 Jared A. Grummer
Jared A. Grummer Luciano J. Avila2
Luciano J. Avila2 Mariana M. Morando
Mariana M. Morando Adam D. Leaché
Adam D. Leaché- 1Department of Biology, Burke Museum of Natural History and Culture, University of Washington, Seattle, WA, United States
- 2Centro Nacional Patagónico–Consejo Nacional de Investigaciones Científicas y Técnicas, Puerto Madryn, Argentina
Hybridization is an evolutionary process that can generate diverse outcomes, such as reinforcing species boundaries, generating new species, or facilitating the introgression of locally-adapted alleles into new genomic backgrounds. Liolaemus is a highly diverse clade of South American lizards with ~260 species and as many as ten new species are described each year. Previous Liolaemus studies have detected gene flow and introgression among species using phylogenetic network methods and/or through comparisons of nuclear and mitochondrial DNA patterns, yet no study has systematically studied hybrid zones between Liolaemus species. Here, we compared three hybrid zones between four species in the Liolaemus fitzingerii group of lizards in Central Argentina where two species, L. melanops and L. xanthoviridis, each hybridize with two other species (L. shehuen and L. fitzingerii). We sampled three transects that were each ~120 km in length and sequenced both mitochondrial and genome-wide SNP data for 267 individuals. In our analyses of nuclear DNA, we also compared bi-allelic SNPs to phased alleles (50 bp RAD loci). Population structure analyses confirmed that boundaries separating species are sharp, and all clines are <65 km wide. Cline center estimates were consistent between SNPs and phased alleles, but cline width estimates were significantly different with the SNPs producing wider estimates. The mitochondrial clines are narrower and shifted 4–20 km southward in comparison to the nuclear clines in all three hybrid zones, indicating that either each of the species has sex-biased dispersal (males northward or females southward), the population densities are unequal, or that the hybrid zones are moving north over time. These comparisons indicate that some patterns of hybridization are similar across hybrid zones (mtDNA clines all narrower and shifted to the south), whereas cline width is variable. Hybridization in the L. fitzingerii group is common and geographically localized; further studies are needed to investigate whether hybrid zones act as hard species boundaries or promoters of speciation through processes such as reinforcement. Nonetheless, this study provides insights into both biotic and abiotic mechanisms helping to maintain species boundaries within the speciose Liolaemus system.
1. Introduction
Hybridization, or interbreeding between distinct populations, has captivated evolutionary biologists for nearly two centuries (Darwin, 1862; Harrison, 1993). It can be a means of transferring adaptive genetic diversity between lineages (Chhatre et al., 2018; Hanemaaijer et al., 2018), forming hybrid swarms and potentially collapsing lineages (Pritchard and Edmands, 2013), or conversely, creating new species through hybrid speciation (e.g., Rieseberg et al., 1995). Hybridization is indeed common across the tree of life, with documentation in 10% of animal species and 25% of plant species (Mallet, 2005). Within Tetrapods, hybridization is particularly common in squamate reptiles—the lizards and snakes (Jančúchová-Lásková et al., 2015). Given the diverse roles that hybridization can play in shaping patterns of diversity, it is important to deepen our understanding of this process in natural systems.
Hybrid zones can provide detailed information about the evolutionary and ecological interactions between species, and replicated hybrid zones offer the additional advantage of investigating the repeatability of evolutionary processes (McKinnon and Rundle, 2002). Replicate transects across a single hybrid zone can offer insights into the extrinsic and intrinsic factors that govern the dynamics of a hybrid zone (Brelsford and Irwin, 2009; Zieliński et al., 2019; Westram et al., 2021, e.g.,). Replicate hybrid zones have mainly been studied in fish, which typically show a high amount of variability of introgression rates and genomic divergence between different hybrid zones. In Xiphophorus swordtail fish, Culumber et al. (2011) found that linkage disequilibrium and Hardy-Weinberg equilibrium estimates varied substantially across seven transects. Nolte et al. (2009) also found little correlation in genomic differentiation between two hybrid zones of sculpin fish (Cottus). And similarly, hybridization rates were found to vary considerably across ten topminnow (Fundulus) replicate hybrid zones (Duvernell and Schaefer, 2014). These differences identified across replicate hybrid zones are typically ascribed to distinct environments that characterize each hybrid zone (Aboim et al., 2010). In this study, we investigate replicate hybrid zones in a species group of liolaemid lizards. Here, we use the term “replicate” not in the statistical sense, but to indicate that one species hybridizes with more than one other distinct species and thus represents “evolutionary replicates” given that the process of hybridization has occurred multiple times in distinct geographic areas.
The genus Liolaemus (family Liolaemidae) is a particularly diverse clade with ~260 species and 5–10 new species described each year (http://www.reptile-database.org/). Some authors have recently posited that hybridization may be one of the factors responsible for generating the exceptional diversity within this clade, particularly when compared to its sister clade Phymaturus that only has 48 species (Olave et al., 2018, 2020; Morando et al., 2020). Indeed, several studies have detected or suggested hybridization in disparate Liolaemus groups including the lineomaculatus series (Breitman et al., 2011) and leopardinus clade (Esquerré et al., 2019), the darwinii, kriegi, and petrophilus complexes (Morando et al., 2004; Feltrin, 2013; Medina et al., 2014), and the chiliensis and fitzingerii groups (Avila et al., 2006; Grummer et al., 2018; Araya-Donoso et al., 2019).
In most cases, hybridization is inferred through incongruence of mitochondrial and nuclear phylogenies and/or morphological species designations, given the contrasting inheritance modes of the two genomes (Ballard and Whitlock, 2004). Furthermore, instances of hybridization are typically localized to areas where two distinct populations or species come into contact, “hybrid zones.” It has been suggested that hybridization can play two important roles within Liolaemus: (1) increasing genetic and adaptive diversity following population bottlenecks, and (2) limiting specialization to maintain a generalist phenotype that is better suited to heterogeneous and unstable habitats, such as those in southern South America (Morando et al., 2020; Olave et al., 2020). Although hybridization is suspected to be relatively common in Liolaemus, detailed examinations of hybrid zones using thorough transect sampling and genomic data analyses are lacking.
Hybrid zones form at the interface between two distinct populations and in some cases are best described as “clines,” which represent transitions in observed character states between distinct populations (Barton and Hewitt, 1985). Clines inferred from different characters that share the same center are said to be coincident, and those that share the same shape/width are said to be concordant. Clines and contact zones are often formed in ecotones where two distinct habitats fuse (Leaché and Cole, 2007). These contact zones typically occur in one area between species and therefore offer a single perspective into the evolutionary process. However, some species complexes have established themselves into loosely formed “rings” (or perhaps more aptly, horseshoes) around unsuitable habitat, where species grade into each other at contact zones, but the forms are reproductively isolated where the “ring” closes (e.g., Ensatina salamanders, Moritz et al., 1992; Phylloscopus warblers, Irwin et al., 2001). In other conceptually related instances, “mosaic” hybrid zones can be formed when individuals from distinct species repeatedly come into contact with each other across the landscape (e.g., Helianthus sunflowers; Rieseberg et al., 1999). In all of these cases, replicate hybrid zones are formed where one species participates in hybridization in >1 geographic area.
In hybrid zones, neutral and selected markers will respond to hybridization in distinct manners. For instance, because nDNA is biparentally inherited and mtDNA maternally inherited in vertebrates (Ballard and Whitlock, 2004), sex-biased dispersal can be seen by comparing nuclear and mitochondrial patterns in hybrid zones (but see Bonnet et al., 2017 for alternative explanations). Furthermore, many mitochondrial genes code for proteins involved in the electron transport chain and ATP production, making the whole mitochondrial genome subject to selection via linkage. Thus, a beneficial mitochondrial haplotype may sweep to fixation in both populations via selection and gene flow in the hybrid zone. However, some authors have argued for the neutral evolution of the mitochondrial genome with respect to phenotype in some systems (e.g., Rohwer et al., 2001). Assuming that mtDNA is neutrally evolving allows for the estimation of hybrid zone movement, because neutral markers geographically lag behind non-neutral markers (McGuire et al., 2007). When a hybrid zone moves due to an invading population, the neutral mitochondrial haplotypes will be left in the wake of the invading species (Rohwer et al., 2001). Differing selection pressures and inheritance patterns of nuclear and mitochondrial genomes mean that cline shape and geographic center may in fact be distinct from one another in a given hybrid zone (e.g., Leaché et al., 2017). Depending on the concordance or discordance between nuclear and mitochondrial clines, an inference can be made about hybrid zone movement, the dispersal behavior of the two sexes, or differential selection between nuclear and mitochondrial genomes in the hybrid zone.
Here, we investigated hybrid zones in the Liolaemus fitzingerii species group through genome-wide single nucleotide polymorphism (SNP) data. The twelve species belonging to this group are distributed throughout the Patagonian shrub-steppe of central Argentina (Avila et al., 2006, 2008, 2010). However, a phylogenetic analysis using genome-wide SNP data and dense geographic sampling of individuals only found support for six distinct genetic groups, suggesting that species diversity in the group could be overestimated (Grummer, 2017). Nonetheless, the four species studied here are supported by both morphological and SNP data. Two putative contact zones were previously discovered through genomic analyses: one between L. melanops and L. shehuen, and a second between L. xanthoviridis and L. fitzingerii (Figure 1; Grummer, 2017). These contact zones are further supported by color polymorphism data and the co-occurrence of mtDNA sequences (cytochrome B from different species in single populations (Morando and Avila, personal communication). Although multiple lines of evidence support the presence of these hybrid zones, nothing is known regarding their geographic arrangements and limits and therefore the biotic and abiotic processes maintaining them. We studied hybrid zones in the L. fitzingerii species group using transect sampling to contrast patterns in both nuclear and mitochondrial genomes through population structure estimation, phylogenetic inference, cline analysis, and network analyses. Our aim is to provide an understanding of evolutionary processes at a fine-scale where the ranges of species come into contact, providing insights into the nature of speciation in a system where species boundaries are porous and blurry.
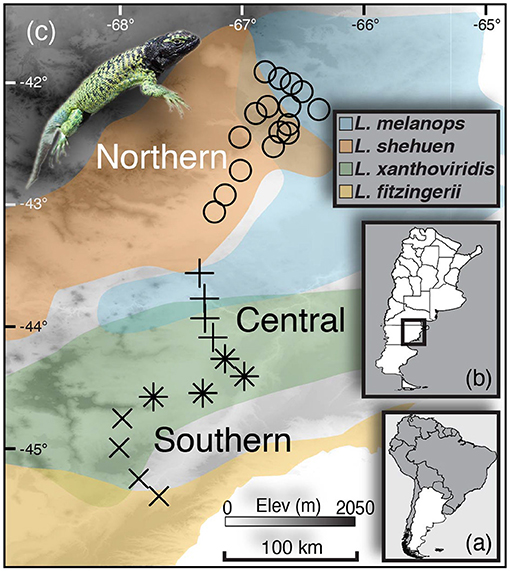
Figure 1. Sampling map with insets showing (A) Argentina and (B) the provincial borders within Argentina and where the study took place (C). Transect sampling localities for the Northern hybrid zone are shown as circles, whereas the “Central” and “Southern” transects are shown as +s and Xs, respectively (the localities marked with asterisks/stars were analysed in both Central and Southern transects). Colors on the map reflect population boundaries as determined by genome-wide SNP data in Grummer (2017) that largely correspond to the species Liolaemus melanops (blue), L. shehuen (orange), L. xanthoviridis (green), and L. fitzingerii (yellow).
2. Materials and Methods
2.1. Bioethics
All research specimens were collected by hand using methods approved by the University of Washington Office of Animal Welfare (IACUC protocol number 4249-01) and in accordance with provincial permits from the Argentinean Dirección de Fauna y Flora Silvestre.
2.2. Sampling and DNA Extraction
All voucher specimens and tissues were deposited into the LJAMM-CNP herpetology collection in the Centro Patagónico Nacional (IPEEC-CONICET), Puerto Madryn, Chubut, Argentina. DNA was extracted from tissue (tails tips and liver) through a salt (NaCl) extraction method (MacManes, 2013). Prior to library sequencing preparation, we discarded samples that had low DNA concentration or had degraded genomic DNA that lacked high molecular weight DNA.
2.2.1. Northern Hybrid Zone
During January and December of 2015, we collected 169 individuals from 17 distinct localities in Rio Negro and Chubut provinces (Figure 1; Supplementary Table 1). Sampling was performed every ~15–20 km along the transect.
2.2.2. Central and Southern Hybrid Zones
In December 2015, we collected 120 individuals from 13 distinct localities in Chubut province (Figure 1; Supplementary Table 2). Analyses revealed that what was assumed to be a single hybrid zone in the southern transect in fact represented two hybrid zones (see Results), so we therefore broke up this single transect into a northern (“Central”) and southern (“Southern”) transect (further detail below).
2.3. DNA Sequence Preparation
2.3.1. Nuclear DNA
We generated a nuclear dataset with the double digestion restriction site-associated DNA sequencing (ddRADseq) approach (Peterson et al., 2012). Genomic DNA was digested with two enzymes, SbfI (8 bp recognition sequence [5′ CCTGCAGG 3′], “rare cutter”; New England Biolabs, Ipswich, MA) and MspI (4 bp recognition sequence [5′ CCGG 3′], “common cutter”; New England Biolabs, Ipswich, MA). Unique barcoded primers were ligated to all genomic DNA fragments to enable multiplex sequencing. Genomic DNA fragments between ~365 and 465 bp (415–515 bp after ligating barcoded oligonucleotides) were size-selected with a Blue Pippin DNA fragment size selector (Sage Science, MA, USA). Samples with distinct barcodes were pooled in multiples of eight and unique indexes were applied to each pool using PCR with NEB Phusion Taq polymerase (New England Biolabs Inc., MA, USA) and the following thermocycler conditions: 98° for 0:30, (98° for 0:10, 58° for 0:30, 72° for 0:30) × 12 cycles, and a final 10:00 extension at 72°C. The amplified pools were multiplexed (up to 160 individuals per sequencing lane, some runs with individuals from other studies) and sequenced on Illumina HiSeq 2500 and 4000 machines (Illumina Inc., CA, USA) with 50 bp single-end reads at the University of California Berkeley's QB3 Vincent J. Coates sequencing facility. After de-multiplexing, each read contained 39 bp of sequenced genomic DNA.
2.3.2. Mitochondrial DNA
We targeted a fragment of the cytochrome B (cytB) gene to sequence for all individuals and contrast with patterns observed from the nuclear genome. Two sets of primers were used, an “external” pair that amplified an ~800 bp fragment, and an “internal” pair that amplified a ~360 bp fragment; primer sequences are given in Morando et al. (2003). Twenty-three μL of Tankara EmeraldAmp GT PCR Master Mix (Takara Bio USA, Inc.; Mountain View, CA, USA) were mixed with 2 μL genomic DNA, and amplified with the following thermocycler conditions: 95°C for 5:00, (95° for 0:45, 55° for 0:30, 72° for 1:00) × 35 cycles, with a final 10:00 extension at 72°C. If individuals did not amplify for the larger fragment, we attempted to amplify the smaller fragment with the internal primers. PCR products were sent to Genewiz (South Plainfield, NJ, USA) where they were purified and sequenced in both forward and reverse directions.
2.4. DNA Dataset Assemblies
2.4.1. ddRAD Bioinformatics and Dataset Assembly
Raw sequence reads were processed to generate “clusters” (e.g., loci) and identify SNPs with the program pyRAD v3.0.66 (Eaton, 2014). After demultiplexing individuals using their unique adapter and barcode sequences, pyRAD uses VSEARCH (Rognes et al., 2016) and MUSCLE (Edgar, 2004) to cluster and align reads into loci. Raw sequence reads were discarded if they had ≥4 bp with a Phred quality score <20. Reads were clustered within individuals and then across individuals with clustering thresholds of 90, 92, and 95%, and we ultimately chose 92% to minimize the number of paralogs while not over-splitting homologous loci given the sequence divergence across populations (Ilut et al., 2014; de Oca et al., 2017). We used a minimum depth of coverage of 10 for all loci. We set the paralog filter in pyRAD to 90%, meaning that up to 90% of individuals at a site can be heterozygous (e.g., be represented by two alleles with an IUPAC ambiguity code), as we expect many heterozygous positions to be due to shared ancestry (e.g., homology) and not due to fixed paralogs differences. We set the missing data threshold at 25%, meaning that ≥75% of individuals had data at each locus. All other parameters in pyRAD were left at their default settings.
2.4.2. Unlinked SNPs vs. Sequence Data
Unlinked SNPs can generate a maximum of four alleles per locus, but are more commonly bi-allelic with only two alleles. However, considering all variant and invariant sites together can greatly increase the number of distinct alleles at a locus. This richer allelic information might offer higher precision in delimiting population boundaries and/or inferring admixture proportions vs. SNPs, so we analyzed both datasets in parallel for comparison. PyRAD generates a “.alleles” file that contains allelic sequence data (e.g., two alleles per individual) for all loci that met all quality and assembly parameters; sequences need not be 39 bp, as indels can cause loci to be >39 bp. It is from these loci that biallelic SNPs are extracted. These ≥39 bp RAD loci were then coded as alleles (e.g., “microhaplotypes”), two per individual. We generated a custom Python script to parse the “.alleles” file into a file formatted for the program Structure (Pritchard et al., 2000), where any non-N difference at a site between alleles constituted a unique and new allele. This dataset (herein termed “alleles”) was then analyzed in parallel to the unlinked SNPs dataset to compare the power of each to identify population boundaries, admixture proportions, and clines.
2.4.3. mtDNA Dataset Assembly
Raw sequence data (“.ab1” chromatograms) from both sequencing directions were made into contigs and hand-edited in Geneious v10 (Biomatters; Auckland, New Zealand). Consensus sequences were exported as .fasta sequences and aligned with Clustal2 (Larkin et al., 2007) in Mesquite v3.2 (Maddison and Maddison, 2017). Liolaemus cuyanus was included as an outgroup to root phylogenetic trees used in cline analyses (see below).
2.5. Geographic Cline Analyses
We estimated clines for both nuclear and mitochondrial datasets to identify the geographic interface between populations, and to contrast cline patterns between markers with different inheritance patterns. To generate transect distances along a single axis between sampling localities of each hybrid zone, we first calculated pairwise distances between every sampling locality as the great circle distance with latitude-longitude coordinates in the R package Fossil (Vavrek and Vavrek, 2012) with the function “earth.dist.” We note that this method does not consider topography when calculating distances. We then used classical multidimensional scaling through principal coordinates analysis to reduce the pairwise matrix of distances between each locality into a single distance value for each locality that retained the original overall pairwise distance structure (as in Gompert et al., 2010). This ordination represents sampling locations along a single axis where kilometer (km) 0 was converted to represent the northern-most sampling site of each transect.
2.5.1. nDNA Clines
We used Structure v2.3 (Pritchard et al., 2000; Falush et al., 2007) and the Evanno method (Evanno et al., 2005) to identify the number of populations (k) in each hybrid zone. Analyses on the Northern hybrid zone dataset were run for 250,000 generations following a 75,000 generation burn-in period with five replicates of each k value of 1–5. Because of a higher number of loci (see Results below), the Central + Southern hybrid zones dataset was run for 300,000 generations with 100,000 burn-in generations, also with five replicate runs of k 1–5.
After identifying the optimal k value, we used Structure to determine the admixture proportions (Q) of all individuals and therefore of each sampling locality. Q estimates from five replicate Structure runs were combined through CLUMPP (Jakobsson and Rosenberg, 2007) and were then used to generate geographic clines. For the combined Central + Southern hybrid zones, the Evanno et al. (2005) method selected k = 3 (Supplementary Figure 1A), where a central population intergrades with two distinct populations, one to the north and another to the south. This larger Central + Southern hybrid zone was therefore split into two separate hybrid zones, where the northern hybrid zone was designated as localities A–I and the southern hybrid zone as localities F-M (Figure 1). The northern half of our bigger southern transect will be referred to as the “Central” hybrid zone, and the southern portion of the southern transect will be referred to as the “Southern” hybrid zone.
With the use of the Q proportions and geographic locations as described above, we estimated geographic maximum likelihood clines, including cline centers and cline widths, in the R package Hzar (Derryberry et al., 2014). Cline models were tested with minimum and maximum values fixed to the observed data, without allowing exponential tails on both sides of the cline. The cline fit analysis was run for 200,000 generations and a burn-in of 40,000 generations, from which the maximum likelihood parameter estimates of the cline were generated. The best-fit cline model (along with 95% confidence interval) was then plotted as a function of geographic distance along the transect.
2.5.2. mtDNA Clines
Because mtDNA is haploid and non-recombining, haplotype frequencies were calculated in terms of the relative proportions of the distinct parental haplotypes found at each sample location. We used both tree-based and network-based approaches to determine haplotype frequencies at each sampling locality. For the tree-based approach, we used jModelTest v2.1.7 (Guindon and Gascuel, 2003; Darriba et al., 2012) to determine the optimal DNA substitution model (HKY + Γ for all datasets), which was then used to estimate maximum likelihood trees in RAxML v8.2 (Stamatakis, 2014) with 100 bootstrap iterations. For each hybrid zone, we calculated haplotype frequencies as the proportion of individuals in each locality that belonged to the “northern” clade, resulting in haplotype frequencies ranging from 0 to 1.
Our second approach was analogous to the tree-based approach, but instead was network-based. We inferred minimum-spanning networks (Bandelt et al., 1999) using the program PopART (http://popart.otago.ac.nz), and divided the network into two groups on the edge (branch) with the highest number of sequence substitutions. As in the tree-based approach, we determined haplotype frequencies by calculating the proportion of individuals from each locality that were in each of the two major groups. Cline analysis was performed with these frequencies using the same methodology as in the nDNA cline estimates.
Because we were interested in contrasting evolutionary patterns in the mitochondrial vs. nuclear genomes, we quantitatively tested how different the cline estimates were for these two datasets. To do so, we constrained the cline estimate of the nuclear data to have either the cline center or cline width that was inferred from the mtDNA, and then estimated the log likelihood of the constrained clines (for both alleles and unlinked SNPs datasets). With the maximum likelihood estimate and number of free parameters in the model, we were able to estimate AIC scores for each cline (with the “hzar.AIC.hzar.cline” function). A difference in AIC score >2 between unconstrained and constrained cline estimates indicated a significant difference between the two genomes.
3. Results
After we removed individuals with poor genomic DNA or sequence data quality and filtered loci based on the parameters above, the nuclear datasets consisted of 151 individuals (2,814–15,963 loci) in the Northern hybrid zone, 73 individuals in the Central and 61 in the Southern hybrid zones (586–13,835 loci). After combining across individuals, the datasets consisted of 1,295 and 2,436 loci in the Northern and Central + Southern hybrid zones, respectively. We removed individuals from a single locality in the Northern hybrid zone because our analyses showed it to be geographically outside (to the east) of the hybrid zone. We also removed a single locality from analysis from the Central hybrid zone because this locality was represented by a single individual. Samples per locality ranged from 3 to 13 in the Northern hybrid zone with an average of 7.8, a range of 3–15 with an average of 9.1 in the Central hybrid zone, and a range of 3–11 with an average of 7.6 individuals in the Southern transect localities (Supplementary Tables 1, 2). In the mitochondrial dataset (832 base pairs), the Northern transect was represented by 146 individuals, whereas the Central and Southern transects had 75 and 59 individuals, respectively (Supplementary Tables 3, 4). Individuals in both transects displayed a high level of morphological variation across both age and localities (Figure 2). The 16 localities in the Northern transect had an average sample size of 8.75 and ranged from 2 to 13 individuals; the average number of mitochondrial samples per locality in the Central and Southern transects ranged from 2 to 15 ( = 9.38) and 2 to 13 ( = 8.00), respectively.

Figure 2. Ventral and dorsal photos of males from localities sampled in the Northern (i) and Central + Southern (ii) hybrid zones. Letters and numbers indicate transect sampling points in Figure 4. Photos were not available for individuals from localities M–P in the northern transect.
3.1. Population Identification
The numbers of unlinked biallelic SNPs used in the Northern and Central + Southern transects were 1,295 (mean number of loci per individual = 1,140) and 2,436 (mean number of loci per individual = 2,173), respectively. Coding the nuclear loci into alleles, which retains all of the SNP variation at each locus, resulted in an average of 6.6, 4.7, and 4.5 alleles per locus, with maximum number of alleles of 32, 28, and 28 for Northern, Central, and Southern transects, respectively (Figure 3).
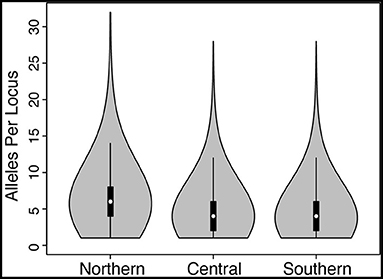
Figure 3. Violin plots showing the number of alleles per locus when the sequence data were coded into alleles (vs. unlinked SNPs) for Northern, Central, and Southern hybrid zones. Density is shown by width of the “violin,” whereas box plots inside depict the mean (white dot), first and third quartiles (black boxes), and 1.5× the inter-quartile range (vertical lines).
3.1.1. Northern Hybrid Zone
The Evanno et al. (2005) method favored two populations (k = 2) with the unlinked SNPs and alleles datasets alike (Supplementary Figure 2). The interface between the two populations is sharp and occurs on the eastern edge of the Somuncura Plateau (Figure 4A).
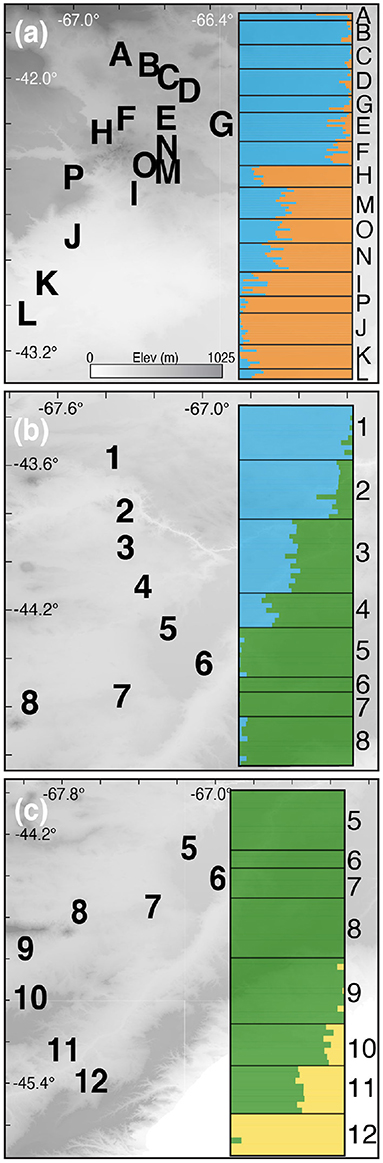
Figure 4. STRUCTURE results and hybrid zone locations for the Northern (A), Central (B), and Southern (C) hybrid zones. Latitude and longitude are shown, and the elevational gradient is the same in all maps. Note that populations 5–8 are used in both Central (B) and Southern (C) transects.
3.1.2. Central and Southern Hybrid Zones
Estimates of the optimal k value via the Evanno et al. (2005) method were in conflict: the unlinked SNPs dataset favored four populations, whereas the alleles dataset supported three (Supplementary Figure 1). Visualizing the results of k = 4 revealed that the fourth inferred population is almost completely confined to individuals in the northern-most sampling locality (“1”; Figure 4; Supplementary Figure 3). The k = 4 result doesn't make biological sense and is in conflict with the results from the alleles dataset, so we therefore focused on the k = 3 results for the larger Central + Southern transect. Visualizing the k = 3 result revealed a “sandwich” hybrid zone in which individuals from the center of the transect (roughly equivalent to the described species Liolaemus xanthoviridis) hybridize with two distinct populations—one to the north (L. melanops) and one to the south (L. fitzingerii; Figures 4B,C). Furthermore, the northern population in the Central hybrid zone is the same “species,” L. melanops, that constitutes the northern populations of the Northern transect (Figure 1; Supplementary Figure 4).
3.2. Clines
3.2.1. Northern Hybrid Zone
Cline width estimates were 30.13 and 35.27 km for the alleles and unlinked SNPs datasets, respectively (Table 1). Estimates of cline centers from the two nuclear datasets were ~0.5 km different from one another in the northern hybrid zone (Table 1). The inferred admixture (Q) proportions were more extreme for the alleles dataset, providing admixture estimates closer to 1 or 0 at the opposite ends of the transect (Figure 5A). In terms of calculating haplotype frequencies from the mitochondrial data, the phylogeny, and network were in 100% agreement (Supplementary Figure 5). When mitochondrial and nuclear clines are compared, the mitochondrial cline is shifted ~7 km to the south of the nDNA clines and is ~13 km narrower at 20.64 km (Table 1; Figure 5). When the nuclear data were inferred under the constraint of the mitochondrial cline center or width estimates, the position of the center, but not the width, was inferred to be significantly different (Table 2).
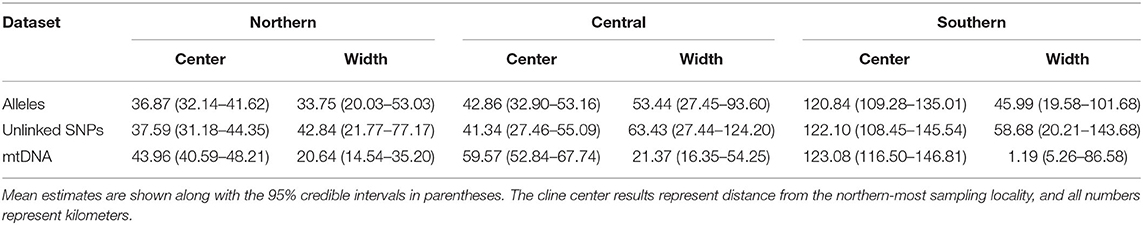
Table 1. Cline analysis results from Hzar for Northern, Central, and Southern hybrid zones.
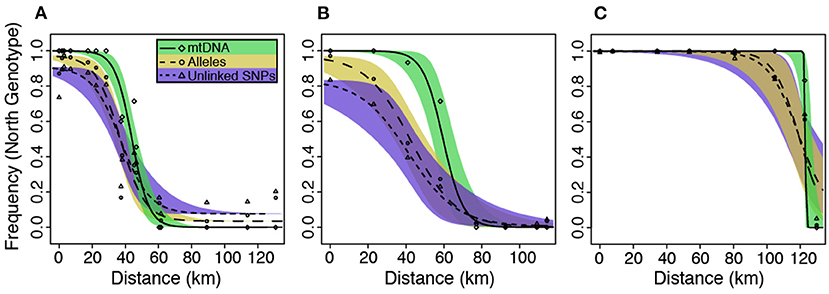
Figure 5. Hzar maximum likelihood cline estimates and 95% credible intervals estimated from both nDNA and mtDNA datasets for the (A) Northern, (B) Central, and (C) Southern hybrid zones. The x-axis represents distances from the northern-most sampling locality.
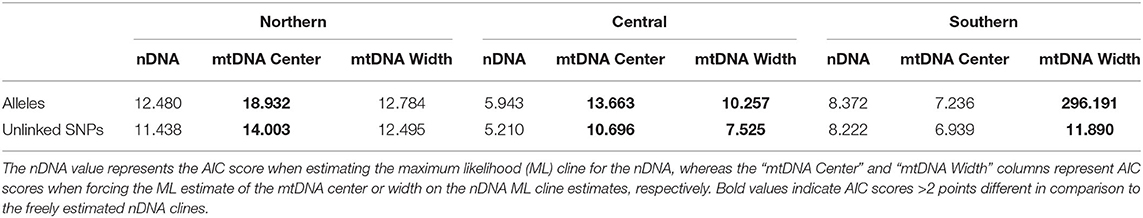
Table 2. AIC scores for genetic clines from Hzar analyses.
3.2.2. Central and Southern Hybrid Zones
Central. As in the Northern hybrid zone, admixture proportions inferred with the alleles dataset were more extreme than the unlinked SNPs dataset (Figure 5B). The cline center inferred from the nDNA is ~40 km to the south of the northern-most sampling locality, and ~45 km wide (Table 1). As in the Northern hybrid zone, the haplotype frequencies calculated from the mtDNA data were identical between phylogeny and network approaches (Supplementary Figure 6). The nDNA clines are in stark contrast to the mtDNA cline, whose center is ~20 km to the south and less than half as wide as the nDNA clines (21.37 km). When the nDNA was constrained to fit the mtDNA cline center and width, the clines estimated from both data types were significantly different from each other in both of these characteristics (Table 2).
Southern. Cline center estimates were only 0.43 km different between alleles and unlinked SNP datasets. However, the alleles cline width estimate was ~10 km narrower (27.42 vs. 37.14 km; Table 1). In comparison to the mitochondrial genealogies inferred for the other two hybrid zones, the phylogeny of the Southern transect individuals did not contain two strongly supported clades (Supplementary Figure 7). However, two distinct groups were inferred in the network that corresponded to a division created by the longest branch in the phylogeny. In contrast to the other two transects, the clines estimated in the Southern transect were in the very southern portion of the transect (Figure 5C). The mtDNA cline center was ~4 km to the south and much narrower (1.19 km) when compared to the nDNA clines (Table 1; Figure 5C). Constraining the nDNA cline center to the mtDNA estimate strangely led to an improvement in model score, however, the nDNA cline widths were significantly wider than the mtDNA cline width (Table 2).
4. Discussion
Our study marks the first in-depth study of hybrid zone dynamics within Liolaemus, a clade where hybridization is widespread and potentially fundamental to its evolutionary history. The arrangement of three geographically sequential hybrid zones in the L. fitzingerii species group, a group in which hybridization appears to be common, is unusual and provides a valuable system for analyzing hybridization in a replicated fashion. In the north, L. melanops hybridizes with L. shehuen and L. xanthoviridis, and in the south, L. xanthoviridis hybridizes with both L. melanops and L. fitzingerii. Analyses revealed similarities shared across all three hybrid zones: mitochondrial clines are (1) steeper compared to nuclear clines, (2) displaced to the south of the nuclear clines, and (3) significantly different from nuclear clines in terms of cline center and/or width. Our results indicate that hybridization is common in the L. fitzingerii species group and the hybrid zones are well-defined. Although hybridization is common and is a potential mechanism for generating extensive phenotypic variation in the L. fitzingerii species group (Figure 2), we did not test whether hybridization enhances speciation (through a mechanism, such as reinforcement) as some authors have hypothesized because it is outside the scope of this paper (e.g., Olave et al., 2018; Morando et al., 2020).
4.1. Hybridization and Species Boundaries in Liolaemus
In spite of considerable progress over the past few decades, much remains to be understood about phylogeography and systematics of southern hemisphere taxa (Beheregaray, 2008). Knowledge on the taxonomic and phylogenetic diversity of Patagonian lizards specifically is incomplete, leaving room for many future studies (Brito, 2010; Diniz-Filho et al., 2013). These uncertainties manifest taxonomically and result in many species “groups” and “complexes” whose geographic distributions, and species limits, are not clearly defined. The results here indicate that hybrid zones are clearly defined in the L. fitzingerii group, and that in spite of many instances of interspecific hybridization, species are clear entities outside of contact zones.
Character clines in hybrid zones can vary substantially in shape—broad vs. narrow—and different shapes can provide insights into the evolutionary processes maintaining hybrid zones. A recent meta-analysis of animal hybrid zones (McEntee et al., 2020) provides some context for interpreting the mtDNA and nuclear cline widths estimated in the L. fitzingerii group. Across a variety of taxa, hybrid zone cline widths range from 10 m to >3,000 km (McEntee et al., 2020). In lizards (n = 95 cline estimates in McEntee et al., 2020), the reported range is ~30–190 km with a left-skewed distribution—20% of the values are <1 km and 90% are <60 km. The hybrid zones in the L. fitzingerii group were estimated to be ~35–65 km wide with nuclear data, or ~1–20 km wide with mitochondrial data (Figure 5). Accordingly, the cline widths in the L. fitzingerii group appear to be “typical” in relation to other lizard species. We would expect much more variance in cline estimates across hybrid zones if a cline was maintained solely by selection, as opposed to a balance between dispersal and selection (Barton and Hewitt, 1985). The observation that both cline width and shape do not vary substantially between hybrid zones indicates that dispersal of parental genotypes into the contact zone is offset by selection against heterozygotes. In the L. fitzingerii species group, the strengths of selection and gene flow seem to be within the same order of magnitude, and similar to those seen in other squamate species (Mallet et al., 1990; McEntee et al., 2020).
4.2. Nuclear vs. Mitochondrial Clines
Geographic cline analyses revealed that the mitochondrial cline center is displaced to the south of the nuclear cline in all three hybrid zones. Furthermore, nuclear and mitochondrial clines were significantly different from each other in cline center and/or width in all three hybrid zones. Observing significantly different clines between nuclear and mtDNA is not necessarily unexpected, given that a variety of biotic and evolutionary processes can generate discordance between nuclear and mitochondrial DNA (Bonnet et al., 2017). These two genomes have different modes of inheritance (unipartental vs. biparental), recombination (mtDNA lacks recombination), and are subject to different selection pressures (Ballard and Whitlock, 2004). Additionally, the amount of gene flow between populations within a species and demographic factors affecting levels of allele “surfing” can mitigate introgression at contact zones and further complicate characterizations of hybrid zone dynamics (Petit and Excoffier, 2009).
Discordance between nuclear and mitochondrial genomes and their estimated clines can be generated by two classes of processes affecting the mitochondrial genome: selective (e.g., positive selection for the introgressing mitochondrial genome or negative pleiotropic selection on many nuclear loci) and neutral processes involving sex-related asymmetries, such as interspecific mate preference (females of taxon a preferring males of taxon b while no such preference occurs in females of taxon b), sex-biased dispersal, or differences in hybrid survival by sex (Funk and Omland, 2003; Bonnet et al., 2017). In Liolaemus lizards, males leave their natal ground to establish home ranges, whereas females disperse shorter distances (Kacoliris et al., 2009), arguing that sex-biased dispersal could result in a southerly shifted mtDNA via northward migration of juvenile males from the southern population into the northern population. Additionally, a southerly shifted mtDNA cline could also result from a southward migration of females from the northern population into the southern population; these two hypotheses are not mutually exclusive. Thus, although sex-biased dispersal, asymmetric mating preferences, or differential survival rates of hybrid offspring can lead to mito-nuclear discordance (Bonnet et al., 2017), we are unable to determine the relative strengths of these processes here.
A second reason for the discordant mt- and nDNA clines is that these hybrid zones could be moving. Many empirical studies have documented moving hybrid zones over time (reviewed in Buggs, 2007). Hybrid zones can move when selection against hybrids is genetically countered by dispersal of parental forms into the hybrid zone (tension zone model; Barton and Hewitt, 1985), or when a change in the external environment causes selection along a gradient to generate fitness differences (May et al., 1975). When an environmental gradient moves (e.g., as the result of a change in climate), geographic ranges and hybrid zones can shift with it (e.g., Leaché et al., 2017). As geographic ranges shift, asymmetric introgression from the expanding species into the stationary one will cause neutral markers to geographically trail behind non-neutral markers (McGuire et al., 2007). In particular, asymmetric introgression of the mitochondrial genome and its discordance with nuclear markers has been used to deduce a moving hybrid zone (Rohwer et al., 2001). Although hybrid zone movement over time can be inferred from discordant mt- and nDNA clines sampled from a single time-point, the most convincing cases of hybrid zone movement come from studies with replicated sampling efforts over time (e.g., Carling and Zuckerberg, 2011; Taylor et al., 2014; Leaché et al., 2017). A lagging cline inferred from the putatively neutral mtDNA that is following the leading edge of an expanding population further suggests northward range expansion of L. shehuen, L. xanthoviridis, and L. fitzingerii.
Concluding that a hybrid zone moves because a trailing mtDNA cline has been observed assumes that the mitochondrial gene(s) under study is/are neutrally evolving in these species, which might not be true. Indirect selection on mtDNA through differential selection of the heterogametic sex (e.g., Haldane's Rule) or direct selection via cyto-nuclear incompatibilities would also impair the effective movement of mtDNA across the hybrid zone (Dasmahapatra et al., 2002). This leads to a third reason for mt-nDNA cline discordance, which is differential selection on the two genomes (Bonnet et al., 2017). If strong enough positive selection was working on any site in the mitochondrial genome, that mitochondrial haplotype could sweep through the population (due to linkage) and the cline would not lag behind as expected for a neutral marker. Although we did not explicitly test for selection, it is unlikely to affect our results because loci under selection would likely be in the minority of our dataset. Nonetheless, we agree with Dasmahapatra et al. (2002) in that “asymmetry of introgression, or lack of introgression of molecular markers, is relatively unconvincing evidence either for or against hybrid zone movement.”
We performed our population structure and cline analyses on two nuclear datasets, one where a single SNP was randomly selected from each RAD locus, and another that used all invariant and variant sites present at each locus recoded into alleles (“alleles”). The alleles dataset provided many more alleles per locus than the unlinked SNPs dataset, with 6.6, 4.7, and 4.5 alleles per locus for the alleles dataset in the Northern, Central, and Southern transects, respectively, whereas the unlinked SNPs datasets contained only bi-allelic loci. Although Structure plots between the two datasets were qualitatively similar (results not shown), admixture proportions (Q) were more “intermediate” for the unlinked SNPs dataset, meaning that the Q values weren't as extreme as in the alleles dataset. This can be seen in the cline estimates (Figure 5), where the frequency of the northern genotype for the alleles dataset reached closer to 0.0 and 1.0. A similar pattern is seen in the cline width estimates (Table 1), where the widths estimated for two of the three transects from the alleles dataset were narrower by ~5–10 km. These narrower cline estimates, and more extreme Q estimates, are almost certainly due to the increased information content associated with higher allelic richness in the alleles dataset. It is not possible to determine which dataset produced more accurate cline parameters without conducting a thorough simulation study where the true cline parameters are known. However, we suspect that the “alleles” data has the advantage over the bi-allelic SNP analysis because it uses all of the variation present in the data.
4.3. Replicated Hybrid Zones
In this study, two species—L. melanops and L. xanthoviridis—are each involved in two hybrid zones. First, L. melanops hybridizes with L. shehuen in the Northern hybrid zone as well as with L. xanthoviridis in the Central hybrid zone (Figure 1). In the Northern hybrid zone, the interface of L. melanops and L. shehuen occurs on the eastern edge of the Somuncura Plateau, a geological feature that is ~25 million years old (Kay et al., 2007) and reaches an elevation of ~1,600 m. That this geologic feature is at the interface of two populations is perhaps not surprising, however, L. shehuen individuals are found both below (to the east) and on top of this plateau. The elevation imposed by this plateau does seem to form a western barrier for L. melanops, which is found in lower elevation Patagonian shrub-steppe habitats to the east and south of the plateau. In fact, elevation explains 32% of the variance in admixture proportions (Q) between these two species (Supplementary Figure 8). Assuming equal dispersal capabilities of L. melanops individuals throughout the range of this species, the narrower cline width in the north (~32 vs. 45 km) qualitatively implies stronger selection in the Northern hybrid zone. This evidence implies that exogenous selection (e.g., environmental differences) is a potential mechanism maintaining L. melanops and L. shehuen as distinct species. The boundary between L. melanops and L. xanthoviridis corresponds with the Chubut River, which is a large river and seasonally >100 m wide in this area. Although the divergence between these two species appears to be allopatric, our genetic data show that the Chubut River is in fact a porous boundary.
Second, in a similarly replicated fashion, Liolaemus xanthoviridis hybridizes in two separate areas: to the north with L. melanops and to the south with L. fitzingerii. The nDNA cline width in the north with L. melanops is ~45 km, whereas it is ~32 km wide in the hybrid zone with L. fitzingerii. Assuming these hybrid zones are best modeled as tension zones that are a balance of dispersal and selection, narrower clines could be the result of two non-mutually exclusive causes: reduced dispersal abilities, or stronger selection. In Liolaemus generally, we do not have good estimates of dispersal (but see Frutos and Belver, 2007 and Camargo et al., 2013 for some estimates), especially when trying to compare differing dispersal abilities between species in the L. fitzingerii group. In terms of selection, the narrower cline seen in the Southern hybrid zone does not seem to be the result of sexual selection via interspecific mating and a higher disparity in body sizes because both taxa are large-bodied (male max SVL = 94 vs. 102 mm for L. xanthoviridis and L. fitzingerii; Etheridge, 2000). The narrower cline in this hybrid zone, however, might be due to exogenous (environmental) causes. Liolaemus fitzingerii is found in loosely formed sand dunes dominated by the mesquite bush Prosopis denudans, whereas L. xanthoviridis occurs in the hardpan Patagonian shrub-steppe habitat.
5. Conclusions
In this study, we were able to compare multiple hybrid zones across Liolaemus lizards in central Argentina. Hybridization appears to be common in the L. fitzingerii group, and the hybrid zones are narrow and geographically localized. Liolaemus melanops hybridizes with two species, and the hybrid zone in the north (with L. shehuen) is significantly narrower than in the south (with L. xanthoviridis), likely due to the environmental gradient (i.e., change in elevation and vegetation) posed by the Somuncura Plateau. Nonetheless, other hybrid zones in this group have formed in the absence of any obvious physical barriers, suggesting that other ecological or intrinsic factors may be playing a role in maintaining species as distinct entities. The discordance between mitochondrial and nuclear cline estimates suggests sex-biased dispersal, divergent selection across genomes, or movement of these hybrid zones over time. Re-sampling these hybrid zones in the future may help tease apart these alternative hypotheses. Lastly, although hybridization has generated novel genotypes and morphological variation in hybrid zones, it is unclear whether hybridization has reinforced species boundaries or promoted speciation within the L. fitzingerii group. This research has provided a fine-scale understanding of hybrid zone dynamics within the Liolaemus fitzingerii group, with implications not only for other Liolaemus species and Patagonian taxa more broadly, but for hybrid zone systems globally.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA670250.
Ethics Statement
All research methods were approved by the University of Washington Office of Animal Welfare (IACUC protocol number 4249-01) and in accordance with provincial permits from the Argentinean Dirección de Fauna y Flora Silvestre.
Author Contributions
JG performed the field sampling, DNA data collection, analyses, and wrote the paper. LA performed the field sampling and provided the financial support. MM and AL edited the manuscript and provided the financial support. All authors designed the research.
Funding
This research was funded in part by a National Science Foundation Doctoral Dissertation Improvement Grant (DEB-1500933) and UW Study Abroad fellowship to JG, ANPCyT-FONCYT PICT 2015-1252 (LA), and CONICET-PIP 2012-336 (MM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. Analyses were facilitated though the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system at the University of Washington. Thanks to C. Pérez and T. Avila for assistance in sample collection during the December 2015 trip.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.624109/full#supplementary-material
References
Aboim, M., Mavárez, J., Bernatchez, L., and Coelho, M. (2010). Introgressive hybridization between two Iberian endemic cyprinid fish: a comparison between two independent hybrid zones. J. Evol. Biol. 23, 817–828. doi: 10.1111/j.1420-9101.2010.01953.x
Araya-Donoso, R., Torres-Pérez, F., Véliz, D., and Lamborot, M. (2019). Hybridization and polyploidy in the weeping lizard Liolaemus chiliensis (Squamata: Liolaemidae). Biol. J. Linn. Soc. 128, 963–974. doi: 10.1093/biolinnean/blz145
Avila, L., Morando, M., and Sites, J. (2006). Congeneric phylogeography: hypothesizing species limits and evolutionary processes in patagonian lizards of the Liolaemus boulengeri group (Squamata: Liolaemini). Biol. J. Linn. Soc. 89, 241–275. doi: 10.1111/j.1095-8312.2006.00666.x
Avila, L. J., Morando, M., and Sites J. W. Jr. (2008). New species of the Iguanian lizard genus Liolaemus (Squamata, Iguania, Liolaemini) from Central Patagonia, Argentina. J. Herpetol. 42, 186–196. doi: 10.1670/06-244R2.1
Avila, L. J., Pérez, C. H. F., Morando, M., and Sites J. W. Jr., (2010). A new species of Liolaemus (Reptilia: Squamata) from southwestern Rio Negro province, Northern Patagonia, Argentina. Zootaxa 2434, 47–59. doi: 10.11646/zootaxa.2434.1.4
Ballard, J. W. O., and Whitlock, M. C. (2004). The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744. doi: 10.1046/j.1365-294X.2003.02063.x
Bandelt, H. J., Forster, P., and Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Barton, N. H., and Hewitt, G. M. (1985). Analysis of hybrid zones. Annu. Rev. Ecol. Syst. 16, 113–148. doi: 10.1146/annurev.es.16.110185.000553
Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the southern hemisphere. Mol. Ecol. 17, 3754–3774. doi: 10.1111/j.1365-294X.2008.03857.x
Bonnet, T., Leblois, R., Rousset, F., and Crochet, P. A. (2017). A reassessment of explanations for discordant introgressions of mitochondrial and nuclear genomes. Evolution 71, 2140–2158. doi: 10.1111/evo.13296
Breitman, M. F., Avila, L. J., Sites, J. W., and Morando, M. (2011). Lizards from the end of the world: phylogenetic relationships of the Liolaemus lineomaculatus section (Squamata: Iguania: Liolaemini). Mol. Phylogenet. Evol. 59, 364–376. doi: 10.1016/j.ympev.2011.02.008
Brelsford, A., and Irwin, D. E. (2009). Incipient speciation despite little assortative mating: the yellow-rumped Warbler hybrid zone. Evolution 63, 3050–3060. doi: 10.1111/j.1558-5646.2009.00777.x
Brito, D. (2010). Overcoming the linnean shortfall: data deficiency and biological survey priorities. Basic Appl. Ecol. 11, 709–713. doi: 10.1016/j.baae.2010.09.007
Buggs, R. (2007). Empirical study of hybrid zone movement. Heredity 99, 301–312. doi: 10.1038/sj.hdy.6800997
Camargo, A., Werneck, F. P., Morando, M., Sites, J. W., and Avila, L. J. (2013). Quaternary range and demographic expansion of Liolaemus darwinii (Squamata: Liolaemidae) in the monte desert of Central Argentina using bayesian phylogeography and ecological niche modelling. Mol. Ecol. 22, 4038–4054. doi: 10.1111/mec.12369
Carling, M. D., and Zuckerberg, B. (2011). Spatio-temporal changes in the genetic structure of the Passerina bunting hybrid zone. Mol. Ecol. 20, 1166–1175. doi: 10.1111/j.1365-294X.2010.04987.x
Chhatre, V. E., Evans, L. M., DiFazio, S. P., and Keller, S. R. (2018). Adaptive introgression and maintenance of a trispecies hybrid complex in range-edge populations of populus. Mol. Ecol. 27, 4820–4838. doi: 10.1111/mec.14820
Culumber, Z., Fisher, H., Tobler, M., Mateos, M., Barber, P., Sorenson, M., et al. (2011). Replicated hybrid zones of xiphophorus swordtails along an elevational gradient. Mol. Ecol. 20, 342–356. doi: 10.1111/j.1365-294X.2010.04949.x
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Darwin, C. (1862). Notes on the causes of cross and hybrid sterility. Correspond. Charles Darwin 10, 700–711.
Dasmahapatra, K. K., Blum, M. J., Aiello, A., Hackwell, S., Davies, N., Bermingham, E. P., et al. (2002). Inferences from a rapidly moving hybrid zone. Evolution 56, 741–753. doi: 10.1111/j.0014-3820.2002.tb01385.x
de Oca, A. N. M., Barley, A. J., Meza-Lázaro, R. N., García-Vázquez, U. O., Zamora-Abrego, J. G., Thomson, R. C., et al. (2017). Phylogenomics and species delimitation in the knob-scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddradseq data reveal a substantial underestimation of diversity. Mol. Phylogenet. Evol. 106, 241–253. doi: 10.1016/j.ympev.2016.09.001
Derryberry, E. P., Derryberry, G. E., Maley, J. M., and Brumfield, R. T. (2014). HZAR: hybrid zone analysis using an r software package. Mol. Ecol. Resour. 14, 652–663. doi: 10.1111/1755-0998.12209
Diniz-Filho, J. A. F., Loyola, R. D., Raia, P., Mooers, A. O., and Bini, L. M. (2013). Darwinian shortfalls in biodiversity conservation. Trends Ecol. Evol. 28, 689–695. doi: 10.1016/j.tree.2013.09.003
Duvernell, D. D., and Schaefer, J. F. (2014). Variation in contact zone dynamics between two species of Topminnows, Fundulus notatus and F. olivaceus, across isolated drainage systems. Evol. Ecol. 28, 37–53. doi: 10.1007/s10682-013-9653-z
Eaton, D. A. (2014). PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30, 1844–1849. doi: 10.1093/bioinformatics/btu121
Edgar, R. C. (2004). Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Esquerré, D., Ramírez-Álvarez, D., Pavón-Vázquez, C. J., Troncoso-Palacios, J., Garín, C. F., Keogh, J. S., et al. (2019). Speciation across mountains: phylogenomics, species delimitation and taxonomy of the Liolaemus leopardinus clade (Squamata, Liolaemidae). Mol. Phylogenet. Evol. 139:106524. doi: 10.1016/j.ympev.2019.106524
Etheridge, R. (2000). A review of lizards of the Liolaemus wiegmannii group (Squamata, Iguania, Tropiduridae), and a history of morphological change in the sand-dwelling species. Herpetol. Monogr. 14, 293–352. doi: 10.2307/1467049
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Falush, D., Stephens, M., and Pritchard, J. K. (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol. Ecol. Notes 7, 574–578. doi: 10.1111/j.1471-8286.2007.01758.x
Feltrin, N. (2013). Conservadurismo o divergencia de nicho filogentico: especies patagonicas del grupo petrophilus (Squamata: Liolaemus) como caso de estudio (Ph.D. thesis), Universidad Nacional de Córdoba, Córdoba, Argentina.
Frutos, N., and Belver, L. C. (2007). Dominio vital de liolaemus koslowskyi etheridge 1993 (Iguania: Liolaemini) en el noroeste de la provincia de La Rioja, Argentina. Cuad. Herpetol. 21, 83–92. Available online at: https://core.ac.uk/download/pdf/301027745.pdf
Funk, D. J., and Omland, K. E. (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial dna. Annu. Rev. Ecol. Evol. Syst. 34, 397–423. doi: 10.1146/annurev.ecolsys.34.011802.132421
Gompert, Z., Lucas, L. K., Fordyce, J. A., Forister, M. L., and Nice, C. C. (2010). Secondary contact between Lycaeides idas and L. melissa in the rocky mountains: extensive admixture and a patchy hybrid zone. Mol. Ecol. 19, 3171–3192. doi: 10.1111/j.1365-294X.2010.04727.x
Grummer, J. A. (2017). Evolutionary history of the Patagonian Liolaemus fitzingerii species group of lizards (Ph.D. thesis), University of Washington, Seattle, WA, United States.
Grummer, J. A., Morando, M. M., Avila, L. J., Sites J. W. Jr., and Leaché, A. D. (2018). Phylogenomic evidence for a recent and rapid radiation of lizards in the Patagonian Liolaemus fitzingerii species group. Mol. Phylogenet. Evol. 125, 243–254. doi: 10.1016/j.ympev.2018.03.023
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Hanemaaijer, M. J., Collier, T. C., Chang, A., Shott, C. C., Houston, P. D., Schmidt, H., et al. (2018). The fate of genes that cross species boundaries after a major hybridization event in a natural mosquito population. Mol. Ecol. 27, 4978–4990. doi: 10.1111/mec.14947
Ilut, D. C., Nydam, M. L., and Hare, M. P. (2014). Defining loci in restriction-based reduced representation genomic data from nonmodel species: sources of bias and diagnostics for optimal clustering. Biomed. Res. Int. 2014:675158. doi: 10.1155/2014/675158
Irwin, D. E., Bensch, S., and Price, T. D. (2001). Speciation in a ring. Nature 409, 333–337. doi: 10.1038/35053059
Jakobsson, M., and Rosenberg, N. A. (2007). Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Jančúchová-Lásková, J., Landová, E., and Frynta, D. (2015). Are genetically distinct lizard species able to hybridize? A review. Curr. Zool. 61, 155–180. doi: 10.1093/czoolo/61.1.155
Kacoliris, F. P., Williams, J. D., De Arcaute, C. R., and Cassino, C. (2009). Home range size and overlap in Liolaemus multimaculatus (Squamata: Liolamidae) in Pampean coastal dunes of Argentina. South Am. J. Herpetol. 4, 229–234. doi: 10.2994/057.004.0305
Kay, S. M., Ardolino, A., Gorring, M., and Ramos, V. (2007). The somuncura large igneous province in Patagonia: interaction of a transient mantle thermal anomaly with a subducting slab. J. Petrol. 48, 43–77. doi: 10.1093/petrology/egl053
Larkin, M. A., Blackshields, G., Brown, N., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal w and clustal x version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Leaché, A. D., and Cole, C. J. (2007). Hybridization between multiple fence lizard lineages in an ecotone: locally discordant variation in mitochondrial DNA, chromosomes, and morphology. Mol. Ecol. 16, 1035–1054. doi: 10.1111/j.1365-294X.2006.03194.x
Leaché, A. D., Grummer, J. A., Harris, R. B., and Breckheimer, I. K. (2017). Evidence for concerted movement of nuclear and mitochondrial clines in a lizard hybrid zone. Mol. Ecol. 26, 2306–2316. doi: 10.1111/mec.14033
MacManes, M. (2013). Available online at: http://dx.doi.org/10.6084/m9.figshare.658946. Figshare 5.
Maddison, W. P., and Maddison, D. (2017). Mesquite: A Modular System for Evolutionary Analysis. version 3.2. Available online at: http://mesquiteproject.org
Mallet, J. (2005). Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237. doi: 10.1016/j.tree.2005.02.010
Mallet, J., Barton, N., Lamas, G., Santisteban, J., Muedas, M., and Eeley, H. (1990). Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in heliconius hybrid zones. Genetics 124, 921–936. doi: 10.1093/genetics/124.4.921
May, R. M., Endler, J. A., and McMurtrie, R. E. (1975). Gene frequency clines in the presence of selection opposed by gene flow. Am. Nat. 109, 659–676. doi: 10.1086/283036
McEntee, J. P., Burleigh, J. G., and Singhal, S. (2020). Dispersal predicts hybrid zone widths across animal diversity: implications for species borders under incomplete reproductive isolation. Am. Nat. 196, 000–000. doi: 10.1086/709109
McGuire, J. A., Linkem, C. W., Koo, M. S., Hutchison, D. W., Lappin, A. K., Orange, D. I., et al. (2007). Mitochondrial introgression and incomplete lineage sorting through space and time: phylogenetics of crotaphytid lizards. Evolution 61, 2879–2897. doi: 10.1111/j.1558-5646.2007.00239.x
McKinnon, J. S., and Rundle, H. D. (2002). Speciation in nature: the threespine stickleback model systems. Trends Ecol. Evol. 17, 480–488. doi: 10.1016/S0169-5347(02)02579-X
Medina, C. D., Avila, L. J., Sites J. W. Jr., and Morando, M. (2014). Multilocus phylogeography of the patagonian lizard complex Liolaemus kriegi (Iguania: Liolaemini). Biol. J. Linn. Soc. 113, 256–269. doi: 10.1111/bij.12285
Morando, M., Avila, L. J., Baker, J., Sites J. W. Jr., and Ashley, M. (2004). Phylogeny and phylogeography of the Liolaemus darwinii complex (Squamata: Liolaemidae): evidence for introgression and incomplete lineage sorting. Evolution 58, 842–861. doi: 10.1111/j.0014-3820.2004.tb00416.x
Morando, M., Avila, L. J., and Sites, J. W. (2003). Sampling strategies for delimiting species: genes, individuals, and populations in the Liolaemus elongatus-kriegi complex (Squamata: Liolaemidae) in Andean-Patagonian South America. Syst. Biol. 52, 159–185. doi: 10.1080/10635150390192717
Morando, M., Medina, C. D., Minoli, I., Pérez, C. H. F., Sites, J. W., and Avila, L. J. (2020). “Diversification and evolutionary histories of Patagonian Steppe lizards,” in Lizards of Patagonia, eds M. Morando, and L. J. Avila (Cham: Springer), 217–254. doi: 10.1007/978-3-030-42752-8_9
Moritz, C., Schneider, C. J., and Wake, D. B. (1992). Evolutionary relationships within the Ensatina eschscholtzii complex confirm the ring species interpretation. Syst. Biol. 41, 273–291. doi: 10.1093/sysbio/41.3.273
Nolte, A., Gompert, Z., and Buerkle, C. (2009). Variable patterns of introgression in two sculpin hybrid zones suggest that genomic isolation differs among populations. Mol. Ecol. 18, 2615–2627. doi: 10.1111/j.1365-294X.2009.04208.x
Olave, M., Avila, L. J., Sites, J. W. Jr., and Morando, M. (2018). Hybridization could be a common phenomenon within the highly diverse lizard genus Liolaemus. J. Evol. Biol. 31, 893–903. doi: 10.1111/jeb.13273
Olave, M., Marín, A. G., Avila, L. J., Sites, J. W., and Morando, M. (2020). “Disparate patterns of diversification within Liolaemini lizards,” in Neotropical Diversification: Patterns and Processes, V. Rull and A. Carnaval (Cham: Springer), 765–790. doi: 10.1007/978-3-030-31167-4_28
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., and Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo snp discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. doi: 10.1371/journal.pone.0037135
Petit, R. J., and Excoffier, L. (2009). Gene flow and species delimitation. Trends Ecol. Evol. 24, 386–393. doi: 10.1016/j.tree.2009.02.011
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Pritchard, V. L., and Edmands, S. (2013). The genomic trajectory of hybrid swarms: outcomes of repeated crosses between populations of Tigriopus californicus. Evolution 67, 774–791. doi: 10.1111/j.1558-5646.2012.01814.x
Rieseberg, L. H., Van Fossen, C., and Desrochers, A. M. (1995). Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 375, 313–316. doi: 10.1038/375313a0
Rieseberg, L. H., Whitton, J., and Gardner, K. (1999). Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152, 713–727. doi: 10.1093/genetics/152.2.713
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rohwer, S., Bermingham, E., and Wood, C. (2001). Plumage and mitochondrial DNA haplotype variation across a moving hybrid zone. Evolution 55, 405–422. doi: 10.1111/j.0014-3820.2001.tb01303.x
Stamatakis, A. (2014). Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Taylor, S. A., White, T. A., Hochachka, W. M., Ferretti, V., Curry, R. L., and Lovette, I. (2014). Climate-mediated movement of an avian hybrid zone. Curr. Biol. 24, 671–676. doi: 10.1016/j.cub.2014.01.069
Westram, A. M., Faria, R., Johannesson, K., and Butlin, R. (2021). Using replicate hybrid zones to understand the genomic basis of adaptive divergence. Mol. Ecol. doi: 10.1111/mec.15861. [Epub ahead of print].
Keywords: SNP, admixture, lizard, population, cline, Patagonia (Argentina), ddRAD
Citation: Grummer JA, Avila LJ, Morando MM and Leaché AD (2021) Four Species Linked by Three Hybrid Zones: Two Instances of Repeated Hybridization in One Species Group (Genus Liolaemus). Front. Ecol. Evol. 9:624109. doi: 10.3389/fevo.2021.624109
Received: 30 October 2020; Accepted: 28 April 2021;
Published: 25 May 2021.
Edited by:
Michael G. Campana, Smithsonian Conservation Biology Institute (SI), United StatesCopyright © 2021 Grummer, Avila, Morando and Leaché. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jared A. Grummer, Z3J1bW1lckB6b29sb2d5LnViYy5jYQ==
†Present address: Jared A. Grummer, Department of Zoology, Biodiversity Research Centre, University of British Columbia, Vancouver, BC, Canada