 Carolina Osuna-Mascaró
Carolina Osuna-Mascaró Rafael Rubio de Casas
Rafael Rubio de Casas Jacob B. Landis
Jacob B. Landis Francisco Perfectti
Francisco Perfectti- 1Departamento de Genética, Universidad de Granada, Granada, Spain
- 2Research Unit Modeling Nature, Universidad de Granada, Granada, Spain
- 3Departamento de Ecología, Universidad de Granada, Granada, Spain
- 4Department of Botany and Plant Sciences, University of California, Riverside, Riverside, CA, United States
- 5Section of Plant Biology and the L.H. Bailey Hortorium, School of Integrative Plant Science, Cornell University, Ithaca, NY, United States
Background and Summary
Erysimum (Brassicaceae) is a genus of more than 200 species (Al-Shehbaz, 2012). It is widely distributed in the Northern Hemisphere and has been the focus of active research in ecology, evolution, and genetics (Gómez and Perfectti, 2010; Gómez, 2012; Valverde et al., 2016). Despite long-standing interest in Erysimum, its taxonomy has yet to be properly established, partly due to a complex and reticulated evolutionary history that renders phylogenetic reconstructions highly challenging (Ancev, 2006; Marhold and Lihová, 2006; Abdelaziz et al., 2014; Gomez et al., 2014; Moazzeni et al., 2014; Züst et al., 2020).
The Baetic Mountains (South-Eastern Iberia) are among the most critical glacial refugia in Europe. The waxing and waning of plant populations following climatic fluctuations have likely complicated the distribution and genetic variation of extant diversity in this region. Isolation and posterior secondary contact between taxa may have favored hybridization and introgression (Médail and Diadema, 2009). The Erysimum species that inhabit these mountains have been a particularly fruitful system for plant evolutionary ecology [e.g., Gómez et al., 2006, 2008; Gómez and Perfectti, 2010; Gómez, 2012; Valverde et al., 2016]. However, the relationships among these species remain unresolved, hampering comparative and evolutionary studies. Genome duplications, incomplete lineage sorting, and hybridization have compromised the phylogenetic reconstructions within Erysimum (Marhold and Lihová, 2006; Osuna-Mascaró, 2020). Additionally, clarifying this group's complex evolution requires extensive genomic resources, which are currently being produced but are mostly lacking.
The fast development of high-throughput sequencing technologies has led to a rapid increase in genomic and transcriptomic for many plant species (Dong et al., 2004; Duvick et al., 2007; Sundell et al., 2015; Boyles et al., 2019). However, obtaining complete genome sequencing remains a challenge with large, repetitive-DNA enriched genomes. Transcriptome sequencing is comparatively more accessible, providing a relatively cheap and fast method to obtain large amounts of functional genomic data (Timme et al., 2012; Yang and Smith, 2013; Wickett et al., 2014; Léveillé-Bourret et al., 2017). Accordingly, global initiatives such as the 1,000 plants (1KP) project have generated transcriptomic resources for over 1,000 plant species (Matasci et al., 2014; Leebens-Mack et al., 2019). In addition, the use of RNA-Seq could be useful in obtaining complete chloroplast genomes in a reliable and accessible way, making possible the use of complete molecules in phylogenomic analyses (Smith, 2013; Osuna-Mascaró et al., 2018; Morales-Briones et al., 2021).
Here, we report the annotation of 18 floral transcriptomes assembled de novo from total RNA-Seq libraries and nine chloroplast genomes from seven Erysimum species inhabiting the Baetic Mountains. The chloroplast genomes were assembled from total RNA-Seq data following a previously-validated reference assemble approach (Osuna-Mascaró et al., 2018). The data presented here represent reliable genomic resources for transcriptomic, proteomic, and phylotranscriptomic studies. These data contribute to the ecological and genetic resources available for Brassicaceae in general and the genus Erysimum in particular, being the only genomic resources for these species coming from flower buds.
Methods
Generation of the Datasets
We sampled flower buds at the same development stage (completely developed non-open buds) from three different populations of Erysimum mediohispanicum, E. nevadense, E. popovii, and E. baeticum, four populations of E. bastetanum, and one population of E. lagascae, and E. fitzii (see Supplementary Table 1 for details). We stored the samples in liquid nitrogen and maintained them in an ultra-freezer (−80°C) until RNA extraction. Then, we extracted RNA from the buds under highly sterile conditions. The buds were snap-frozen in liquid nitrogen and ground with mortar and pestle. We used the Qiagen RNeasy Plant Mini Kit, following the manufacturer's protocol, to extract total RNA and their quality and quantity were checked using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States) and with an Agilent 2100 Bioanalyzer system (Agilent Technologies Inc). Library preparation and RNA sequencing were conducted by Macrogen Inc. (Seoul, Korea). We used rRNA-depletion (Ribo-Zero) for mRNA enrichment and to avoid sequencing rRNAs. Library preparation was performed using the TruSeq Stranded Total RNA LT Sample Preparation Kit (Plant). The sequencing of the 18 libraries was carried out using the Hiseq 3000-4000 sequencing protocol and TruSeq 3000-4000 SBS Kit v3 reagent, following a paired-end 150 bp strategy on the Illumina HiSeq 4000 platform. A summary of sequencing statistics appears in Supplementary Table 2.
Data Processing and Transcriptome Analyses
We analyzed the fastq files for each library using FastQC v 0.11.5 (Andrews, 2010). Then, we trimmed the adapters using cutadapt v 1.1540 (Martin, 2011), specifying the “-b” option for trimming the adapters in 5′ and 3′ and the “-n” option to search repeatedly for the adapter sequences (28 iterations). This option ensures that the correct adapters were detected by searching in loops until any adapter match is found or until the specified number of rounds is reached. Following, we trimmed the reads by quality using Sickle v 1.3341 (Joshi and Fass, 2011), using the “pe” option for paired-end reads and the “-t” to use Illumina quality values (see https://github.com/najoshi/sickle). This trimming software uses sliding-window analyses and quality and length thresholds to cut and discard the reads that do not fit the selected threshold values. We specified the “pe” option for paired-end reads and the “-t” to use Illumina quality values, setting a threshold quality value of Q20 (see https://github.com/najoshi/sickle). After trimming, we used FastQC (Andrews, 2010) again to verify the trimming efficiency. The summary of the number of reads after the quality trimming is represented in Supplementary Table 3.
To assemble contigs from the resulting high-quality cleaned reads, we followed a de novo approach using Trinity v 2.8.4 (Grabherr et al., 2011), due to the absence of an available published assembled genome for Erysimum at the time of the analyses. Each library was normalized in silico before assembly to validate and reduce the number of reads using the “insilico_read_normalization.pl” function in Trinity (Haas et al., 2013). Then we used the parameter “min_kmer_cov 2” to eliminate single-occurrence k-mers that are heavily enriched in sequencing errors, following Haas et al. (2013). Thus, only k-mers that occur more than once were considered for contigs. Candidate open reading frames (ORF) within transcript sequences were predicted and translated using TransDecoder v 5.2.0 (Haas et al., 2013). We performed functional annotation of Trinity transcripts with ORFs using Trinotate v 3.0.1 (Haas, 2015), an annotation suite designed for automatic functional annotation of de novo assembled transcriptomes. Sequences were searched against UniProt (UniProt Consortium, 2014), using SwissProt databases (Bairoch and Apweiler, 2000) (with BLASTX and BLASTP searching and an e-value cutoff of 10). We then used the Pfam database (Bateman et al., 2004) to annotate protein domains for each predicted protein sequence. We also annotated the transcripts using the databases eggnog (Jensen et al., 2007), GO (Gene Ontology Consortium, 2004), and Kegg (Kanehisa and Goto, 2000). We obtained between 104K and 382K different Trinity transcripts after assembling, producing between 66K and 235K Trinity isogenes. The total assembled bases ranged from 92 Mbp (in Em21 population of E. mediohispanicum) to 319 Mbp (in En10 population of E. nevadense). The summary statistics of the assembled transcriptomes appear in Supplementary Table 4. Among the annotated unigenes, the highest proportion was annotated using BLASTX search against the SwissProt reference database, and the number of genes annotated ranges between 71,606 (E. nevadense, En12) and 197,069 (E. baeticum, Ebb10); mean value 146,314.35. The unigenes from the assembled sequences using different databases are shown in Supplementary Table 5.
Lastly, we used BUSCO v 2.0 (Seppey et al., 2019) to validate the quality of all the assemblies, using the plant database brassicales_odb10.2019-11-20. Overall, a high level of single-copy orthologous retrieval was noted for the 18 assemblies, as shown in Figure 1. Specifically, we found a completeness ratio ranging from 72.29 to 85.62% for E. popovii, from 73.06 to 83.26% for E. nevadense, from 71.60 to 84.65% to E. mediohispanicum, from 63. 12 to 86.74% for E. bastetanum, from 79.31 to 72.5% for E. baeticum, a completeness ratio of 86.46% for E. lagascae, and 75.07% for E. fitzii. The least complete case was for E. bastetanum, Ebt13, exhibiting a 63.12% ratio, with 8% missing orthologs and a 28.88% partial completeness.
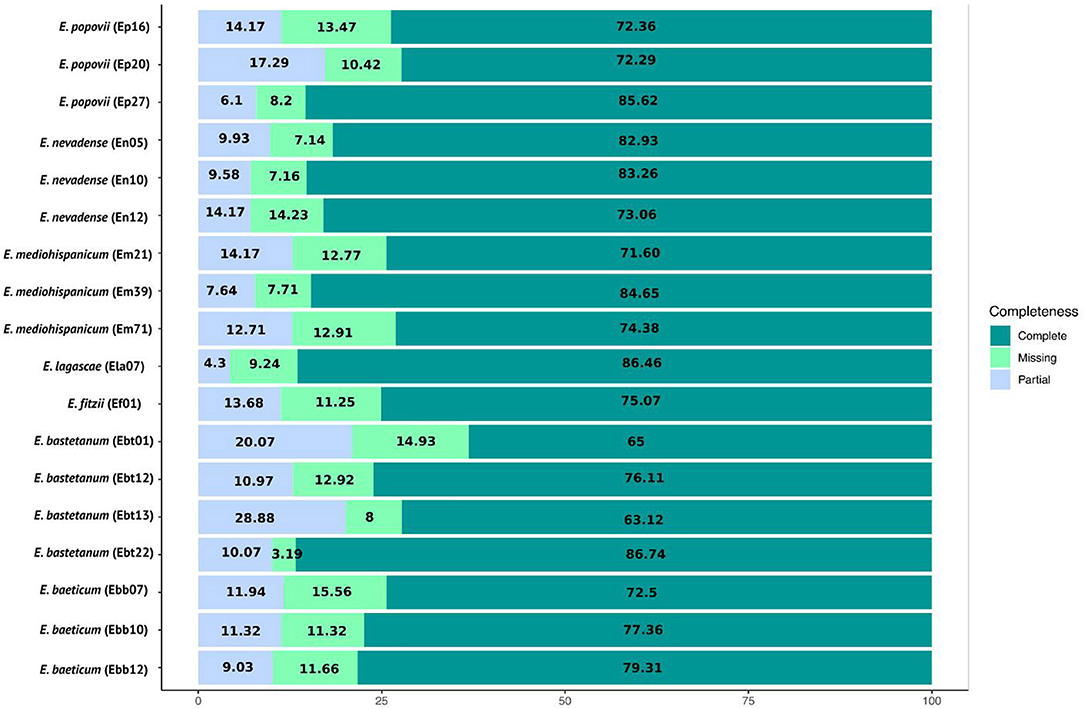
Figure 1. BUSCO assessment results for the 18 assembled transcriptomes.
The transcriptome annotations, the set of assembled unigenes and their annotations, and the predicted amino acid sequences can be found in Data citation 2, Data citation 3, and Data citation 4, respectively.
Chloroplast Genome Assembly and Annotation
We assembled the chloroplast genome from nine Erysimum RNA-Seq libraries. Specifically, we assembled E. bastetanum (Ebt01, Ebt10, Ebt12, Ebt22), E. fitzii (Ef), E. lagascae (Ela07), and E. popovii (Ep16, Ep20, Ep27). Our team (Osuna-Mascaró et al., 2018) previously assembled the remaining chloroplast genomes (the ones corresponding to E. baeticum, E. mediohispanicum, and E. nevadense RNA-Seq libraries). Here, we used a reference assembly approach, using Geneious R.11 (Kearse et al., 2012) with the A. thaliana chloroplast genome as reference (NC_000932.1 (Sato et al., 1999)). This method has been previously validated with the chloroplast genomes of E. mediohispanicum, E. nevadense, and E. baeticum (Osuna-Mascaró et al., 2018). We annotated the chloroplast genomes using cpGAVAS (Liu et al., 2012). The annotations were manually curated using Geneious R.11 (Kearse et al., 2012). All transfer RNA sequences (tRNA) encoded in the chloroplast genomes were verified using tRNAscan-SE 2.0 (Schattner et al., 2005) and ARAGORN v1.2.38 (Laslett and Canback, 2004) with the default search settings. The annotation summary is presented in Supplementary Table 6. We obtained almost complete chloroplast genomes, with some missing genes detected (Supplementary Table 7). Most of the missing genes were related to photosynthesis such as the group of subunits of NADH-dehydrogenase (e.g., ndhA), genes with conserved open reading frame (e.g., ycf1, and ycf5), or genes related with subunit od Acetyl-CoA carboxylase (aacD), or the elongation factor (tuf).
The assemblies of chloroplast genomes and their annotations are shown in Data citation 5. Chloroplast genome resources including trn's, rrn's, mrn's, genes, tRNA validation results, and annotation report files are shown in Data citation 6. We uploaded the nine chloroplast genome sequences to GenBank with accession numbers showed in Data citation 7.
Time-Calibrated Phylogeny Reconstruction
We aligned the chloroplast sequences using MAFFT v.7 with default parameters (Katoh and Standley, 2013). Then, we reconstructed a time-calibrated phylogeny using Beast 2.0 (Bouckaert et al., 2014), with Arabidopsis thaliana chloroplast genome sequence (NC_000932.1 (Sato et al., 1999)) as an outgroup. We made three different partitions: one for coding regions, one for non-coding regions, and the last for the third positions of the coding regions, for which substitutions are synonymous. We calibrated using an average mutation rate reported for synonymous sites of chloroplast genes of seed plants (1.2–1.7 × 109 substitutions/site/year) (Graur and Li, 2000) applying it for the partition of the third position region. In addition, we included a timed-calibration obtained from the literature. In Moazzeni et al. (2014) the divergence of Western European Erysimum species was estimated in the middle Pleistocene (2.43 – 0.74 Mya, using a fast substitution rate; or 8.48 – 2.15 Mya, using a slow substitution rate). Here, we used the average of these dating intervals (2.43 and 2.15 Mya) as a calibration point (2.29 Mya). The Bayesian search for tree topologies and node ages was conducted during 20,000,000 generations in BEAST using a strict clock model and a Yule process as prior. MCMC was sampled every 1,000 generations, discarding a burn-in of 10%. We checked the MCMC trace files generated using Tracer v1.6.1 (Rambaut et al., 2014). The time-calibrated phylogeny is shown in Figure 2.
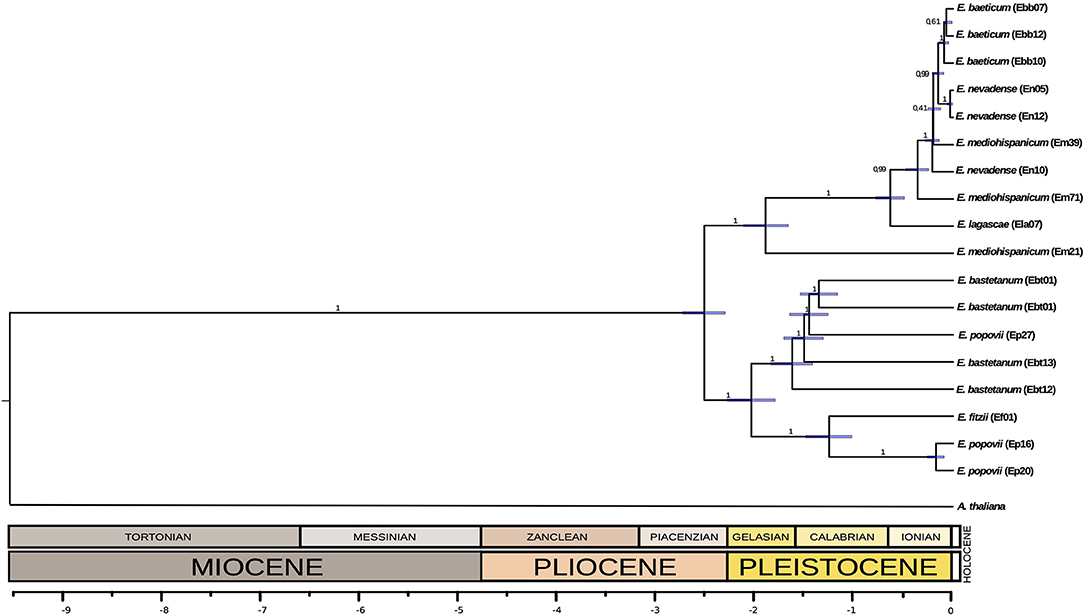
Figure 2. A time-calibrated phylogeny for the chloroplast DNA of the different populations of the Erysimum species analyzed here. Note the reticulated position of some populations, probably due to hybridization events.
Data Records
The raw sequence read data for all the transcriptomes were deposited in the NCBI Sequence Read Archive (Data citation 1).
Furthermore, for the free download of the generated data, we have created a project on figshare containing: the assembled transcriptomes (Data citation 2), the transcriptome annotations (Data citation 3), the set of assembled unigenes, their annotations, and the predicted amino acid sequences (Data citation 4), the chloroplast genomes assemblies and their annotations (Data citation 5), and chloroplast genomic resources including trn's, rrn's, mrn's, genes, trna validation results, and annotation report files (Data citation 6). The chloroplast genome sequences were deposited in GenBank. The accession numbers can be found in Data citation 7.
Data Citations
Data citation 1: NCBI Sequence Read Archive, BioProject PRJNA607615 under the following accession numbers: E. popovii: Ep27 (SRX7756239), Ep20 (SRX7756238), Ep16 (SRX7756237); E. lagascae: Ela07 (SRX7756236); E. fitzii: Ef01 (SRX7756235); E. bastetanum: Ebt22 (SRX7756234), Ebt13 (SRX7756233), Ebt12 (SRX7756232), Ebt01 (SRX7756231), and BioProject PRJNA473238 under the following accession numbers: E. baeticum: Ebb12 (SRX4130243), Ebb10 (SRX4130242), Ebb07 (SRX4130235); E. mediohispanicum: Em39 (SRX4130241), Em71 (SRX4130240), Em21 (SRX4130233); E. nevadense: En12 (SRX4130237), En10 (SRX4130236), En05 (SRX4130234).
Data citation 2: Osuna-Mascaró, C., de Casas, R. R., Landis, J.B., & Perfectti, F., figshare https://doi.org/10.6084/m9.figshare.11877786.v3 (2020).
Data citation 3: Osuna-Mascaró, C., de Casas, R. R., Landis, J.B., & Perfectti, F., figshare https://doi.org/10.6084/m9.figshare.11866389.v3 (2020).
Data citation 4: Osuna-Mascaró, C., de Casas, R. R., Landis, J.B., & Perfectti, F., figshare https://doi.org/10.6084/m9.figshare.11873937.v1 (2020).
Data citation 5: Osuna-Mascaró, C., de Casas, R. R., Landis, J.B., & Perfectti, F., figshare https://doi.org/10.6084/m9.figshare.11881656.v2 (2020).
Data citation 6: Osuna-Mascaró, C., de Casas, R. R., Landis, J.B., & Perfectti, F., figshare https://doi.org/10.6084/m9.figshare.11881419.v2 (2020).
Data citation 7: Chloroplast genome sequences deposited in GenBank under the following accession numbers:
E. bastetanum: Ebt01 (MT150122), Ebt12 (MT150121), Ebt13 (MT150114), Ebt22 (MT150115); E. fitzii: Ef01 (MT150118); E. lagascae: Ela07 (MT150116); E. popovii: Ep16 (MT150117), Ep20 (MT150119), Ep27 (MT150120).
Usage Notes
Erysimum is a genus for which phylogenetic relationships have not yet been fully established. Therefore, the primary use of this dataset will likely lie in molecular evolution analyses aimed at disentangling the taxonomy and biogeography of these and related species. Moreover, since the primary data are transcriptomes, they could be useful in plant evo-devo and physiological studies. They can also be incorporated into comparative studies aimed at identifying the differential expression of the genes expressed in the tissues sequenced in this work (i.e., flower buds). Although we expect our dataset to be essentially free of contamination, caution is advised when using this Data Report as we did not filter the reads for the presence of alien (i.e., bacterial or fungi) sequences.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
COM, RR, and FP conceived and designed the study. COM analyzed the data, with the help of FP and JL and wrote the first draft. The final version of the M.S. was redacted with the contribution of all the authors.
Funding
Funding was provided by the Spanish Ministry of Science and Competitiveness (CGL2016- 79950-R; CGL2017-86626-C2-2-P), including FEDER funds. This research was also funded by the Consejería de Economía, Conocimiento, Empresas y Universidad, and European Regional Development Fund (ERDF), ref. SOMM17/ 6109/UGR and A-RNM-505-UGR18. COM was supported by the Ministry of Economy and Competitiveness (BES-2014-069022).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Modesto Berbel Cascales and José M. Gómez for their help in sampling and DNA/RNA extractions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.620601/full#supplementary-material
References
Abdelaziz, M., Munoz-Pajares, A. J., Lorite, J., Herrador, M. B., Perfectti, F., and Gómez, J. M. (2014). Phylogenetic relationships of “Erysimum” (Brassicaceae) from the Baetic Mountains (SE Iberian Peninsula). Anales del Jardín Botánico de Madrid 71:5. doi: 10.3989/ajbm.2377
Al-Shehbaz, I. A. (2012). A generic and tribal synopsis of the Brassicaceae (Cruciferae). Taxon 61, 931–954. doi: 10.1002/tax.615002
Ancev, M. (2006). Polyploidy and hybridization in Bulgarian Brassicaceae: distribution and evolutionary role. Phytol. Balcanica 12, 357–366.
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed September 26, 2020).
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucl. Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucl. Acids Res. 32(suppl_1), D138–D141. doi: 10.1093/nar/gkh121
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Boyles, R. E., Brenton, Z. W., and Kresovich, S. (2019). Genetic and genomic resources of sorghum to connect genotype with phenotype in contrasting environments. Plant J. 97, 19–39. doi: 10.1111/tpj.14113
Dong, Q., Schlueter, S. D., and Brendel, V. (2004). PlantGDB, plant genome database and analysis tools. Nucl. Acids Res. 32(suppl_1), D354–D359. doi: 10.1093/nar/gkh046
Duvick, J., Fu, A., Muppirala, U., Sabharwal, M., Wilkerson, M. D., Lawrence, C. J., et al. (2007). PlantGDB: a resource for comparative plant genomics. Nucl. Acids Res. 36(suppl_1), D959–D965. doi: 10.1093/nar/gkm1041
Gene Ontology Consortium (2004). The Gene Ontology (GO) database and informatics resource. Nucl. Acids Res. 32(suppl_1), D258–D261. doi: 10.1093/nar/gkh036
Gómez, J. M. (2012). Herbivory reduces the strength of pollinator-mediated selection in the Mediterranean herb Erysimum mediohispanicum: consequences for plant specialization. Am. Naturalist 162, 242–256. doi: 10.1086/376574
Gómez, J. M., Bosch, J., Perfectti, F., Fernández, J. D., Abdelaziz, M., and Camacho, J. P. M. (2008). Association between floral traits and rewards in Erysimum mediohispanicum (Brassicaceae). Ann. Botany 101, 1413–1420. doi: 10.1093/aob/mcn053
Gomez, J. M., Munoz-Pajares, A. J., Abdelaziz, M., Lorite, J., and Perfectti, F. (2014). Evolution of pollination niches and floral divergence in the generalist plant Erysimum mediohispanicum. Ann. Botany 113, 237–249. doi: 10.1093/aob/mct186
Gómez, J. M., and Perfectti, F. (2010). Evolution of complex traits: the case of Erysimum corolla shape. Int. J. Plant Sci. 171, 987–998. doi: 10.1086/656475
Gómez, J. M., Perfectti, F., and Camacho, J. P. M. (2006). Natural selection on Erysimum mediohispanicum flower shape: insights into the evolution of zygomorphy. Am. Naturalist 168, 531–545. doi: 10.1086/507048
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Graur, D., and Li, W. H. (2000). Fundamentals of molecular evolution. Dynamics 20, 160–229. doi: 10.1017/s0016672300030032
Haas, B. J. (2015). Trinotate: Transcriptome Functional Annotation and Analysis. Available online at: http://trinotate.github.io (accessed September 26, 2020).
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protocols 8, 1494–1512. doi: 10.1038/nprot.2013.084
Jensen, L. J., Julien, P., Kuhn, M., von Mering, C., Muller, J., Doerks, T., et al. (2007). eggNOG: automated construction and annotation of orthologous groups of genes. Nucl. Acids Res. 36(suppl_1), D250–D254. doi: 10.1093/nar/gkm796
Joshi, N. A., and Fass, J. N. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. Available online at: https://github.com/najoshi/sickle (accessed September 26, 2020).
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucl. Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Leebens-Mack, J. H., Barker, M. S., Carpenter, E. J., Deyholos, M. K., Gitzendanner, M. A., Graham, S. W., et al. (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Léveillé-Bourret, É., Starr, J. R., Ford, B. A., Moriarty Lemmon, E., and Lemmon, A. R. (2017). Resolving rapid radiations within angiosperm families using anchored phylogenomics. Systematic Biol. 67, 94–112. doi: 10.1093/sysbio/syx050
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 13, 1–7. doi: 10.1186/1471-2164-13-715
Marhold, K., and Lihová, J. (2006). Polyploidy, hybridization and reticulate evolution: lessons from the Brassicaceae. Plant Systematics Evol. 259, 143–174. doi: 10.1007/s00606-006-0417-x
Martin, M. (2011). Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Matasci, N., Hung, L. H., Yan, Z., Carpenter, E. J., Wickett, N. J., Mirarab, S., et al. (2014). Data access for the 1,000 Plants (1KP) project. Gigascience 3, 2047–217X. doi: 10.1186/2047-217X-3-17
Médail, F., and Diadema, K. (2009). Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 36, 1333–1345. doi: 10.1111/j.1365-2699.2008.02051.x
Moazzeni, H., Zarre, S., Pfeil, B. E., Bertrand, Y. J., German, D. A., Al-Shehbaz, I. A., et al. (2014). Phylogenetic perspectives on diversification and character evolution in the species-rich genus Erysimum (Erysimeae; Brassicaceae) based on a densely sampled ITS approach. Botanical J. Linnean Soc. 175, 497–522. doi: 10.1111/boj.12184
Morales-Briones, D. F., Kadereit, G., Tefarikis, D. T., Moore, M. J., Smith, S. A., Brockington, S. F., et al. (2021). Disentangling sources of gene tree discordance in phylogenomic data sets: testing ancient hybridizations in amaranthaceae sl. Systematic Biol. 70, 219–235. doi: 10.1093/sysbio/syaa066
Osuna-Mascaró, C. (2020). Hybridization as an Evolutionary Driver for Speciation: A Case in the Southern European Erysimum species. (Ph.D. Thesis), Universidad de Granada, Granada, Spain.
Osuna-Mascaró, C., de Casas, R. R., and Perfectti, F. (2018). Comparative assessment shows the reliability of chloroplast genome assembly using RNA-seq. Sci. Rep. 8:17404. doi: 10.1038/s41598-018-35654-3
Rambaut, A., Suchard, M. A., Xie, D., and Drummond, A. J. (2014). Tracer v1. 6. Avaialble online at: http://beast.community/tracer.html (accessed September 26, 2020).
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., and Tabata, S. (1999). Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290. doi: 10.1093/dnares/6.5.283
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucl. Acids Res. 33(suppl_2), W686–W689. doi: 10.1093/nar/gki366
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). “BUSCO: assessing genome assembly and annotation completeness,” in Gene Prediction (New York, NY: Humana), 227–245. doi: 10.1007/978-1-4939-9173-0_14
Smith, D. R. (2013). RNA-Seq data: a goldmine for organelle research. Briefings Funct. Genom. 12, 454–456. doi: 10.1093/bfgp/els066
Sundell, D., Mannapperuma, C., Netotea, S., Delhomme, N., Lin, Y. C., Sjödin, A., et al. (2015). The Plant genome integrative explorer resource: PlantGen IE.org. New Phytol. 208, 1149–1156. doi: 10.1111/nph.13557
Timme, R. E., Bachvaroff, T. R., and Delwiche, C. F. (2012). Broad phylogenomic sampling and the sister lineage of land plants. PLoS ONE 7:e29696. doi: 10.1371/journal.pone.0029696
UniProt Consortium. (2014). UniProt: a hub for protein information. Nucl. Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Valverde, J., Gómez, J. M., and Perfectti, F. (2016). The temporal dimension in individual-based plant pollination networks. Oikos 125, 468–479. doi: 10.1111/oik.02661
Wickett, N. J., Mirarab, S., Nguyen, N., Warnow, T., Carpenter, E., Matasci, N., et al. (2014). Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. 111, E4859–E4868. doi: 10.1073/pnas.1323926111
Yang, Y., and Smith, S. A. (2013). Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genom. 14:328. doi: 10.1186/1471-2164-14-328
Keywords: transcriptomes, de novo assembly, phylotranscriptomic, Brassicaceae, chloroplast, time calibrated phylogeny
Citation: Osuna-Mascaró C, Rubio de Casas R, Landis JB and Perfectti F (2021) Genomic Resources for Erysimum spp. (Brassicaceae): Transcriptome and Chloroplast Genomes. Front. Ecol. Evol. 9:620601. doi: 10.3389/fevo.2021.620601
Received: 23 October 2020; Accepted: 18 March 2021;
Published: 13 April 2021.
Edited by:
Diego F. Morales-Briones, University of Minnesota Twin Cities, United StatesReviewed by:
Tatiana Arias, Tecnológico de Antioquia, ColombiaLiming Cai, University of California, Riverside, United States
Copyright © 2021 Osuna-Mascaró, Rubio de Casas, Landis and Perfectti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Perfectti, ZnBlcmZlY3RAdWdyLmVz; Carolina Osuna-Mascaró, Y2lvbUB1Z3IuZXM=