 Carlee A. Resh1
Carlee A. Resh1 Matthew P. Galaska2
Matthew P. Galaska2 Kasey C. Benesh1
Kasey C. Benesh1 Jonathan P. A. Gardner3,4
Jonathan P. A. Gardner3,4 Kai-Jian Wei4
Kai-Jian Wei4 Ruo-Jin Yan4†
Ruo-Jin Yan4† Andrew R. Mahon1*
Andrew R. Mahon1*- 1Department of Biology, Central Michigan University, Mount Pleasant, MI, United States
- 2Cooperative Institute for Climate, Ocean, and Ecosystem Studies, University of Washington and National Oceanic and Atmospheric Administration Pacific Marine Environmental Lab, Seattle, WA, United States
- 3School of Biological Sciences, Victoria University of Wellington, Wellington, New Zealand
- 4Key Laboratory of Freshwater Animal Breeding, Ministry of Agriculture, College of Fisheries, Huazhong Agricultural University, Wuhan, China
The introduction and subsequent range expansion of the Northern snakehead (Channa argus: Channidae, Anabantiformes) is one of a growing number of problematic biological invasions in the United States. This harmful aquatic invasive species is a predatory freshwater fish native to northeastern Asia that, following deliberate introduction, has established itself in multiple water basins in the eastern United States, as well as expanding its range into the Midwest. Previous work assessed the population structure and estimated the long-term effective population sizes of the populations present in the United States, but the source of the initial introduction(s) to the U.S. remains unidentified. Building on earlier work, we used whole genome scans (2b-RAD genomic sequencing) to analyze single nucleotide polymorphisms (SNPs) from C. argus to screen the genomes of these invasive fish from United States waters and from three sites in their native range in China. We recovered 2,822 SNP loci from genomic DNA extracted from 164 fish sampled from the eastern United States and Arkansas (Mississippi River basin), plus 30 fish sampled from three regions of the Yangtze River basin in China (n = 10 individuals per basin). Our results provide evidence supporting the Yangtze River basin in China, specifically the Bohu and/or Liangzi lakes, is a likely source of the C. argus introductions in multiple regions of the U.S., including the Lower Hudson River basin, Upper Hudson River basin and Philadelphia (Lower Delaware River basin). This information, in conjunction with additional sampling from the native range, will help to determine the source(s) of introduction for the other U.S. populations. Additionally, this work will provide valuable information for management to help prevent and manage future introductions into United States waterways, as well as aid in the development of more targeted strategies to regulate established populations and inhibit further spread.
Introduction
Invasive species are usually environmentally harmful and economically expensive, yet deliberate and non-intentional introductions continue to occur at an increasing rate (Leung et al., 2002; Lodge et al., 2006; Pimentel, 2005). These invasions contribute to environmental change, loss of biodiversity, and threaten human health and the economy in many regions of the world (Nuñez and Pauchard, 2009). In the United States alone, invasive species control costs are estimated to be over $100 billion annually (Pimentel, 2005). The cost of invasive species control increases as the abundance of a non-native species increases over time and once an invasive species has become established, eradication becomes extremely unlikely, and long-term management is required. Therefore, knowledge of population histories of invasive species is extremely important, particularly when early detection and rapid responses to invasions can still be effective (Early et al., 2016). Genomic biosurveillance, including genomics and other cutting edge molecular technologies such as whole genome scans, have advanced to levels where they are capable of rapidly informing management agencies and stakeholders about the invasive organisms in both their native and non-native habitats (e.g., Hamelin and Roe, 2019). In particular, the development of datasets from high throughput sequencing technologies that include genome-wide single nucleotide polymorphisms (SNPs) can provide fine scale, cost efficient, information about the invasion process (Cristescu, 2015; Hamelin and Roe, 2019).
The Northern Snakehead (Channa argus) is a harmful aquatic invasive fish with multiple established populations in the eastern United States and Arkansas (Resh et al., 2018; Fuller et al., 2020). The native range of the species is China, eastern Russia, and portions of North Korea, and it is primarily found in the Yangtze River drainage (Yan et al., 2018). Channa argus is a voracious predator, matures rapidly, and can withstand a wide range of environmental conditions, including surviving up to 4 days out of water (Orrell et al., 2005; Fuller et al., 2020). These characteristics, as well the fish’s high fecundity and parental care exhibited by both parents for several weeks after fry hatch, have contributed to its ability to spread and establish populations throughout the United States since first being observed approximately 20 years ago (Fuller et al., 2020). Once a population of an invading species becomes established in a region, the logistics and costs of eradication become exponentially higher than at pre-establishment. Therefore, as well as it being imperative to understand the population dynamics of C. argus to prevent further spread within the United States, it is equally important to identify the source(s) of introduction prevent further introductions into the United States.
In our previous work, we identified five genetically distinct populations of C. argus present in the United States that were the result of at least two separate introductions (Wegleitner et al., 2016; Resh et al., 2018). However, the source or sources of the C. argus invasion(s) remain unknown. The addition of samples from three sites in the Yangtze River basin (Yan et al., 2018), part of the native range of C. argus, to our previous datasets provide an opportunity to build on this work and potentially discover the location of source populations of C. argus in the U.S. Identifying the sources of the invasions of non-native species helps to identify invasion routes and vectors, which will allow for more targeted management plans to be implemented that aim to prevent trade and reduce the risk of further introduction of this harmful aquatic invasive species (Harris et al., 2016). Additionally, knowledge of the invasive species’ native environment provides information about factors that regulate its distribution and population, which in turn, will enable better predictions about potential expansion of the invasive species in its introduced range (Geller et al., 2010). Therefore, the goal of this study is to expand on our previous 2b-RAD sequencing data from fish collected from a portion of the native range of C. argus, to begin to determine the source(s) of the C. argus introduction(s) in the United States. The populations in the United States are admixed, so it is not possible to determine how many introductions have occurred, but previous studies provide evidence for at least two separate introductions (Wegleitner et al., 2016; Resh et al., 2018). As a result, we hypothesize that individuals from at least one of the introduction events originated from the Yangtze River basin in China. An understanding of source population information is important to manage existing populations and reduce the risk of future introductions of C. argus into the United States. This study presents a methodological advance in analyzing populations of invasive species using cutting edge genomic technology and provides a novel suite of data that can be directly used by management stakeholders. The resulting data from this study will aid in that goal because they will contribute to more effective predictions of potential expansion and the development of biological control options.
Materials and Methods
Sample Collection and Preparation
Sequence data from Resh et al. (2018) were used for this study. Genomic DNA was extracted from the fin clips of C. argus from the eastern United States and Arkansas (Mississippi River basin) using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, United States), following manufacturer’s protocols (Figure 1; full collection information in Supplementary Table 1). Additionally, fin clips were collected from 30 C. argus individuals from three lakes in the native range of C. argus that are part of the Yangtze River basin in China: Bohu Lake (n = 10), Liangzi Lake (n = 10), and Poyang Lake (n = 10) (Figure 1). Genomic DNA was extracted from the fin clips using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, United States), following manufacturer’s protocols.
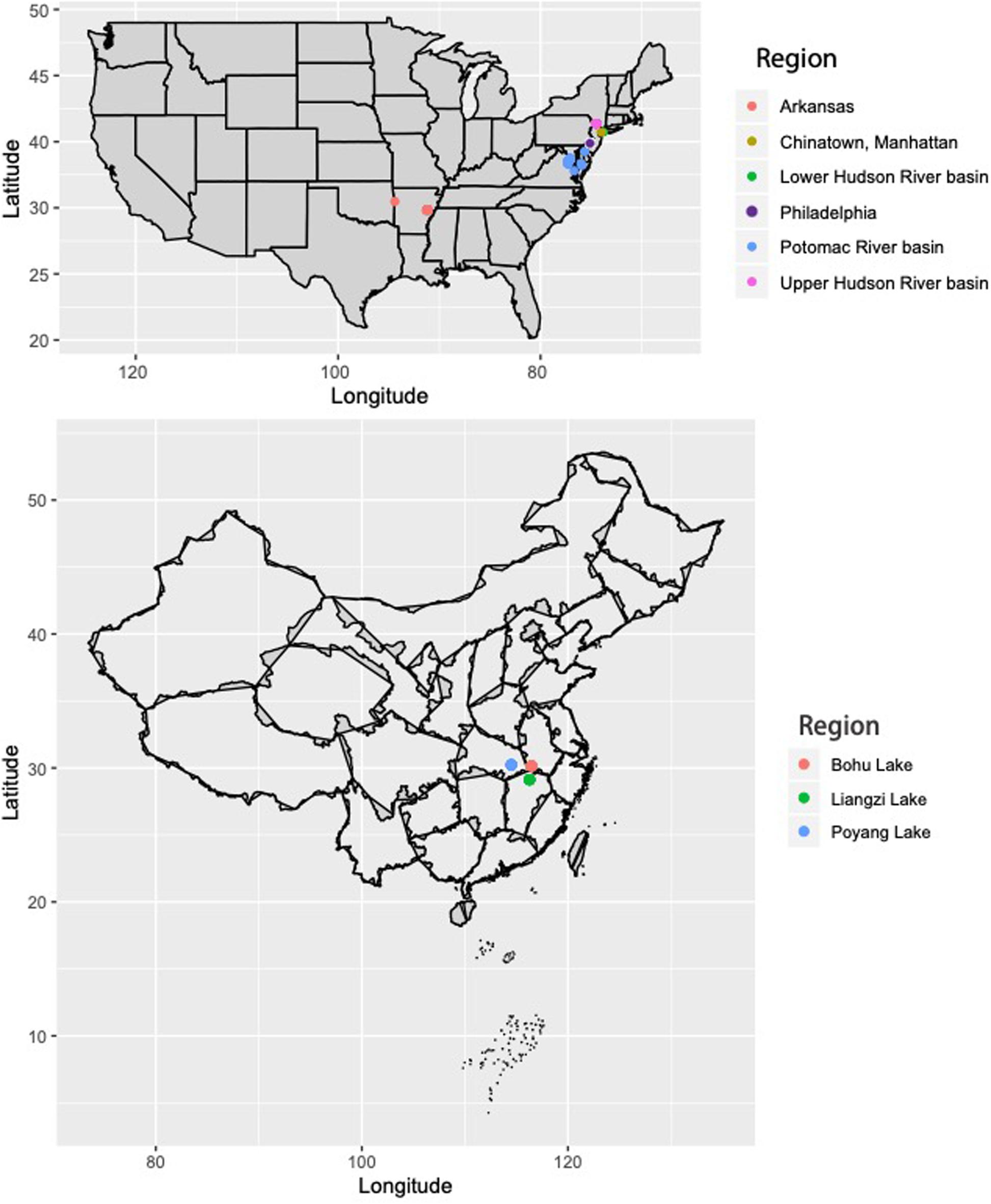
Figure 1. Collection locations for Channa argus individuals in the United States and China. For detailed map of U.S. collections, see Resh et al. (2018) and Supplementary Table 1.
2b-RAD Data Collection
Genomic DNA was prepared based on the 2b-RAD protocol from Wang et al. (2012), with the restriction enzyme AlfI. A one quarter (1/4) reduction scheme using ligation adaptors (NC/NN) was chosen based on the approximate genome size of C. argus (616–861 Mb, Gregory, 2021), as well as to target approximately 2,500 SNP loci. Samples were dual barcoded with unique combinations and then sequenced to generate 50 bp single end reads at the Genomics and Cell Characterization Core (University of Oregon, OR, United States) on an Illumina Hi-Seq 4000 using v4 chemistry.
Data Analyses
The raw Illumina reads were demultiplexed by sample, quality filtered, and the AlfI recognition sites were extracted using Dr. Eli Meyer’s scripts (Oregon State University)1. The software package Stacks v.2.41 created a custom de novo reference file using the pipeline denovo_map.pl within Stacks v2.41 to align the raw Illumina reads and assign to unique stacks (Catchen et al., 2011, 2013). Assignment of homozygotic loci was allowed to have a maximum variance of 1% and heterozygotic loci assignment required a minimum of 25% variance. Loci had to occur in 75% or more individuals within a sampling locality and be present in at least 2 localities to be processed. If these requirements were not met, then the loci were discarded prior to analyses.
Principal components analyses (PCAs) were performed by locality on the dataset using the Analysis of Ecological Data: Exploratory and Euclidean Methods in Environmental Sciences (ade4) v1.7-13 package (Dray and Dufour, 2007) in the R v3.6.1 statistical program (R Core Team, 2015) and the top principal components were compared. Discriminant Analysis of Principal Components (DAPC) in the Adegenet v2.1.1 package (Jombart and Ahmed, 2011) in R was used to analyze the SNP data to determine group (population) membership across localities (i.e., collection locations of each sample). Adegenet v2.1.1 conducts a series of PCAs on the dataset and then performs a Discriminant Analysis on all the retained principal components. The optimal number of clusters (K), which estimates the number of populations, is identified through Bayesian information criterion (BIC) likelihood values from retained principal components. Visualization of these analyses was performed in the Adegenet v2.1.1 package (Jombart and Ahmed, 2011).
Population structure and potential admixture were assessed using the Landscape and Ecological Associations (LEA) v1.8.1 package in R (Frichot and François, 2015). The number of ancestral populations, K, was estimated with the cross-entropy criterion and least squares estimates (Frichot and François, 2015).
Summary statistics were generated for the samples from the nine localities and genetic differentiation was analyzed using the HIERFSTAT v0.04-30 package in R (Goudet, 2005). Summary statistics included the private alleles at each locality, expected heterozygosity, observed heterozygosity, and the inbreeding coefficient (Fis), with the variance and standard error values for each statistic included. Additionally, Bartlett tests were conducted using the HIERFSTAT v0.04-30 package for each locality to determine if the expected and observed heterozygosities differed (Goudet, 2005). Pairwise genetic distances (Fst) were estimated and bootstrapping was performed over the pairwise Fst values to generate 97% confidence intervals (Nei, 1987; Goudet, 2005).
Lastly, we calculated the migration rates as a proxy for how closely related introduced and native localities may have been, using the diveRsity v1.9.90 package in R (Keenan et al., 2013). DiveRsity calculates genetic diversity and differentiation statistics (Nm), as well as bootstrapped 95% confidence intervals, for pairwise locality comparisons and allows for calculation of directional migration levels among localities.
Results
In total, 52,409 single nucleotide polymorphic loci were recovered from 194 C. argus individuals. After quality filtering of the data and subsequent SNP calling, 2,822 independent SNP loci were retained in the final dataset.
The results of the principal components analysis (PCA) by locality indicate the genetic similarity between the native Bohu Lake and Liangzi Lake populations, and the introduced Philadelphia (Lower Delaware River basin) and Lower Hudson River basin populations (Figure 2). In contrast, the PCA (Figures 2A,C) identified the Poyang Lake population as being separate from the other two native Chinese populations, as well as the introduced U.S. populations. These results indicate that there is genetic dissimilarity between the Poyang Lake individuals and the fish of the other two native localities, despite the three native range sites being part of the same river basin. Additionally, Poyang Lake individuals were identified as being genetically differentiated from the introduced U.S. populations.
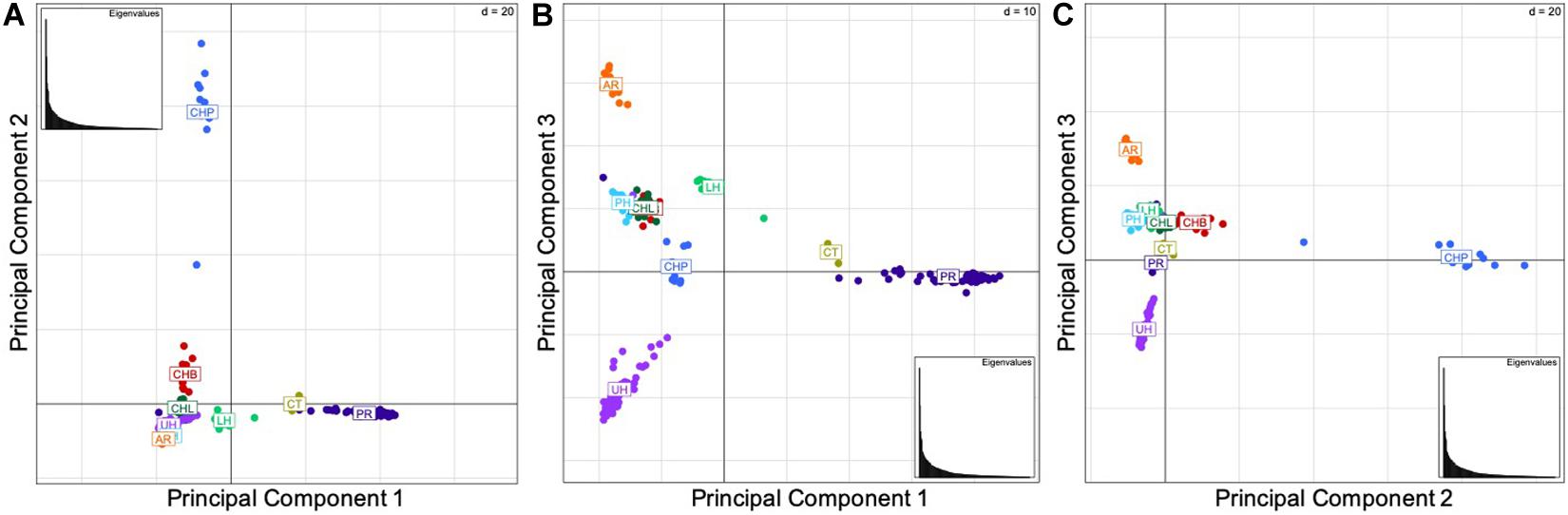
Figure 2. (A) Components 1 and 2, (B) Components 1 and 3, (C) Components 2 and 3. Principal component analyses by locality. The abbreviations are as follows: CHB, Bohu Lake, China; CHL, Liangzi Lake, China; CHP, Poyang Lake, China; AR, Arkansas; CT, Chinatown, Manhattan; LH, Lower Hudson River basin; PH, Philadelphia; PR, Potomac River basin; and UH, Upper Hudson River basin. The three principal components explain 46.49% of the variation in the data set.
Results of the DAPC analyses indicated six geographically and genomically distinct clusters or populations of C. argus (K = 6, Figure 3 and Supplementary Figure 11). Cluster 1 contained 10 individuals and consisted of 100% of the fish collected from the Poyang Lake. Cluster 2 contained 58 individuals, 98% (58 of 59) of the fish that were collected from the Upper Hudson River basin. Cluster 3 contained 18 individuals: 100% of the fish collected from Arkansas. Cluster 4 contained 22 individuals: 1.9% (1 of 54) of the fish collected from the Potomac River basin and 100% of the fish collected from Philadelphia. Cluster 5 contained 53 individuals: 98% of the fish collected from the Potomac River basin (53 of 54). Cluster 6 contained 34 individuals: 100% of the fish collected from Bohu Lake and Liangzi Lake, 1.7% (1 of 59) of the fish collected from the Upper Hudson River basin, 100% of the fish collected from the Lower Hudson River basin, and 100% of the fish from the Chinatown, Manhattan fish market.
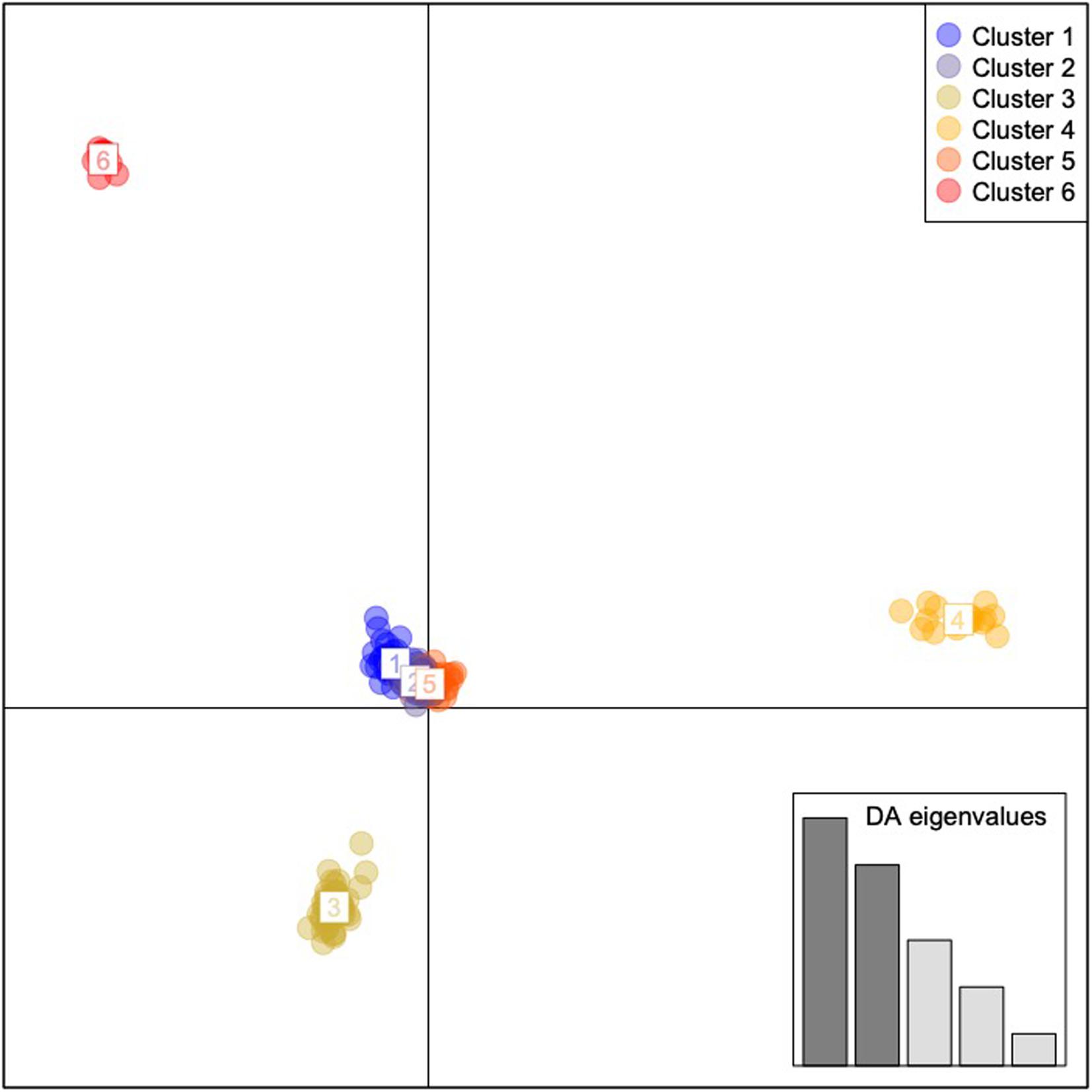
Figure 3. Discriminant Analysis of Principal Components for Channa argus single nucleotide polymorphism data. Cluster 1: all of the fish collected from the Bohu and Liangzi Lakes in China, 1.7% (1 of 59) of the fish collected from the Upper Hudson River basin, all of the fish collected from the Lower Hudson River basin, and all of the fish from the Chinatown, Manhattan fish market. Cluster 2: 1.9% (1 of 54) of the fish collected from the Potomac River basin and all of the fish collected from Philadelphia. Cluster 3: 98% of the fish collected from the Potomac River basin (53 of 54). Cluster 4: all of the fish collected from Arkansas. Cluster 5: 98% (58 of 59) of the fish that were collected from the Upper Hudson River basin. Cluster 6: All individuals sampled from Poyang Lake, China.
The results of the admixture analyses in LEA also supported the existence of six populations (K = 6, Figure 4 and Supplementary Figure 2). Each of the populations was partially admixed, although the relative amount of admixture varied among the populations. The fish from Bohu Lake and Liangzi Lake were statistically from the same population. These two lakes share ancestral genotypes with the fish from Poyang Lake, the Lower Hudson River basin, Chinatown Manhattan, Philadelphia, and Arkansas populations, as well as one individual in both the Upper Hudson River and Potomac River basins. In contrast, the main ancestral genotype of the rest of the fish from the Upper Hudson River and Potomac River is rare in the Bohu Lake and Liangzi Lake populations.
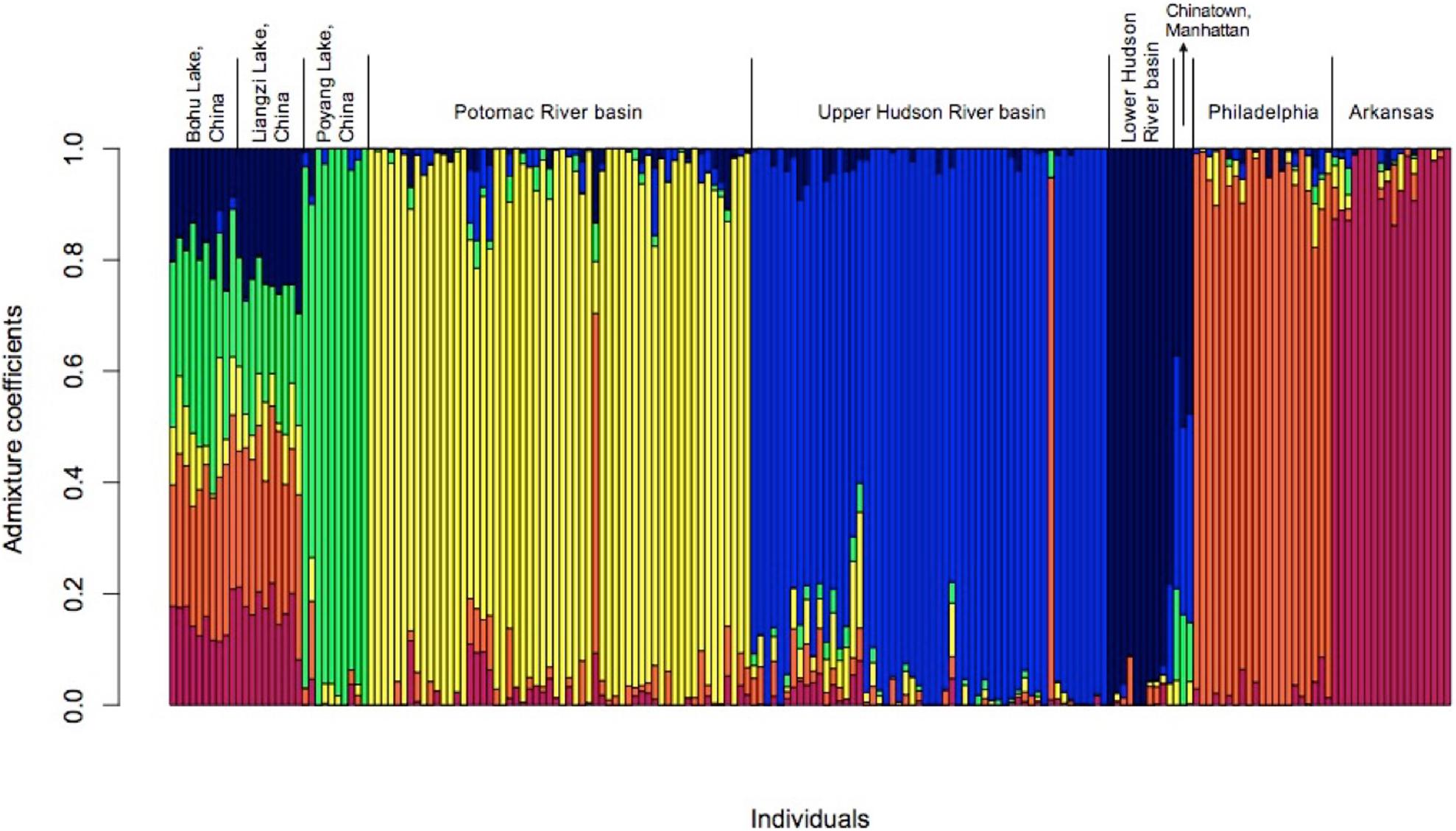
Figure 4. Admixture for C. argus populations (k = 6) in the United States when compared to three native range populations from central China.
Summary statistics of genetic diversity and genetic distances between the putative C. argus populations (Tables 1–3) revealed that each population contained private alleles. The Poyang Lake population contained the most private alleles and the Upper Hudson River population the least (337 and 60, respectively).
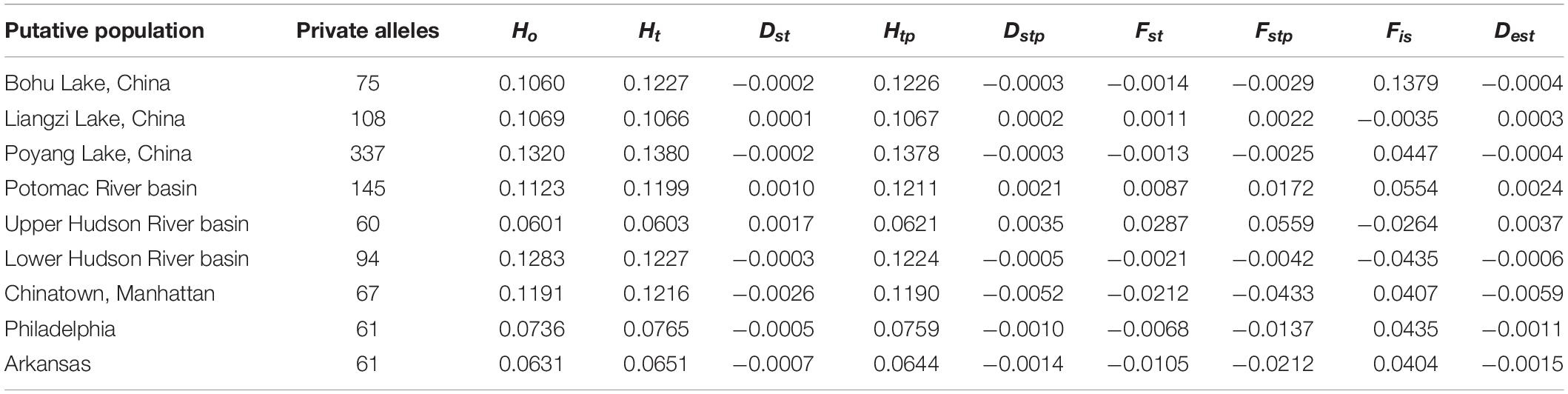
Table 1. Summary statistics for single nucleotide polymorphism locus variation at each collection site.
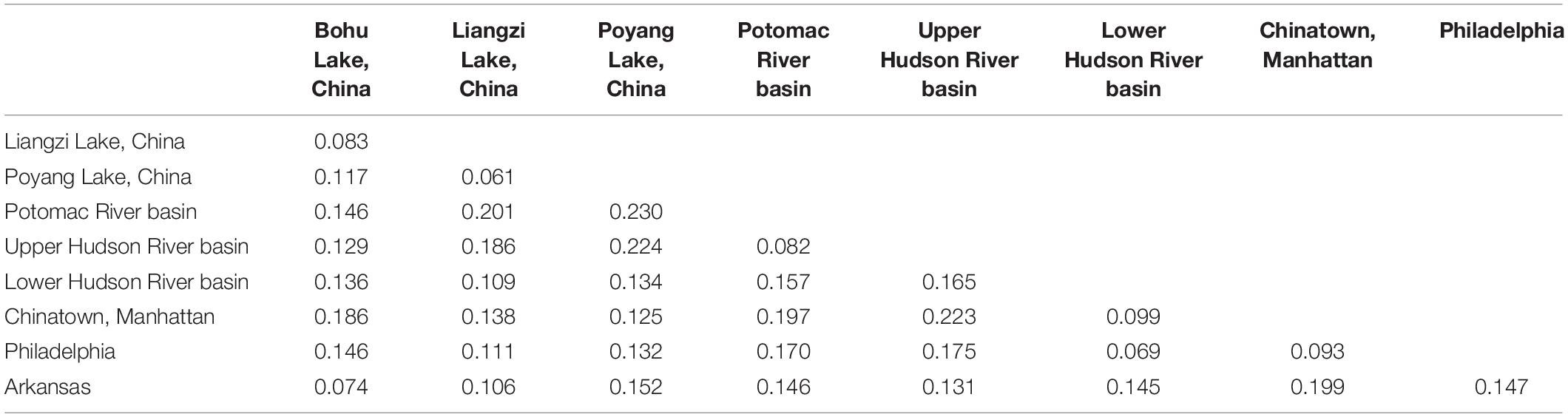
Table 2. Pairwise genetic distances (Fst) between Channa argus putative populations.
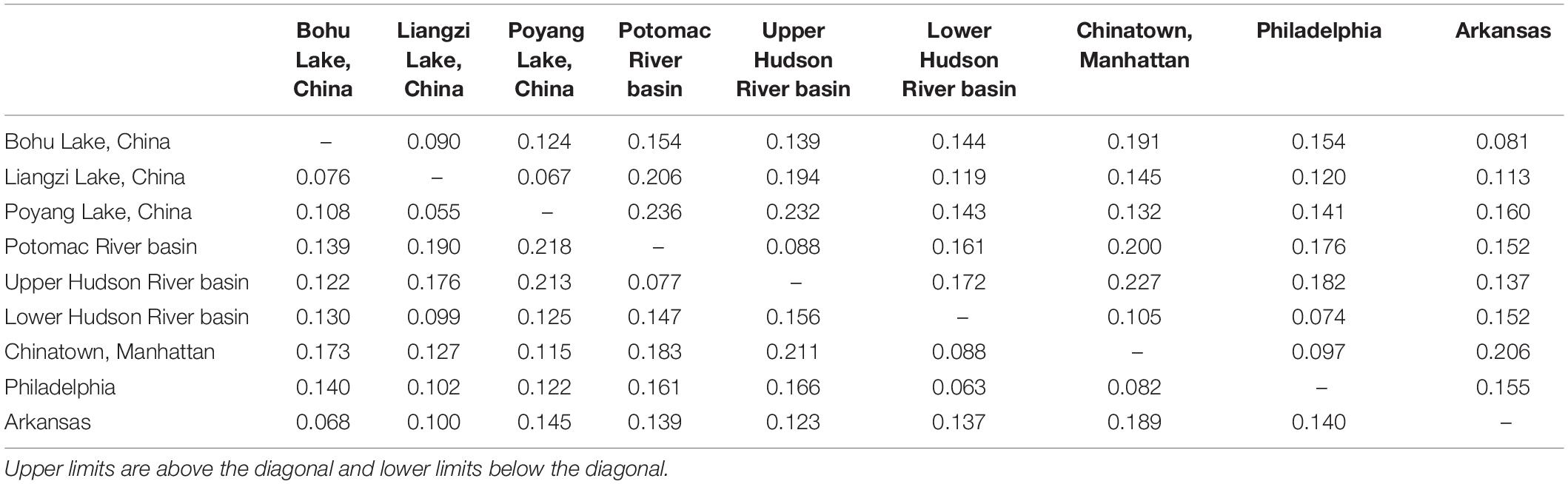
Table 3. Upper and lower confidence interval limits for pairwise genetic distances (Fst) between Channa argus putative populations.
The observed heterozygosity was lower than expected heterozygosity for the Potomac River basin (Ho: 0.112 and He: 0.120; p = 0.044), Arkansas (Ho: 0.063 and He: 0.065; p = 0.003), and Poyang Lake (Ho: 0.132 and He: 0.138; p = 8.28e–8) populations. Observed heterozygosity was higher than expected heterozygosity for the Lower Hudson River basin population (Ho: 0.128 and He: 0.123; p = 2.2e–16). The observed heterozygosity did not deviate from expected values for Bohu Lake (Ho: 0.106 and He: 0.123; p = 0.386), Upper Hudson River basin (Ho: 0.060 and He: 0.060; p = 0.379), and Philadelphia (Ho: 0.074 and He: 0.077; p = 0.255). Observed heterozygosity was significantly different than expected heterozygosity for Liangzi Lake (Ho: 0.107 and He: 0.107; p = 3.49e–10) and the fishes that came from the Chinatown, Manhattan fish market (Ho: 0.120 and He: 0.122; p = 2.2e–16). The inbreeding coefficient was positive for all putative populations except Liangzi Lake and the Upper and Lower Hudson River basins. Bohu Lake had the highest inbreeding coefficient at 0.1379. The two localities that had the smallest genetic distance (0.061) were Poyang Lake and Liangzi Lake within the native range of C. argus. In contrast, the largest pairwise genetic distance value occurred between Poyang Lake and the Upper Hudson River basin populations (0.224).
The relative directional migration rates between the localities are shown as a network (Figures 5, 6). Each node represents a locality, and arrows indicate the direction of gene flow, with the relative strength of the flow indicated by the bootstrap support value, as well as the shading and thickness of each connecting line. For the first analysis (Figure 5) the three native lake localities in China were grouped together, and for second (Figure 6) the three native lake localities were analyzed separately. In both cases, there was evidence of gene flow between the native populations in China and the introduced Philadelphia and Lower Hudson River basin populations. The relative directional migration levels were relatively higher between the populations of Bohu Lake and Liangzi Lake vs. Philadelphia (Nm = 0.26). The Lower Hudson River basin vs. Liangzi Lake and Bohu Lake populations had Nm values of 0.31 and 0.27, respectively, showing evidence of gene flow between the localities. While these values do not represent migration between the sites or waterways, they are instead indicative of human-mediated transfer of fish and this information is still valuable because the data show how the fish were related to one another before the introductions occurred, thereby helping identify putative source populations.
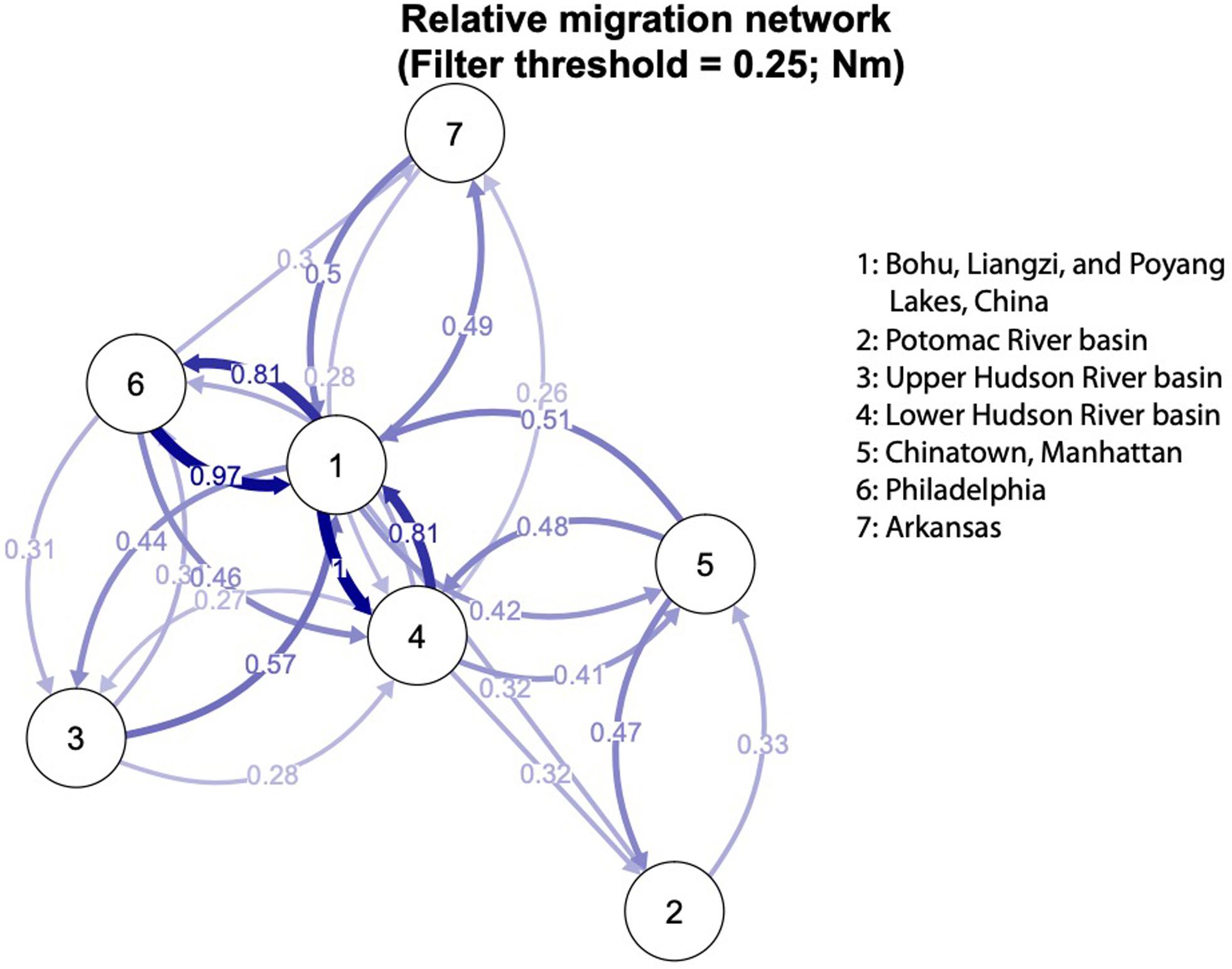
Figure 5. Relative migration network of C. argus putative populations.
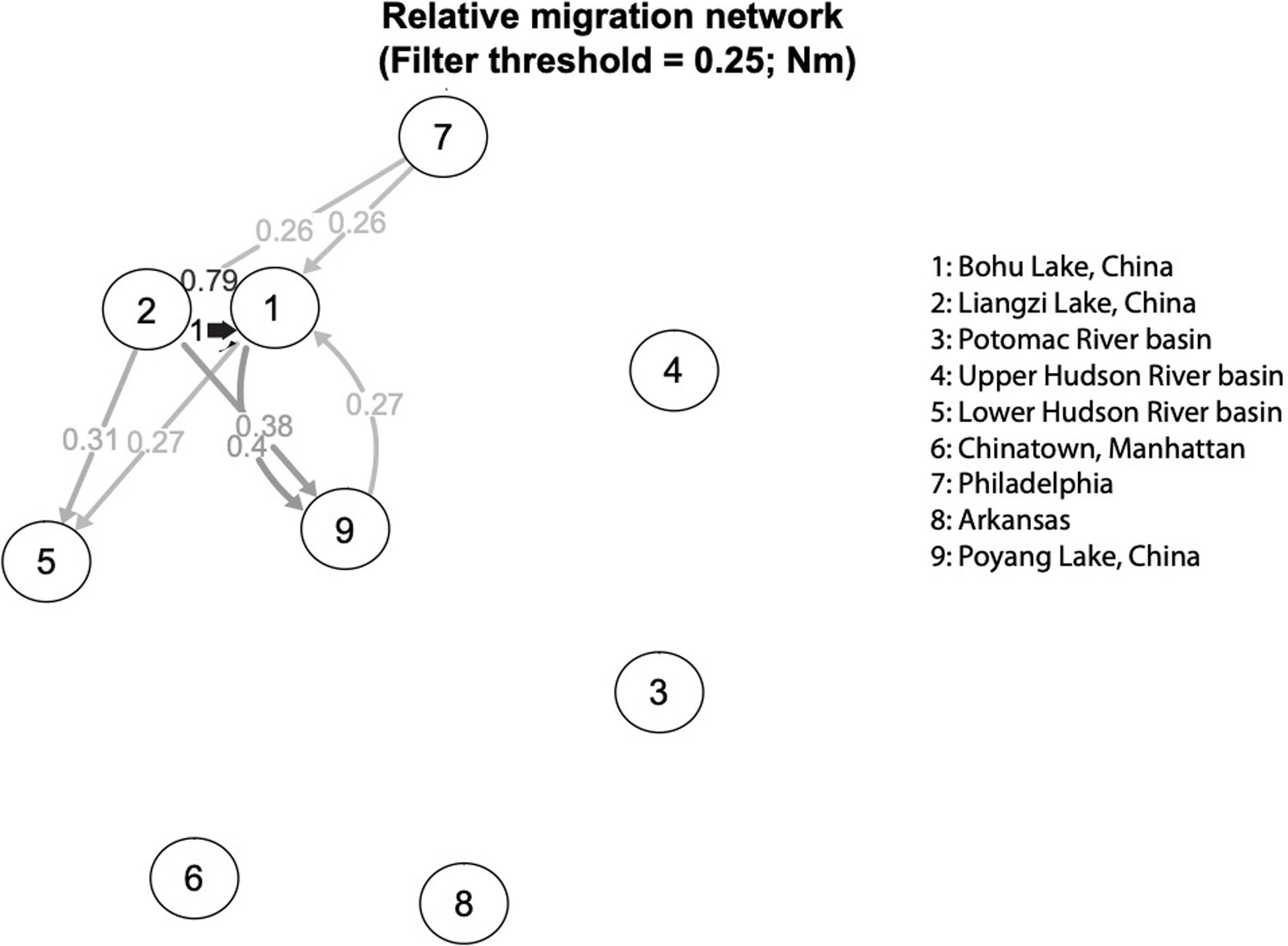
Figure 6. Relative migration network of C. argus putative populations, assessing each Chinese sampling locality separately.
Discussion
The main goal of this study was to use cutting edge technology in the form of whole genome scans to analyze genomic data from Northern snakehead individuals (C. argus) from the introduced populations in the United States, as well as a portion of their native range, to attempt to determine the source of the North American C. argus introductions. The genomic data generated in this study, with the inclusion of the new samples from the native range in China, support the presence of six genetically distinct populations of C. argus recovered from the United States and central China. While five of the U.S. populations are still extant, the Upper Hudson River basin population was successfully eradicated in 2008–2009 (Aquatic Nuisance Species [ANS] Task Force, 2014). The addition of genomic data from fishes collected from the new collection locations within the native range of C. argus may provide a number of benefits to management agencies, as well as to ecological and environmental research in the field.
Identification of source populations is crucial for effective management because information about source(s) provides insight into the invasion pathway(s) and mode(s) of introduction, which will aid in effective development of strategies to prohibit natural and human mediated transport in the introduced ranges (Collins et al., 2002; Casso et al., 2019). For instance, Austin et al. (2011) used a multi-locus genomic dataset from both the native and introduced ranges to determine the origin, mode, and tempo of the invasion of a scincid lizard (Carlia) that was introduced to Guam, the Northern Marianas, and Palau islands. Additionally, source information can aid in the effective development and implementation of biological control agents. For example, the predatory ladybird beetle, Cryptolaemus montrouzieri, native to Australia, is a widely used biological control agent that has been introduced to over 64 countries/territories to control over 16 pest species over the last century (Kairo, 2013). Li et al. (2019) showed that pronounced genetic differentiation has occurred between the sampled populations from both native and introduced ranges of C. montrouzieri, which may impact the efficiency and invasion potential of this important biological control agent. This source information aids in the discovery of cryptic species in biological control agent populations, which is important for minimizing unpredicted non-target effects so as to maximize biological control efficiency, as well as providing potential new biological control agents (Paterson et al., 2016; Smith et al., 2017).
The addition of genomic data from the native range of an invasive species is beneficial for ecological and evolutionary studies because it allows researchers to compare the invasive species’ response to its native and introduced environments and determine invasion success (Hierro et al., 2005; Bock et al., 2015; Martin et al., 2016). For example, Tepolt and Palumbi (2015) provided evidence that local adaptation in the native range of the European green crab (Carcinus maenas) may have facilitated the spread of its invasion through multiple introductions in North America. Invasive species’ adaptations can be better understood with the addition of source population information, which will greatly improve predictions of invasion risk and development of effective management strategies (Lodge et al., 2006; Tepolt, 2014). Future studies should also consider using the cost efficient and advanced genomic SNP data to investigate loci under selection that would allow us to better understand the genetic basis of adaptation and invasion success.
The likely sources of the introduced C. argus in Philadelphia, the Lower Hudson River basin, and Upper Hudson River basin are the Bohu and/or Liangzi lakes, part of the Yangtze River basin in central China. This was supported by our analyses that found significant genetic similarity among the fish of the native Bohu and Liangzi lakes populations and the introduced Philadelphia, Lower and Upper Hudson River basin populations. In contrast, the Potomac River basin and Arkansas populations share less genetic similarity with the Bohu and Liangzi lakes populations, and did not cluster together in any analyses. However, that does not eliminate the possibility of those Chinese lakes being the source of those introductions, as well, they are simply not supported by the current dataset. Channa argus is a freshwater fish, so after introduction, the individuals likely experienced reproductive isolation, which led to genetic divergence, and thus could account for the genetic structure recovered in those two introduced populations.
In its native range in China, C. argus is an important aquaculture species due to its rapid growth rate, strong resistance to disease, and ease of culture in ponds (Yan et al., 2014, 2018). While beneficial in China as an important source of food, these characteristics have likely contributed to the invasion success of C. argus in the United States and elsewhere. Channa argus has a large native range that includes China, Russia and Korea (Courtenay and Williams, 2004). One caveat is that our study only included individuals from three source localities that are all from a similar region in China, and therefore our confidence in assigning true source population identity is low unless a high degree of genetic similarity is observed between Chinese and American fishes. Nonetheless, the results of this study are important because they provide evidence that the Bohu and/or Liangzi lakes are the likely sources of at least some of the C. argus introductions into the United States. In future studies, additional sampling from other sites in the native range beyond central China and the Yangtze River system could help to determine the source(s) of introduction for the other fish in the eastern United States and Arkansas.
Invasive species management is a complex issue that involves ecological, economic, and cultural factors, which often conflict with each other, and thus make decision making difficult (Maguire, 2004). However, the ability to opportunistically obtain samples ideally covering the whole native range and then apply novel genomic methods to compare SNPs from both native and non-native populations, provides a new way for managers to view fine scale population-level data for invasions. Channa argus was imported to the United States because it is a popular valuable food source in China, and it was likely introduced into United States waterways by intentional release. Due to its recognition as a species with considerable potential to cause environmental and economic damage, importation and cross-border transport of live individuals was prohibited in the United States in 2002 when it was listed under the Lacey Act. However, despite this regulation, C. argus is still sold in areas of the United States where its possession is illegal, illustrating its value as a food source, as well as the possibility of continued live transport in the United States (Fuller et al., 2020). Additionally, C. argus is popular for recreational fishing in Meadow Lake, New York, and throughout the Potomac River, and thus contributes to the recreational fishing industry. While C. argus may have economic value because of its popularity as a source of food and recreational fishing interest, it has great potential to spread and become established throughout the United States, which will negatively impact native aquatic communities (Fuller et al., 2020). These competing interests illustrate the importance of the results presented here.
To our knowledge, this is one of the first studies using restriction-site-associated digestion sequencing to identify the source of an invasive introduction. This represents a technological advance beyond traditional genetic barcoding and microsatellite analyses, again using modern genomic methods, for analyzing fine-scale links between populations of species in their native and invaded ranges. These results demonstrate that RAD sequencing is an effective method for identifying the source of invasive introductions. This study also demonstrates the power of RAD methods in comparison to traditional microsatellite or mtDNA investigations to resolve fine scale structure (e.g., Wegleitner et al., 2016). Source population information provides insight into invasion pathways and modes of introduction, and therefore is important for management agencies. It will allow for the development of more targeted strategies to prevent further transport of these fishes to the United States. Additionally, it will enable researchers to begin to determine potential sources for biological control for this, and other, harmful aquatic invasive species.
Data Availability Statement
The data presented in this study are deposited in the NCBI BioSample Database under accession numbers SAMN16703399 through SAMN16703428.
Ethics Statement
Ethical review and approval was not required for the animal study because: we did not collect or use live animals (only museum tissues) for our genomic work. Thus, no IACUC approval was required.
Author Contributions
AM conceived and designed the experiments. JG, K-JW, and R-JY provided tissues from Chinese sampling locations. CR completed the laboratory work and drafted the manuscript. CR and MG completed the data analyses. AM finalized and submitted the manuscript on behalf of all co-authors. All authors edited subsequent drafts and approved prior to submission.
Funding
This study was funded through internal awards to AM from the College of Science and Engineering at Central Michigan University. Additionally, CR was funded (stipend and tuition) through the Earth and Ecosystem Sciences Ph.D. program at Central Michigan University. This publication is partially funded by the Joint Institute for the Study of the Atmosphere and Ocean (JISAO) under NOAA Cooperative Agreement NA15OAR4320063, Contribution No. 2021-1123 and PMEL Contribution No. 5197.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michael Flaherty, Josh Newhard, Joe Love, Melissa Cohen, Allen Temple, Paul Overbeck, Gabriela Hogue, John Odenkirk, and Justin Homan for contributing C. argus tissue samples used in this study. We also thank Tian-Xi Fu and Hai-Ping Chen for assistance with sample collection of the Chinese tissue samples. Additionally, we also thank the Central Michigan University College of Science and Engineering and the Department of Biology for fellowship and assistantship support to CR.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.575599/full#supplementary-material
Footnotes
References
Aquatic Nuisance Species [ANS] Task Force (2014). Report to congress. 38. http://www.anstaskforce.gov (accessed on March 12, 2020).
Austin, C. C., Rittmeyer, E. N., Oliver, L. A., Andermann, J. O., Zug, G. R., Rodda, G. H., et al. (2011). The bioinvasion of Guam: inferring geographic origin, pace, pattern and process of an invasive lizard (Carlia) in the Pacific using multi-locus genomic data. Biol. Invasions 13, 1951–1967. doi: 10.1007/s10530-011-0014-y
Bock, D. G., Caseys, C., Cousens, R. D., Hahn, M. A., Heredia, S. M., Hübner, S., et al. (2015). What we still don’t know about invasion genetics. Mol. Ecol. 24, 2277–2297. doi: 10.1111/mec.13032
Casso, M., Turon, X., and Pascual, M. (2019). Single zooids, multiple loci: independent colonisations revealed by population genomics of a global invader. Biol. Invasions 21, 3575–3592. doi: 10.1007/s10530-019-02069-8
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A., and Cresko, W. A. (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. doi: 10.1111/mec.12354
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W., and Postlethwait, J. H. (2011). Stacks: building and genotyping Loci de novo from short-read sequences. G3 (Bethesda) 1, 171–182. doi: 10.1534/g3.111.000240
Collins, T. M., Trexler, J. C., Nico, L. G., and Rawlings, T. A. (2002). Genetic diversity in a morphologically conservative invasive taxon: multiple introductions of swamp eels to the southeastern United States. Conserv. Biol. 16, 1024–1035. doi: 10.1046/j.1523-1739.2002.01182.x
Courtenay, W. Jr., and Williams, J. D. (2004). Snakeheads (Pisces, Channidae): A Biological Synopsis and Risk Assessment. Washington, DC: United States Department of the Interior.
Cristescu, M. E. (2015). Genetic reconstructions of invasion history. Mol. Ecol. 24, 2212–2225. doi: 10.1111/mec.13117
Dray, S., and Dufour, A. (2007). The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20.
Early, R., Bradley, B. A., Dukes, J. S., Lawler, J. J., Olden, J. D., Blumenthal, D. M., et al. (2016). Global threats from invasive alien species in the twenty-first century and national response capacities. Nat. Commun. 7:12485. doi: 10.1038/ncomms12485
Frichot, E., and François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929. doi: 10.1111/2041-210X.12382
Fuller, P. L., Benson, A. J., Nunez, G., Fusaro, A., and Neilson, M. (2020). Channa Argus (Cantor, 1842). Gainesville, FL: U.S. Geological Survey, Nonindigenous Aquatic Species Database.
Geller, J. B., Darling, J. A., and Carlton, J. T. (2010). Genetic perspectives on marine biological invasions. Ann. Rev. Mar. Sci. 2, 367–393. doi: 10.1146/annurev.marine.010908.163745
Goudet, J. (2005). HIERFSTAT, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186. doi: 10.1111/j.1471-8278
Gregory, T. R. (2021). Animal Genome Size Database. Available online at: http://www.genomesize.com
Hamelin, R. C., and Roe, A. D. (2019). Genomic biosurveillance of forest invasive alien enemies: a story written in code. Evol. App. 13, 95–115.
Harris, A., Page, T. J., Fotedar, S., Duffy, R., and Snow, M. (2016). Molecular identification of the precise geographic origins of an invasive shrimp species in a globally significant Australian biodiversity hotspot. Biol. Invasions 19, 463–468. doi: 10.1007/s10530-016-1311-2
Hierro, J. L., Maron, J. L., and Callaway, R. M. (2005). A biogeographical approach to plant invasions: the importance of studying exotics in their introduced and native range. J. Ecol. 93, 5–15. doi: 10.1111/j.1365-2745.2004.00953.x
Jombart, T., and Ahmed, I. (2011). Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Kairo, M. (2013). Cryptolaemus montrouzieri (Mulsant) (Coccinellidae: Scymninae): a review of biology, ecology, and use in biological control with particular reference to potential impact on non-target organisms. CAB Reviews 8, 1–20. doi: 10.1079/PAVSNNR20138005
Keenan, K., McGinnity, P., Cross, T. F., Crozier, W. W., and Prodöhl, P. A. (2013). Diversity: an Rpackage for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 4, 782–788. doi: 10.1111/2041-210X.12067
Leung, B., Lodge, D. M., Finnoff, D., Shogren, J. F., Lewis, M. A., and Lamberti, G. (2002). An ounce of prevention or a pound of cure: bioeconomic risk analysis of invasive species. Proc. Biol. Sci. 269, 2407–2413. doi: 10.1098/rspb.2002.2179
Li, H. S., Heckel, G., Huang, Y. H., Fan, W. J., Ślipiński, A., and Pang, H. (2019). Genomic changes in the biological control agent Cryptolaemus montrouzieri associated with introduction. Evol. Appl. 12, 989–1000. doi: 10.1111/eva.12774
Lodge, D. M., Williams, S., MacIsaac, H. J., Hayes, K. R., Leung, B., Reichard, S., et al. (2006). Biological invasions: recommendations for U.S. policy and management. Ecol. Appl. 16, 2035–2054.
Maguire, L. A. (2004). What can decision analysis do for invasive species management? Risk Anal. 24, 859–868. doi: 10.1111/j.0272-4332.2004.00484.x
Martin, M. D., Olsen, M. T., Samaniego, J. A., Zimmer, E. A., and Gilbert, M. T. P. (2016). The population genomic basis of geographic differentiation in North American common ragweed (Ambrosia artemisiifolia L.). Ecol. Evol. 6, 3760–3771. doi: 10.1002/ece3.2143
Nuñez, M. A., and Pauchard, A. (2009). Biological invasions in developing and developed countries: does one model fit all? Biol. Invasions 12, 707–714. doi: 10.1007/s10530-009-9517-1
Orrell, T. M., Weigt, L., and Murdy, E. O. (2005). The northern snakehead Channa argus (Anabantomorpha: Channidae), a non-indigenous fish species in the Potomac River. USA. Proc. Biol. Soc. Wash. 118, 407–415.
Paterson, I. D., Mangan, R., Downie, D. A., Coetzee, J. A., Hill, M. P., Burke, A. M., et al. (2016). Two in one: cryptic species discovered in biological control agent populations using molecular data and crossbreeding experiments. Ecol. Evol. 6, 6139–6150. doi: 10.1002/ece3.2297
Pimentel, D. (2005). Aquatic nuisance species in the New York State Canal and Hudson River systems and the Great Lakes Basin: an economic and environmental assessment. Environ. Manage. 35, 692–702. doi: 10.1007/s00267-004-0214-7
R Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Project for Statistical Computing.
Resh, C. A., Galaska, M. P., and Mahon, A. R. (2018). Genomic analyses of Northern snakehead (Channa argus) populations in North America. PeerJ 6:e4581. doi: 10.7717/peerj.4581
Smith, L., Cristofaro, M., Bon, M.-C., De Biase, A., Petanoviæ, R., and Vidoviæ, B. (2017). The importance of cryptic species and subspecific populations in classic biological control of weeds: a North American perspective. BioControl 63, 417–425. doi: 10.1007/s10526-017-9859-z
Tepolt, C. K. (2014). Adaptation in marine invasion: a genetic perspective. Biol. Invasions 17, 887–903. doi: 10.1007/s10530-014-0825-8
Tepolt, C. K., and Palumbi, S. R. (2015). Transcriptome sequencing reveals both neutral and adaptive genome dynamics in a marine invader. Mol. Ecol. 24, 4145–4158. doi: 10.1111/mec.13294
Wang, S., Meyer, E., McKay, J. K., and Matz, M. V. (2012). 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat. Methods 9, 808–810. doi: 10.1038/nmeth.2023
Wegleitner, B., Tucker, A., Chadderton, W. L., and Mahon, A. R. (2016). Identifying the genetic structure of introduced populations of northern snakehead (Channa argus) in Eastern USA. Aquat. Invasions 11, 199–208. doi: 10.3391/ai.2016.11.2.09
Yan, R.-J., Wei, K.-J., Guo, X.-Z., Zhang, G.-R., Gardner, J. P. A., Yang, R.-B., et al. (2014). Isolation and characterization of nineteen novel polymorphic microsatellite loci for the northern snakehead Channa argus. Conserv. Genet. Resour. 6, 621–623. doi: 10.1007/s12686-014-0155-2
Yan, R.-J., Zhang, G.-R., Guo, X.-Z., Ji, W., Chen, K.-C., Zou, G.-W., et al. (2018). Genetic diversity and population structure of the northern snakehead (Channa argus Channidae: Teleostei) in central China: implications for conservation and management. Conserv. Genet. 19, 467–480. doi: 10.1007/s10592-017-1023-x
Keywords: population genomics, invasive species, RADseq, molecular ecology, Northern snakehead (Channa argus)
Citation: Resh CA, Galaska MP, Benesh KC, Gardner JPA, Wei K-J, Yan R-J and Mahon AR (2021) Using Genomics to Link Populations of an Invasive Species to Its Potential Sources. Front. Ecol. Evol. 9:575599. doi: 10.3389/fevo.2021.575599
Received: 23 June 2020; Accepted: 18 January 2021;
Published: 05 February 2021.
Edited by:
Ana Novoa, Institute of Botany (ASCR), CzechiaReviewed by:
Giacomo Bernardi, University of California, Santa Cruz, United StatesAna Filipa Filipe, Centro de Investigacao em Biodiversidade e Recursos Geneticos (CIBIO-InBIO), Portugal
Copyright © 2021 Resh, Galaska, Benesh, Gardner, Wei, Yan and Mahon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew R. Mahon, bWFob24yYUBjbWljaC5lZHU=
†Present address: Ruo-Jin Yan, College of Energy, Soochow University, Suzhou, China