 Vikas Kumar
Vikas Kumar Inderjeet Tyagi†
Inderjeet Tyagi† Kaomud Tyagi
Kaomud Tyagi- Centre for DNA Taxonomy, Molecular Systematics Division, Zoological Survey of India, Kolkata, India
The gut microbiomes of insects were known to have great impact on their physiological properties for survival like nutrition, behavior, and health. In nature, spiders are one of the main predators of insects, and yet their gut microbiomes remain unclear. It is important to explore the gut microbiomes of spiders in the wild to gain an insight on the host–bacterial relationship. Here, we studied the diversity and structure of gut bacterial communities of seven spider species belonging to two families, i.e., Thomisidae and Oxyopidae, from different states of India. Our data revealed a total of 16 bacterial phyla with Proteobacteria as the predominant group in Thomisidae and Firmicutes in Oxyopidae. The core bacterial communities in the spider guts include the genera of Acinetobacter, Staphylococcus, Corynebacterium, Cutibacterium, and Pseudomonas. The genus Paraclostridium was observed for the first time and only in one spider species, i.e., Peucetia viridans. Our data also indicated a higher gut bacterial community similarity between spider species belonging to Thomisidae as compared to those belonging to Oxyopidae bacteria. Furthermore, PICRUSt2 analysis predicted the presence of nine active functional metabolic pathways related to the metabolism of fatty acids and sugar, degradation of organic compounds, and biosynthesis of vitamin E.
Introduction
The gut microbiome significantly impacts the several metabolic activities of arthropods and helps us to understand their diversification, adaptation to novel habitat, and evolutionary patterns (Esposti and Romero, 2017). The abundance of insects is dependent on their countless relationships with bacterial communities that were known for multiple metabolic activities like diet-nutrient upgradation, endosymbiosis, digestive aids, mating, and reproductive systems (Klepzig et al., 2009; Engel and Moran, 2013). Advanced sequencing techniques like next-generation sequencing (NGS) have led researchers to explore the gut bacterial communities in honeybee (Anjum et al., 2018), mosquito (Muturi et al., 2017), and moths (Snyman et al., 2016), along with their potential role in body metabolism and evolutionary patterns. However, very little is known about the structure of spider gut bacterial communities and their potential metabolic functions. Spiders are the most diverse group of the class Arachnida, which are widely distributed across the globe and play a crucial role in maintaining the ecological balance through a prey–predator relationship (Michalko and Pekár, 2015; World Spider Catalog, 2020).
Available literature revealed the presence of endosymbionts like Wolbachia, Cardinium, Spiroplasma, Rickettsia, and Rickettsiella, in the different spider hosts, also affecting spider reproduction due to induced variation in sex ratio (Goodacre et al., 2006; Vanthournout et al., 2011; Zhang et al., 2017, 2018). On the other hand, there are limited studies on the gut bacterial communities in different families of spiders (Rivera et al., 2017; Macías-Hernández et al., 2018; Hu et al., 2019; Kennedy et al., 2020; Sheffer et al., 2020). To predict the host–bacterial relationship, it is a fundamental step to explore the gut bacterial communities of spiders. Spiders can be used as a good study model for host bacteria relationships due to their unique feeding behavior (extracellular digestion), which involves multiple steps like prey location, prey pulling, venom injection, and prey immobilization through, for example, silk threads (Foelix, 2011).
In the present study, we made an effort to decipher the gut bacterial communities using seven spider species obtained from different locations. Out of seven, four spider species (Epidius parvati, Xysticus himalayensis, Thomisus unidentatus, Camaricus formosus) of the family Thomisidae were collected from Kerala, West Bengal, Rajasthan, and Assam states of India, respectively. Three species of the family Oxyopidae, Peucetia viridans, and Oxyopes javanus were collected from Odisha state and Oxyopes birmanicus from Assam state of India (Table 1). In line with this, we also made an effort to differentiate the gut bacterial communities present in these two spider families based on statistical approaches along with their predicted functional roles in spider body metabolism through PICRUSt2 analysis.

Table 1. Collection details of seven spider species belong to two families Thomisidae and Oxyopidae.
Materials and Methods
Specimen Collection, DNA Isolation, and 16S rRNA Amplicon Sequencing
At the fields, twenty specimens of each spider species were collected using either sweep net collection, pitfall trap, or hand picking method. After 8 h of collection, the individual specimen was stored in a separate vial filled with 90% alcohol and transferred to a restricted lab space using a 4°C portable chamber. The collected specimens were first rinsed with PBS solution and later with MilliQ water thrice to remove surface impurities.
Total DNA was extracted from the spider gut contents using the DNeasy Blood and Tissue Kit (Qiagen, Cat. No. 69506, Germany) following the standard protocol, and the DNA extracts of twenty specimens for each spider species were pooled for further analysis. The quantity and quality of the extracted DNA were checked by Qubit 2.0 Fluorometer (Q32866, Thermo Fisher) and agarose gel electrophoresis (Cell BioScience Alphalmager MINI) respectively. The bacterial 16S rRNA V3–V4 regions were amplified using the primer sets, 341F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The 25-μL reaction mixture includes 1 μl each of forward and reverse primer, 0.5 μl of Taq DNA polymerase, 1 μl of dNTPs, 2.5 μl of 10 × buffer, and a DNA template and adjusted by Milli-Q water. The thermal cycle includes 5 min at 98°C followed by 35 cycles of 30 s at 98°C (denaturation), 45 s at 53°C (annealing), and 72°C for 45 s (elongation), and 7 min at 72°C (final extension). The PCR products were then checked by agarose gels electrophoresis prior to Illumina Miseq sequencing (PHIXGEN, Pvt. Ltd., Gurugram, India). The generated raw reads were submitted to The National Center for Biotechnology Information (NCBI) GenBank Portal under the BioProject ID PRJNA638522 with accession numbers SAMN15656778 to SAMN15656784.
Downstream (Bioinformatics) and Statistical Analysis
The paired-end reads were merged using FLASH version 1.2.7 (Magoc and Salzberg, 2011) and were subjected to quality filtering (Q30), trimming, denoising, and chimera removal in DADA2 (Callahan et al., 2016) pipeline using QIIME2 (ver. 2019.10) (Bolyen et al., 2019). The quality-filtered reads were clustered into OTUs and taxonomically classified with a 99% similarity threshold using the SILVA database in the QIIME q2-feature classifier plugin (release 132).
The output files of QIIME2, i.e., taxonomy table, OTU table, and phylogenetic tree, along with the metadata file were used for further analysis in MicrobiomeAnalyst (Dhariwal et al., 2017) under the MDP (Marker Data Profiling) module. A total of 5039 OTUs was obtained which, on singleton removal and filtering, i.e., low variance features (10% removal-based interquantile range) and low abundance features (minimum count 4, prevalence in samples 20%), result in 965 OTUs for further data analysis. The rarefaction to the minimum library size, i.e., 57635, has also been carried out using total sum scaling (TSS).
Alpha diversity measures (Observed, Chao1, Shannon, and Simpson) based on t-test/ANOVA statistical methods were used to calculate the diversity and richness of these two families. Principal component analysis (Bray–Curtis index) using the PERMANOVA statistical method and hierarchal clustering (Unweighted Unifrac Distance measure) based on Ward’s clustering algorithm was used to decipher the beta diversity analysis. To explore the differential abundance analysis between these two families, we performed DESeq2 analysis in MicrobiomeAnalyst with cut off 0.05 (Supplementary Table S1).
The number of shared and unique OTUs between the two families of spiders was represented in a Venn diagram using jvenn software (Bardou et al., 2014). The heat tree of both the spider families, i.e., Thomisidae and Oxyopidae, were constructed and visualized using the Metacoder software package (Foster et al., 2017) in R (ver., 3.6.1) (R Core Team, 2020).
Functional Predictions
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt21) (Langille, 2018) analysis based on 16S rRNA sequences were used to predict the functional profile of the gut bacterial community of two families of the spider. The functional profile of the bacterial community was predicted with the help of an inbuilt KEGG database (Kanehisa and Goto, 2000; Kanehisa, 2019; Kanehisa et al., 2019). The prediction accuracy of PICRUSt2 was evaluated based on the Nearest Sequenced Taxon Index (NSTI), where the lower the value, the higher the prediction accuracy. STAMP (Parks et al., 2014) software was used for the statistical verifications/corrections.
Results
Rarefaction and Distribution of OTUs
A total number of 569815 paired-end sequences ranging from minimum 57,635 to maximum 101,779 were generated after pre-processing steps like quality filtering, trimming, and chimera removal. The obtained sequences were clustered into 5039 OTUs at 99% identity clusters, which further reduced to 965 OTUs for downstream analysis after the removal of singletons, low variance, and low abundance features, etc. Furthermore, Venn analysis revealed 5039 OTUs, of which 16% of the annotated OTUs was shared between both spider families, whereas 35% OTUs in Oxyopidae and 48% OTUs in Thomisidae were reported as unique (Figure 1A). Further, the core microbiome analysis revealed that 16% of shared OTUs between both spider families comprise the members of the phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes (Figure 1B). In addition to this, these phyla also distributed along with phylum Cyanobacteria in 35% unique OTUs in case of Oxyopidae and in 48% unique OTUs with phylum Deinococcus _Thermus in Thomisidae. Moreover, within the spider families, 13 OTUs were shared by the three spider species, i.e., Peucetia viridans (AA 2450), Oxyopes javanus (AA830), and Oxyopes birmanicus (AA932) belonging to family Oxyopidae (Figure 1C), and 103 OTUs were shared by four spider species, i.e., Epidius parvati (AA1457), Xysticus himalayensis (AA2136), Thomisus unidentatus (AA564), and Camaricus formosus (AA768) belonging to family Thomisidae (Figure 1D).
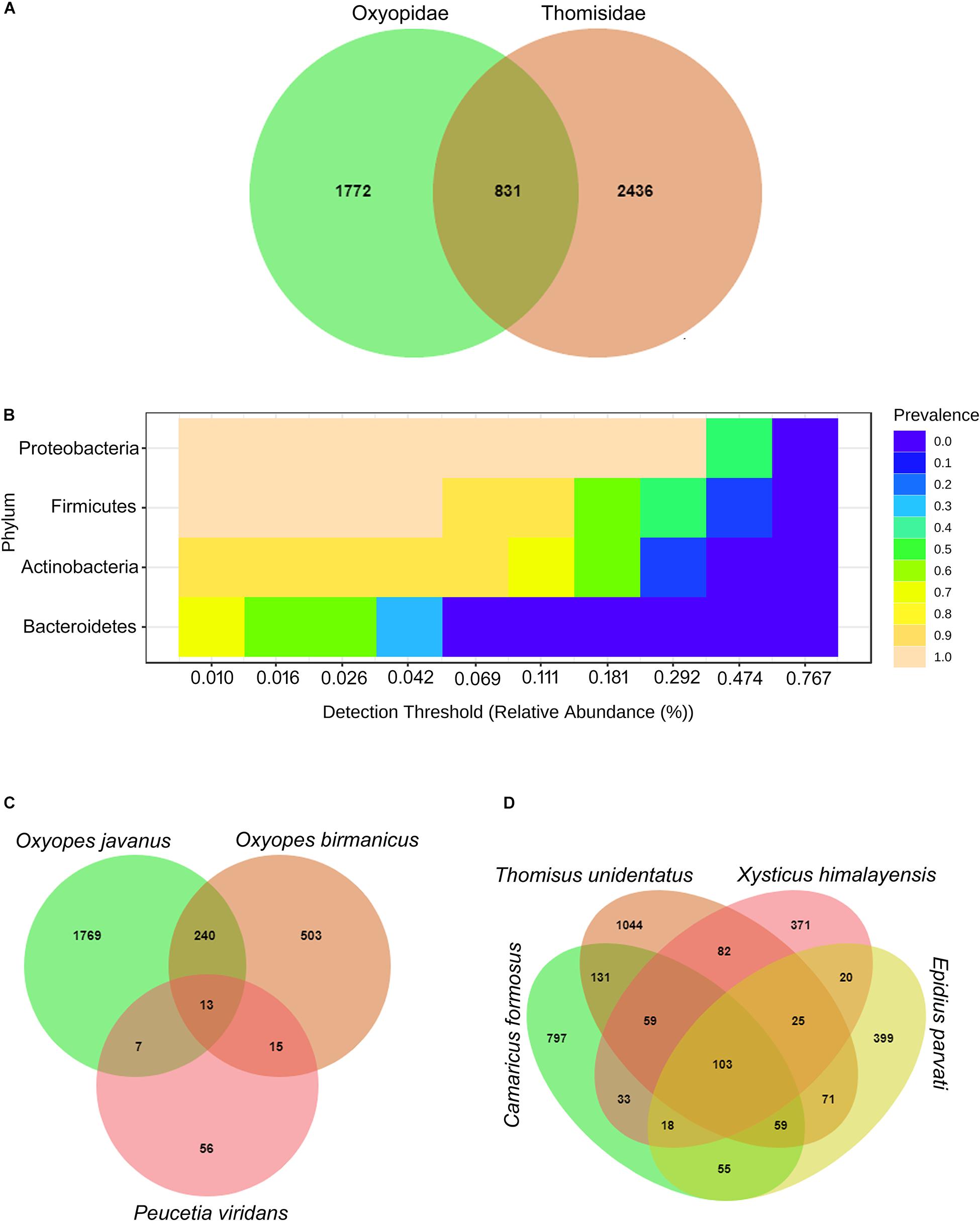
Figure 1. (A) Venn diagram representing the shared and unique OTUs between spider family Oxyopidae and Thomisidae. (B) Phylum comprising the core microbiome. Venn diagram representing the shared and unique OTUs (C) within the family Oxyopidae and (D) within the family Thomisidae.
In Supplementary Figure S1, a rarefaction curve for all spider species belonging to two families reaches saturation, which reveals that sufficient sequencing depth has been achieved for downstream analysis which is further used for deciphering the community richness (Alpha and Beta diversity).
Bacterial Community Structure of Spider Guts
The taxonomic classification of clustered OTUs (99% similarity based on SILVA database up to genus level) reveals the presence of bacterial communities belonging to 16 phyla comprising 35 classes, 77 orders, 150 families, and 320 genera. At the phylum level, Proteobacteria was the most abundant, ranging from 40 to 77% in family Thomisidae while in the case of Oxyopidae, it becomes the second most abundant phylum with abundance ranging from 31 to 37% (Figure 2A). Phylum Firmicutes ranks second in abundance ranging from 5 to 32% in Thomisidae while it ranks first in Oxyopidae, ranging from 17 to 68%. Moreover, Phylum Actinobacteria followed by Bacteroidetes ranked 3rd and 4th in phylum abundance in both the spider families with a combined contribution of 21 and 25% respectively (Figures 2B,C). Hence, these four phyla, i.e., Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes, contribute 98% to the total bacterial diversity in both the families of spider.
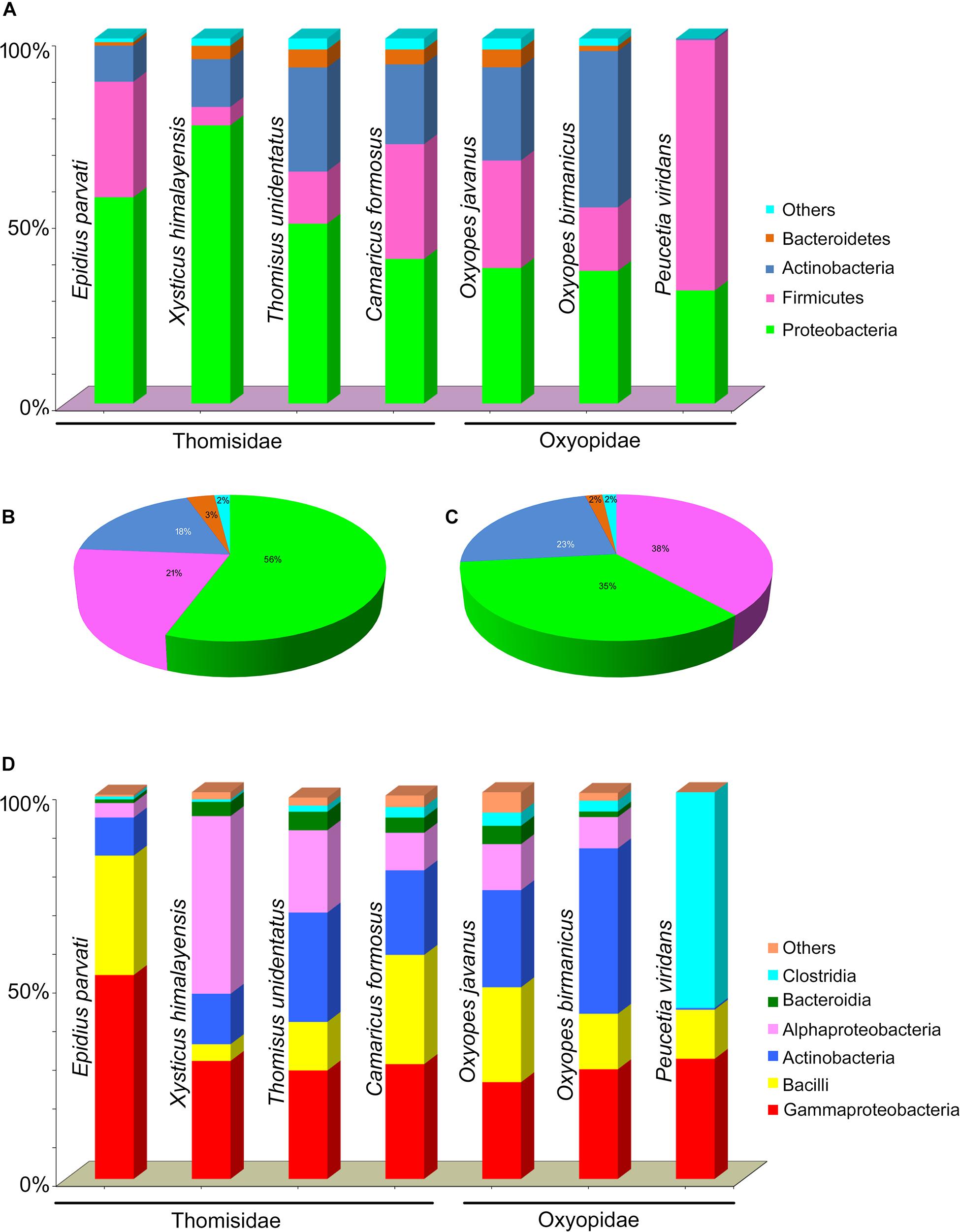
Figure 2. (A) Percentage abundance (bar chart) of phyla in seven spider species. Pie chart of percentage abundance in two spider families (B) Thomisidae and (C) Oxyopidae. (D) Percentage abundance (bar chart) of classes in seven spider species.
Out of 35, five classes, i.e., Gammaproteobacteria, Alphaproteobacteria, Bacilli, Actinobacteria, and Bacteroidia, were present in the gut of the seven spider species. Moreover, an exceptionally high abundance of class Clostridia in Peucetia viridans (Oxyopidae) was observed (Figure 2D).
Heat tree analysis (Figures 3A,B) reveals that the order like Pseudomonadales, followed by Bacillales, Clostridiales, Micrococcales, Enterobacteriales, Propionibacteriales, Corynebacteriales, Lactobacillales, etc., dominated the gut bacterial diversity. However, in spider families, i.e., Oxyopidae and Thomisidae, the following observation in order abundance has been made: order Pseudomonadales and Bacillales was the most abundant order with percentage contribution in the range of 13–43 and 4–25% respectively. On the other hand, the percentage contribution of order Clostridiales in spider family Oxyopidae was greater than that of Thomisidae. Overall, it contributed to the range of 1–56% with major contribution in Peucetia viridans (Oxyopidae) while in the rest of the six spider species it contributed only 1–3%. The order Micrococcales contributed 37% in the family Thomisidae and 24% in Oxyopidae while Propionibacteriales contributed around 17% in Thomisidae and 22% in Oxyopidae and Corynebacteriales contributed 16% in Thomisidae and 19% in Oxyopidae (Supplementary Table S2).
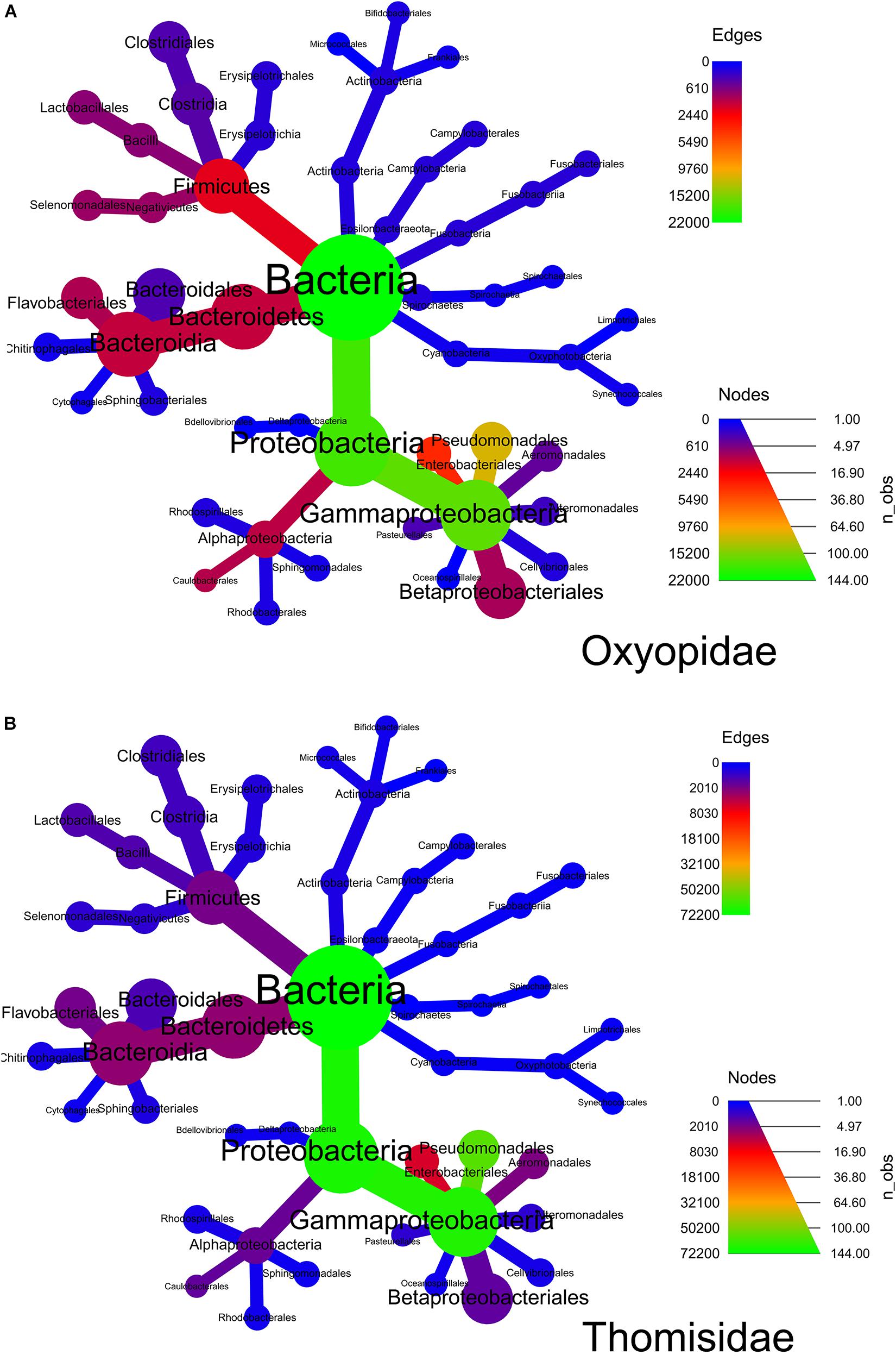
Figure 3. Heat tree analysis to decipher the abundance of bacterial orders in two spider families, i.e., (A) Oxyopidae and (B) Thomisidae. The heat tree was plotted in R software.
At the genus level, differences in the gut bacterial community of seven spider species were observed. Genera like Acinetobacter, Paraclostridium, Sphingomonas, Staphylococcus, Bacillus, Corynebacterium, Cutibacterium, Pseudomonas, and Micrococcus were observed as the top nine genera with different percentage contributions toward the gut microbiome of spider species. Furthermore, it was observed that Acinetobacter contributed to the range of 10–41% in family Thomisidae while 0–18% in Oxyopidae with a minimum 0% contribution in Peucetia viridans. Genera like Staphylococcus, Corynebacterium, Cutibacterium, and Micrococcus were present only in six spider species except for Peucetia viridans and contributed in the range of 3–16, 2–14, 2–16, and 1–6%, respectively, to the total bacterial diversity in six spider species. The only genus that was observed in the gut of all the spider species was Pseudomonas which contributed to the range of 1–13% of the total gut bacterial diversity. On the other hand, genus like Sphingomonas was present only in three spider species, i.e., two spider species (AA2136 and AA564) of family Thomisidae and one spider species (AA830) of family Oxyopidae, while genus like Bacillus was observed only in four spider species with a major contribution in two spider species of family Thomisidae and a minor contribution in two spider species of family Oxyopidae. Furthermore, the genus Paraclostridium (56%) is observed in only one species of the family Oxyopidae Peucetia viridans (Supplementary Table S3).
Alpha Diversity Metrics
To decipher the bacterial community diversity within the spider species, alpha diversity analysis was performed with the help of richness and diversity measures like Chao1, Observed, Shannon, and Simpson. From the results obtained, it was observed that the values for Chao1, Observed, Shannon, and Simpson measures lie in the range of 3–17, 3–17, 0.64–1.38, and 0.38–0.70, respectively (Supplementary Table S3). Moreover, the results for all the diversity measures are non-significant (p-value > 0.05). In the spider family Thomisidae, Camaricus formosus was the least diverse and Thomisus unidentatus was the most diverse while within the family Oxyopidae Peucetia viridans and Oxyopes javanus were the least and most diverse respectively in terms of gut bacterial diversity. Furthermore, interfamily comparison revealed that species belonging to the family Thomisidae were more diverse (Chao1 and Observed: 13–17; Shannon: 0.82–1.34; Simpson: 0.38–0.69) than species belonging to family Oxyopidae (Chao1 and Observed: 3–16; Shannon: 0.64–1.38; Simpson: 0.43–0.70) (Figure 4 and Supplementary Figure S2).
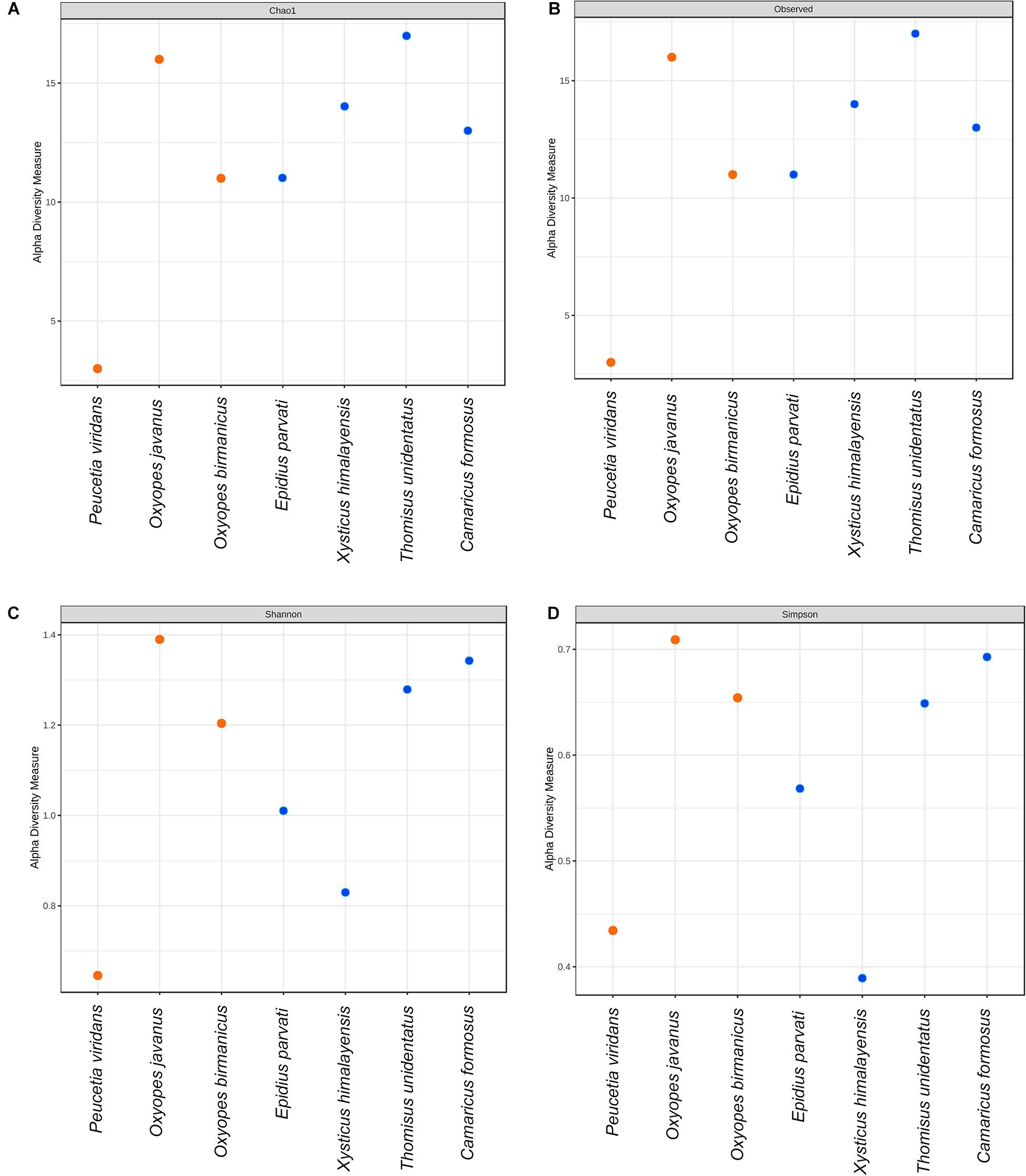
Figure 4. Alpha diversity metrics based on (A) Chao1, (B) Observed, (C) Shannon, and (D) Simpson diversity measures.
Beta-Diversity Metrics
The PCoA analyses based on Bray–Curtis index, weighted Unifrac method, and unweighted Unifrac distance method with PERMANOVA as statistical methods were carried out to reveal the variation in the gut microbiota of seven spider species. Bray–Curtis index (Figure 5A)-based analysis revealed that there is a non-significant (p-value 0.19; R2 = 0.28; F-value = 1.98) variation in the gut bacterial diversity of spider species belonging to families Thomisidae and Oxyopidae. Similarly, the PERMANOVA test for the weighted Unifrac (F-value: 2.4256; R2: 0.32666; p-value < 0.085) and unweighted Unifrac (F-value: 1.3321; R2: 0.21037; p-value < 0.26) distance methods were also non-significant.
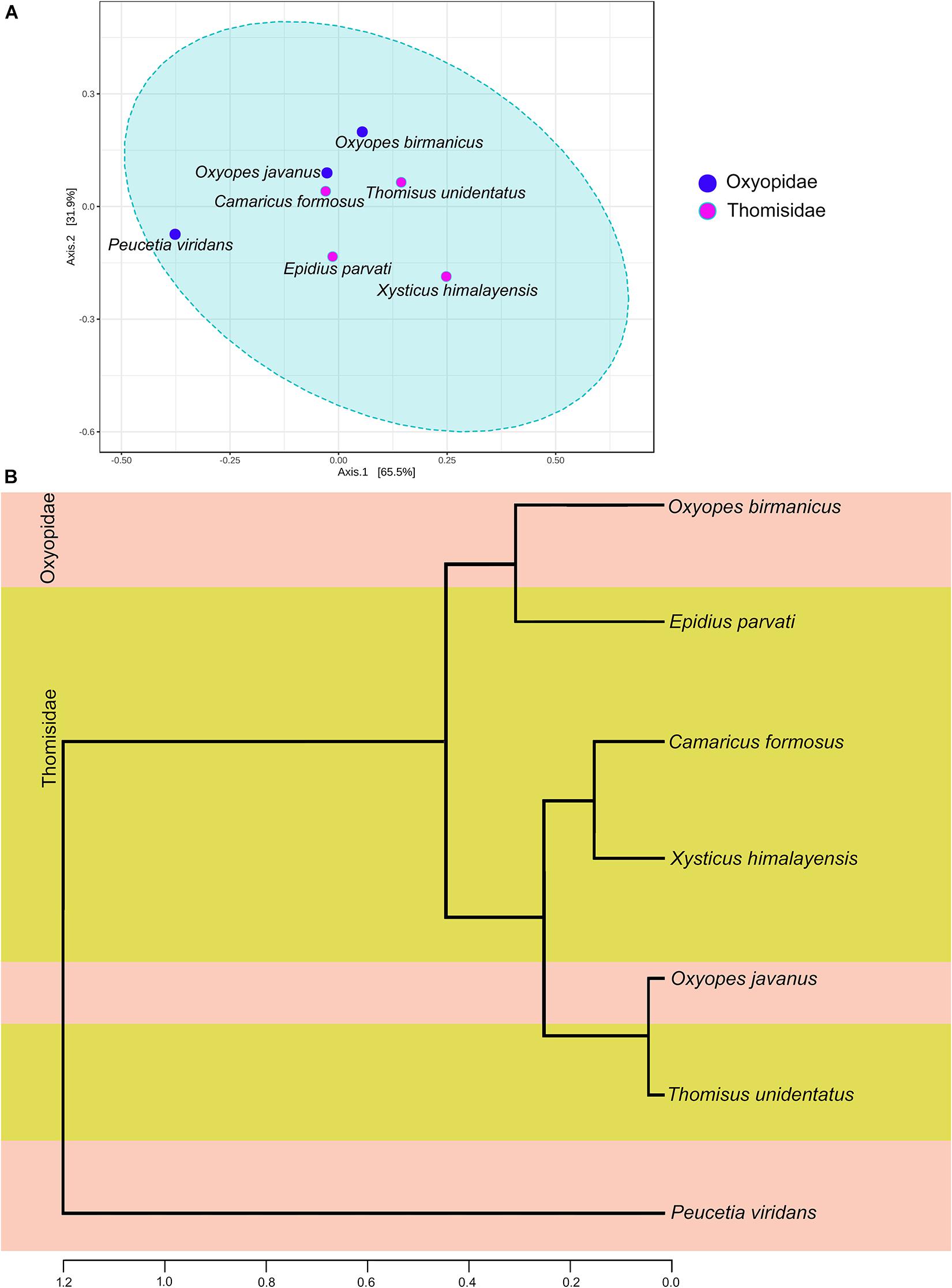
Figure 5. Beta diversity analysis (A) PCoA plot based on the Bray–Curtis index with the PERMANOVA statistical method. (B) Dendrogram analysis based on unweighted Unifrac distance measure.
Dendrogram analysis based on the unweighted Unifrac distance measure and Ward’s linkage clustering algorithm reveal the phylogenetic relationship between the gut bacterial diversity of seven spider species (Figure 5B). The dendrogram was clustered into two clades: clade I contains only one species, i.e., Peucetia viridans, while clade II with the rest of the six spider species was further branched out (Figure 5B). From clade II, it was observed that gut bacterial diversity of spider species Oxyopes javanus belonging to family Oxyopidae was closer to the gut bacterial diversity of spider species Thomisus unidentatus of the family Thomisidae while that of Oxyopes birmanicus was closer to the gut microbiome of Epidius parvati. On the other hand, gut bacterial diversity of the other two spider species, i.e., Camaricus formosus and Xysticus himalayensis belonging to family Thomisidae was similar to each other. The placement of Peucetia viridans in clade I of dendrogram was due to the high abundance of genus Paraclostridium, which greatly influences the gut bacterial community in this spider species.
Differential Abundance Analysis
To explore the differential abundance of the gut microbiota of seven species between the two families, we have carried out the DEseq2 analysis in MicrobiomeAnalyst with default parameters (p-value = 0.05). This analysis identified a group of 8 OTUs that were significantly more abundant in these two families (Figure 6 and Supplementary Table S1). These OTUs represent the genera Paraclostridium, Lysinibacillus, Enterococcus, Pseudocitrobacter, Pseudomonas, and Bacteroides. The family Oxyopidae has the relative high abundance of the genus Paraclostridium and phylum Firmicutes than family Thomisidae. The genera Enterococcus and Lysinibacillus of Firmicutes, Pseudocitrobacter, and Bacteroides of Proteobacteria are more abundant in family Thomisidae than Oxyopidae, whereas the genus Pseudomonas (Proteobacteria) is more abundant in family Oxyopidae than Thomisidae. This result indicated that the members of the family Thomisidae showed the closer gut bacterial community resemblance than the members of the family Oxyopidae.
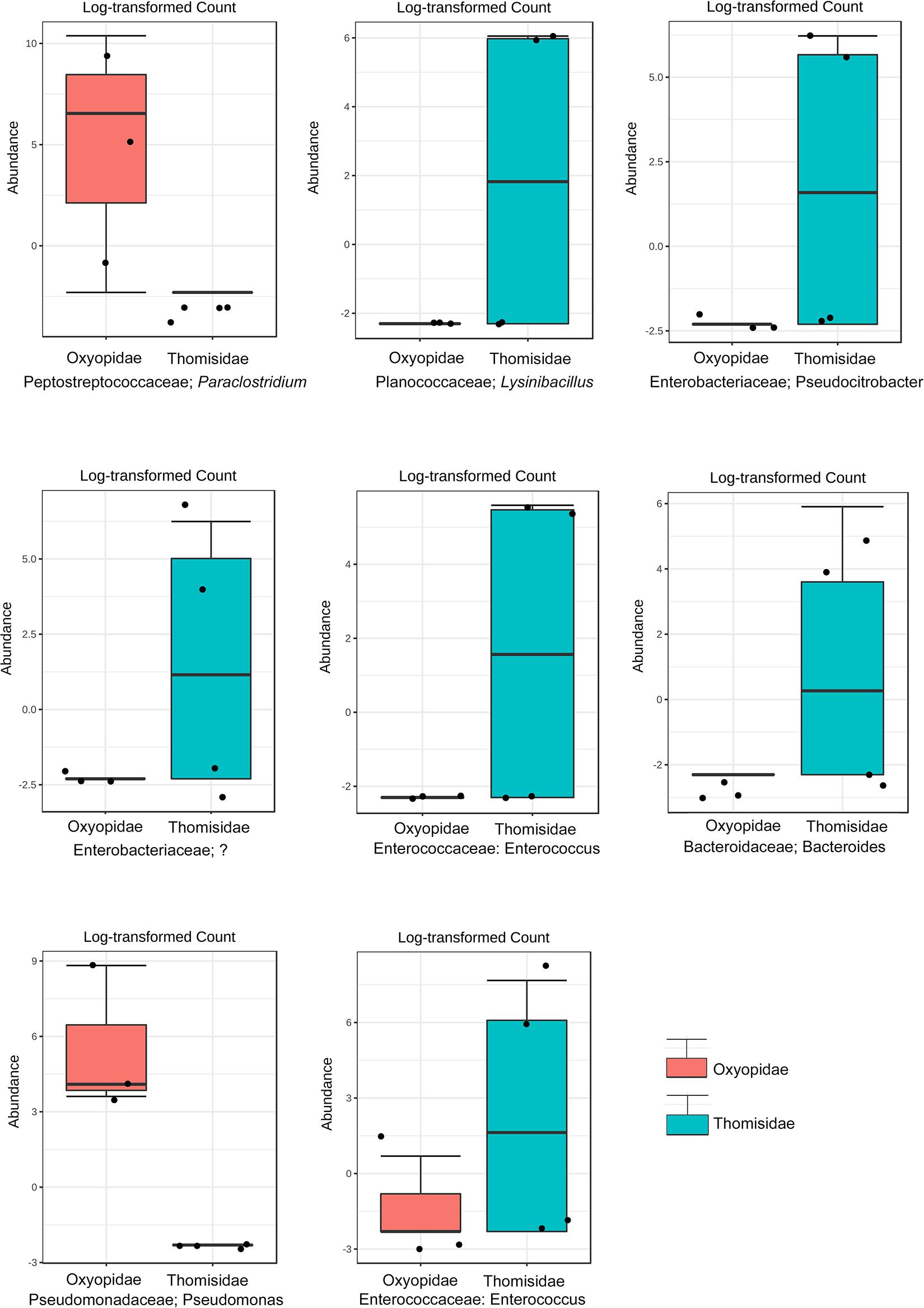
Figure 6. Significant OTUs obtained from differential abundance analysis DESeq2.
Comparative Functional Metabolic Pathway Analysis
PICRUSt2 analysis was carried out to predict the functional metabolic pathways present in the seven spider species belonging to two families, i.e., Oxyopidae and Thomisidae (p < 0.05, Kruskal–Wallis H-test, with Tukey–Kramer post hoc test, Benjamini–Hochberg FDR correction for multiple comparisons, and corrected p-values were used in Stamp software) (Supplementary Figures S3–S5). From the results obtained (Figure 7), it was observed that nine active pathways related to fatty acid metabolism, organic compound degradation, sugar metabolism, and vitamin E biosynthesis (tocopherols) are common between these two families. The scatter plot indicated that these predicted pathways are more active in Thomisidae than Oxyopidae (Supplementary Figures S3, S4). Further, the heat map showed two clades: clade 1 includes the three species of the family Thomisidae (Thomisus unidentatus, Epidius parvati, Xysticus himalayensis), and clade 2 includes all the members of family Oxyopidae and one species of the family Thomisidae (Camaricus formosus). Thomisus unidentatus and Epidius parvati are closer in terms of their metabolic functional activity as they share pathways like sucrose degradation III, 8-amino-7-oxononanoate biosynthesis, and glycolysis III, while Xysticus himalayensis is a little different because of the high involvement in the aromatic biogenic amine degradation and fatty acid oxidation. The species Camaricus formosus is more close to the members of the family Oxyopidae rather than Thomisidae due to high involvement in sucrose degradation. Oxyopes birmanicus and O. javanus are closer and share pathways like sucrose degradation III, 8-amino-7-oxononanoate biosynthesis, aromatic biogenic amine degradation, and pyrimidine deoxyribonucleotide de novo biosynthesis, whereas the pyrimidine deoxyribonucleotide de novo biosynthesis pathway is more prominent in Peucetia viridans.
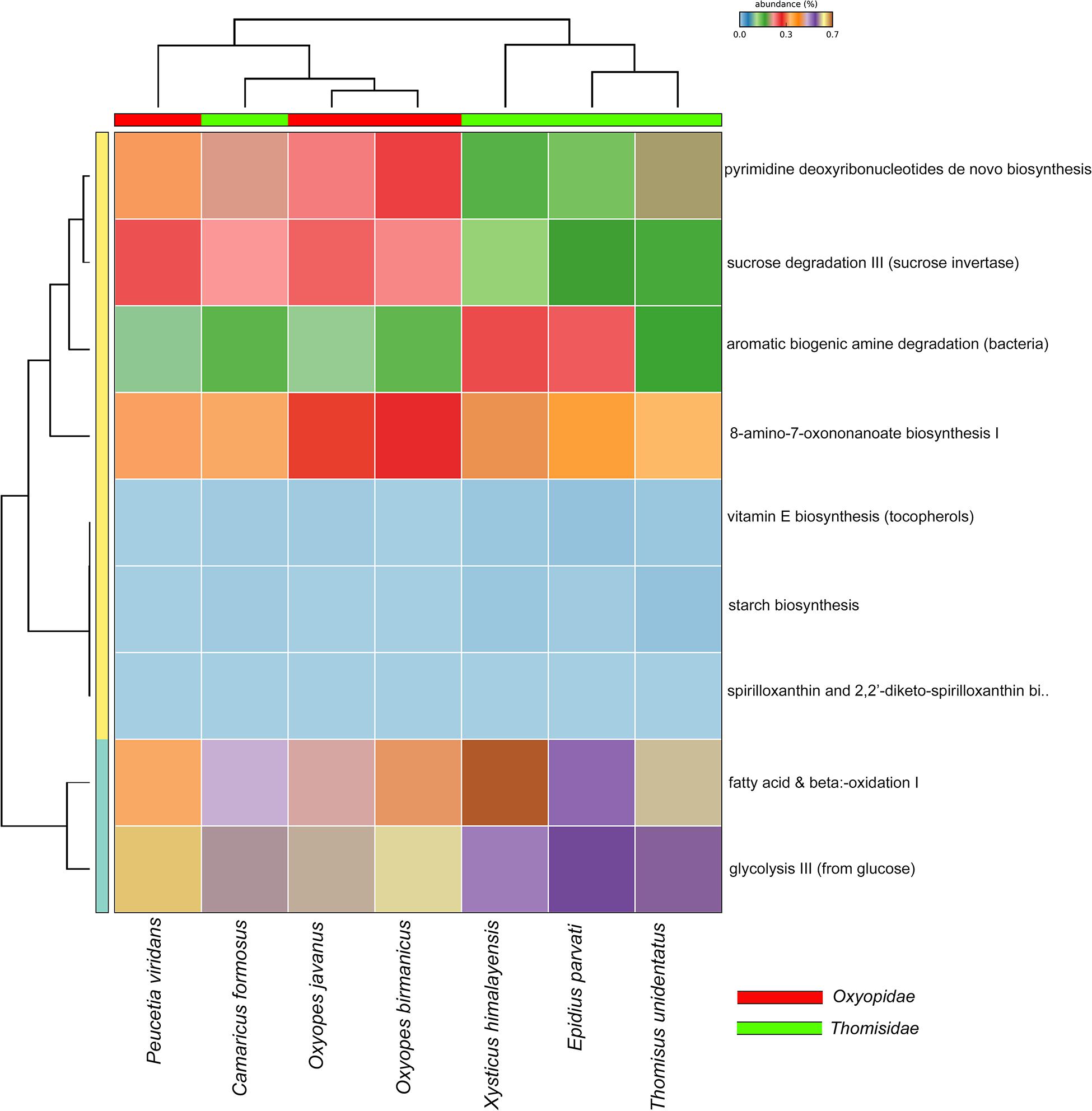
Figure 7. Bar plot based on PICRUSt2 analysis to predict the functional metabolic profile in both the spider families.
Discussion
The present work represents the comprehensive comparative studies on the gut bacterial diversity of seven spider species directly collected from a field belonging to two families, i.e., Oxyopidae and Thomisidae. Our study detected 5039 OTUs, of which 16% of the annotated OTUs represent the core bacterial composition between these two families. The core microbiome in these two families consists the members of phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes which play a vital role in nutrient and energy metabolism (Hu et al., 2019). Moreover, these are also involved in activities like fatty acids and sugar metabolism, degradation of organic compounds, and biosynthesis of vitamin cofactors like vitamin E (tocopherols) as revealed from the PICRUSt2 analysis. The gut bacterial communities of Peucetia viridans were clustered apart from other spider species, which might be related to their feeding preferences or geographical location or abnormal ingestion of prey before capturing (Suenami et al., 2019). The Proteobacteria is a major dominant phylum in family Thomisidae and Firmicutes in family Oxyopidae. Further, the high abundance of Firmicutes in family Oxyopidae is due to the presence of Paraclostridium genus (56%) (phylum Firmicutes) in Peucetia viridans. Similar results, i.e., high abundance of phylum Proteobacteria, were also observed in the gut for the wide range of arthropods including spiders (Hu et al., 2019), butterflies (Chen et al., 2016), bugs (Hammer et al., 2017), bees (Engel et al., 2012; Engel and Moran, 2013), and moths (Ruokolainen et al., 2016).
At the genus level, significant changes in the gut bacterial community were observed and genera like Acinetobacter, Staphylococcus, Corynebacterium, Cutibacterium, and Pseudomonas contribute differently to the gut microbiome of six spider species. Peucetia viridans showed the high abundance of the genus Paraclostridium. The results obtained were not in agreement with the previous studies of spider gut microbiome where Burkholderia, Rickettsiella, Ochrobactrum, Rhodoplanes, etc. (Hu et al., 2019), as well as Staphylococcus (Rivera et al., 2017), contributed mainly to the gut bacterial diversity, based on which it can be concluded that the spiders lack the core gut bacterial community and their gut microbiome majorly depends on the prey feeding and environmental linkages (Kennedy et al., 2020). Moreover, lack of endosymbionts in the seven spider species suggested that the gut bacterial community was offering protection against endosymbionts and other pathogens as observed in the other insect gut (Engel et al., 2012; Moran, 2015). The genera Acinetobacter and Pseudomonas were known for functional activities like degradation of amylose, cellulose, lipids, etc., in the gut of different arthropods (Engel et al., 2012; Briones-Roblero et al., 2017). Moreover, Acinetobacter was also known for the phenol degradation (Van Dexter and Boopathy, 2019) in termite gut while Sphingomonas and Pseudomonas were known for insecticide degradation (Itoh et al., 2018). Members of bacterial genus, i.e., Cutibacterium were involved in the carbohydrate fermentation and lactase to pyruvate conversion in the infant gut and also known for causing inflammation (Perry and Lambert, 2006). The members of this genus were also reported as the resident gut microbiota in different insects (Wang et al., 2011; Zucchi et al., 2012). The genus Corynebacterium was one of the most widely distributed genera in the gut microbiota of insects (Engel et al., 2012), and it was known for multiple functional activities like organic molecule degradation and amino-acid metabolism, which is also evident from the PICRUSt2 analysis, and it was also observed as a symbiont in the gut of T. infestans (Durvasula et al., 2008). Considering genus Paraclostridium which is the most abundant and observed only in one spider species, i.e., Peucetia viridans, earlier it was reported in the gut of earthworm (Meier et al., 2018) and Egyptian mongoose (H. ichneumon) (Pereira et al., 2020) and was known for the fermentation of amino acids. In addition to the functional metabolic potential, the member of the genus Paraclostridium was known to release neurotoxins that target mainly mosquitoes like Anopheles (Contreras et al., 2019) and Aedes (Qureshi et al., 2014). However, the functional metabolic role or relationships of Paraclostridium within spiders are unclear as it was reported for the first time, which need to be further investigated.
Furthermore, the variation in the bacterial abundance in terms of the genus in the gut of seven spider species might be due to the different prey and environmental habitats (Hu et al., 2019). Moreover, Kennedy et al. (2020), in their studies, revealed that prey greatly influenced the gut microbiome in spider species, which undergoes temporal fluctuations based on prey diet. It may be attributed to the stress induced on spiders which are reared and fed with fixed prey diet. Moreover, to better understand the core gut bacterial communities in spider, extensive sampling from different environmental habitats has been required.
Conclusion
This study unveiled the diversity, structure, and predicted functions of bacterial communities in the gut of seven spider species belonging to two different families, i.e., Oxyopidae and Thomisidae collected from the wild. No significant differences in the gut bacterial communities were observed between the studied spider families or spider species except the spider Peucetia viridans. The classified gut bacteria were predicted to be related to fatty acid and sugar metabolisms, organic compound degradation, and vitamin E biosynthesis. The genus Paraclostridium was observed for the first time in spider (Peucetia viridans), which open up the new scope for the researchers to further explore the relationship of this particular genus with that of host specimens.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA638522.
Author Contributions
KT, IT, and VK: conceptualization, data generation, amplicon analysis, and manuscript writing. VK and KC: project supervision and financial assistance. All authors reviewed the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All the authors are thankful to Director of Zoological Survey of India for the encouragement and financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.588102/full#supplementary-material
Footnotes
References
Anjum, S. I., Shah, A. H., Aurongzeb, M., Kori, J., Azim, M. K., Ansari, M. J., et al. (2018). Characterization of gut bacterial flora of Apis mellifera from north-west Pakistan. Saudi J. Biol. Sci. 25, 388–392. doi: 10.1016/j.sjbs.2017.05.008
Bardou, P., Mariette, J., Escudié, F., Djemiel, C., and Klopp, C. (2014). jvenn: an interactive Venn diagram viewer. BMC Bioinform. 15, 293–300. doi: 10.1186/1471-2105-15-293
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Briones-Roblero, I., Hernańdez-Garcıa, J. A., Gonzalez-Escobedo, R., Soto-Robles, L. V., RiveraOrduña, F. N., and Zuñiga, G. (2017). Structure and dynamics of the gut bacterial microbiota of the bark beetle, Dendroctonus rhizophagus (Curculionidae: Scolytinae) across their life stages. PLoS One 12:e0175470. doi: 10.1371/journal.pone.0175470
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, B., Beng-Soon, T., Sun, C., Hu, S., Lu, X., Wilhelm, B., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Contreras, E., Masuyer, G., Qureshi, N., Chawla, S., Dhillon, H. S., Lee, H. L., et al. (2019). A neurotoxin that specifically targets Anopheles mosquitoes. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-10732-w
Dhariwal, A., Chong, J., Habib, S., King, I., Agellon, L. B., and Xia, J. (2017). MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188. doi: 10.1093/nar/gkx295
Durvasula, R. V., Sundaram, R. K., Kirsch, P., Hurwitz, I., Crawford, C. V., Dotson, E., et al. (2008). Genetic transformation of a Corynebacterial symbiont from the chagas disease vector Triatoma infestans. Exp. Parasitol. 119, 94–98. doi: 10.1016/j.exppara.2007.12.020
Engel, P., Martinson, V. G., and Moran, N. A. (2012). Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acadm. Sci. U.S.A. 109, 11002–11007. doi: 10.1073/pnas.1202970109
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects-diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Esposti, M. D., and Romero, E. M. (2017). The functional microbiome of arthropods. PLoS One 12:e0176573. doi: 10.1371/journal.pone.0176573
Foster, Z. S. L., Sharpton, T. J., and Grunwald, N. J. (2017). Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 13:e1005404. doi: 10.1371/journal.pcbi.1005404
Goodacre, S. L., Martin, O. Y., Thomas, C. F. G., and Hewitt, G. M. (2006). Wolbachia and other endosymbiont infections in spiders. Mol. Ecol. 15, 517–527. doi: 10.1111/j.1365-294X.2005.02802.x
Hammer, T. J., Janzen, D. H., Hallwachs, W., Jaffe, S. P., and Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proc. Natl. Acadm. Sci. U.S.A. 114, 9641–9646. doi: 10.1073/pnas.1707186114
Hu, G., Zhang, L., Yun, Y., and Peng, Y. (2019). Taking insight into the gut microbiota of three spider species: no characteristic symbiont was found corresponding to the special feeding style of spiders. Ecol. Evol. 9, 8146–8156. doi: 10.1002/ece3.5382
Itoh, H., Hori, T., Sato, Y., Nagayama, A., Tago, K., Hayatsu, M., et al. (2018). Infection dynamics of insecticide-degrading symbionts from soil to insects in response to insecticide spraying. ISME J. 12, 909–920. doi: 10.1038/s41396-017-0021-9
Kanehisa, M. (2019). Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. doi: 10.1002/pro.3715
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K., and Tanabe, M. (2019). New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595. doi: 10.1093/nar/gky962
Kennedy, S. R., Tsau, S., Gillespie, R., and Krehenwinkel, H. (2020). Are you what you eat? A highly transient and prey-influenced gut microbiome in the grey house spider Badumna longinqua. Mol. Ecol. 29, 1001–1015. doi: 10.1111/mec.15370
Klepzig, K. D., Adams, A. S., Handelsman, J., and Raffa, K. F. (2009). Symbioses: a key driver of insect physiological processes, ecological interactions, evolutionary diversification, and impacts on humans. Environ. Entomol. 38, 67–77. doi: 10.1603/022.038.0109
Langille, M. G. I. (2018). Exploring linkages between taxonomic and functional profiles of the human microbiome. mSystems 3:e00163-17. doi: 10.1128/mSystems.00163-17
Macías-Hernández, N., Athey, K., Tonzo, V., Wangensteen, O. S., Arnedo, M., and Harwood, J. D. (2018). Molecular gut content analysis of different spider body parts. PLoS One 13:e0196589. doi: 10.1371/journal.pone.0196589
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Meier, A. B., Hunger, S., and Drake, H. L. (2018). Differential engagement of fermentative taxa in gut contents of the earthworm Lumbricus terrestris. Appl. Environ. Microbiol. 84, 1–17. doi: 10.1128/AEM.01851-17
Michalko, R., and Pekár, S. (2015). The biocontrol potential of Philodromus (Araneae, Philodromidae) spiders for the suppression of pome fruit orchard pests. Biol. Control 82, 13–20. doi: 10.1016/j.biocontrol.2014.12.001
Moran, N. A. (2015). Genomics of the honey bee microbiome. Curr. Opin. Insect. Sci. 10, 22–28. doi: 10.1016/j.cois.2015.04.003
Muturi, E. J., Ramirez, J. L., Rooney, A. P., and Kim, C. H. (2017). Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl. Trop. D 11:e0005377. doi: 10.1371/journal.pone.005377
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Pereira, A. C., Bandeira, V., Fonseca, C., and Cunha, M. V. (2020). Egyptian mongoose gut microbiota: Taxonomical and functional differences across sex and age classes. Microorganisms 8:392. doi: 10.20944/preprints202002.0306.v1
Perry, A. L., and Lambert, P. A. (2006). Propionibacterium acnes. Lett. Appl. Microbiol. 42, 185–188. doi: 10.1111/j.1472-765X.2006.01866.x
Qureshi, N., Chawla, S., Likitvivatanavonga, S., Ham, L. L., and Gill, S. S. (2014). The cry toxin operon of Clostridium bifermentans subsp. malaysia is highly toxic to Aedes larval Mosquitoes. Appl. Environ. Microbiol. 80, 5689–5696. doi: 10.1128/AEM.01139-14
R Core Team (2020). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. Available online at: http://www.R-project.org/
Rivera, P., Stork, R., and Hug, A. (2017). A first look at the microbial community of Rabidosa rabida, a Wolf spider in searcy. Arkansas. J. Ark Acad. Sci. 71, 51–55.
Ruokolainen, L., Ikonen, S., Makkonen, H., and Hanski, I. (2016). Larvalgrowth rate is associated with the composition of the gut microbiotain the glanville fritillary butterfly. Oecologia 181, 895–903.
Sheffer, M. M., Uhl, G., Prost, S., Lueders, T., Urich, T., and Bengtsson, M. M. (2020). Tissue-and Population-Level Microbiome analysis of the wasp spider Argiope bruennichi identified a novel dominant bacterial symbiont. Microorganisms 8:8. doi: 10.3390/microorganisms8010008
Snyman, M., Gupta, A. K., Bezuidenhout, C. C., Claassens, S., and Van den Berg, J. (2016). Gut microbiota of Busseola fusca (Lepidoptera: Noctuidae). World J. Microb. Biot. 32:115. doi: 10.1007/s11274-016-2066-8
Suenami, S., Nobu, M. K., and Miyazaki, R. (2019). Community analysis of gut microbiota in hornets, the largest eusocial wasps, Vespa mandarinia and V. simillima. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-46388-1
Van Dexter, S., and Boopathy, R. (2019). Biodegradation of phenol by Acinetobacter tandoii isolated from the gut of the termite. Environ. Sci. Pollut. Res. 26, 34067–34072. doi: 10.1007/s11356-018-3292-4
Vanthournout, B., Swaegers, J., and Hendrickx, F. (2011). Spiders do notescape reproductive manipulations by Wolbachia. BMC Evol. Biol. 11:15. doi: 10.1186/1471-2148-11-15
Wang, Y., Gilbreath, T. M., Kukutla, P., Yan, G., and Xu, J. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. doi: 10.1371/journal.pone.024767
World Spider Catalog (2020). Version 20. Natural History Museum Bern. Available online at: http://wsc.nmbe.ch (accessed June 15, 2020).
Zhang, L., Yun, Y., Hu, G., and Peng, Y. (2018). Insights into the bacterial symbiont diversity in spiders. Ecol. Evol. 8, 4899–4906. doi: 10.1002/ece3.4051
Zhang, L. H., Zhang, G. M., Yun, Y. L., and Peng, Y. (2017). Bacterial community of a spider, Marpiss magister (Salticidae). 3Biotech 7:371. doi: 10.1007/s13205-017-0994-0
Keywords: gut microbiome, spiders, Oxyopidae, Thomisidae, arthropods
Citation: Kumar V, Tyagi I, Tyagi K and Chandra K (2020) Diversity and Structure of Bacterial Communities in the Gut of Spider: Thomisidae and Oxyopidae. Front. Ecol. Evol. 8:588102. doi: 10.3389/fevo.2020.588102
Received: 28 July 2020; Accepted: 23 September 2020;
Published: 29 October 2020.
Edited by:
Peter Convey, British Antarctic Survey (BAS), United KingdomReviewed by:
Antonio Carapelli, University of Siena, ItalyWen Chyin Yew, Northumbria University, United Kingdom
Copyright © 2020 Kumar, Tyagi, Tyagi and Chandra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaomud Tyagi, a3VtdWQudHlhZ2k1QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work