited by low taxon sampling (which is also biased toward some parasitic groups) and by the technical challenges inherent in obtaining a large quantity of genetic information from a single nematode with a small body size. In such cases, the use of whole mitochondrial genome (mitogenome) sequences is one alternative that has been widely applied to many nematode branches where relationships were unclear. Here we review the use of the mitogenome thus far in nematode phylogenomics, its challenges and advantages, and point out areas for future work.
Nematode Mitochondrial Genomes
Nematode mitogenomes are similar to those of other animals in many respects, but have a few unusual features including high variation in conservation of gene order across major branches and the occasional presence of multiple chromosomes. The nematode mitogenome is usually a single, circular molecule (exceptions discussed below) ranging in size from 12 to 22 kb and containing 36 (sometimes 37) genes: 12 (or 13) protein-coding genes (cox1-cox3, cytb, nad1-nad6, nad4L, atp6, and rarely atp8), 2 ribosomal RNA (lrRNA and srRNA), and 22 tRNA genes. The atp8 gene, which is found in most other metazoan mitogenomes (except the parasitic platyhelminth clade Neodermata; Egger et al., 2017), is usually absent in nematode mitogenomes, although it does appear in Trichinella spp. and Trichuris spp. (Lavrov and Brown, 2001; Liu et al., 2012a, b, 2013; Mohandas et al., 2014; Hawash et al., 2015). Nematode mitochondrial genes can be coded on one strand (which is the case for all chromadorean nematodes except for some Plectus species; Kim et al., 2017) or both strands (a general feature of enoplean nematodes) (Hyman et al., 2011). Nematode mitogenomes usually show a strong nucleotide compositional bias toward A and T, which together account for between 63.7% [Longidorus vineacola (Kim et al., 2014; Palomares-Rius et al., 2017)] and 85.4% of total nucleotides in the genome [Radopholus similis (Jacob et al., 2009)]. This compositional bias in nematodes is due to a high propensity for using T-rich codons (having more than two Ts in a triplet) and unequal usage of synonymous codons with a bias against C-rich codons in the third codon position in protein-coding genes (Park et al., 2011; Palomares-Rius et al., 2017). Nucleotide compositional bias (negative AT skew and positive GC skew; Kim et al., 2014) and variable gene order (substantial gene rearrangement, particularly in enoplean species) should be taken into account when designing studies and selecting analytical methods for nematode mitogenomes.
One of the major strengths of the mitogenome as a phylogenetic marker comes from the fact that its 36 genes have different substitution rates (Saccone et al., 1999), some with high rates that trace relatively recent splits, and some with slow rates that are useful for elucidating deeper relationships. In essence, the mitogenome can provide resolution for an enormously broad range of phylogenetic depths, from shallow divergence times between populations of a single species to deep divergence within an entire phylum. For instance, on the shallow end of the scale, mitogenomes have permitted the estimation of a divergence time of 47,000 generations between West African and Papua New Guinean populations of Wuchereria bancrofti (a filarial nematode that constitutes a serious human parasite; Ramesh et al., 2012). Across another short timeframe, mtDNA genes have been used to compare population divergence of Ascaris roundworms (a common human parasite) within individual hosts and among households (Anderson et al., 1995). On the deep end of the scale, mitogenomes have contributed to resolving relationships among major clades within various metazoan phyla (Park et al., 2007 [Platyhelminthes]; Min and Park, 2009 [Rotifera]), and a mitochondrial phylogeny has even been attempted for Metazoa as a whole (Bernt et al., 2013). Mitogenomes often prove useful for elucidating formerly intractable phylogenies, clarifying the relationships within phylogenetically difficult groups where rapid radiations made other markers ineffective (Yu et al., 2007; Miya and Nishida, 2014). Other advantages of the mitogenome include its size, which is large enough to contain a lot of informative characters yet small enough to be analyzed on an ordinary desktop computer; its haploidy and maternal inheritance (with the exception of the doubly uniparental inheritance in some bivalve species; see Breton et al., 2007), which give it a short coalescence time (Moore, 1995); its low rate of recombination (but see Piganeau et al., 2004 and Tsaousis et al., 2005); and the unambiguous orthology of its encoded genes (Wolstenholme, 1992; Nadler and De Len, 2011). In addition to sequence information, mitogenome gene order (which we will discuss in more detail later) is also sometimes useful in phylogenetics as corroborative evidence (Boore, 1999).
The mitogenome has some drawbacks as well [reviewed in Ballard and Whitlock (2004) and Hoelzer (1997)], and before choosing to use mtDNA for phylogenetic research, consideration should be given to the needs of a particular project. For instance, a known drawback is that the mitogenome can reflect only mitochondrial evolutionary history, which is not necessarily the same as species evolutionary history (Nichols, 2001). Since the mitochondria are essentially inherited as a single locus (for some cases of mitochondrial recombination, see Ladoukakis and Zouros, 2001; Burzyński et al., 2003), its individual genes are not statistically independent samples for the purposes of estimating phylogenies, and a mitogenome-based tree may not capture the same broad perspective that could be achieved from using multiple unlinked markers. However, the difference between species trees and mitogenome trees is often ignored without dire consequences. Another consideration that should be taken into account is whether the focal taxa exhibit female philopatry and male dispersal, which cause mtDNA to evolve differently than nuclear DNA (Hoelzer, 1997). Although this can be a disadvantage in phylogenetics it can be an advantage in other ways, since the differences between mtDNA and nuclear DNA evolution can be used to discover previously unknown, sex-biased dispersal patterns (Prugnolle and de Meeus, 2002). Sex differences in dispersal behavior are found in many vertebrates and in at least a few parasitic nematode species (Grewal et al., 1993); however, dispersal behaviors of most nematode species have never been studied. Finally, a current logistical constraint in using mitogenomes is the number of species available in public databases. Although the number of sequenced nematode mitogenomes has risen to 200 species, Nematoda is still extremely underrepresented considering the taxonomic diversity of the phylum, and taxon sampling is strongly biased toward chromadorean species (Table 1; there are 172 chromadorean and 28 enoplean species in GenBank as of December 2019).
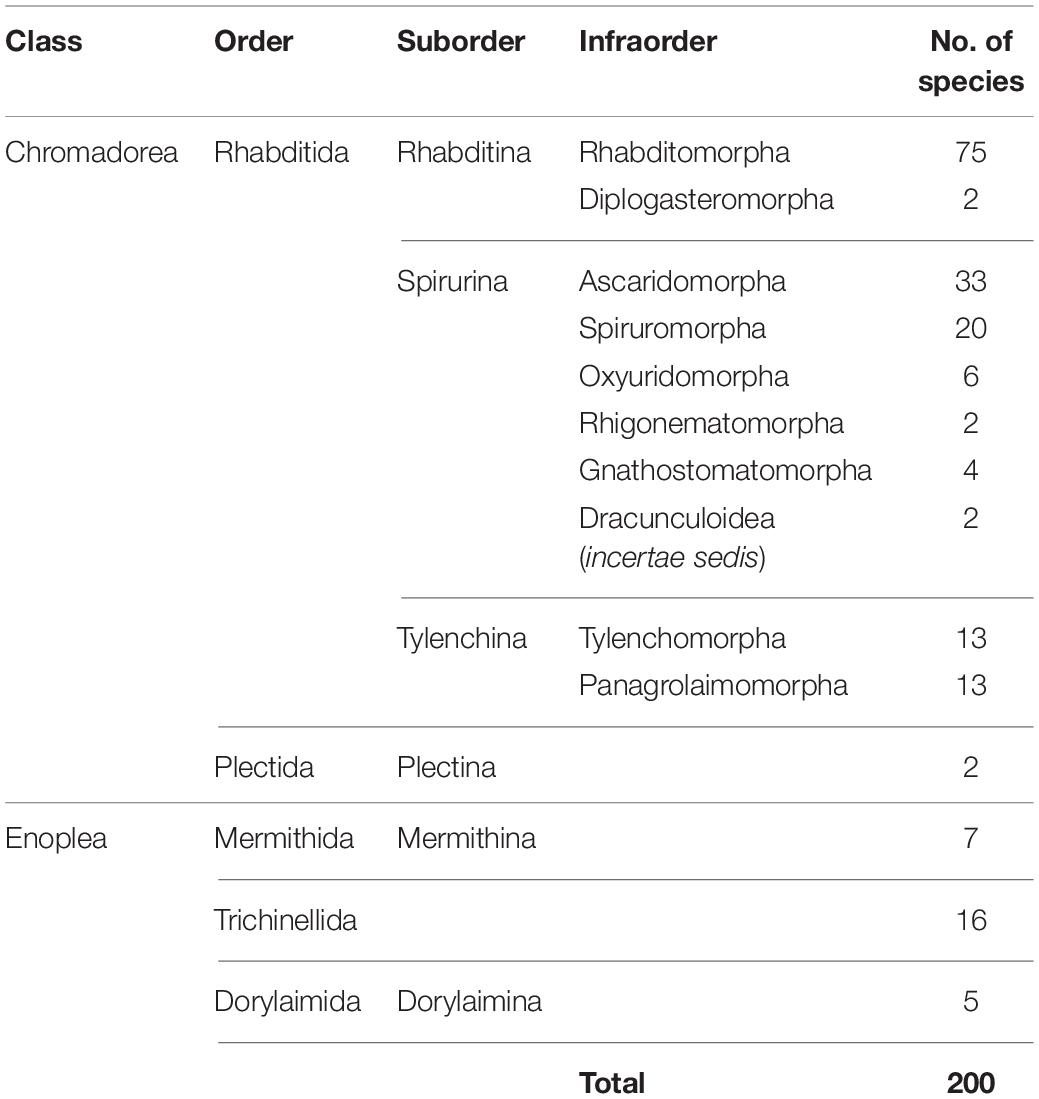
Table 1. Number of complete mitochondrial genomes of nematode species available on GenBank (as of December 2019).
Discordance Between Mitogenome and Nuclear Gene Phylogenies
For the majority of nematode clades, mitogenome analyses have yielded results similar to nuclear gene trees (which are mostly SSU rDNA trees). In addition, mitochondrial data have often clarified relationships where there was poor resolution by nuclear DNA sequence data or where there was disagreement between morphological characters and genetic data. However, sometimes mtDNA data have strongly supported different tree topologies from SSU rDNA-based phylogenies (Figure 1). The following sections describe some of these conflicts. These areas represent key places that future researchers could focus on by incorporating additional taxa and/or independent genetic markers.
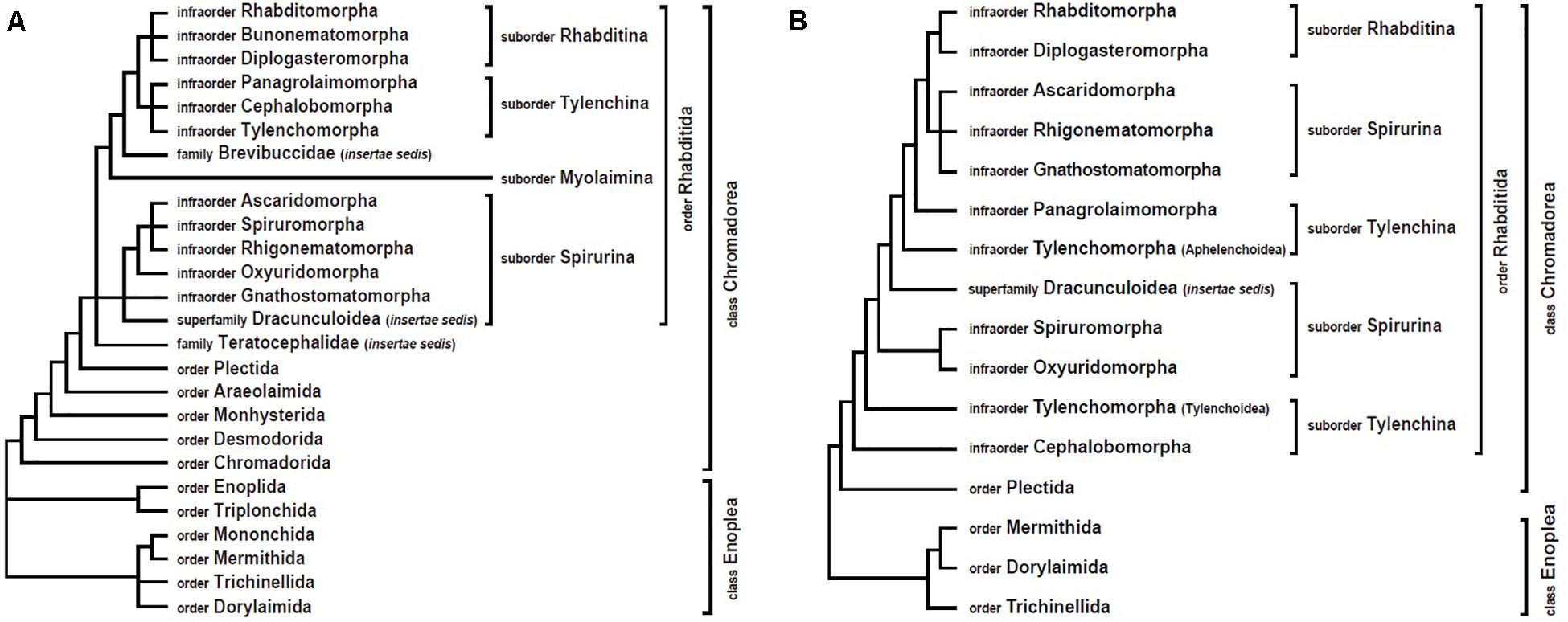
Figure 1. Comparison of nematode phylogenetic relationships derived from nuclear versus mitochondrial analyses. (A) Overview of SSU rDNA gene tree (based on De Ley and Blaxter, 2002, 2004). (B) Overview of mitogenome tree, constructed with amino acid sequences and maximum likelihood methods (based on Kim et al., 2017).
Non-monophyly of Suborder Spirurina
In the first phylum-wide molecular phylogeny of nematodes (Blaxter et al., 1998), five major clades (clades I–V) were identified. Among these, clade III grouped together four animal parasitic orders (Oxyurida, Spirurida, Rhigonematida, and Ascaridida, with Oxyurida being sister to the others). Members of clade III have major impacts on human and animal health, and understanding their phylogenetic relationships to non-parasitic nematodes can inform our understanding of the evolution of zooparasitism. In the revised nematode classification system based on SSU rDNA data (De Ley and Blaxter, 2002, 2004), the four groups within clade III were each treated as infraorders (Oxyuridomorpha, Spiruromorpha, Rhigonematomorpha, and Ascaridomorpha) and placed in the suborder Spirurina along with two additional groups, Gnathostomatomorpha and Dracunculoidea (incertae sedis) (Figure 1A). Clade III was again widely accepted as monophyletic (and called Group 8) in a subsequent phylum-wide SSU rDNA analysis that included many more species (Holterman et al., 2006; van Megen et al., 2009). However, recent mitogenome phylogenies strongly support non-monophyly of clade III (suborder Spirurina) (Kang et al., 2009; Park et al., 2011; Kim et al., 2014, 2017, 2020). Specifically, in mtDNA trees, the members of clade III are split into two separate groups: Rhigonematomorpha is nested within Ascaridomorpha, forming a sister clade to Rhabditomorpha and Diplogasteromorpha, while the remaining clade III members, Oxyuridomorpha and Spiruromorpha, form a separate group (Kim et al., 2017, 2020; Figure 1B). Relationships among oxyuridomorphs, spiruromorphs and dracunculoids have varied depending on the type of mitogenome datasets used (nucleotide versus amino acid sequence datasets) and/or the analytic methods applied (Bayesian versus maximum likelihood). More comprehensive taxon sampling and/or additional genetic markers will be needed to address conflicts between mtDNA trees and nuclear gene trees.
Non-monophyly of Suborder Tylenchina
The suborder Tylenchina (class Chromadorea) contains a broad array of fungivores, bacteriovores, plant feeders, insect associates/parasites, some vertebrate parasites, and certain plant parasites that cause devastating agricultural losses (e.g., Heterodera glycines). Phylogenetic assessment of this suborder using SSU rDNA recognized Tylenchina as monophyletic, with four infraorders: Tylenchomorpha, Panagrolaimomorpha, Cephalobomorpha, and Drilonematomorpha (De Ley and Blaxter, 2002, 2004). In contrast however, mitogenome trees consistently depict Tylenchina as non-monophyletic (Kim et al., 2015, 2017, 2020). In mitochondrial analyses, Aphelenchoidea (one of the Tylenchomorpha superfamilies) and Panagrolaimomorpha are situated separately from Tylenchoidea (Tylenchomorpha) and Cephalobomorpha (Figure 1B).
Within Tylenchina, the infraorder Tylenchomorpha is also depicted differently in SSU trees versus mitogenome trees. In a recent nuclear gene tree, Aphelenchoidea members were split: Aphelenchoididae, Parasitaphelenchidae, and Seinuridae were nested within Panagrolaimomorpha (not including Steinernematidae) and were separated from Aphelenchidae, which was sister to the remaining members of Tylenchomorpha (van Megen et al., 2009). In contrast, mitogenome trees have consistently depicted Aphelenchoidea as being monophyletic, positioned away from other plant-parasitic Tylenchoidea members (Tylenchomorpha) and located inside a novel clade containing species from some non-Tylenchomorpha infraorders: Rhabditomorpha, Panagrolaimomorpha, Diplogasteromorpha, Rhigonamtomorpha, Gnathostomatomorpha, and Ascaridomorpha (Kim et al., 2015, 2017, 2020). Thus, mitochondrial phylogenies strongly support Tylenchomorpha as non-monophyletic.
Finally, another contribution of mitochondrial phylogenetics is strong support for the novel clade mentioned above (Rhabditomorpha, Panagrolaimomorpha, Diplogasteromorpha, Rhigonematomorpha, Gnathostomatomorpha, and Ascaridomorpha) with Aphelenchoidea placed within this group. In this clade, the position of Diplogasteromorpha is nested within Rhabditomorpha, and Rhigonematomorpha is sister to the heterakoid species (Ascaridomorpha) (Kim et al., 2014, 2016, 2017). The close relationship between rhigonematomorphs and ascaridomorphs is consistent with SSU rDNA trees, but those trees only produced very weak support for this ascaridomorph-rhigonematomorph association (Smythe et al., 2006; Nadler et al., 2007; van Megen et al., 2009).
Gene Order as a Phylogenetic Tool in Nematodes
Mitochondrial gene order, i.e., how genes are arranged on the mitochondrial chromosome, is sometimes used as a data source for phylogenetic reconstruction (Boore and Brown, 1998; Fritzsch et al., 2006) or to confirm findings based on mitochondrial DNA sequences. In nematodes it is not generally advisable to rely on gene order alone to infer phylogenies, and doing so could be quite misleading. However, in situations where the placement of a clade is uncertain based on molecular phylogenies (for example, when results differ depending on analytical methods or markers), it can sometimes be helpful to compare the gene order patterns of the taxon in question with its potential sister clades.
In nematodes, syntenic comparisons should be done with a clear understanding of the gene order trends of the focal group (Supplementary Figure S1). Gene order is so variable in the Enoplea nematodes that even closely related congeneric species may have extremely different gene orders (Tang and Hyman, 2007; Hyman et al., 2011), rendering mitochondrial gene order useless as a phylogenetic character among the thus-far sequenced members of this group. On the other extreme, some members of Rhabditomorpha, Ascaridomorpha, Diplogasteromorpha, Panagrolaimomorpha, and Aphelenchoidea (Tylenchomorpha) are so nearly identical in gene order (Kim et al., 2016, 2020) that inferring their relationships based on gene order is likewise impossible. The difference in gene arrangement between Chromadorea and its basal group Enoplea also makes rooting the tree problematic when inferring chromadorean gene-order phylogenies.
Despite these caveats, gene order can be a useful tool in most of the Chromadorea nematodes, where it is moderately conserved. Here, most groups that are known to be monophyletic have similar within-group gene orders, varying by a few tRNA translocations (Park et al., 2011; Kim et al., 2017). Disregarding the tRNA genes, 71 out of 72 Rhabditomorpha species, 25 out of 30 Ascaridomorpha species, two Diplogasteromorpha species, 3 out of 4 Aphelenchoidea species and 6 out of 12 Panagrolaimomorpha species share the same gene order; this has been interpreted as additional evidence of their close relationship (Supplementary Figure S1). However, certain idiosyncratic exceptions occur in Ascaridia columbae, A. galli, Heterakis beramporia, H. dispar, H. gallinarum, Rhabditophanes sp., Parastrongyloides trichosuri, Strongyloides stercoralis, S. papillosus, S. ratti, S. venezuelensis, Heterorhabditis bacteriophora, rhigonematomorphs, and gnathostomatomorphs, which have gene arrangements that do not match the gene orders of their closest relatives.
Differences in the positions of tRNA genes are the most common types of gene order changes among Chromadorea nematodes. In this respect nematodes are similar to some other metazoans like arthropods (specifically, isopods and endopterygote insects), where tRNA translocations are much more common than protein gene or rRNA gene translocations (Dowton and Austin, 1999; Shao et al., 2001; Kilpert and Podsiadlowski, 2006). The mechanisms behind tRNA translocations are believed to be duplication and deletion (Macey et al., 1997; Song et al., 2016). Translocations of tRNA genes probably accumulate more quickly because of their small size; for this reason, designating tRNA genes as hypermobile and excluding them from gene order phylogenies is one possible way of dealing with the difference in rearrangement rate between tRNA genes and other genes when assessing nematode relationships (Kim et al., 2017).
Novel Insights and Promising Future Areas of Research
Beyond its phylogenetic utility, the sequencing and study of nematode mitogenomes has the potential to produce other interesting biological insights. In rare instances, some nematode species with multiple circular chromosomes have been sequenced (Armstrong et al., 2000; Hunt et al., 2016; Phillips et al., 2016; Kim et al., 2018). The discovery of multiple mitochondrial chromosomes makes nematodes one of only four known animal phyla with such unusual mitogenome architecture (the other three are dicyema, arthropods, and rotifers; Watanabe et al., 1999; Suga et al., 2008; Shao et al., 2012). In a recent study describing the existence of two distinct mitochondrial chromosomes in the nematode Ruizia karukerae, small-scale sequence differences (i.e., a single non-synonymous nucleotide substitution) between duplicated copies of cox2 gene on each of the two chromosomes suggest that the process of chromosome multiplication might be at an early stage in this group (Kim et al., 2018). It is interesting to note that nematode species with multipartite mitochondrial chromosomes are so far all parasitic (Globodera ellingtonae, G. pallida, G. rostochiensis [plant parasitic]; Rhabditophanes sp. KR3021, R. karukerae [animal parasitic]), but there is not enough evidence yet to conclude that parasitic lifestyles are correlated with multipartite mitogenomes. Since the mechanisms and processes by which multiple mitochondrial chromosomes arise and are maintained are almost completely unknown (not just for nematodes but also for any other phylum), nematode mitochondrial genomics could conceivably help uncover the mechanisms leading to these unusual genome structures.
Another interesting contribution from mitogenomes is the discovery of the striking difference in the degree of gene order conservation between two major nematode lineages, Enoplea and Chromadorea. As mentioned previously, the Enoplea display substantial gene rearrangement even among closely related species, while members of Chromadorea show far less rearrangement (Kim et al., 2017). The driving forces behind non-conserved gene orders in animals are not well understood, and nematodes could offer the opportunity to study how and why gene rearrangements occur more frequently in some clades than others (Zou et al., 2017). In general, gene rearrangements should be selected against because a random rearrangement is likely to disrupt genes and interfere with transcription: this is especially true for mitochondrial genes, which are tightly spaced together and do not have large non-coding regions between each gene (Wolstenholme, 1992; Boore, 1999). Various hypotheses have been used to explain Enoplea’s unusual diversity of gene patterns (Hyman et al., 2011; Zou et al., 2017). One reason for high levels of gene rearrangement can be clade age (and since Enoplea is basal to Chromadorea it therefore might contain ancestral types of gene orders); however, extreme variation within enoplean genera and even species (in other words, high variation over short divergence times) makes this an unlikely explanation (Tang and Hyman, 2007; Hyman et al., 2011). In insects, which also have different within-phylum degrees of gene rearrangement and conservation, it was initially thought that a parasitic lifestyle might promote a high frequency of rearrangement (Dowton and Austin, 1999) but additional evidence from other arthropods failed to support this idea (Shao et al., 2001; Castro et al., 2002; Silvestre and Arias, 2006). Enoplea contains both parasitic and free-living nematodes, which would make this an ideal group for an independent test of the parasitism hypothesis, but first enough mitogenomes of free-living Enoplea species will need to be sequenced.
Finally, the distribution of nematodes in an infinite diversity of habitat types and niches also allows for testing various interesting hypotheses about mitogenome evolution. For example, recent work comparing endoparasitic and ectoparasitic nematodes has uncovered a correlation between the size of nematodes mitogenomes and the local temperature of their niches, suggesting that higher metabolic rate selects for higher replication efficiency, and therefore smaller mtDNA genomes (Lagisz et al., 2013). Other work has used nematode phylogeny to examine the evolution of different modes of reproduction, and has discovered that the evolution of a parthenogenic (self-reproducing) mode of reproduction often accompanies the evolution of plant parasitism (Denver et al., 2011; this is probably because plant-parasitic nematodes encounter potential mates more rarely). Since nematodes are represented in nearly every conceivable habitat type, from tundra soil to the internal organs of tropical animals, they have the potential to provide many opportunities for investigating links between ecology and genome evolution. Essential to such research is a clear and well-supported understanding of nematode phylogeny, to which mitogenomes can continue to contribute.
Conclusion
The mitochondrial genome is a useful tool for nematode phylogenetics and has been widely applied to resolve uncertainty within this group. Specific instances of non-agreement between mitochondrial and nuclear gene trees warrant additional investigation. Gene order can be used with discretion as a source of phylogenetic information in nematodes. The diversity within nematode mitogenome architecture, its variable rate of gene rearrangement, and the representation of nearly every kind of lifestyle and habitat ecology within nematodes make this phylum an exciting area for addressing questions about mitogenome evolution.
Data Availability Statement
All data for this study is contained within the manuscript.
Author Contributions
EK wrote the manuscript. J-KP directed and wrote the manuscript. TK assessed differences between nuclear and mitochondrial trees and created the figures and table. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a RP-Grant 2019 of Ewha Womans University to EK and by a grant from the Marine Biotechnology Program (Nos. 20170431 and 20170488) funded by the Ministry of Oceans and Fisheries and the Basic Science Research Program through the National Research Foundation of Korea (NRF) by the Ministry of Science, ICT & Future Planning (NRF-2015R1A4A1041997) to J-KP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00250/full#supplementary-material
FIGURE S1 | Mitochondrial gene orders of 191 nematode species, arranged in a phylogenetic tree based on whole mitogenome sequences (see main Figure 1B). Groups of species with identical gene orders are combined on one line. Protein-coding and rRNA genes are represented by colored boxes labeled with gene names, and tRNA genes are represented by circles containing their single-letter amino acid codes assigned by the International Union of Pure and Applied Chemistry. The two leucine and two serine tRNA genes are labeled as L1 (trnL1-NAG), L2 (trnL2-YAA), S1 (trnS1-NCU), and S2 (trnS2-NGA). Non-coding regions are not included. All genes are transcribed in the same direction except those underlined by black bars, which are transcribed in the reverse direction (e.g., nad2 in Plectus species). All sequences were obtained from GenBank.
References
Anderson, R. C. (2000). Nematode Parasites of Vertebrates: Their Development and Transmission. New York, NY: CABI.
Anderson, T. J. C., Romero-Abal, M. E., and Jaenike, J. (1995). Mitochondrial DNA and Ascaris microepidemiology: the composition of parasite populations from individual hosts, families and villages. Parasitology 110, 221–229. doi: 10.1017/s003118200006399x
Armstrong, M. R., Blok, V. C., and Phillips, M. S. (2000). A multipartite mitochondrial genome in the potato cyst nematode, Globodera pallida. Genetics 154, 181–192.
Ballard, J. W. O., and Whitlock, M. C. (2004). The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744. doi: 10.1046/j.1365-294x.2003.02063.x
Bernt, M., Bleidorn, C., Braband, A., Dambach, J., Donath, A., Fritzsch, G., et al. (2013). A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenet. Evol. 69, 352–364. doi: 10.1016/j.ympev.2013.05.002
Blaxter, M., and Koutsovoulos, G. (2015). The evolution of parasitism in Nematoda. Parasitology 142(Suppl. 1), S26–S39. doi: 10.1017/S0031182014000791
Blaxter, M. L., De Ley, P., Garey, J. R., Liu, L. X., Scheldeman, P., Vierstraete, A., et al. (1998). A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75. doi: 10.1038/32160
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Boore, J. L., and Brown, W. M. (1998). Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 8, 668–674. doi: 10.1016/s0959-437x(98)80035-x
Breton, S., Beaupré, H. D., Stewart, D. T., Hoeh, W. R., and Blier, P. U. (2007). The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23, 465–474. doi: 10.1016/j.tig.2007.05.011
Burzyński, A., Zbawicka, M., Skibinski, D. O., and Wenne, R. (2003). Evidence for recombination of mtDNA in the marine mussel Mytilus trossulus from the Baltic. Mol. Biol. Evol. 20, 388–392. doi: 10.1093/molbev/msg058
Castro, L. R., Austin, A. D., and Dowton, M. (2002). Contrasting rates of mitochondrial molecular evolution in parasitic diptera and hymenoptera. Mol. Biol. Evol. 19, 1100–1113. doi: 10.1093/oxfordjournals.molbev.a004168
De Ley, P., and Blaxter, M. L. (2002). “Systematic position and phylogeny,” in The Biology of Nematodes, ed. D. L. Lee (New York, NY: Taylor & Francis), 1–30. doi: 10.1201/b12614-2
De Ley, P., and Blaxter, M. L. (2004). A new system for Nematoda: combining morphological characters with molecular trees, and translating clades into ranks and taxa. Nematol. Monogr. Perspect. 2, 633–653.
Denver, D. R., Clark, K. A., and Raboin, M. J. (2011). Reproductive mode evolution in nematodes: insights from molecular phylogenies and recently discovered species. Mol. Phylogenet. Evol. 61, 584–592. doi: 10.1016/j.ympev.2011.07.007
Dowton, M., and Austin, A. D. (1999). Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the hymenoptera. Mol. Biol. Evol. 16, 298–309. doi: 10.1093/oxfordjournals.molbev.a026111
Egger, B., Bachmann, L., and Fromm, B. (2017). Atp8 is in the ground pattern of flatworm mitochondrial genomes. BMC Genomics 18:414. doi: 10.1016/j.ympev.2015.02.013
Fritzsch, G., Schlegel, M., and Stadler, P. F. (2006). Alignments of mitochondrial genome arrangements: applications to metazoan phylogeny. J. Theor. Biol. 240, 511–520. doi: 10.1016/j.jtbi.2005.10.010
Golden, A. M. (1971). “Classification of the genera and higher categories of the order Tylenchida (Nematoda),” in Plant Parasitic Nematodes, Vol. 1, eds B. M. Zuckerman, W. F. Mai, and R. A. Rohde (New York, NY: Academic Press), 191–232. doi: 10.1016/b978-0-12-782201-3.50014-9
Grewal, P. S., Selvan, S., Lewis, E. E., and Gaugler, R. (1993). Male insect-parasitic nematodes: a colonizing sex. Experientia 49, 605–608. doi: 10.1007/bf01955173
Hawash, M. B. F., Andersen, L. O., Gasser, R. B., Stensvold, C. R., and Nejsum, P. (2015). Mitochondrial genome analyses suggest multiple Trichuris species in humans, baboons, and pigs from different geographical regions. PLoS Negl. Trop. Dis. 9:e0004059. doi: 10.1371/journal.pntd.0004059
Hoelzer, G. A. (1997). Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees revisited. Evolution 51, 622–626. doi: 10.1111/j.1558-5646.1997.tb02451.x
Holterman, M., van der Wurff, A., van den Elsen, S., van Megen, H., Bongers, T., Holovachov, O., et al. (2006). Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown Clades. Mol. Biol. Evol. 23, 1792–1800. doi: 10.1093/molbev/msl044
Hugot, J. P., Baujard, P., and Morand, S. (2001). Biodiversity in helminths and nematodes as a field of study: an overview. Nematology 3, 199–208. doi: 10.1163/156854101750413270
Hunt, V. L., Tsai, I. J., Coghlan, A., Reid, A. J., Holroyd, N., Foth, B. J., et al. (2016). The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat. Genet. 48, 299–307. doi: 10.1038/ng.3495
Hyman, B. C., Lewis, S. C., Tang, S., and Wu, Z. (2011). Rampant gene rearrangement and haplotype hypervariation among nematode mitochondrial genomes. Genetica 139, 611–615. doi: 10.1007/s10709-010-9531-3
Jacob, J. E., Vanholme, B., Van Leeuwen, T., and Gheysen, G. (2009). A unique genetic code change in the mitochondrial genome of the parasitic nematode Radopholus similis. BMC Res. Notes 2:192. doi: 10.1186/1756-0500-2-192
Kang, S., Sultana, T., Eom, K. S., Park, Y. C., Soonthornpong, N., Nadler, S. A., et al. (2009). The mitochondrial genome sequence of Enterobius vermicularis (Nematoda: Oxyurida)—an idiosyncratic gene order and phylogenetic information for chromadorean nematodes. Gene 429, 87–97. doi: 10.1016/j.gene.2008.09.011
Kilpert, F., and Podsiadlowski, L. (2006). The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genomics 7:241. doi: 10.1186/1471-2164-7-241
Kim, J., Kern, E., Kim, T., Sim, M., Kim, J., Kim, Y., et al. (2017). Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within Chromadorea. Mol. Phylogenet. Evol. 107, 90–102. doi: 10.1016/j.ympev.2016.10.010
Kim, J., Lee, S. H., Gazi, M., Kim, T., Jung, D., Chun, J. Y., et al. (2015). Mitochondrial genomes advance phylogenetic hypotheses for Tylenchina (Nematoda: Chromadorea). Zool. Scr. 44, 446–462. doi: 10.1111/zsc.12112
Kim, T., Kern, E., Park, C., Nadler, S. A., Bae, Y. J., and Park, J.-K. (2018). The bipartite mitochondrial genome of Ruizia karukerae (Rhigonematomorpha, Nematoda). Sci. Rep. 8:7482. doi: 10.1038/s41598-018-25759-0
Kim, T., Kim, J., Cho, S., Min, G. S., Park, C., Carreno, R. A., et al. (2014). Phylogeny of Rhigonematomorpha based on the complete mitochondrial genome of Rhigonema thysanophora (Nematoda: Chromadorea). Zool. Scr. 43, 289–303. doi: 10.1111/zsc.12047
Kim, T., Kim, J., Nadler, S. A., and Park, J.-K. (2016). The complete mitochondrial genome of Koerneria sudhausi (Diplogasteromorpha: Nematoda) supports monophyly of Diplogasteromorpha within Rhabditomorpha. Curr. Genet. 62, 391–403. doi: 10.1007/s00294-015-0536-4
Kim, T., Lee, Y., Kil, H.-J., and Park, J.-K. (2020). The mitochondrial genome of Acrobeloides varius (Cephalobomorpha) confirms non-monophyly of Tylenchina (Nematoda). PeerJ 8:e9108. doi: 10.7717/peerj.9108
Kumar, S., Koutsovoulos, G., Kaur, G., and Blaxter, M. (2012). Toward 959 nematode genomes. Worm 1, 42–50. doi: 10.4161/worm.19046
Ladoukakis, E. D., and Zouros, E. (2001). Direct evidence for homologous recombination in mussel (Mytilus galloprovincialis) mitochondrial DNA. Mol. Biol. Evol. 18, 1168–1175. doi: 10.1093/oxfordjournals.molbev.a003904
Lagisz, M., Poulin, R., and Nakagawa, S. (2013). You are where you live: parasitic nematode mitochondrial genome size is associated with the thermal environment generated by hosts. J. Evol. Biol. 26, 683–690. doi: 10.1111/jeb.12068
Lavrov, D. V., and Brown, W. M. (2001). Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics 157, 621–637.
Liu, G.-H., Gasser, R. B., Nejsum, P., Wang, Y., Chen, Q., Song, H.-Q., et al. (2013). Mitochondrial and nuclear ribosomal DNA evidence supports the existence of a new Trichuris species in the endangered françois’ leaf-monkey. PLoS One 8:e66249. doi: 10.1371/journal.pone.0066249
Liu, G.-H., Gasser, R. B., Su, A., Nejsum, P., Peng, L., Lin, R.-Q., et al. (2012a). Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl. Trop. Dis. 6:e1539. doi: 10.1371/journal.pntd.0001539
Liu, G.-H., Wang, Y., Xu, M.-J., Zhou, D.-H., Ye, Y.-G., Li, J.-Y., et al. (2012b). Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae). Infect. Genet. Evol. 12, 1635–1641. doi: 10.1016/j.meegid.2012.08.005
Macey, J. R., Larson, A., Ananjeva, N. B., Fang, Z., and Papenfuss, T. J. (1997). Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 14, 91–104. doi: 10.1093/oxfordjournals.molbev.a025706
Min, G. S., and Park, J.-K. (2009). Eurotatorian paraphyly: revisiting phylogenetic relationships based on the complete mitochondrial genome sequence of Rotaria rotatoria (Bdelloidea: Rotifera: Syndermata). BMC Genomics 10:533. doi: 10.1186/1471-2164-10-533
Miya, M., and Nishida, M. (2014). The mitogenomic contributions to molecular phylogenetics and evolution of fishes: a 15-year retrospect. Ichthyol. Res. 62, 29–71. doi: 10.1007/s10228-014-0440-9
Mohandas, N., Pozio, E., La Rosa, G., Korhonen, P. K., Young, N. D., Koehler, A. V., et al. (2014). Mitochondrial genomes of Trichinella species and genotypes—a basis for diagnosis, and systematic and epidemiological explorations. Int. J. Parasitol. 44, 1073–1080. doi: 10.1016/j.ijpara.2014.08.010
Moore, W. S. (1995). Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49, 718–726. doi: 10.1111/j.1558-5646.1995.tb02308.x
Nadler, S. A., Carreno, R. A., Mejía-Madrid, H., Ullberg, J., Pagan, C., Houston, R., et al. (2007). Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology 134, 1421–1442. doi: 10.1017/s0031182007002880
Nadler, S. A., and De Len, G. P. P. (2011). Integrating molecular and morphological approaches for characterizing parasite cryptic species: implications for parasitology. Parasitology 138, 1688–1709. doi: 10.1017/s003118201000168x
Nichols, R. (2001). Gene trees and species trees are not the same. Trends Ecol. Evol. 16, 358–364. doi: 10.1016/s0169-5347(01)02203-0
Palomares-Rius, J. E., Cantalapiedra-Navarrete, C., Archidona-Yuste, A., Blok, V. C., and Castillo, P. (2017). Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Sci. Rep. 7:41813. doi: 10.1038/srep41813
Park, J.-K., Kim, K. H., Kang, S., Kim, W., Eom, K. S., and Littlewood, D. T. J. (2007). A common origin of complex life cycles in parasitic flatworms: evidence from the complete mitochondrial genome of Microcotyle sebastis (Monogenea: Platyhelminthes). BMC Evol. Biol. 7:11. doi: 10.1186/1471-2148-7-11
Park, J.-K., Sultana, T., Lee, S. H., Kang, S., Kim, H. K., Min, G. S., et al. (2011). Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics 12:392. doi: 10.1186/1471-2164-12-392
Phillips, W. S., Brown, A. M. V., Howe, D. K., Peetz, A. B., Blok, V. C., Denver, D. R., et al. (2016). The mitochondrial genome of Globodera ellingtonae is composed of two circles with segregated gene content and differential copy numbers. BMC Genomics 17:706. doi: 10.1186/s12864-016-3047-x
Piganeau, G., Gardner, M., and Eyre-Walker, A. (2004). A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 21, 2319–2325. doi: 10.1093/molbev/msh244
Poinar, G. O. (1991). “Nematoda and Nematomorpha,” in Ecology and Classification of North American Freshwater Invertebrates, eds J. H. Thorp and A. P. Covich (New York, NY: Academic Press), 255–295. doi: 10.1016/b978-012690647-9/50010-7
Prugnolle, F., and de Meeus, T. (2002). Inferring sex-biased dispersal from population genetic tools: a review. Heredity 88, 161–165. doi: 10.1038/sj.hdy.6800060
Ramesh, A., Small, S. T., Kloos, Z. A., Kazura, J. W., Nutman, T. B., Serre, D., et al. (2012). The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 183, 32–41. doi: 10.1016/j.molbiopara.2012.01.004
Ruehle, J. L. (1967). Distribution of Plant-Parasitic Nematodes Associated with Forest Trees of the World. Asheville, NC: US Department of Agriculture.
Saccone, C., De Giorgi, C., Gissi, C., Pesole, G., and Reyes, A. (1999). Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene 238, 195–209. doi: 10.1016/s0378-1119(99)00270-x
Semprucci, F., Frontalini, F., Sbrocca, C., du Châtelet, E. A., Bout-Roumazeilles, V., Coccioni, R., et al. (2015). Meiobenthos and free-living nematodes as tools for biomonitoring environments affected by riverine impact. Environ. Monit. Assess. 187:251. doi: 10.1007/s10661-015-4493-7
Shao, R., Campbell, N. J. H., Schmidt, E. R., and Barker, S. C. (2001). Increased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol. Biol. Evol. 18, 1828–1832. doi: 10.1093/oxfordjournals.molbev.a003970
Shao, R., Zhu, X.-Q., Barker, S. C., and Herd, K. (2012). Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol. Evol. 4, 1088–1101. doi: 10.1093/gbe/evs088
Sikora, R. A., Coyne, D., Hallman, J., and Timper, P. (2005). Plant Parasitic Nematodes in Subtropical and Tropical Agriculture. Boston, MA: CABI.
Silvestre, D., and Arias, M. C. (2006). Mitochondria tRNA gene translocations in highly eusocial bees. Genet. Mol. Biol. 29, 572–575. doi: 10.1590/s1415-47572006000300030
Smythe, A. B., Holovachov, O., and Kocot, K. M. (2019). Improved phylogenomic sampling of free-living nematodes enhances resolution of higher-level nematode phylogeny. BMC Evol. Biol 19:121. doi: 10.1186/s12862-019-1444-x
Smythe, A. B., Sanderson, M. J., and Nadler, S. A. (2006). Nematode small subunit phylogeny correlates with alignment parameters. Syst. Biol. 55, 972–992. doi: 10.1080/10635150601089001
Song, F., Li, H., Shao, R., Shi, A., Bai, X., Zheng, X., et al. (2016). Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 6:25725. doi: 10.1038/srep25725
Suga, K., Mark Welch, D. B., Tanaka, Y., Sakakura, Y., and Hagiwara, A. (2008). Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 25, 1129–1137. doi: 10.1093/molbev/msn058
Tang, S., and Hyman, B. C. (2007). Mitochondrial genome haplotype hypervariation within the isopod parasitic nematode Thaumamermis cosgrovei. Genetics 176, 1139–1150. doi: 10.1534/genetics.106.069518
Tsaousis, A. D., Martin, D. P., Ladoukakis, E. D., Posada, D., and Zouros, E. (2005). Widespread recombination in published animal mtDNA sequences. Mol. Biol. Evol. 22, 925–933. doi: 10.1093/molbev/msi084
van Megen, H., van den Elsen, S., Holterman, M., Karssen, G., Mooyman, P., Bongers, T., et al. (2009). A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 11, 927–950. doi: 10.1163/156854109x456862
Watanabe, K. I., Bessho, Y., Kawasaki, M., and Hori, H. (1999). Mitochondrial genes are found on minicircle DNA molecules in the mesozoan animal Dicyema. J. Mol. Biol. 286, 645–650. doi: 10.1006/jmbi.1998.2523
Wolstenholme, D. R. (1992). Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216. doi: 10.1016/s0074-7696(08)62066-5
Yu, L., Li, Y.-W., Ryder, O. A., and Zhang, Y.-P. (2007). Analysis of complete mitochondrial genome sequences increases phylogenetic resolution of bears (Ursidae), a mammalian family that experienced rapid speciation. BMC Evol. Biol. 7:198. doi: 10.1186/1471-2148-7-198
Zou, H., Jakovlić, I., Chen, R., Zhang, D., Zhang, J., Li, W. X., et al. (2017). The complete mitochondrial genome of parasitic nematode Camallanus cotti: extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genomics 18:840. doi: 10.1186/s12864-017-4237-x
Keywords: Chromadorea, Enoplea, Nematoda, mito-nuclear discordance, mitogenome, mitochondrial gene arrangement, phylogeny
Citation: Kern EMA, Kim T and Park J-K (2020) The Mitochondrial Genome in Nematode Phylogenetics. Front. Ecol. Evol. 8:250. doi: 10.3389/fevo.2020.00250
Received: 13 December 2019; Accepted: 09 July 2020;
Published: 04 August 2020.
Edited by:
Eric Pante, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Jonathan J. Fong, Lingnan University, ChinaSergei Subbotin, California Department of Food and Agriculture, United States
Copyright © 2020 Kern, Kim and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joong-Ki Park, amtwYXJrQGV3aGEuYWMua3I=